DOI:
10.1039/D4DT01608D
(Paper)
Dalton Trans., 2024,
53, 15801-15814
Taming 2,2′-biimidazole ligands in trivalent chromium complexes†
Received
3rd June 2024
, Accepted 4th July 2024
First published on 6th July 2024
Abstract
Complete or partial replacement of well-known five-membered chelating 2,2′-bipyridine (bipy) or 1,10-phenanthroline (phen) ligands with analogous didentate 2,2′-biimidazole (H2biim) provides novel perspectives for exploiting the latter pH-tuneable bridging unit for connecting inert trivalent chromium with cationic partners. The most simple homoleptic complex [Cr(H2biim)3]3+ and its stepwise deprotonated analogues are only poorly soluble in most solvents and their characterization is limited to some solid-state structures, in which the pseudo-octahedral [CrN6] units are found to be intermolecularly connected via peripheral N–H⋯X hydrogen bonds. Moreover, the associated high-energy stretching N–H vibrations drastically quench the targeted near infrared (NIR) CrIII-based phosphorescence, which makes these homoleptic building blocks incompatible with the design of molecular-based luminescent assemblies. Restricting the number of bound 2,2′-biimidazole ligands to a single unit in the challenging heteroleptic [Cr(phen)2(Hxbiim)](1+x)+ (x = 2–0) complexes overcomes the latter limitations and allows (i) the synthesis and characterization of these [CrN6] chromophores in the solid state and in solution, (ii) the stepwise and controlled deprotonation of the bound 2,2′-biimidazole ligand and (iii) the implementation of Cr-centered phosphorescence with energies, lifetimes and quantum yields adapted for using the latter chromophores as sensitizers in promising ‘complex-as-ligand’ strategies.
Introduction
The lack of first-order orbital momentum in pseudo-octahedral trivalent chromium complexes makes isotropic and spin-only [CrX6] units ideal partners for programming, tuning and rationalizing magnetic coupling with neighbouring paramagnetic d-block1–5 and f-block6–10 cations. In this context, many of the chromium-containing heterometallic assemblies exploit the kinetically inert [Cr(CN)6]3− synthon, which behaves as a ‘complex-as-ligand’ connecting cationic metals via cyanide bridges in solid state materials.1–10 The realization that, beyond magnetic properties, both lanthanide-based light downshifting11,12 and light upconversion13,14 can be boosted by close Cr(III) sensitizers/emitters resulted in some active search for novel synthetic strategies, leading to CrIII–LnIII molecular pairs (Ln(III) is a trivalent lanthanide cation) beyond serendipitous co-crystallization processes.15 One approach involves self-assembly processes with segmental multisite ligands where labile CrII precursors are selectively recognized by didentate binding units while LnIII is caught by adjacent tridentate sites. The subsequent CrII to CrIII oxidation provides kinetically inert heterometallic triple-stranded CrIII–LnIII and CrIII–LnIII–CrIII helicates with remarkable photophysical properties,16,17 among which is the programming of the first molecular-based energy transfer light-upconversion (ETU) process.13
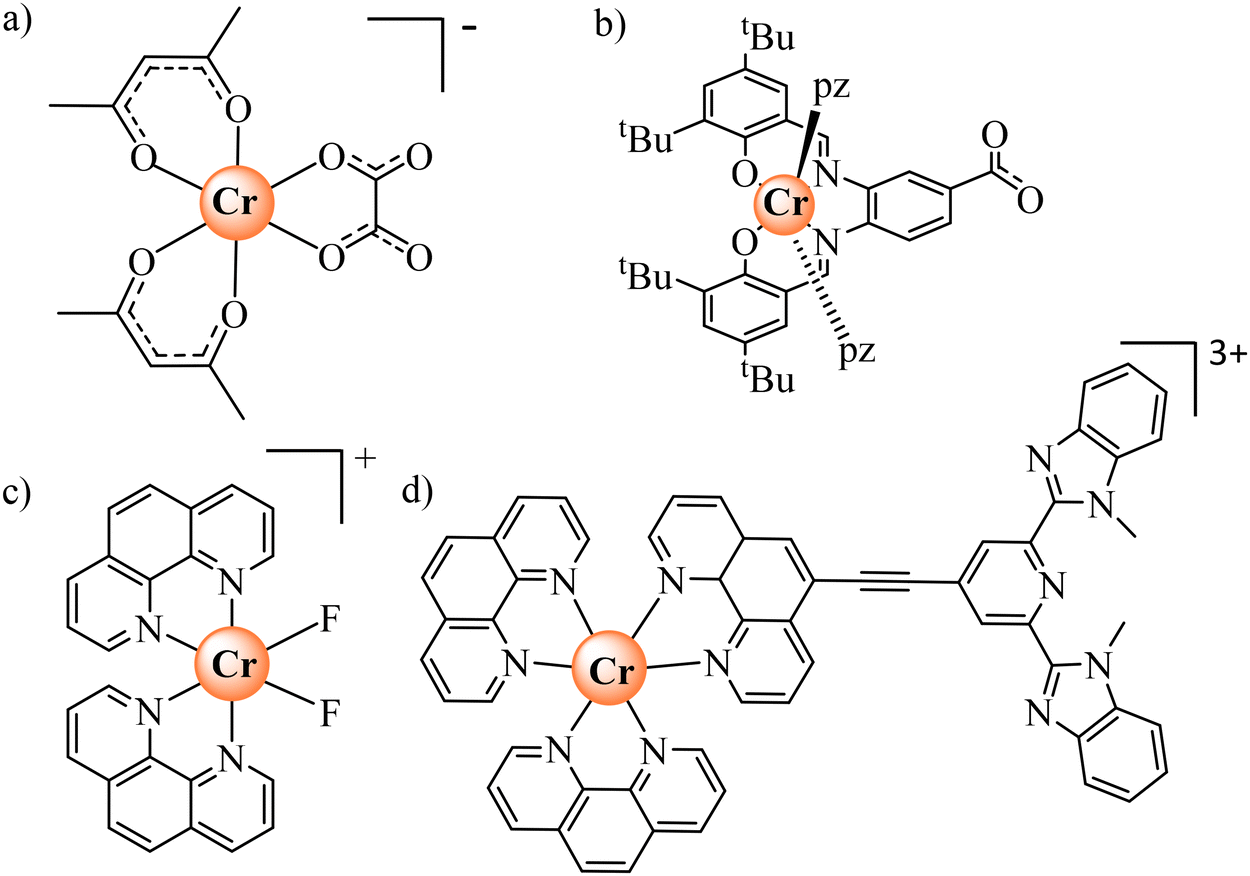
A more versatile synthetic strategy for incorporating open-shell [CrX6] chromophores as tuneable and operable sensitizers in multimetallic (supra)molecular architectures involves extending the ‘complex-as-ligand’ strategy, originally used for introducing [Cr(CN)6]3− into multimetallic coordination polymers.1–10,18 Taking advantage of kinetically-controlled ligand exchange processes around inert Cr(III), a few heteroleptic six-coordinate complexes could be prepared, in which an oxalate (Scheme 1a),12 a phenyl-carboxylate (Scheme 1b),19 a difluoride (Scheme 1c)20 or an extended ethyne-bis(benzimidazole)pyridine (Scheme 1d)21 acted as a bridging unit between the Cr(III)-based complex-as-ligand unit and some adjacent d-block or f-block partners. However, the molecular aspects of their association processes in solution remain elusive and only solid-state crystal data support the physicochemical analyses. Considering the recent recognition that strong-field [CrN6] chromophores are ideal for maximizing phosphorescence quantum yields, emission lifetimes and sensitization in ‘molecular rubies’,22–24 there is clearly a need for the design of novel [CrN6] analogues working as complex-as-ligand, but using more accessible and reliable bridging units.
 |
| | Scheme 1 Kinetically-inert heteroleptic six-coordinate CrIII complexes used as complex-as-ligand when preparing multimetallic assemblies.12,18–21 | |
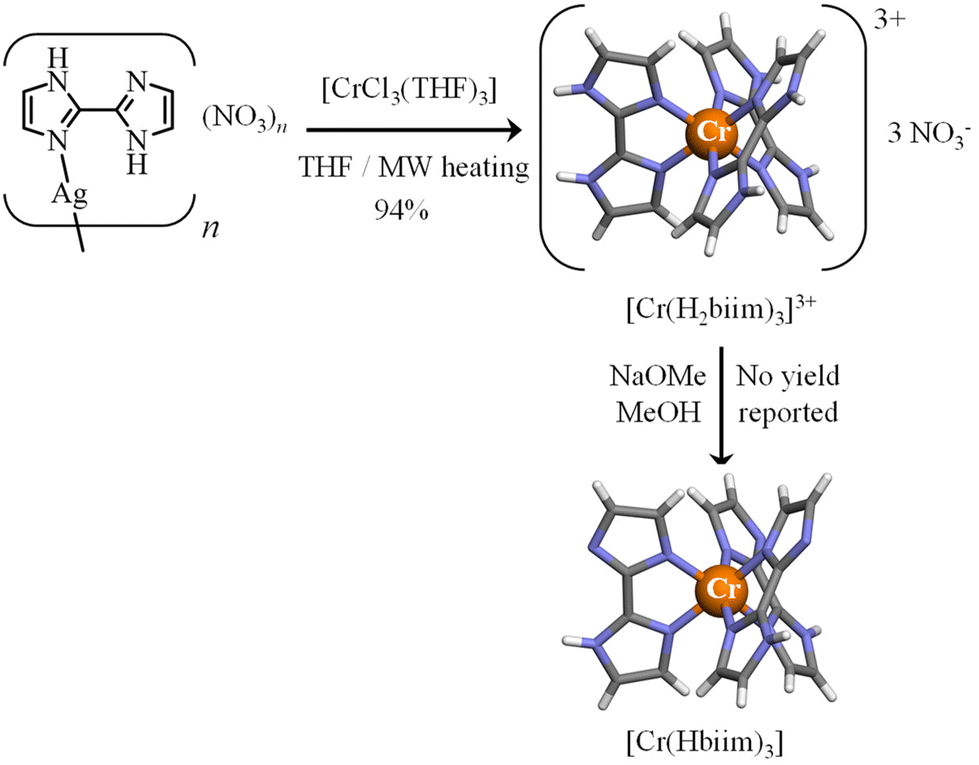
With this in mind, the 2,2′-biimidazole ligand (H2biim, Scheme 2a)25 is famous for working as a versatile bridging ligand after its binding to a metallic cation (Scheme 2b). In its protonated form, it can either form hydrogen bonds for sensing anions (mode A in Scheme 2b)26,27 or connect other cations in a linear way (mode B in Scheme 2b).28 More often,29 multimetallic assemblies are obtained after stepwise deprotonation of the bound 2,2′-biimidazole ligand to give bridging Hbiim− (modes C and D in Scheme 2b)30 and biim2− scaffolds (modes E–I in Scheme 2b).31–34 Surprisingly, the studies dealing with the complexation of inert CrIII centers by potentially bridging 2,2′-biimidazole are undervalued, probably due to the synthetic difficulties associated with the synthesis of these poorly soluble and pH-sensitive complexes.35–38 To the best of our knowledge, only the molecular structures of [Cr(H2biim)3]3+,36 [Cr(H2biim)2(Hbiim)]2+ (ref. 37) and [Cr(Hbiim)3]36 have been characterized in the solid state following poorly reproducible and serendipitous crystallization from intricate mixtures of ligands and metals in the presence of various amounts of counter-anions and/or solvent molecules. A recent synthetic improvement, which used anhydrous THF under microwave heating, gave [Cr(H2biim)3](NO3)3 in 94% yield (Fig. 1).35
 |
| | Scheme 2 a) Successive acid–base equilibria of fully protonated 2,2′-biimidazole (H4biim2+) with pKa values measured in DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 7:3 (I = 0.1 M).25 (b) Different coordination modes encountered in the literature for H2biim (A26,27 and B28), Hbiim− (C and D)30 and biim2− (E–I).31–34 :H2O = 7:3 (I = 0.1 M).25 (b) Different coordination modes encountered in the literature for H2biim (A26,27 and B28), Hbiim− (C and D)30 and biim2− (E–I).31–34 | |
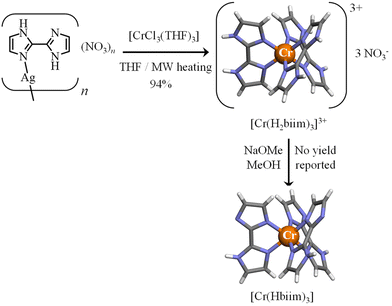 |
| | Fig. 1 Synthesis of [Cr(H2biim)3]3+ (ref. 35) and [Cr(Hbiim)3].36 The molecular structures of the complexes are those found in the crystal structures of [Cr(H2biim)3](NO3)3 (CCDC-603707) and [Cr(Hbiim)3]·C6H6·2H2O (CCDC-603730).36 Color code: C = grey, N = blue, H = white, and Cr = orange. | |
Beyond the detailed descriptions of (i) sophisticated hydrogen-bonding networks in which the latter homoleptic complexes are a part and (ii) some expected axial flattening produced by didentate five-membered chelating 2,2′-biimidazole bound to CrIII,36–38 no effort has been focused on the thermodynamic deprotonation processes and the associated control of the photophysical properties. Moreover, to the best of our knowledge, no attempt to prepare heteroleptic [L2Cr(H2biim)]3+ has been made, while related systems with inert 4d and 5d metal ions have been designed regularly to access the two crucial pKa values for their use as complex-as-ligand (Table S1 in the ESI†).25,39–47 In order to provide new perspectives for exploiting 2,2′-biimidazole as a bridging ligand between photophysically-active CrIII and promising open-shell lanthanides, we report here on the molecular structures and photophysical properties of the accessible and isolable homoleptic complexes [Cr(H2biim)3]3+, [Cr(Hbiim)3] and [Cr(Hbiim)2(biim)]− in solution and in the solid state. The second part proposes the synthesis of the unprecedented heteroleptic [(phen)2Cr(H2biim)]3+ complex with its detailed acid–base and photophysical properties, which become accessible in the solid state and in solution.
Results and discussion
Preparation and structures of the homoleptic [Cr(Me2biim)3]3+, [Cr(H2biim)3]3+, [Cr(Hbiim)3] and [Cr(Hbiim)2(biim)]− complexes
2,2′-Biimidazole (H2biim) was synthesized from ammonium acetate and glyoxal under aqueous conditions with moderate yield.48 Its methyl derivative 1,1′-dimethyl-2,2′-bi-1H-imidazole (Me2biim) could be obtained by deprotonation, followed by methylation with methyl iodide (Fig. 2, top).49
 |
| | Fig. 2 Synthesis of ligands 2,2′-biimidazole (H2biim) and 1,1′-dimethyl-2,2′-bi-1H-imidazole (Me2biim) and their homoleptic [Cr(H2biim)3]3+, [Cr(Hbiim)3], [Cr(Hbiim)2(biim)]− and [Cr(Me2biim)3]3+ complexes. The molecular structures of the metallic complexes are those found in the associated crystal structures. Color code: C = grey, N = blue, H = white, and Cr = orange. The counter-ions and hydrogen atoms (for Me2biim ligands) are omitted for clarity. | |
The original literature synthesis of [Cr(H2biim)3](NO3)3 involved the reaction of CrCl3·3THF with [Ag(H2biim)]NO3 in MeOH36 or in THF35 (Fig. 1) because the formation of highly insoluble AgCl drove the reaction to completion. To simplify the procedure, the intermediate [Ag(H2biim)]NO3 was not isolated in this work, but was formed in situ by mixing AgNO3 and H2biim in MeOH. Then CrCl3·3THF was added into the resulting solution of [Ag(H2biim)]NO3. The purification method was identical to that used in ref. 35 and [Cr(H2biim)3](NO3)3 was isolated in good yield (Fig. 2). Recrystallization by vapor diffusion of Et2O into a methanolic solution provided crystals suitable for X-ray diffraction, the structural resolution of which at 120 K confirmed the crystal structure previously reported for [Cr(H2biim)3](NO3)3 at 290 K (Fig. 2, Tables S2–S3 and Fig. S1†).36 The synthesis of [Cr(Hbiim)3] was first described by Gruia et al.,36 where they used a stochiometric amount of NaOMe to deprotonate [Cr(H2biim)3]3+ in MeOH. However, the complete insolubility of the formed neutral [Cr(Hbiim)3] complex did not allow any reproducible recrystallization techniques. To overcome this problem, a methanolic solution of [Cr(H2biim)3]3+ was treated in this work with vapor diffusion of an excess of volatile triethylamine, the limited pKa of which (10.74)50 prevented any double deprotonation of the bound Hbim ligand and finally gave crystals of [Cr(Hbiim)3] with good yield (61%) and in a reproducible way (Fig. 2, Tables S4–S6 and Fig. S2†). Further deprotonation of [Cr(Hbiim)3] could not be obtained by Gruia et al.,36 but some partial reports of analogs of [Cr(biim)3]3−, i.e. Ba1.5[Co(biim)3]41 and K3[Ru(biim)3]51 that were obtained using harsh basic conditions (aqueous NaOH 2 M/BaCl2 and tBuOK 1.2 M in MeOH, respectively) have been noted. Consequently, [Cr(Hbiim)3] was dissolved in an excess of aqueous NaOH (0.5 M) until a clear yellow solution was formed. The addition of a concentrated solution of PPh4Cl immediately resulted in the formation of an orange precipitate. Recrystallization from MeOH by vapor diffusion of tBuOMe provided block-shaped crystals of [Cr(Hbiim)2(biim)]PPh4(CH3OH) suitable for X-ray diffraction analysis in a good yield (66%) (Fig. 2, Tables S7–S9 and Fig. S3†). A detailed geometrical analysis of these homoleptic complexes (Appendix 2 in the ESI†) concludes that the [Cr(Hxbiim)3]n+ units display standard pseudo-octahedral arrangements of the six bound nitrogen donor atoms, with a compression along the pseudo-C3 axis due to the 79.6–80.4° ligand bite angles (Table A2-1 in Appendix 2†), which is characteristic of five-membered chelating polyaromatic ligands23 as reported for related [Cr(bipy)3]3+ (bipy = 2,2′-bipyridine, 79.1°)52 and [Cr(phen)3]3+ (phen = 1,10-phenanthroline, 81.0°) complexes.53 The Cr–N distances in [Cr(Hxbiim)3]n+ do not vary drastically (2.028–2.037 Å, compared with averages of 2.042 Å for [Cr(bipy)3]3+ and 2.051 Å for [Cr(phen)3]3+) and do not show obvious correlations with the degree of deprotonation of the bound ligand. Due to the chelation of semi-rigid didentate polyaromatic ligands, some standard trigonal distortions characterize all these complexes (Table A2-1 in Appendix 2†).
The main difference between [Cr(bipy)3]3+ and [Cr(phen)3]3+ on one hand, and the family of [Cr(Hxbiim)3]n+ complexes, is associated with the bound didentate 2,2′-biimidazole ligands which may act as N–H hydrogen-bond donors when they are protonated and as N− hydrogen-bond acceptors when they are deprotonated. For the fully protonated [Cr(H2biim)3](NO3)3 complex, the weak N–H⋯O hydrogen bonds observed between the bound ligand and the nitrate counter-anion (Fig. A2-2†) are responsible for the circa 400 cm−1 decreases of the N–H stretching frequency in the vibrational spectrum with respect to the free ligand (ν(NH) ≈ 3000 cm−1 in Fig. S14 and S18-top, Table S29†). The formation of strong intermolecular N–H⋯N hydrogen bonds observed in the deprotonated complexes [Cr(Hbiim)3] (Fig. A2-4†) and [Cr(Hbiim)2(biim)]PPh4 (Fig. A2-6†) further weakens the N–H bond force constants and stepwise shifts the associated stretching frequency toward ν(NH) ≈ 2400 cm−1 (Fig. S15 and S18 center, Table S30†) and ν(NH) ≈ 2300 cm−1 (Fig. S16 and S18 bottom, Table S31†), respectively. In this context, the replacement of hydrogen atoms with methyl groups to give [Cr(Me2biim)3](CF3SO3)3via the Kane-Maguire synthetic strategy (Fig. 2) maintains the pseudo-octahedral structure of the [CrN6] core (Tables S16–S17 and Fig. S7†),54 but it limits intermolecular hydrogen bonds in the crystal structure (Fig. A2-10 in Appendix 2†).
The spectroscopic properties of the ligands H2biim and Me2biim and the chromium complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 could be recorded in methanol or in acetonitrile. Unfortunately, the neutral complex [Cr(Hbiim)3] is not soluble enough in common organic solvents to perform reliable measurements in solution. The electrospray ionization mass spectrometry (ESI-MS) spectra (Fig. S19–S38 and Tables S33–S35†) show complicated mixtures in the gas phase containing various amounts of intact 1:3 complexes with variable degrees of deprotonation ([Cr(Hxbim)3]n+) together with (i) partial ligand dissociation to give 1:2 complexes ([Cr(Hxbim)2]n+ + H2biim) and (ii) metal reduction into Cr(II)-based systems. All peaks could be identified from high-resolution MS spectra with a special emphasis on the detection of the ‘full’ deprotonated [Cr(Hbiim)2(biim)]− anion (negative mode) for [Cr(Hbiim)2(biim)]PPh4 (Fig. S25 and Table S34†). It is concluded that these deprotonatable complexes suffer from the ionization process and exist as fragmented mixtures in the gas phase.
Photophysical properties of the homoleptic [Cr(Me2biim)3]3+, [Cr(H2biim)3]3+, [Cr(Hbiim)3] and [Cr(Hbiim)2(biim)]− complexes
The absorption (Fig. 3 and 4a) and emission (Fig. 4b and S39†) spectra of the ligands H2biim and Me2biim and the complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 could be recorded in MeOH. Unfortunately, the neutral complex [Cr(Hbiim)3] is not soluble enough to perform spectroscopy except for solid-state investigations (Fig. S40†). The UV parts of the absorption spectra are dominated by intraligand-centered π* ← π transitions (ILCT), which are split and red-shifted upon coordination to Cr3+ in the associated complexes. Additional LMCT (Ligand-to-Metal Charge Transfer) transitions are known to also contribute within the 330–400 nm range for these [CrN6] chromophores (Fig. 3).29,55–57 The spin-allowed, but parity forbidden ligand-field Cr(4T2 ← 4A2) transition (intensity 10 < ε < 100 M−1 cm−1, highlighted in Fig. 3b) can be detected as a minor contribution to the low-energy tail of the charge transfer bands.58–63 Careful spectral deconvolutions are required (Fig. S41–S43 and Tables S36–S39†) to safely assign E(4T2), i.e., the energy of the Cr(4T2) excited level with respect to the ground state E(4A2) = 0 (Table 1, column 2).
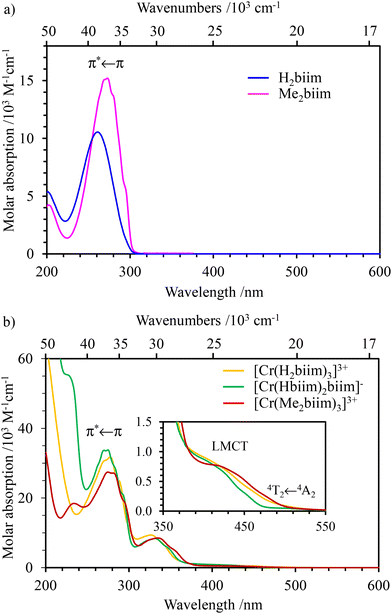 |
| | Fig. 3 UV-visible absorption spectra of (a) non-coordinated ligands H2biim (purple) and Me2biim (dark blue) and (b) complexes [Cr(H2biim)3](NO3)3 (orange), [Cr(Hbiim)2(biim)]PPh4 (green) and [Cr(Me2biim)3](CF3SO3)3 (red) in MeOH at 293 K. | |
 |
| | Fig. 4 NIR (a) absorption spectra of [Cr(H2biim)3](NO3)3 (water) and [Cr(Me2biim)3](CF3SO3)3 (acetonitrile) and (b) emission (λexc = 330 nm) spectra of complexes [Cr(H2biim)3](NO3)3 (orange), [Cr(Hbiim)2(biim)]PPh4 (green) and [Cr(Me2biim)3](CF3SO3)3 (red) in MeOH at 293 K. | |
Table 1 Energy of intrashell d–d transitions (in MeOH solution for E(4T2) and in the solid state for E(2T1) and E(2E)), ligand-field strength Δ (eqn (1)) and Racah parameters B (eqn (2)) and C (eqn (3)) for [Cr(H2biim)3](NO3)3, [Cr(Hbiim)2(biim)]PPh4, [Cr(Me2biim)3](CF3SO3)3, and [Cr(phen)2(H2biim)]3+ and for closely related pseudo-octahedral [CrIIIN6] chromophoresa
| Complex |
E(4T2)/cm−1 |
E(2T1)/cm−1 |
E(2E)/cm−1 |
Δ/cm−1 |
B/cm−1 |
C/cm−1 |
E(4T2) − E(2E)/cm−1 |
Δ/B |
C/B |
Ref. |
 |
| [Cr(H2biim)3]3+ |
20268 |
14376 |
13717 |
20268 |
717 |
2845 |
6551 |
28.3 |
4.0 |
This work |
| [Cr(Hbiim)3] |
— |
14263 |
13670 |
— |
— |
— |
— |
— |
— |
This work |
| [Cr(Hbiim)2(biim)]− |
19956 |
14096 |
13482 |
19956 |
686 |
2828 |
6474 |
29.1 |
4.1 |
This work |
| [Cr(Me2biim)3]3+ |
21026 |
14164 |
13538 |
21026 |
712 |
2779 |
7488 |
29.6 |
3.9 |
This work |
| [Cr(phen)2(H2biim)]3+ |
23202 |
14381 |
13723 |
23202 |
766 |
2697 |
9479 |
30.3 |
3.5 |
This work |
| [Cr(bpy)3]3+ |
23400 |
14450 |
13800 |
23400 |
765 |
2730 |
9600 |
30.6 |
3.6 |
60
|
| [Cr(phen)3]3+ |
22075 |
14451 |
13736 |
22075 |
779 |
2700 |
8339 |
28.3 |
3.5 |
53
|
| [Cr(tpy)2]3+ |
18750 |
13584 |
12953 |
18750 |
790 |
2512 |
5797 |
23.7 |
3.2 |
61
|
| [Cr(ddpd)2]3+ |
22990 |
13550 |
12903 |
22990 |
756 |
2419 |
10087 |
30.4 |
3.2 |
23
|
| [Cr(dqp)2]3+ |
24937 |
13864 |
13405 |
24937 |
656 |
2791 |
11532 |
38.0 |
4.3 |
24
|
| [Cr(dpc)2]+ |
19200 |
— |
9370 |
19200 |
470 |
1880 |
9830 |
40.9 |
4.0 |
62
|
| [Cr(CN)6]3− |
26600 |
— |
12400 |
26600 |
480 |
2800 |
14200 |
55.4 |
5.8 |
63
|
| [Cr(bik)3]3+ |
23094 |
14771 |
14044 |
23094 |
804 |
2737 |
9050 |
28.7 |
3.4 |
57
|
| [Cr(bim)3]3+ |
21008 |
14859 |
14104 |
21008 |
781 |
2842 |
6904 |
26.9 |
3.6 |
57
|
| [Cr(bie)3]3+ |
20747 |
14837 |
14124 |
20747 |
754 |
2902 |
6623 |
27.5 |
3.8 |
57
|
Interestingly, for pseudo-octahedral d3 complexes, E(4T2) provides a straightforward estimation of the ligand-field splitting (eqn (1)),64,65 which covers a narrow 19956 ≤ Δ ≤ 21026 cm−1 range for [Cr(H2biim)3]3+, [Cr(Hbiim)2(biim)]− and [Cr(Me2biim)3]3+ in solution (Table 1).
As established for the programming of spin crossover processes while tuning the ligand-field strength,66 the replacement of didentate 2,2′-bipyridine (bpy) or 1,10-phenanthroline (phen), made of two connected 6-membered heterocyclic rings, with 2,2′-biimidazole (H2biim), made of two connected five-membered heterocyclic rings, is accompanied by an increase of the trigonal distortion in their pseudo-octahedral complexes. This is measured by a stepwise increase in the θ angular distortion (θ[Cr(phen)3]3+ = 47.5° < θ[Cr(bpy)3]3+ = 63.6° < θ[Cr(Me2biim)3]3+ = 80.4° computed with eqn (A2-2) and gathered in Table A2-1, see Appendix 2†), which results in a concomitant stepwise reduction of the ligand-field strength Δ[Cr(bpy)3]3+ ≈ Δ[Cr(phen)3]3+ ≈ 22500 cm−1 > Δ[Cr(Me2biim)3]3+ ≈ 20500 cm−1 > Δ[Cr(tpy)2]3+ = 18750 cm−1 (Table 1).
The interelectronic repulsion is estimated using the Racah parameters B (eqn (2)) and C (eqn (3)), which requires the energy of the lowest doublet levels E(2E) and E(2T1) to be accessible (Fig. 5).64,65
| |  | (2) |
| | 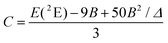 | (3) |
 |
| | Fig. 5 Jablonski diagram of homoleptic [Cr(H2biim)3]3+ or [Cr(Me2biim)3]3+ complexes showing the antenna effect upon UV excitation at 330 nm. The modeling of the energies of the Cr levels is taken from ref. 64. | |
Assuming an Oh symmetry, the ground state absorption bands Cr(2E ← 4A2) and Cr(2T1 ← 4A2) have very weak intensities (0.05 < ε < 1 M−1 cm−1) due to the breaking of both parity and spin conservation rules. These transitions could be detected in the NIR domain of the solid-state absorption spectra of the four investigated complexes (Fig. S40†), while related solution data could be recorded only for the most soluble [Cr(H2biim)3](NO3)3 (c ≥ 10−2 M in water) and [Cr(Me2biim)3](CF3SO3)3 (c ≥ 10−2 M in acetonitrile) complexes (Fig. 4a). Introducing E(4T2), E(2E) and E(2T1) into eqn (1)–(3) provides the Racah parameters B and C collected in Table 1. Interestingly, 686 ≤ B ≤ 717 cm−1 found for [Cr(H2biim)3](NO3)3, [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 points to a global increase of the nephelauxetic effect when didentate 2,2′-bipyridine or 1,10-phenanthroline type ligands bound to Cr3+ (765 ≤ B ≤ 790 cm−1) are replaced with 2,2′-biimidazole type ligands.
For absorption spectra recorded in solution (Fig. 4a), it is possible to calculate the radiative rate constant of the emissive levels 2E and 2T1 using the Strickler–Berg eqn (4)67,68, which is derived from Einstein's relationship for spontaneous emission (Table 2, column 2 and Table S40†).69
| | 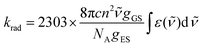 | (4) |
Table 2 Experimental Cr(2E) lifetimes (τ) and radiative (krad) and non-radiative (knon-rad) relaxation rate constants and intrinsic quantum yields (ϕintrinsiccomplex = krad/(krad + knon-rad)) of chromium complexes
| Complex |
k
rad/s−1 |
τ
293 K, Ara/μs |
k
293 K, Arnon-rada/103 s−1 |
ϕ
intrinsiccomplex 293 K, Ar |
τ
293 K, airb/μs |
k
293 K, airnon-radb/103 s−1 |
ϕ
intrinsiccomplex 293 K, air |
τ
77 Kc/μs |
k
77 Knon-rad/s−1 |
ϕ
intrinsiccomplex 77 K |
Ref. |
|
Recorded in degassed solution at room temperature.
Recorded in air-equilibrated solution at room temperature.
Recorded in a frozen solvent mixture at 77 K.
In MeOH.
In MeOH/H2O.
In CH3CN.
In CH3CN/C2H5CN.
In H2O.
In H2O/DMSO.
In aq. HClO4/MeOH.
In aq. HCl.
|
| [Cr(H2biim)3]3+ |
75(4) |
0.47(2)d |
2120(106) |
3(1) × 10−5 |
0.44(2)d |
2260(113) |
3(1) × 10−5 |
2950(150)e |
263(13) |
2.0(6) × 10−1 |
This work |
| [Cr(Hbiim)2(biim)]− |
— |
0.178(9)d |
— |
— |
0.141(7)d |
— |
— |
2780(140)e |
— |
— |
This work |
| [Cr(Me2biim)3]3+ |
73(4) |
4.28(4)d |
234(12) |
3(1) × 10−4 |
3.31(9)d |
302(1) |
2.4(8) × 10−4 |
3005(150)e |
260(13) |
2.2(7) × 10−1 |
This work |
| [Cr(phen)2(H2biim)]3+ |
65(3) |
38(1)f |
26(7) |
2.5(8) × 10−3 |
14(1)f |
71(7) |
9(3) × 10−4 |
2940(140)g |
|
1.9(6) × 10−1 |
This work |
| [Cr(bpy)3]3+ |
182 |
74h |
13.3 |
1.4(4) × 10−2 |
52h |
19 |
9(3) × 10−3 |
5000i |
18 |
9(3) × 10−1 |
54, 58 and 70–72 |
|
|
39d |
25.5 |
7(2) × 10−3 |
29d |
34 |
5(2) × 10−3 |
6500j |
≈0 |
≈1 |
| [Cr(phen)3]3+ |
319 |
356h |
2.49 |
1.1(4) × 10−1 |
74h |
13 |
2.4(7) × 10−2 |
2100g |
157 |
6.7(2) × 10−1 |
53, 54, 58 and 70–73 |
|
|
34d |
29.1 |
1.1(3) × 10−2 |
22d |
45 |
7(2) × 10−3 |
5300j |
≈0 |
≈1 |
|
|
224f |
4.15 |
7(2) × 10−2 |
37f |
27 |
1.2(3) × 10−2 |
|
|
|
|
|
270k |
3.38 |
9(3) × 10−2 |
|
|
|
|
|
|
| [Cr(tpy)2]3+ |
— |
— |
— |
— |
0.14f |
— |
— |
540i |
— |
— |
61, 74 and 75 |
|
|
|
|
|
|
|
|
670g |
— |
|
| [Cr(dqp)2]3+ |
30 |
1270h |
0.76 |
4(1) × 10−2 |
83h |
12 |
2.5(8) × 10−3 |
3070i |
296 |
9(3) × 10−2 |
24
|
|
|
2140f |
0.44 |
6(2) × 10−2 |
31f |
32 |
9(3) × 10−4 |
|
296 |
9(3) × 10−2 |
| [Cr(CN)6]3− |
97 |
— |
— |
— |
0.12i |
8330 |
1.2(4) × 10−5 |
3950i |
156 |
4(1) × 10−1 |
72
|
| [Cr(bik)3]3+ |
31 |
209f |
4.75 |
7(2) × 10−3 |
128f |
7.8 |
4(1) × 10−3 |
8220g |
91 |
2.6(8) × 10−1 |
57
|
| [Cr(bim)3]3+ |
37 |
131f |
7.60 |
5(2) × 10−3 |
73f |
14 |
2.7(8) × 10−3 |
6750g |
111 |
2.5(8) × 10−1 |
57
|
| [Cr(bie)3]3+ |
60 |
7f |
143 |
4(1) × 10−4 |
8f |
125 |
5(2) × 10−4 |
5250g |
130 |
3(1) × 10−1 |
57
|
Here c is the speed of light in vacuum (cm s−1), n is the refractive index of the solvent, NA is the Avogadro number (mol−1), gGS is the degeneracy of the ground state (g(4A2) = 4), gES is the degeneracy of the excited state (g(2T1) = 6 and g(2E) = 4), ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) is the barycenter of the transition in the wavenumber (cm−1) and
is the barycenter of the transition in the wavenumber (cm−1) and  is the area under the absorption spectrum of each transition (M−1 cm−2, Fig. S44 and S45†). The radiative rate constants of the emissive Cr(2E) excited level estimated for [Cr(H2biim)3]3+ (krad = 75(4) s−1) and [Cr(Me2biim)3] (krad = 73(4) s−1) are smaller than those reported for [Cr(bpy)3]3+ (krad = 182 s−1) and [Cr(phen)3]3+ (krad = 319 s−1, Table 2),58,69–73 but fall within the expected range for pseudo-octahedral [CrN6] chromophores (Table S40†).70–77 Upon room-temperature ligand-based excitation at 330 nm in solution, the complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 show the expected downshifted NIR spin–flip Cr(2E → 4A2) phosphorescence at 730–740 nm together with weak shoulders corresponding to Cr(2T1 → 4A2) within the 700–710 nm domain (Fig. 4b and 5). The latter dual emission disappears at 77 K (Fig. S39†) as a result of the depopulation of the high-energy Cr(2T1) level, and a single band Cr(2E → 4A2) contributes to phosphorescence. Very similar results are observed in the solid state upon 330 nm excitation for the four complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)3], [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 (Fig. S40b†), which ultimately demonstrate (i) the expected negligible Stokes shifts affecting the spin–flip Cr(2T1,2E ↔ 4A2) transitions (Fig. S46†) and (ii) the efficient sensitization of the spin–flip phosphorescence by all the accessible ligand-based excited states (Fig. S47†).
is the area under the absorption spectrum of each transition (M−1 cm−2, Fig. S44 and S45†). The radiative rate constants of the emissive Cr(2E) excited level estimated for [Cr(H2biim)3]3+ (krad = 75(4) s−1) and [Cr(Me2biim)3] (krad = 73(4) s−1) are smaller than those reported for [Cr(bpy)3]3+ (krad = 182 s−1) and [Cr(phen)3]3+ (krad = 319 s−1, Table 2),58,69–73 but fall within the expected range for pseudo-octahedral [CrN6] chromophores (Table S40†).70–77 Upon room-temperature ligand-based excitation at 330 nm in solution, the complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 show the expected downshifted NIR spin–flip Cr(2E → 4A2) phosphorescence at 730–740 nm together with weak shoulders corresponding to Cr(2T1 → 4A2) within the 700–710 nm domain (Fig. 4b and 5). The latter dual emission disappears at 77 K (Fig. S39†) as a result of the depopulation of the high-energy Cr(2T1) level, and a single band Cr(2E → 4A2) contributes to phosphorescence. Very similar results are observed in the solid state upon 330 nm excitation for the four complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)3], [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 (Fig. S40b†), which ultimately demonstrate (i) the expected negligible Stokes shifts affecting the spin–flip Cr(2T1,2E ↔ 4A2) transitions (Fig. S46†) and (ii) the efficient sensitization of the spin–flip phosphorescence by all the accessible ligand-based excited states (Fig. S47†).
Upon pulsed laser excitation at 355 nm at 77 K in frozen MeOH/H2O solutions, the characteristic lifetime of the emissive Cr(2E) levels for [Cr(H2biim)3](NO3)3 (τ77 K = 3.0(1) ms), [Cr(Hbiim)2(biim)]PPh4 (τ77 K = 2.7(1) ms) and [Cr(Me2biim)3](CF3SO3)3 (τ77 K = 3.0(1) ms) tends toward the radiative lifetime τrad = 13.4(7) ms, in agreement with minor non-radiative vibrational quenching constants at this temperature for rigid triple-helical units, as similarly reported for [Cr(bpy)3]3+ and [Cr(phen)3]3+ (Table 2 column 8). At room temperature, the Cr(2E) lifetimes drastically drop below the microsecond range for [Cr(H2biim)3](NO3)3 and [Cr(Hbiim)2(biim)]PPh4, which possess high-energy N–H oscillators, while the decrease of the lifetime for the methylated analogue [Cr(Me2biim)3](CF3SO3)3 (τ293 K, Ar = 4.28(4) μs) is less dramatic and mirrors those reported for [Cr(bpy)3]3+ and [Cr(phen)3]3+ (Table 2 and Fig. S48–S56†). Consequently, the non-radiative rate constant k293 K, Arnon-rad = 1/τ293 K, Ar − krad = 2.1(1) × 106 s−1 measured for [Cr(H2biim)3](NO3)3 in MeOH corresponds to the largest vibrational quenching process along the series of [Cr(N∩N)3]3+ chromophores collected in Table 2. Finally, the presence of 3O2 in solution has only a minor effect on the lifetime, indicating that quenching by oxygen is not a major contributor to energy relaxation in these systems (τ293 K, air in Table 2, column 6).
Acid–base properties of homoleptic [Cr(H2biim)3]3+, [Cr(Hbiim)3] and [Cr(Hbiim)2(biim)]− complexes
In order to extract the acid–base thermodynamic constants connecting [Cr(H2biim)3]3+ with its successive deprotonated forms [Cr(H2biim)2(Hbiim)]2+ (Ka1), [Cr(H2biim)(Hbiim)2]+ (Ka2), [Cr(Hbiim)3] (Ka3) and [Cr(Hbiim)2(biim)]− (Ka4), we performed two successive pH-metric titrations of aqueous solutions of [Cr(H2biim)3](NO3)3 using NaOH as the titrant. An initial addition of 0.2 equivalents of HNO3 ensured that the complex exists initially in its fully protonated form [Cr(H2biim)3]3+ (Fig. 6). The pH curve profile is different from the standard pH-metric titration expected for a weak polyacid with a strong base due to the concomitant precipitation of insoluble [Cr(Hbiim)3] (Fig. 6). It is therefore not possible to extract the searched pKa values and one can only estimate qualitatively pKa1, pKa2 < 7. Moreover, the preservation of [Cr(Hbiim)3] as a precipitate in the presence of a large excess of base indicates its negligible deprotonation for pH ≤ 12 in water and pKa4 > 12.
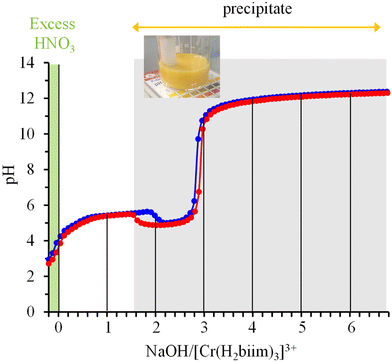 |
| | Fig. 6 Reproducible acid–base titrations (blue trace for titration 1 and red trace for titration 2) of a solution of [Cr(H2biim)3](NO3)3 + 0.2 eq. HNO3 with NaOH (water, 293 K) highlighting the formation of an insoluble yellow precipitate. | |
Preparation and structures of the heteroleptic [Cr(phen)2(Hxbiim)](1+x)+ (x = 2–0) complexes
In order to limit the number of bound deprotonatable ligands and the concomitant formation of insoluble neutral complexes, we followed the Kane-Maguire synthetic strategy for preparing inert heteroleptic [Cr(phen)2(H2biim)]3+ (Fig. 7).54 Taking advantage of the trans influence,78 the complexation of 2.0 eq. of 1,10-phenanthroline (phen) to CrCl3 yields almost quantitatively cis-[Cr(phen)2Cl2]+ under reducing conditions for catalyzing Cr(III)/Cr(II) exchange processes and ligand-exchange dynamics.53 The replacement of inert Cr–Cl bonds with labile Cr–OSO2CF3 bonds79–81 was performed under soft conditions by using Ag(O3SCF3) instead of an excess of triflic acid.24 The addition of H2biim finally provided [Cr(phen)2(H2biim)](CF3SO3)3 in moderate yield (37%, Fig. 7). Subsequent deprotonation with aqueous NaOH under stoichiometric conditions gave [Cr(phen)2(Hbiim)](CF3SO3)2, whereas the use of an excess of base yielded the doubly deprotonated complex [Cr(phen)2(biim)](CF3SO3). X-ray quality monocrystals could be grown by slow diffusion of diethyl ether into solutions of the complexes either in acetonitrile to give [Cr(phen)2(H2biim)](CF3SO3)3(H2O)0.25 (Fig. 7 and Tables S18–S20†) or in methanol to provide [Cr(phen)2(biim)](CF3SO3)(CH3OH)1.5 (Fig. 7 and Tables S24–S26†). X-ray quality prisms of [Cr(phen)2(Hbiim)](CF3SO3)2(H2O)0.5 (Fig. 7 and Tables S21–S23†) were isolated from a cooled (4 °C) aqueous solution.
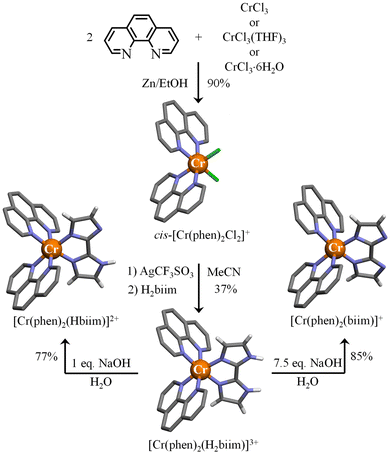 |
| | Fig. 7 Synthesis of the heteroleptic cis-[Cr(phen)2Cl2]+ (CCDC-1865022),53 [Cr(phen)2(H2biim)]3+, [Cr(phen)2(Hbiim)]2+ and [Cr(phen)2(biim)]+ complexes. The molecular structures of the metallic complexes are those found in the associated crystal structures. Color code: C = grey, N = blue, H = white, and Cr = orange. The counter-ions and hydrogen atoms (for 1,10-phenanthroline ligands) are omitted for clarity. | |
The molecular structures of the three [Cr(phen)2(Hxbiim)](1+x)+ cations (x = 2–0) display pseudo-octahedral [CrN6] chromophores with bond lengths and trigonal distortions typical of chromium complexes bound by three didentate 5-membered chelate polyaromatic ligands (Appendix 3†), similar to the discussion in the previous section for the homoleptic analogues [Cr(H2biim)3]3+, [Cr(Hbiim)3] and [Cr(Hbiim)2(biim)]− (see Appendix 2†). For the heteroleptic complexes, one notices that the Cr–N bond lengths are shorter for the bound biimidazole ligands compared to those of the bound phen ligands (dCr–N(biim) < dCr–N(phen), Table 3). The contraction of the dCr–N(biim) bond lengths upon stepwise deprotonation can be assigned to the increased basicity of the bound biimidazole ligand. The compensating longer dCr–N(phen) bond lengths result from the reduced charge borne by the central chromium metal. In terms of intermolecular hydrogen bonds, the bound protonated H2biim ligand in [Cr(phen)2(H2biim)](CF3SO3)3(H2O)0.25 acts as a NH donor for acceptor oxygen atoms of triflate counter-anions and interstitial water molecules. For the mono-deprotonated bound Hbiim− ligand in [Cr(phen)2(Hbiim)](CF3SO3)2(H2O)0.5, intermolecular hydrogen bonds between two adjacent complexes through N–H⋯N bonds are observed (Fig. A3-3†), as previously described for [Cr(Hbiim)3] (Fig. A2-4†). Finally, the totally deprotonated bound biim2− ligand in [Cr(phen)2(biim)](CF3SO3)(CH3OH)1.5 is not involved in hydrogen bonding.
Table 3 Comparison of the average Cr–N bond lengths (![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) Cr–N)a of complexes [Cr(phen)2(H2biim)](CF3SO3)3(H2O)0.25, [Cr(phen)2(Hbiim)](CF3SO3)2(H2O)0.5 and [Cr(phen)2(biim)](CF3SO3)(CH3OH)1.5 in their crystal structures
Cr–N)a of complexes [Cr(phen)2(H2biim)](CF3SO3)3(H2O)0.25, [Cr(phen)2(Hbiim)](CF3SO3)2(H2O)0.5 and [Cr(phen)2(biim)](CF3SO3)(CH3OH)1.5 in their crystal structures
| Complex |
Cr–N/Å |
Cr–N(phen)/Å |
Cr–N(biim)/Å |
|
The standard deviations refer to deviations from the computed averages.
|
| [Cr(phen)2(H2biim)](CF3SO3)3 |
2.04(2) |
2.054(3) |
2.024(7) |
| [Cr(phen)2(Hbiim)](CF3SO3)2 |
2.05(2) |
2.062(6) |
2.018(0) |
| [Cr(phen)2(biim)]CF3SO3 |
2.05(4) |
2.07(1) |
1.997(5) |
In the IR spectra of the heteroleptic complexes, the O–H stretching vibrations of co-crystallized water or methanol molecules involved in hydrogen bonding (3500 ≤ νOH ≤ 2500 cm−1) hinder a straightforward interpretation of N–H stretching bands associated with the bound Hxbiim ligands (x = 2–0; Fig. S57†). On the other hand, and as previously mentioned for the related homoleptic complexes, the ESI-MS spectra recorded in acetonitrile do not vary significantly with the degree of protonation of the bound 2,2′-biimidazole ligands for the different [Cr(phen)2(Hxbiim)](1+x)+ cations (x = 2–0, Fig. S58†). High-resolution ESI-MS analyses confirm the formation of [Cr(phen)2(Hbiim)]2+ as the major gas-phase cation, regardless of the degree of protonation of the bound 2,2′-biimidazole ligand in the selected complex (Table S41 and Fig. S58–62†).
Acid–base properties of the heteroleptic [Cr(phen)2(H2biim)]3+ complex
The successive deprotonation of bound 2,2′-biimidazole in [Cr(phen)2(H2biim)]3+ has been quantitatively studied by pH metric titrations in water at fixed ionic strength (0.1 M KNO3, Fig. 8a). The two successive pH jumps (5 ≤ pH ≤ 8 and 9 ≤ pH ≤ 11) are accompanied by concomitant abrupt changes in colors from yellow ([Cr(phen)2(H2biim)]3+) to dark orange ([Cr(phen)2(Hbiim)]2+) and finally to dark brown ([Cr(phen)2(biim)]+, Fig. 8a). The associated occupancy factors 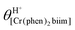 experimentally obtained with eqn (5) (red circles in Fig. 8b) display a two-step binding isotherm typical of the anti-cooperative successive fixation of two protons according to equilibria (6)–(7) and modeled with eqn (8). The best fits for the two successive deprotonation steps of [Cr(phen)2(H2biim)]3+ (black trace in Fig. 8b) correspond to pKa1 = 4.67(3) and pKa2 = 8.59(11), which are approximately eight orders of magnitude more acidic than those measured for free 2,2′-biimidazole (pKa1 = 12.31 and pKa2 = 16.33),25
experimentally obtained with eqn (5) (red circles in Fig. 8b) display a two-step binding isotherm typical of the anti-cooperative successive fixation of two protons according to equilibria (6)–(7) and modeled with eqn (8). The best fits for the two successive deprotonation steps of [Cr(phen)2(H2biim)]3+ (black trace in Fig. 8b) correspond to pKa1 = 4.67(3) and pKa2 = 8.59(11), which are approximately eight orders of magnitude more acidic than those measured for free 2,2′-biimidazole (pKa1 = 12.31 and pKa2 = 16.33),25| |  | (5) |
| |  | (6) |
| |  | (7) |
| | 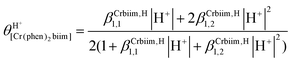 | (8) |
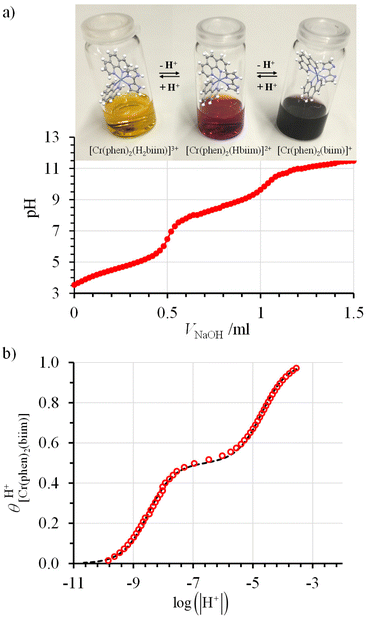 |
| | Fig. 8 (a) Titration of 51 mg (52 μmol) of [Cr(phen)2(H2biim)](CF3SO3)3 (10 mL aqueous KNO3 0.1 M, c = 5.2 mmol L−1) with NaOH 0.1 N highlighting the color changes and (b) associated binding isotherm depicted as plots of experimental (red circles, eqn (5)) and fitted (dashed black trace, eqn (8)) occupancy factors as a function of log(|H+|). | |
Compared with [CoIII(en)2(H2biim)]3+ (pKa1 = 5.5 and pKa2 = 9.9; ionic radius = 0.545 Å),41 the one order of magnitude lower pKa measured for [CrIII(phen)2(H2biim)]3+ (ionic radius = 0.615 Å) suggests that the {CrIII(phen)2}3+ scaffold (with respect to {CoIII(en)2}3+) better stabilizes the negative charges brought by the bound deprotonated 2,2′-biimidazole ligand. Consequently, upon successive deprotonation, one can reasonably predict the appearance of low energy (phen)π* ← (Hxbiim)π (x = 2–0) intramolecular ligand-to-ligand charge transfer (LLCT) bands in the absorption spectra of [CrIII(phen)2(Hxbiim)](1+x)+ upon deprotonation. These transitions are confirmed by TD-DFT calculations (Fig. A4-2 to A4-10 and Tables A4-6 to A4-8 in Appendix 4†) and indeed observed in solution (Fig. 9).
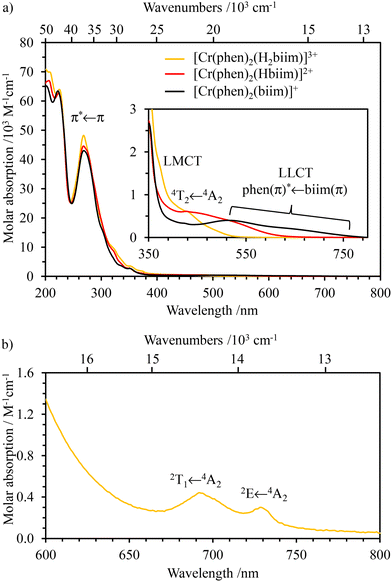 |
| | Fig. 9 Absorption spectra of (a) [Cr(phen)2(H2biim)]3+ (orange trace), [Cr(phen)2(Hbiim)]2+ (red trace) and [Cr(phen)2(biim)]+ (black trace) in CH3CN (c ≈ 10−5 mol L−1) and (b) [Cr(phen)2(H2biim)]3+ in CH3CN (c ≈ 10−2 mol L−1). LMCT = ligand to metal charge transfer and LLCT = ligand to ligand charge transfer. | |
Finally, taking advantage of the pH-dependence of the absorption spectra, the determination of the pKa values in water at ionic strength close to zero (I ≈ 0) could be carried out with the help of spectrophotometry to give pKa1 = 3.9(1) and pKa2 = 7.8(1) (Fig. S63†), which are close to those determined at I = 0.1 M (KNO3).
Photophysical properties of the heteroleptic [Cr(phen)2(Hxbiim)](1+x)+ (x = 2–0) complexes
The three heteroleptic complexes exhibit similar and intense absorption bands in the UV range (below 350 nm, Fig. 9a), which can be assigned to intraligand π–π* transitions (ILCT) completed by variable amounts of interligand (LLCT), ligand to-metal (LMCT) and metal-to-ligand (MLCT) charge transfer bands (see Appendix 4† for TD-DFT calculations). The spectra differ in the visible domain (350–750 nm, highlighted in Fig. 9a) mainly due to a shift of the interligand (phen)π* ← (Hxbiim)π (x = 0–2, LLCT) transitions toward lower energies upon successive deprotonations (Fig. S64 and Fig. A4-4, A4-7 and A4-9†). This shift is responsible for the color change accompanying the stepwise deprotonation of [Cr(phen)2(H2biim)]3+ (Fig. 8a). According to theoretical TD-DFT calculations (Table 1, entries 1–3) and CASSCF(7,12)/FIC-NEVPT2 (Table A4-2†) and in line with the trend observed in the homoleptic complexes, the ligand-field strength Δ, reminiscent of Cr(4T2 ← 4A2) transitions in octahedral geometry, also decreases with stepwise deprotonation: [Cr(phen)2(H2biim)]3+ (412.9 nm, Δ ≈ 24219 cm−1) > [Cr(phen)2(Hbiim)]2+ (434 nm, Δ ≈ 23042 cm−1) > [Cr(phen)2(biim)]+ (457.8 nm, Δ ≈ 21844 cm−1). This reduction in the ligand-field strength is accompanied by (i) a decrease of the computed total spin density at the chromium center (Fig. A4-1†) which reflects the reduced total positive charge borne by this atom and (ii) a negligible change in the estimated Racah parameters B and C (Table A4-1†). One can thus predict that the stronger interactions with the deprotonated 2,2-biimidazole ligand (Cr–N(biim) is becoming shorter) in [Cr(phen)2(Hxbiim)](1+x)+ (x = 2–0) are more than compensated by the removal of the bound 1,10-phenanthroline ligands (Cr–N(phen) is becoming longer) as exemplified in the molecular structures in the crystalline state (push–pull effect, Table 3).
The NIR absorption spectrum of [Cr(phen)2(H2biim)]3+ (Fig. 9b) exhibits two well-resolved absorption bands that are assigned to the spin–flip transitions Cr(2E ← 4A2) and Cr(2T1 ← 4A2) assuming Oh symmetry and with ε = 0.27 and 0.19 M−1 cm−1, respectively. For the deprotonated derivatives [Cr(phen)2(Hbiim)]2+ and [Cr(phen)2(biim)]+, the larger residual interligand charge transfer bands mask these weak forbidden spin–flip transitions (Fig. S65†). Consequently, Racah parameters B = 766 cm−1 and C = 2697 cm−1 (eqn (2) and (3), Table 1, entry 4) together with the radiative rate constant of krad = 65(3) s−1 (eqn (4), and Table 2, column 1), typical of [CrN6] chromophores, could be estimated only for [Cr(phen)2(H2biim)]3+.
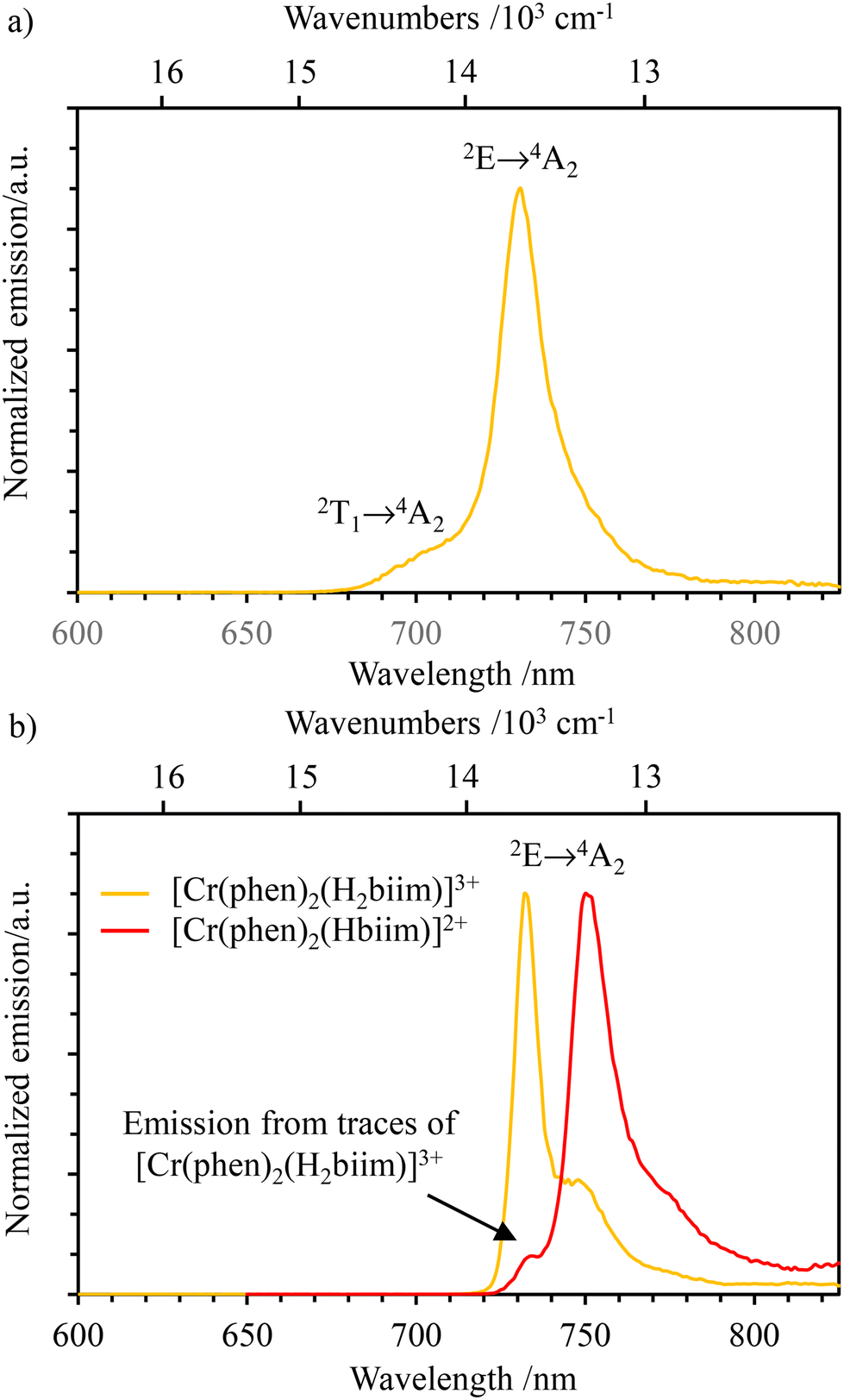
At room temperature in CH3CN, only [Cr(phen)2(H2biim)]3+ is emissive and shows the typical dual Cr(2T1 → 4A2) and Cr(2E → 4A2) emission observed for many Cr(III) complexes (Fig. 10a). In addition to [Cr(phen)2(H2biim)]3+, the NIR emission of the complex [Cr(phen)2(Hbiim)]2+ can be detected at low temperature in frozen solvent mixtures (CH3CN/C2H5CN 6:4 at 77 K, Fig. 10b), whereas [Cr(phen)2(biim)]+ remains non-emissive. Compared with the emission band of [Cr(phen)2(H2biim)]3+ (λmax = 732 nm; = 13661 cm−1), the first deprotonated analog [Cr(phen)2(Hbiim)]2+ shows a red-shifted Cr(2E → 4A2) transition at λmax = 750 nm; = 13333 cm−1 (Fig. 10b). Additionally, the spectrum of [Cr(phen)2(Hbiim)]2+ contains a band foot at 733 nm originating from a small amount of [Cr(phen)2(H2biim)]3+, which is inevitably present in solution due to the proton-transfer equilibrium (9):
| |  | (9) |
 |
| | Fig. 10 Emission spectra (λexc = 350 nm) of (a) [Cr(phen)2(H2biim)]3+ at 293 K in CH3CN and (b) [Cr(phen)2(H2biim)]3+ (orange trace) and [Cr(phen)2(Hbiim)]2+ (red trace) at 77 K in frozen CH3CN/C2H5CN (6:4). | |
Introducing Ka1 and Ka2 gives Kexch = 1.2(2) × 10−4, from which the ratio of the equilibrium concentration  implies contamination of [Cr(phen)2(Hbiim)]2+ by circa 1% with the more emissive (protonated) [Cr(phen)2(H2biim)]3+ complex (red trace in Fig. 10b).
implies contamination of [Cr(phen)2(Hbiim)]2+ by circa 1% with the more emissive (protonated) [Cr(phen)2(H2biim)]3+ complex (red trace in Fig. 10b).
Excited state lifetimes for the NIR emission arising from the Cr(2E) excited state upon excitation at 355 nm were recorded in solution at room temperature and at 77 K for [Cr(phen)2(H2biim)]3+, and only at 77 K for [Cr(phen)2(Hbiim)]2+ (Fig. S67–S70†). In frozen solutions at 77 K, [Cr(phen)2(H2biim)]3+ displays a mono-exponential decay of 2.94 ms, which is typical of Cr(III)-polyimine complexes (Table 2). The emission decay curve of [Cr(phen)2(Hbiim)]2+ could not be fit with a mono-exponential function. Since the complex exists as a 98:1:1 mixture of [Cr(phen)2(Hbiim)]2+, [Cr(phen)2(H2biim)]3+ and [Cr(phen)2(biim)]+ (eqn (9)), one expects multi-exponential decays weighted by the mole fractions and the quantum yields of each contributor. A rough bi-exponential fit (Fig. S70†) is compatible with the experimental decay curves showing a long component (1.77(9) ms), which is reminiscent of [Cr(phen)2(H2biim)]3+, and a short contribution (723(30) μs) which is tentatively assigned to [Cr(phen)2(Hbiim)]2+, for which the smaller energy gap between the emissive doublet state level Cr(2E) and the silent LLCT band probably boosts the efficiency of non-radiative decay (Fig. 11 and Appendix 4†). At room temperature, only the protonated complex [Cr(phen)2(H2biim)]3+ is emissive and it exhibits a mono-exponential emission decay of 38 μs in deaerated CH3CN. This excited state lifetime is orders of magnitude longer than those of the three isolated homoleptic parent complexes [Cr(Hxbiim)3]n+ (Table 2) due to the replacement of two 2,2′-biimidazole ligands with two 1,10-phenanthroline units, which are devoid of high-energy N–H stretching vibrations. The lifetime of [Cr(phen)2(H2biim)]3+ is in the same range as that of [Cr(bpy)3]3+; however, it is one order of magnitude shorter than that of [Cr(phen)3]3+ (Table 2). The emission lifetime is reduced to 14 μs in aerated solution because of some additional quenching via energy transfer to the 3O2 molecules present in solution.
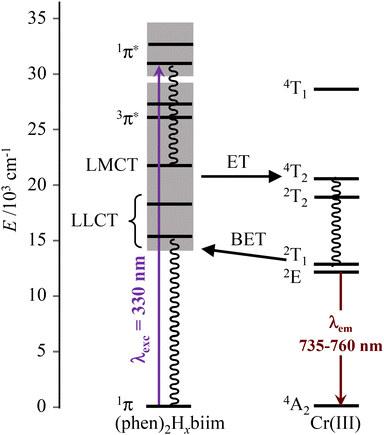 |
| | Fig. 11 Jablonski diagram of heteroleptic [CrIII(phen)2(Hxbiim)](1+x)+ complexes showing the antenna effect upon UV excitation at 330 nm, the existence of low-energy ligand-to-ligand charge transfer excited levels and their potential unfavorable effect on the global quantum yields via back energy transfer (BET) processes. | |
Finally, the global luminescence quantum yield of the complex [Cr(phen)2(H2biim)]3+ upon ligand-based excitation at λexc = 450 nm was determined experimentally using the relative method (Fig. S71†). We found ϕglobalcomplex = 2.8(3) × 10−3 in deaerated acetonitrile and ϕglobalcomplex = 9.5(9) × 10−4 in the presence of dioxygen at room temperature. One further notes that the intrinsic Cr(III)-centered quantum yields calculated with the help of the emission lifetimes τtot(2E) and krad(2E) collected at room temperature (Table 2 and eqn (10)) amount to ϕintrinsiccomplex = 2.5 × 10−3 in deaerated acetonitrile and ϕintrinsiccomplex = 9.1 × 10−4 in aerated acetonitrile.
| |  | (10) |
Since (i) ϕglobalcomplex = ηsens·ϕintrinsiccomplex and (ii) ϕglobalcomplex ≃ ϕintrinsiccomplex, one concludes that the ligand-to-metal sensitization process is close to being quantitative (ηsens ≈ 100%). These values are in the same range as those reported for [Cr(bpy)3]3+ (ϕglobal, no aircomplex = 1.7 × 10−3 in deaerated acetonitrile and ϕglobal, aircomplex = 8.9 × 10−4 in aerated water),70,71,79,80 but four times smaller than that of [Cr(phen)3]3+ (ϕglobal, no aircomplex = 1.2 × 10−2 in deaerated water + 1 M HCl),53,81 which makes [Cr(phen)2(H2biim)]3+ a moderate emitter for a Cr(III) complex.72
Conclusions
In line with the only minor interest attracted by the homoleptic [Cr(Hxbiim)3]n+ during the past few decades,35–38 we confirm here that their low solubility in any common solvent upon stepwise deprotonation, together with their poorly attracting photophysical properties (a short phosphorescence lifetime and negligible intrinsic quantum yield), makes them poorly adapted to be involved in the complex-as-ligand strategy for programming the assemblies of polymetallic optically-active Cr-based complexes. The solution to the problem arises from the successful synthesis of the soluble heteroleptic [Cr(phen)2(Hxbiim)](1+x)+ (x = 2–0) complexes via a modified Kane-Maguire strategy. The bound H2biim ligand can be stepwise deprotonated in solution at pH compatible for further complexation processes with d-block or f-block cations. The deprotonation processes are accompanied by characteristic color changes resulting from the appearance of low-energy intramolecular interligand (phen)π* ← (Hxbiim)π (x = 2–0, LLCT) ligand-to-ligand charge transfer transitions confirmed by theoretical TD-DFT calculations. The latter reversible process makes the putative heterometallic dyads [Cr(phen)2(biim)–Mz+](z+1)+ (Mz+ is an open-shell d- or f-block cation) reminiscent of protonated [Cr(phen)2(H2biim)]3+ in terms of photophysical properties, which paves the way for their use as sensitizers in luminescent polymetallic assemblies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the Swiss National Science Foundation is gratefully acknowledged (grant 200020_207313).
References
- M. Verdaguer, A. Bleuzen, V. Marvaud, J. Vaissermann, M. Seuleiman, C. Desplanches, A. Scuiller, C. Train, R. Garde, G. Gelly, C. Lomenech, I. Rosenman, P. Veillet, C. Cartier and F. Villain, Coord. Chem. Rev., 1999, 190, 1023–1047 CrossRef
 .
.
- R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2002, 1–10 RSC .
- J. M. Herrera, A. Bachschmidt, F. Villain, A. Bleuzen, V. Marvaud, W. Wernsdorfer and M. Verdaguer, Philos. Trans. R. Soc., A, 2008, 366, 127–138 CrossRef CAS PubMed .
- K. S. Pedersen, M. A. Sorensen and J. Bendix, Coord. Chem. Rev., 2015, 299, 1–21 CrossRef CAS .
- Y. Journaux, J. Ferrando-Soria, E. Pardo, R. Ruiz-Garcia, M. Julve, F. Lloret, J. Cano, Y. L. Li, L. Lisnard, P. Yu, H. Stumpf and C. L. M. Pereira, Eur. J. Inorg. Chem., 2018, 228–247 CrossRef CAS .
- F. Hulliger, M. Landolt and H. Vetsch, J. Solid State Chem., 1976, 18, 283–291 CrossRef CAS .
- S. Decurtins, M. Gross, H. W. Schmalle and S. Ferlay, Inorg. Chem., 1998, 37, 2443–2448 CrossRef CAS .
- F. Yan and Z. D. Chen, J. Phys. Chem. A, 2000, 104, 6295–6300 CrossRef CAS .
- H. Z. Kou, S. Gao, B. W. Sun and J. Zhang, Chem. Mater., 2001, 13, 1431–1433 CrossRef CAS .
- R. Lescouëzec, L. M. Toma, J. Vaissermann, M. Verdaguer, F. S. Delgado, C. Ruiz-Perez, F. Lloret and M. Julve, Coord. Chem. Rev., 2005, 249, 2691–2729 CrossRef .
- P. A. Brayshaw, J.-C. G. Bünzli, P. Froidevaux, J. M. Harrowfield, Y. Kim and A. N. Sobolev, Inorg. Chem., 1995, 34, 2068–2076 CrossRef CAS .
- T. Sanada, T. Suzuki, T. Yoshida and S. Kaizaki, Inorg. Chem., 1998, 37, 4712–4717 CrossRef CAS PubMed .
- L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J.-C. G. Bünzli, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2011, 50, 4108–4112 CrossRef CAS PubMed .
- J. Kalmbach, C. Wang, Y. You, C. Forster, H. Schubert, K. Heinze, U. Resch-Genger and M. Seitz, Angew. Chem., Int. Ed., 2020, 39, 18804–18808 CrossRef PubMed .
- R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2002, 1–10 RSC .
- L. Aboshyan-Sorgho, M. Cantuel, S. Petoud, A. Hauser and C. Piguet, Coord. Chem. Rev., 2012, 256, 1644–1663 CrossRef CAS .
- Y. Suffren, B. Golesorkhi, D. Zare, L. Guénée, H. Nozary, S. V. Eliseeva, S. Petoud, A. Hauser and C. Piguet, Inorg. Chem., 2016, 55, 9964–9972 CrossRef CAS PubMed .
- T. Lazarides, G. M. Davies, H. Adams, C. Sabatini, F. Barigelletti, A. Barbieri, S. J. A. Pope, S. Faulkner and M. D. Ward, Photochem. Photobiol. Sci., 2007, 6, 1152–1157 CrossRef CAS PubMed .
- M. Schley, S. Fritzsche, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2010, 39, 4090–4106 RSC .
- T. Birk, K. S. Pedersen, C. A. Thuesen, T. Weyhermueller, M. Scha-Magnusen, S. Piligkos, H. Weihe, S. Mossin, M. Evangelisti and J. Bendix, Inorg. Chem., 2012, 51, 5435–5443 CrossRef CAS PubMed .
- B. Doistau, J.-R. Jiménez, S. Guerra, C. Besnard and C. Piguet, Inorg. Chem., 2020, 59, 1424–1435 CrossRef CAS PubMed .
- S. Otto, M. Grabolle, C. Förtser, C. Kreitner, U. Resh-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS PubMed .
- S. Otto, M. Dorn, C. Förster, M. Bauer, M. Seitz and K. Heinze, Coord. Chem. Rev., 2018, 359, 102–111 CrossRef CAS .
- J.-R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campana and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244–13252 CrossRef PubMed .
- T. Akutagawa and G. Saito, Bull. Chem. Soc. Jpn., 1995, 68, 1753–1773 CrossRef CAS .
- D. J. Mercer and S. J. Loeb, Chem. Soc. Rev., 2010, 39, 3612–3620 RSC .
- S. A. Rommel, D. Sorsche, M. Fleischmann and S. Rau, Chem. – Eur. J., 2017, 23, 18101–18119 CrossRef CAS PubMed .
- R. L. Sang and L. Xu, Eur. J. Inorg. Chem., 2006, 1260–1267 CrossRef CAS .
- P. J. Steel, Coord. Chem. Rev., 1990, 106, 227–265 CrossRef CAS .
- M. Tadokoro and K. Nakasuji, Coord. Chem. Rev., 2000, 198, 205–218 CrossRef CAS .
- M. S. Haddad and D. N. Hendrickson, Inorg. Chem., 1978, 17, 2622–2630 CrossRef CAS .
- M. Barquín, M. J. G. Garmendia and V. Bellido, Transition Met. Chem., 2003, 28, 356–360 CrossRef .
- A. Mayboroda, P. Comba, H. Pritzkow, G. Rheinwald, H. Lang and G. van Koten, Eur. J. Inorg. Chem., 2003, 1703–1710 CrossRef CAS .
- J. N. Leung, H. T. T. Luong and H. V. Huynh, Inorg. Chem., 2023, 62, 4606–4617 CrossRef CAS PubMed .
- M. J. Silvero, W. J. Peláez, P. F. Garcia and G. A. Argüello, RSC Adv., 2014, 4, 15507–15510 RSC .
- L. M. Gruia, F. D. Rochon and A. L. Beauchamp, Can. J. Chem., 2006, 84, 949–959 CrossRef CAS .
- K. Larsson and L. Ohrstrom, CrystEngComm, 2003, 5, 222–225 RSC .
- K. Watanabe, M. Oguni, M. Tadokoro and C. Kobayashi, J. Phys. Chem. B, 2009, 113, 14323–14328 CrossRef CAS PubMed .
- M. Haga, Inorg. Chim. Acta, 1983, 75, 29–35 CrossRef CAS .
- S. Fortin and A. L. Beauchamp, Inorg. Chem., 2001, 40, 105–112 CrossRef CAS PubMed .
- H. Kanno, S. Manriki, E. Yamazaki, S. Utsuno and J. Fujita, Bull. Chem. Soc. Jpn., 1996, 69, 1981–1986 CrossRef CAS .
- R. Sahai, W. R. Murphy and J. D. Petersen, Inorg. Chim. Acta, 1986, 114, 137–140 CrossRef CAS .
- P. Majumdar, S. M. Peng and S. Goswami, J. Chem. Soc., Dalton Trans., 1998, 1569–1574 RSC .
- M. J. Calhorda and A. R. Dias, J. Organomet. Chem., 1980, 197, 291–302 CrossRef CAS .
- D. P. Rillema, R. Sahai, P. Matthews, A. K. Edwards, R. J. Shaver and L. Morgan, Inorg. Chem., 1990, 29, 167–175 CrossRef CAS .
- A. M. Bond and M. Haga, Inorg. Chem., 1986, 25, 4507–4514 CrossRef CAS .
- S. Mardanya, S. Karmakar, M. Bar and S. Baitalik, Dalton Trans., 2015, 44, 21053–21072 RSC .
- M. N. Feng, G. Y. Zhao, H. L. Gao and S. J. Zhang, Aust. J. Chem., 2015, 68, 1513–1517 CrossRef CAS .
- H. M. Zhang, X. G. Yan, J. Zhao, X. L. Yang, Z. Huang, G. J. Zhou and Y. Wu, RSC Adv., 2015, 5, 88758–88766 RSC .
- A. G. Grechin, H. J. Buschmann and E. Schollmeyer, Thermochim. Acta, 2006, 449, 67–72 CrossRef CAS .
- M. Tadokoro, H. Hosoda, T. Inoue, A. Murayama, K. Noguchi, A. Iioka, R. Nishimura, M. Itoh, T. Sugaya, H. Kamebuchi and M. Hagallg, Inorg. Chem., 2017, 56, 8513–8526 CrossRef CAS PubMed .
- K. V. Goodwin, W. T. Pennington and J. D. Petersen, Inorg. Chem., 1989, 28, 2016–2018 CrossRef CAS .
- B. Doistau, G. Collet, E. Acuna Bolomey, V. Sadat-Noorbakhsh, C. Besnard and C. Piguet, Inorg. Chem., 2018, 57, 14362–14373 CrossRef CAS PubMed .
- K. D. Barker, K. A. Barnett, S. M. Connel, J. W. Glaeser, A. J. Wallace, J. Wildsmith, B. J. Herbert, J. F. Wheeler and N. A. P. Kane-Maguire, Inorg. Chim. Acta, 2001, 316, 41–49 CrossRef CAS .
- F. Reichenauer, C. Wang, C. Forster, P. Boden, N. Ugur, R. Baez-Cruz, J. Kalmbach, L. M. Carrella, E. Rentschler, C. Ramanan, G. Niedner-Schatteburg, M. Gerhards, M. Seitz, U. Resch-Genger and K. Heinze, J. Am. Chem. Soc., 2021, 143, 11843–11855 CrossRef CAS PubMed .
- J.-R. Jiménez, S. Miguez-Lago, M. Poncet, Y. T. Ye, C. L. Ruiz, C. M. Cruz, A. G. G. Campana, E. Colacio, C. Piguet and J. M. Herrera, J. Mater. Chem. C, 2023, 11, 2582–2590 RSC .
- A. Benchohra, J. L. Chong, C. M. Cruz, C. Besnard, L. Guénée, A. Rosspeintner and C. Piguet, Inorg. Chem., 2024, 63, 3617–3629 CrossRef CAS PubMed .
- N. Serpone, M. A. Jamieson, M. S. Henry, M. Z. Hoffman, F. Bolletta and M. Maestri, J. Am. Chem. Soc., 1979, 101, 2907–2916 CrossRef CAS .
- N. Serpone and M. Z. Hoffman, J. Chem. Educ., 1983, 60, 853–860 CrossRef CAS .
- A. Hauser, M. Mäder, W. T. Robinson, R. Murugesan and J. Ferguson, Inorg. Chem., 1987, 26, 1331–1338 CrossRef CAS .
- J.-R. Jiménez, B. Doistau, C. Besnard and C. Piguet, Chem. Commun., 2018, 54, 13228–13231 RSC .
- N. Sinha, J.-R. Jiménez, B. Pfund, A. Prescimone, C. Piguet and O. S. Wenger, Angew. Chem., Int. Ed., 2021, 60, 23722–23728 CrossRef CAS PubMed .
- J. J. Alexander and H. B. Gray, J. Am. Chem. Soc., 1968, 90, 4260–4271 CrossRef CAS .
- D. Zare, B. Doistau, H. Nozary, C. Besnard, L. Guénée, Y. Suffren, A.-L. Pelé, A. Hauser and C. Piguet, Dalton Trans., 2017, 46, 8992–9009 RSC .
- Z. Song, P. A. Tanner and Q. L. Liu, J. Phys. Chem. Lett., 2024, 15, 2319–2324 CrossRef CAS PubMed .
- H. Phan, J. J. Hrudka, D. Igimbayeva, L. M. L. Daku and M. Shatruk, J. Am. Chem. Soc., 2017, 139, 6437–6447 CrossRef CAS PubMed .
- S. J. Strickler and R. A. Berg, J. Chem. Phys., 1962, 37, 814–822 CrossRef CAS .
- J.-C. G. Bünzli, A.-S. Chauvin, H. K. Kim, E. Deiters and S. V. Eliseeva, Coord. Chem. Rev., 2010, 254, 2623–2633 CrossRef .
- R. C. Hilborn, Am. J. Phys., 1982, 50, 982–986 CrossRef CAS .
- N. A. P. Kane-Maguire, R. C. Kerr and J. R. Walters, Inorg. Chim. Acta, 1979, 33, L163–L165 CrossRef CAS .
- C. K. Ryu and J. F. Endicott, Inorg. Chem., 1988, 27, 2203–2214 CrossRef CAS .
- L. S. Forster, Chem. Rev., 1990, 90, 331–353 CrossRef CAS .
- M. Isaacs, A. G. Sykes and S. Ronco, Inorg. Chim. Acta, 2006, 359, 3847–3854 CrossRef CAS .
- J. C. Barbour, A. J. I. Kim, E. deVries, S. E. Shaner and B. M. Lovaasen, Inorg. Chem., 2017, 56, 8212–8222 CrossRef CAS PubMed .
- J. F. Endicott, R. B. Lessard, Y. Lei, C. K. Ryu and R. Tamilarasan, ACS Symp. Ser., 1986, 307, 85–103 CrossRef CAS .
- J.-R. Jiménez, M. Poncet, B. Doistau, C. Besnard and C. Piguet, Dalton Trans., 2020, 49, 13528–13532 RSC .
- J.-R. Jiménez, M. Poncet, S. Miguez-Lago, S. Grass, J. Lacour, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Angew. Chem., Int. Ed., 2021, 60, 10095–10102 CrossRef PubMed .
- G. B. Kauffman, J. Chem. Educ., 1977, 54, 86–89 CrossRef CAS .
- A. D. Kirk and G. B. Porter, J. Phys. Chem., 1980, 84, 887–891 CrossRef CAS .
- A. M. McDaniels, H.-W. Tseng, E. A. Hill, N. H. Damrauer, A. K. Rappé and M. P. Shores, Inorg. Chem., 2013, 52, 1368–1378 CrossRef PubMed .
- A. M. McDaniel, H.-W. Tseng, N. H. Damrauer and M. P. Shores, Inorg. Chem., 2010, 49, 7981–7991 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2355642–2355650. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01608d |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Céline
Besnard
ab,
Céline
Besnard









 is the area under the absorption spectrum of each transition (M−1 cm−2, Fig. S44 and S45†). The radiative rate constants of the emissive Cr(2E) excited level estimated for [Cr(H2biim)3]3+ (krad = 75(4) s−1) and [Cr(Me2biim)3] (krad = 73(4) s−1) are smaller than those reported for [Cr(bpy)3]3+ (krad = 182 s−1) and [Cr(phen)3]3+ (krad = 319 s−1, Table 2),58,69–73 but fall within the expected range for pseudo-octahedral [CrN6] chromophores (Table S40†).70–77 Upon room-temperature ligand-based excitation at 330 nm in solution, the complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 show the expected downshifted NIR spin–flip Cr(2E → 4A2) phosphorescence at 730–740 nm together with weak shoulders corresponding to Cr(2T1 → 4A2) within the 700–710 nm domain (Fig. 4b and 5). The latter dual emission disappears at 77 K (Fig. S39†) as a result of the depopulation of the high-energy Cr(2T1) level, and a single band Cr(2E → 4A2) contributes to phosphorescence. Very similar results are observed in the solid state upon 330 nm excitation for the four complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)3], [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 (Fig. S40b†), which ultimately demonstrate (i) the expected negligible Stokes shifts affecting the spin–flip Cr(2T1,2E ↔ 4A2) transitions (Fig. S46†) and (ii) the efficient sensitization of the spin–flip phosphorescence by all the accessible ligand-based excited states (Fig. S47†).
is the area under the absorption spectrum of each transition (M−1 cm−2, Fig. S44 and S45†). The radiative rate constants of the emissive Cr(2E) excited level estimated for [Cr(H2biim)3]3+ (krad = 75(4) s−1) and [Cr(Me2biim)3] (krad = 73(4) s−1) are smaller than those reported for [Cr(bpy)3]3+ (krad = 182 s−1) and [Cr(phen)3]3+ (krad = 319 s−1, Table 2),58,69–73 but fall within the expected range for pseudo-octahedral [CrN6] chromophores (Table S40†).70–77 Upon room-temperature ligand-based excitation at 330 nm in solution, the complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 show the expected downshifted NIR spin–flip Cr(2E → 4A2) phosphorescence at 730–740 nm together with weak shoulders corresponding to Cr(2T1 → 4A2) within the 700–710 nm domain (Fig. 4b and 5). The latter dual emission disappears at 77 K (Fig. S39†) as a result of the depopulation of the high-energy Cr(2T1) level, and a single band Cr(2E → 4A2) contributes to phosphorescence. Very similar results are observed in the solid state upon 330 nm excitation for the four complexes [Cr(H2biim)3](NO3)3, [Cr(Hbiim)3], [Cr(Hbiim)2(biim)]PPh4 and [Cr(Me2biim)3](CF3SO3)3 (Fig. S40b†), which ultimately demonstrate (i) the expected negligible Stokes shifts affecting the spin–flip Cr(2T1,2E ↔ 4A2) transitions (Fig. S46†) and (ii) the efficient sensitization of the spin–flip phosphorescence by all the accessible ligand-based excited states (Fig. S47†).

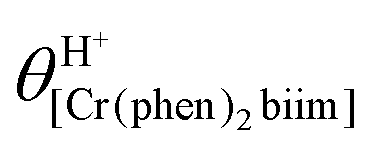 experimentally obtained with eqn (5) (red circles in Fig. 8b) display a two-step binding isotherm typical of the anti-cooperative successive fixation of two protons according to equilibria (6)–(7) and modeled with eqn (8). The best fits for the two successive deprotonation steps of [Cr(phen)2(H2biim)]3+ (black trace in Fig. 8b) correspond to pKa1 = 4.67(3) and pKa2 = 8.59(11), which are approximately eight orders of magnitude more acidic than those measured for free 2,2′-biimidazole (pKa1 = 12.31 and pKa2 = 16.33),25
experimentally obtained with eqn (5) (red circles in Fig. 8b) display a two-step binding isotherm typical of the anti-cooperative successive fixation of two protons according to equilibria (6)–(7) and modeled with eqn (8). The best fits for the two successive deprotonation steps of [Cr(phen)2(H2biim)]3+ (black trace in Fig. 8b) correspond to pKa1 = 4.67(3) and pKa2 = 8.59(11), which are approximately eight orders of magnitude more acidic than those measured for free 2,2′-biimidazole (pKa1 = 12.31 and pKa2 = 16.33),25

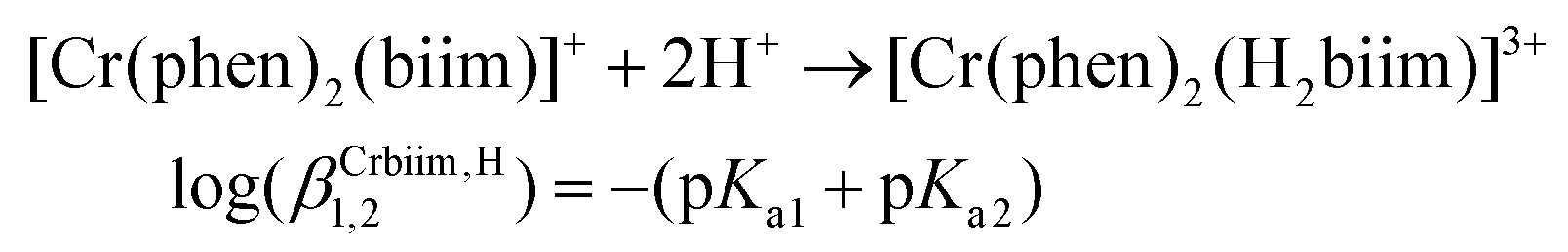




 implies contamination of [Cr(phen)2(Hbiim)]2+ by circa 1% with the more emissive (protonated) [Cr(phen)2(H2biim)]3+ complex (red trace in Fig. 10b).
implies contamination of [Cr(phen)2(Hbiim)]2+ by circa 1% with the more emissive (protonated) [Cr(phen)2(H2biim)]3+ complex (red trace in Fig. 10b).
