
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photosubstitution and photoreduction of a diazido platinum(IV) anticancer complex†
Huayun
Shi‡
 a,
Christian
Ward-Deitrich‡
b,
Fortuna
Ponte
c,
Emilia
Sicilia
*c,
Heidi
Goenaga-Infante
*b and
Peter J.
Sadler
*a
a,
Christian
Ward-Deitrich‡
b,
Fortuna
Ponte
c,
Emilia
Sicilia
*c,
Heidi
Goenaga-Infante
*b and
Peter J.
Sadler
*a
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: P.J.Sadler@warwick.ac.uk
bLGC Limited, National Measurement Laboratory (NML), Queens Road, Teddington, Middlesex TW11 0LY, UK. E-mail: heidi.goenaga-infante@lgcgroup.com
cDepartment of Chemistry and Chemical Technologies, University of Calabria, via Pietro Bucci, 87036 Arcavacata di Rende, Cs, Italy. E-mail: emilia.sicilia@unical.it
First published on 19th July 2024
Abstract
The hyphenation of HPLC with its high separation ability and ICP-MS with its excellent sensitivity, allows the analysis of Pt drugs in biological samples at the low nanomolar concentration levels. On the other hand, LC-MS provides molecular structural confirmation for each species. Using a combination of these methods, we have investigated the speciation of the photoactive anticancer complex diazido Pt(IV) complex trans, trans, trans-[Pt(N3)2(OH)2(py)2] (FM-190) in aqueous solution and biofluids at single-digit nanomolar concentrations before and after irradiation. FM-190 displays high stability in human blood plasma in the dark at 37 °C. Interestingly, the polyhydroxido species [{PtIV(py)2(OH)4} + Na]+ and [{PtIV(py)2(N3)(OH)3} + Na]+ resulting from the replacement of azido ligands, as determined by LC-MS, were the major products after photoirradiation of FM-190 with blue light (463 nm). This finding suggests that such photosubstituted Pt(IV) tri- and tetra-hydroxido species could play important roles in the biological activity of this anticancer complex. Density Functional Theory (DFT) and Time-Dependent DFT (TDDFT) calculations show that these Pt(IV) species arising from FM-190 in aqueous media can be formed directly from a singlet excited state. The results highlight how speciation analysis (metallomics) can shed light on photoactivation pathways for FM-190 and formation of potential excited-state pharmacophores. The ability to detect and identify photoproducts at physiologically-relevant concentrations in cells and tissues will be important for preclinical development studies of this class of photoactivatable platinum drugs.
1. Introduction
Platinum drugs have been widely used in chemotherapy since the FDA approval of cisplatin for testicular cancer treatment in 1978.1,2 Owing to the lack of selectivity and dose-limiting side effects of cisplatin, photoactive Pt(IV) complexes that are more kinetically stable are being developed as a new generation of platinum drugs.3–7 Photoactive Pt(IV) complexes offer the prospect of low toxicity towards normal tissue, and selective cytotoxicity towards tumours when irradiated with spatially-directed light.8–13 Octahedral diam(m)ine dihydroxido diazido Pt(IV) complexes are especially promising for further development. They display a significant stability in the dark under physiological conditions and photocytotoxicity towards a wide range of human cancer cells.5,6cis,trans,cis-[Pt(N3)2(OH)2(NH3)2] was the first reported photoactive anticancer diazido Pt(IV) complex.14 However, the all-trans analogue trans,trans,trans-[Pt(N3)2(OH)2(NH3)2] exhibits improved aqueous solubility, a more intense and ca. 30 nm red-shifted LMCT band, and an enhanced photocytotoxicity.15–17 Replacement of an ammine ligand with pyridine results in trans,trans,trans-[Pt(N3)2(OH)2(NH3)(py)] with high UVA photocytotoxicity.18 Notably, compared with photolabile ammine ligands, no pyridine release is detected upon irradiation. trans,trans,trans-[Pt(N3)2(OH)2(py)2] (FM-190, Fig. 1a) with two pyridine ligands has a promising visible light photocytotoxicity.19 | ||
| Fig. 1 (a) Structure of photoactivatable Pt(IV) complex FM-190 studied in this work. (b) HR-MS and expected structure of the main photoproduct c ([{PtIV(py)2(N3)(OH)3} + Na]+, m/z 469.0562) from FM-190 in water. | ||
The photoactivation of FM-190 in aqueous solution has been investigated previously to provide insights into its mechanism of action.19,20 Pt(II) species and azidyl radicals appear to be the main cytotoxic species produced by such photoactivated diazido Pt(IV) complexes.5 Pt(II) species can bind to DNA and azidyl radicals can attack proteins.5 The formation of Pt(II) species has been monitored by NMR, UV-vis, LC-MS, Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR) spectroscopy, and transient electronic absorption (TEA) spectroscopy.19,20 In addition, photo-substitution of one azide ligand by a H2O/OH molecule has been frequently reported as an initial step in the photoactivation of diazido Pt(IV) complexes.5,6 However, due to the sensitivity limit, accurate qualitative and quantitative analysis of Pt(II) and Pt(IV) species at nanomolar concentrations, which is important for in vivo investigations, cannot be achieved using these methods. DFT calculations can also help to elucidate possible photodecompostion pathways.
Due to its high selectivity, mass spectrometry based on characteristic mass-to-charge ratios (m/z) of analytes and their fragments is used for a wide range of applications.21–23 Inductively coupled plasma-mass spectrometry (ICP-MS) has received intense attention in the development of metal-based drugs as a standalone configuration or coupled to separation techniques, and can be applied in the determination of not only extracellular transformations of metallodrugs, but also their accumulation, distribution, and processing in cells, tissues and organs.24–27 Reverse phase high performance liquid chromatography (RP-HPLC) is a most frequently used separation technique, due to its promising capability for separating a wide range of compounds.28,29 The combination of HPLC and ICP-MS provides the potential for the accurate analysis of the products from interaction of metallodrugs with small biomolecules,30 their binding to proteins by (metallo)-proteomics,31,32 as well as the dark stability and metabolism of metallodrugs in human plasma and cells at nanomolar concentrations.26,33–35
Investigations of classical platinum drugs using HPLC-ICP-MS have been widely reported, including their stability in biofluids, interactions with biomolecules, and adducts within cells, but photoactive diazido Pt(IV) complexes have not yet been fully explored.36–45 Here, we have developed an HPLC-ICP-MS method for Pt analysis at extremely low, physiologically-relevant concentrations, and used HPLC-MS and ESI-HR-MS to identify the speciation of FM-190 in aqueous solution, phosphate-buffered saline (PBS) and human blood plasma in the dark and upon irradiation. We have also optimised the extraction methods for photoproducts of FM-190 in plasma. DFT calculations were carried out to investigate possible mechanisms of photoactivation based on the photoproducts detected by mass spectrometry. This work provides a foundation for the accurate speciation analysis of photoactive Pt(IV) complexes in body fluids, cells and tissues.
2. Results
2.1. HPLC-ICP-MS method development
To achieve reliable quantification of Pt species at biologically relevant nanomolar levels, a detector with high sensitivity and selectivity, such as ICP-MS, is required to be hyphenated to HPLC. HPLC-UV methods were explored as summarised in Table S1,† and the corresponding chromatograms are shown in Fig. S1.† Method-7 was selected as the best choice to reduce the amount of carbon entering ICP-MS, at the same time allowing FM-190 to be eluted from the column. It was then applied in all HPLC-ICP-MS experiments. Using method-7, a linear relationship between concentration and peak area was observed for FM-190 for the concentration range of 0.06 to 17.6 μM with r2 = 0.999 (Fig. S2†). However, when the concentration was lower than 60 nM, no HPLC-UV peak was visible.FM-190 (764.8 nM) prepared in water was injected onto the C18 column, and the separated Pt species sequentially detected by the ICP-MS detector. For the dark samples (Table S2†), a peak at 14.1 min was detected and assigned as FM-190 with purity of 99. 9% (Table S3†). The slight shift in retention time (0.2 min) for the HPLC-ICP-MS is caused by the difference in distance between UV and ICP-MS detectors and the HPLC column (Fig. 2a). Three Pt isotopes (194Pt, 195Pt, and 196Pt) were measured, and similar results were observed for all of them, with signals matching the abundance of these isotopes (Fig. S3†). This experiment was independently repeated twice to ensure the reproducibility of the method. If not stated otherwise, the 195Pt signal from the first repetition is used to represent the result obtained for HPLC-ICP-MS in this paper. FM-190 at lower concentrations of 45.9, 9.2, and 4.6 nM were also measured using the same method (Fig. 2b–d, Table S3†). A single main Pt-peak with the same retention time was detected for all of the samples, highlighting the high sensitivity for detection of ICP-MS, down to low nM concentrations. A linear relationship between concentrations and sum of 195Pt peak areas is observed (Fig. 2e). Notably, the two very small peaks at 4.6 (0.065%) and 8.8 (0.048%) min were only detectable when the FM-190 concentration was 764.8 nM, but not at lower concentration levels (Table S3†).
 | ||
| Fig. 2 195Pt signals of FM-190 in MilliQ water in the dark obtained by HPLC-ICP-MS at (a) 764.8; (b) 45.9; (c) 9.2; and (d) 4.6 nM; and (e) the relationship between the concentration of FM-190 and sum of peak areas of 195Pt signals. | ||
2.2. Photoactivation of FM-190 in water determined by HPLC-ICP-MS
The Pt-containing photoactivation products were also determined by HPLC-ICP-MS. However, to ensure no retention of side products on the C18 column, method-8 was adopted by adding a column washing step (Table S2†). A total of 8 Pt-containing ICP-MS peaks were detected for FM-190 (764.8 nM) in water after 1 h irradiation with blue light (463 nm, Fig. 3a and Table S4†). An approximate 92% decrease in the peak area assigned to FM-190 was observed. These species are stable in solution for 24 h in the dark at 25 °C (Fig. 3b). Notably, when the concentration of FM-190 was decreased to 45.9 and 4.6 nM, only 5 Pt species were detected after irradiation (Fig. 3c–d and Table S4†). Notably, when the concentration of FM-190 was 4.6 nM, the smallest peak detected in the irradiated sample had a concentration of ca. 0.16 nM. A linear relationship was found between the Pt concentration and the sum of all peak areas in all samples, suggesting all Pt containing species are determined, and that quantification of Pt using HPLC-ICP-MS is also reliable for photoactivated samples (Fig. 3e). | ||
| Fig. 3 195Pt signals of FM-190 in MilliQ water after irradiation with blue light (463 nm, 1 h) obtained by HPLC-ICP-MS at (a) 764.8 nM determined immediately, and (b) after samples had stood for 24 h in the dark; (c) 45.9; and (d) 4.6 nM; and (e) the relationship between concentration of FM-190 and sum of peak areas of 195Pt signals. | ||
2.3. Photoactivation of FM-190 in water investigated by LC-MS
In order to identify the species formed from FM-190 in aqueous solution upon irradiation with blue light (463 nm, 1 h, 25 °C), LC-MS was used. For LC-MS, the same methods as those used in HPLC-ICP-MS, method-7 (2% to 5% B in 10 min, maintain 5% B for 5 min) and method-8 (80% B after 18 min), were used for dark and irradiated samples, respectively, to ensure all species were detected (Table S2†). After 1 h irradiation with blue light (463 nm), 6 main species were observed for photoactivated FM-190 (31.8 μM) solutions (Fig. S4†). The retention times of these species are similar to those detected in HPLC-ICP-MS. The detection of more species by HPLC-ICP-MS compared with LC-MS suggests a lower detection limit of the HPLC-ICP-MS method. In addition, the difference in peak intensities between HPLC-ICP-MS and LC-MS arises from the varied ability of these species to form molecular ions. The peak at 13.9 min is assigned to FM-190 itself. Photoproducts are assigned as [{PtIV(py)2(OH)4} + Na]+ (m/z 443.95, a); [{PtIV(py)2(OH)3}2(μ-O2) + Na]+ (m/z 863.04, b); [{PtIV(py)2(N3)(OH)3} + Na]+ (m/z 468.95, c); [{PtIII(py)2(HCOO)(OH)2(H2O)} + Na]+ (m/z 472.94, d); and [{PtII(py)2(HCOO)3} + H]+ (m/z 488.94, e), summarised in Table S5.† Species {PtIII(py)2(OH)2}+ (m/z 386.96) was observed in all peaks, and might be formed during ionisation by release of the corresponding ligands. The isotopic pattern for platinum was observed in all species (Fig. S5†). Notably, the most abundant photoproduct (c) found by LC-MS can be assigned as [{PtIV(py)2(N3)(OH)3} + Na]+, which is a Pt(IV) species from photo-substitution rather than a Pt(II) species resulting from photoreduction. The fraction containing the main Pt(IV) species c was collected to obtain high resolution MS data, determined to be 469.0562 and matches well with calculated value of 469.0559 (Fig. 1b). These results also suggest that the photoproducts remained stable after lyophilisation. Unfortunately, attempts to purify species c were unsuccessful, perhaps due to its reactivity under the conditions used. For 1.3 μM FM-190, only two photoproducts were detected due to the low sensitivity of the LC-MS method compared with HPLC-ICP-MS (Fig. S4c and S6c†). Interestingly, the FM-190 LC-MS peak disappeared after irradiation at this low concentration (1.3 μM), while being still visible when the concentration was higher (31.8 and 6.4 μM).In order to exclude the effect of mobile phases, water and acetonitrile with 0.1% formic acid (FA) were also used as the mobile phases. This is the most common mobile phase combination used in LC-MS for the study on Pt species.5 However, mobile phase composition needs to be evaluated since there is possibility that acetonitrile can become a potential binding partner and an acidic pH can result in ligand release. Similar species with different retention times were determined for photoactivated FM-190 at concentrations of 50 μM using method-10, where acetonitrile was used instead of methanol as mobile phase B (Fig. S7, Tables S6 and S7†). This suggests that the organic mobile phase exerts no significant effects on the detection of these photoproducts.
2.4. DFT calculations
Once the feasibility of ISC processes was established, for the accurate characterization of the involved triplet excited states, their structures were optimized using the TDDFT approximation. Indeed, as singlet excited states generally have a lifetime five orders of magnitude shorter than that of triplet states (10−8vs. 10−3 s), it is reasonable to assume that the primary mechanism for the formation of Pt(II) active species involves an excited triplet state rather than a singlet state. As a consequence, the optimized structures obtained for the triplet states, served as initial points for the optimization within the unrestricted Kohn–Sham formalism (UKS),47,48 known to mitigate instabilities in excited triplet states involving charge transfer. Applying this approach, all the examined triplet excited states collapsed into four distinct states denoted as Ta, Tb, Tc, and Td. Detailed information about the obtained triplet states, namely optimized structures together with the corresponding spin density distributions, spin density values on the Pt centre and N atoms directly bonded to the metal, relative energies calculated with respect to the ground state (GS) and the main bond distances are provided in Fig. 4.
 | ||
| Fig. 4 (a) Structures of the optimised S0 ground state and Ta, Tb, Tc and Td triplet states together with relevant bond lengths (in Å) compared with corresponding GS values. (b) Plot of spin density distribution. (c) Spin density values (SD in a.u.) for Pt and nitrogen atoms directly bonded to the metal centre. (d) Relative energies calculated with respect to the GS. | ||
As is evident, Ta, Tb, and Tc triplet structures exhibit dissociative character towards both azide ligands, while the py ligands remain firmly attached to the Pt centre. However, Td triplet shows two elongated Pt–py bonds, while azides remain in the Pt coordination sphere. This particular triplet state is considered unlikely, since no pyridine release from FM-190 was observed experimentally, and thus excluded from the investigation of photoreaction. The relative energies of the excited triplet states calculated respect to the ground state follow the order Tc < Tb < Ta < Td. As the optimised geometries of the most stable Tc and Tb triplets indicate that such states are highly dissociative for the N3 ligands, with the Pt–N3 distances significantly elongated by 0.5 Å (Pt–N1) and 0.6 Å (Pt–N2) compared to the ground-state geometry, they give rise to the formation of the [PtII(OH)2(py)2] photoproduct. In the Ta triplet state, however, the Pt–N3 bonds are partially elongated and the distance increases by approximately 0.3 Å. Generally, an excited state exhibiting MC character cannot be involved in the emissive processes, but it can play a role as an active species in the release of reactive ligands.49 In this context, the nature of all the intercepted triplet states (3MC) promote ligand photodissociation. Based on these results, formation of photoproducts from FM-190 can be achieved by following two paths. First, the population of Tb and Tc states leads to the formation of the reduced form, [PtII(OH)2(py)2], of FM-190 as shown in Scheme 1. Second, the Ta triplet state could lead to the formation of the aquated forms of the complex. Experimental data show that FM-190 is inert until it is converted to active aquated forms, through the release of N3 ligands. Starting from the Ta state, all the attempts to identify minima and transition states along triplet pathways leading to the formation of mono-aquated and/or bi-aquated species failed. Therefore, it is realistic to postulate that from the complex in its excited triplet state only the reduced photoproduct [PtII(OH)2(py)2] can be generated as a result of the elimination of the two N3˙ radicals.
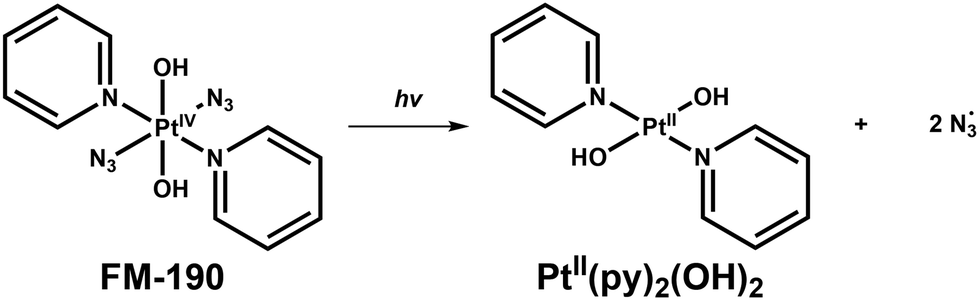 | ||
| Scheme 1 Proposed photoreduction mechanism for the formation of the Pt(II) product based on DFT calculations. | ||
On the contrary, formation of Pt(IV) products, experimentally detected by MS and obtained by substitution of anionic N3− ligands with water molecules, might take place from FM-190 in its singlet ground state, but only when overcoming high energy barriers according to calculations. Further explorations are required to completely reconcile experimental and theoretical results, although probing the possible involvement of excited singlet states will require use of more sophisticated computational strategies.
According to computational analysis, reaction intermediates [PtIV(N3)(H2O)(OH)2(py)2]+, [PtIV(H2O)(OH)3(py)2]+ and the final product [PtIV(OH)4(py)2] are generated along pathways in singlet multiplicity. The proposed mechanism leading to the formation of the intermediates and the final product is shown in Scheme 2, and the calculated energy profile for this process is shown in Fig. 5. Relative energies are calculated with respect to the reference energy of the first formed adduct between FM-190 and two water molecules set as the zero on the energy scale.
 | ||
| Scheme 2 Proposed mechanism for the formation of hydrolysed Pt(IV) products based on DFT calculations. | ||
 | ||
| Fig. 5 DFT-calculated free energy profile in water describing the hydrolysis mechanism of FM-190. Relative energies are in kcal mol−1 and calculated with respect to the ground state energy of FM-190. | ||
According to the proposed mechanism, the first hydrolysis process for the Pt complex occurs following a second-order nucleophilic substitution. One of the N3− ligands is displaced from FM-190 in favor of the coordination of a water molecule, leading to the formation of the intermediate [PtIV(N3)(H2O)(OH)2(py)2]+ that lies 9.6 kcal mol−1 above the reference energy of the corresponding first adduct. The mono-aquated Pt(IV) complex [PtIV(N3)(H2O)(OH)2(py)2]+ is formed by overcoming an energy barrier of 35.6 kcal mol−1. Starting from intermediate [PtIV(N3)(H2O)(OH)2(py)2]+, the reaction can proceed by both deprotonation of the bound water molecule and replacement of the second azide ligand by water. In the former case, overcoming a very low energy barrier, only 1.1 kcal mol−1, allows the formation of the [PtIV(N3)(OH)3(py)2] species, that is calculated to be endergonic by 8.1 kcal mol−1 with respect to reference zero energy of the first adduct is achieved.
The second N3− ligand is replaced with the same mechanism by a second molecule of water, but during the course of the reaction, the transfer of a proton from the coordinated water molecule to the N3− ligand detached in the first step occurs simultaneously. The height of the barrier that is necessary to overcome for such a rearrangement to occur is 35.2 kcal mol−1, while the second [PtIV(H2O)(OH)3(py)2]+ intermediate is destabilised with respect to the initial adduct by 16.0 kcal mol−1. Both kinetics and thermodynamics significantly favor the deprotonation reaction according to experimental findings that assign [{PtIV(py)2(N3)(OH)3}+Na]+ (c) as the main product. Finally, the Pt(IV) species [PtIV(OH)4(py)2] (a) is formed through the protonation of the N3− group present in the reaction environment by the coordinated water molecule. The formation of final product is calculated to be endergonic by 14.4 kcal mol−1 and occurs by overcoming a very low energy barrier of 1.9 kcal mol−1.
The acidity of aqua ligands coordinated to Pt was also estimated computationally. The first acidity constant was calculated for the deprotonation process of the mono-aquated Pt species, [PtIV(N3)(H2O)(OH)2(py)2]+, generated in the first step as reported in Fig. 5. Starting from the bis-aquated Pt species, [PtIV(H2O)2(OH)2(py)2]2+ the second investigated process involves the deprotonation of one water molecule coordinated to the Pt centre. As outlined previously, calculations failed to locate this species owing to the high acidity of the coordinated water molecule, which donates a proton to the released N3− ligand. Finally, the third acidity constant is relative to the deprotonation of the [PtIV(H2O)(OH)3(py)2]+ complex. The estimated values of the acidity constants obtained for the three processes illustrated above are:
 | (1) |
 | (2) |
 | (3) |
The large value of Ka for equation (2) means that the bis-aquated complex is a strong acid, and it dissociates completely as illustrated in Fig. 5, where it is shown that the formation of the [PtIV(H2O)2(OH)2(py)2]2+ species and its deprotonation occur simultaneously.
2.5. Dark stability and photoactivation in plasma and PBS
Blood plasma and PBS were also used as media for the investigation of dark stability and photoactivation of FM-190. FM-190 (7756 nM) in human plasma was incubated at 37 °C for 2 h in the dark, then transferred into a centrifugal filter unit (MWCO 5000) and centrifuged to remove proteins. The solvent was removed and the residue was redissolved in water to make a final 10× diluted solution. The major peak contains 97.7% of the total Pt eluting at ca. 14.2 min indicating the high dark stability of FM-190 in human plasma (Fig. S11a†). For comparison, FM-190 in PBS shows a similar retention time and a major peak Pt of 98.85% (Fig. S11b†). The sum of the Pt peak areas for the sample in plasma is about 95% of that in PBS, suggesting the high efficacy of this extraction method (Table S13†). Another extraction method that precipitates plasma proteins by addition of acetonitrile was also investigated. FM-190 contributes 99.4% of the total Pt (Fig. S12a†). Although slightly larger peaks areas were observed by using this method, but the difference is within 5% (Tables S13 and S14†).Photoproducts from FM-190 (7756 nM) in plasma were also monitored by HPLC-ICP-MS after 1 h incubation at 37 °C in the dark and after 1 h irradiation (463 nm) at 25 °C, and then extracted using the same method as for the dark samples (Fig. S11c–d, S12b, Tables S14 and S15†). About 91% of FM-190 undergoes photoactivation after irradiation. Compared to the photoproducts formed in water, plasma components might interact with those photoproducts, resulting in formation of many small peaks, while the number of the most intense peaks (peak area >10%) decreased to four. The extent of photoactivation in PBS is not as significant as that in plasma and the retention times of those photoproducts are different. Notably, the sum of peak areas determined for irradiated samples is about 12–36% lower than that for the corresponding dark samples.
LC-MS was carried out to identify the species formed by FM-190 after irradiation (Fig. S13–S15†). Due to the complex matrix of plasma and the relatively low sensitivity of the LC-MS method, only the [{PtIV(py)2(N3)(OH)3} + Na]+ (m/z 468.96, c) peak was observed as the photoproduct of FM-190 extracted using different protocols, even when a high concentration of 194.9 μM was used. Pt adducts might be formed with components of plasma, which are not detectable by LC-MS. For comparison, a number of smaller peaks other than m/z 468.96 were observed for samples in PBS due to its relatively simple matrix (Fig. S15†). Compared with the chromatogram obtained by HPLC-ICP-MS, there are some additional peaks in LC-MS attributable to plasma components, which form a complicated matrix.
3. Discussion
In contrast to labile square-planar d8 Pt(II) complexes, low-spin d6 Pt(IV) complexes are usually resistant to hydrolysis, but can undergo reduction in the presence of bio-reductants.8 However, some combinations of ligands (e.g. axial carboxylates) can facilitate hydrolysis.50–52 Identification and quantitative analysis of Pt metabolites in vitro and in vivo at bio-relevant concentrations is important for potential preclinical development of anticancer Pt drugs.27,35,42,43HPLC-UV does not readily identify Pt species and does not allow a quantitative analysis due to the unknown extinction coefficients of each species. LC-MS gives structural information, but is not capable of quantitation owing to the different ionisation abilities among Pt species. Importantly, both detectors are not sensitive enough to detect Pt species at nanomolar concentrations. HPLC-ICP-MS with a high accuracy and sensitivity allows analysis of Pt metabolites extracted from cells and patients. The combination of LC-MS and HPLC-ICP-MS results provides accurate identification and quantitation of Pt samples, which provides a deeper insight into the metabolism of Pt anticancer drugs within biological systems.
Pt(IV) species were determined to be the main photoproducts when the activation light wavelength was 463 nm (blue light). Azide release from FM-190 upon irradiation was observed at the same time as the replacement by hydroxido ligands, with [{PtIV(py)2(N3)(OH)3} + Na]+ (c) assigned by LC-MS as the main photoproduct (Fig. 1b). DFT calculations suggest that it is favourable to form this species as the main product from the reaction between FM-190 and water, directly in the singlet state. The large acidity constant Ka of [PtIV(H2O)2(OH)2(py)2]2+ indicates that it is a strong acid which explains why [{PtIV(py)2(N3)(OH)3} + Na]+ is the main product. Despite DFT calculations indicating that Pt(II) species are the main photoproducts formed during photoactivation, we detected Pt(IV) species that require the overcoming of high energy barriers and unfavourable thermodynamics to be formed. In view of the high dark stability of FM-190 in water, we assume that such reactions might be facilitated by the photoactivation. An alternative mechanism involving photoreduction, followed by ligand substitution on Pt(II) and re-oxidation by any strongly oxidising photoproducts formed seems less likely at low Pt concentrations.
Small amounts of Pt(II) species were also detected by LC-MS, but when the organic solvent ratio increased to 80%, they did not enter the ICP-MS detector. However, according to the difference between the total Pt peak areas of ICP-MS in dark and irradiated samples, these Pt(II) species make only an insignificant contribution to the total Pt after irradiation. The sum of 195Pt peak areas in irradiated samples is even slightly higher (<10%) than that of the corresponding dark samples, which might be caused due to the slight difference in the detection sensitivity of ICP-MS detector for different Pt species. In contrast to irradiation at 463 nm, in the photoactivation of FM-190 by indigo light (420 nm), Pt(II) species were determined to be the main photoproducts.20 These results show that the products from photoactivation of FM-190 with blue light (463 nm) gives more initial products than final products. DFT results also suggest the dissociative character of both azide ligands that can lead to the formation of the photoreduced Pt(II) species, [PtII(OH)2(py)2]. Therefore, photo-substituted Pt(IV) species can be assigned as the initial intermediates from the photoactivation of FM-190 before photoreduction occurs. These are in agreement with a recent study on photo-substitution of FM-190 as the first step of photoactivation using steady-state and laser flash photolysis.53 However, no detailed structural information for photoproducts was reported using those methods. These results also indicate the reason why +4 dominates the oxidization state of Pt in cellular accumulated FM-190 in PC3 cancer cells after irradiation with blue light (465 nm).54
Using methanol rather than acetonitrile as the organic mobile phase did not affect the determination of Pt photoproducts, except for the retention time. Due to the different set-ups of the LC-MS and HPLC-ICP-MS, a small shift in retention times was observed. The parent compound FM-190 served as a reference signal. The retention time of FM-190 determined by UV, MS and ICP-MS under exactly the same conditions (method-7) was 13.9, 14.4 and 14.1 min, respectively (Fig. 6). In theory, a −0.3 min difference between MS and ICP-MS signals can be applied when assigning the Pt peaks detected by ICP. However, due to the different modes of detection, species b–e have a longer retention time in ICP-MS.
 | ||
| Fig. 6 (a) LC UV (detection wavelength 254 nm), MS and ICP-MS (195Pt) signals of FM-190 in MilliQ water obtained using method-7 for dark samples and method-8 for photoactivated samples (463 nm, 1 h). Species a–e are assigned in Table S5 (ESI†). (b) Ratio of Pt species with different retention times in irradiated samples determined using different detectors (assumes all species have same extinction coefficient and ionisation ability). | ||
The three intense sharp ICP-MS Pt peaks a, c and d can be assigned as [{PtIV(py)2(OH)4} + Na]+ (m/z 443.58, a); [{PtIV(py)2(N3)(OH)3} + Na]+ (m/z 468.58, c); and [{PtIII(py)2(HCOO)(OH)2(H2O)} + Na]+ (m/z 472.57, d). Interestingly, only peak c shows an intense absorbance at 254 nm, attributable to its Pt–N3 ligand-to-metal charge-transfer transition. In addition, the broad Pt peak b detected by ICP-MS gives very weak UV and MS signals. According to its MS isotopic pattern, this peak can be assigned to a dinuclear Pt species [{PtIV(py)2(OH)3}2(μ-O2) + Na]+ (m/z 863.04, b), which might explain its intense ICP signal. Due to the high sensitivity of ICP-MS detection mode, analysis of Pt photoproducts at biologically relevant concentrations (low nM levels) only becomes feasible with LC-MS analysis using a higher FM-190 concentration (double-digit μM). Based on these results, a potential photoactivation pathway for FM-190 in aqueous solution with visible light irradiation can be proposed, as shown in Fig. S16.†
An interesting observation is the effect of the concentration of the compound on the number of peaks detected, not only by ICP-MS, but also UV and MS. At low concentrations, photoactivation of FM-190 is less likely to lead to intermolecular interactions in the formation of products compared to high concentrations. Another reason for the missing Pt peaks is their low concentrations, below the detection limit of the method.
The extraction of Pt drugs and their photo-products from biofluids, such as blood plasma, is an essential step in drug development. Two methods, centrifugal filter unit (MWCO 5000) centrifugation and acetonitrile precipitation, were employed to precipitate proteins in plasma to give Pt-containing solutions suitable for HPLC separation. Both methods gave a high recovery for Pt species, with a difference <5% compared to the same samples in PBS. Notably, the sum of peak areas determined for irradiated samples is about 12–36% lower than that for the corresponding dark samples. No Pt species were detectable in plasma samples when the organic mobile phase was increased to 80% for LC-MS. These results indicate that adducts between photoproducts and plasma are either insoluble in water or have a molecular weight >5000 Da.42 Considering the complex matrix of low and high molecular-weight components in plasma, a range of species was detected by LC-MS which complicated the analysis. This problem was simplified using HPLC-ICP-MS, which gives signals only for Pt species.
4. Conclusions
Photoactive Pt(IV) complexes are promising next-generation platinum anticancer drugs due to their high photo-selectivity and novel mechanisms of action. Investigations of photoproducts from diazido Pt(IV) complexes in cells and tissues plays an important role in the exploration of their mechanism of action. In this work, an analytical speciation approach for FM-190 and its photoproducts has been developed using HR-MS, LC-MS and HPLC-ICP-MS. LC-MS and HR-MS were employed to identify these photoproducts. Interestingly [{PtIV(py)2(N3)(OH)3} + Na]+ and [{PtIV(py)2(OH)4} + Na]+ resulting from the replacement of azide by hydroxide were detected by MS and characterised, with [{PtIV(py)2(N3)(OH)3} + Na]+ as the most abundant photoproduct.Our findings highlight the need to investigate further the chemical and biological activity of tri- and tetra-hydroxido Pt(IV) complexes, and their possible role in the photocytotoxicity, where very little knowledge is currently available. The DFT calculations estimate that the first pKa values for these complexes are close to the pH values in the cell cytoplasm and various organelles (ca. 5.6–7.4). It might be expected that a coordinated aqua ligand will be much more reactive towards substitution than a hydroxido ligand. Also axial hydroxido ligands on Pt(IV) tend to stabilise Pt(IV) towards reduction, although the introduction of cis-hydroxido ligands may give rise to new reaction pathways. Studies of the electrochemical properties of such complexes might help to elucidate this.
It will also be interesting to investigate possible adducts of these hydroxide complexes with biological macromolecules such as proteins and oligonucleotides. Depending on the specific geometrical arrangement of the hydroxido ligands (which also needs to be investigated), these tri-and tetra-hydroxido Pt(IV) complexes offer strong H-bond donor and acceptor sites.
By combining the results from LC-MS and HPLC-ICP-MS, species at nanomolar concentration can be separated and assigned to analyse potential photodecomposition pathways for Pt(IV) complexes, which is essential for elucidation of the mechanism of photoactivation for these prodrugs. Both ultrafiltration and protein precipitation methods proved useful for analysis of low molecular-weight Pt species arising from FM-190.
FM-190 displays extremely high stability in human plasma in the dark at 37 °C, with promising ability to undergo photoactivation when exposed to blue light (463 nm). Work is in progress on other related diazido Pt(IV) complexes in this family which can be activated by longer wavelengths of light and penetrate more deeply into tissues. The high separation power of HPLC combined with the high detection capability of ICP-MS provide an important strategy for the speciation analysis of photoactive Pt(IV) complexes and their photoproducts with biomolecules in cells and tissues, such analysis is important for the investigation of the stability and metabolism of platinum drugs in vivo. In future work HPLC-ICP-MS methods which allow higher ratios of organic solvent will be developed to allow exploration of lipophilic photoactive Pt(IV) complexes, for which FM-190 can be used as an internal standard.
Author contributions
H. S., C. W.-D., P. J. S. and H. G.-I. designed the experiments, H. S. and C. W.-D. carried out HPLC-ICP-MS experiments, H. S. also carried out LC-MS and HR-MS experiments. F. P. and E. S. carried out DFT and TDDFT calculations. All authors drafted the script and contributed to the final submission.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC Grant No. EP/P030572/1), Anglo-American Platinum (fellowship for H. S.), the Italian Association for Cancer Research, AIRC (ID 25578 for F. P.), University of Calabria, and Department of Science Innovation and Technology (DSIT) for funding. We thank Dr Lijiang Song for assistance with HR-MS.References
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- C. Zhang, C. Xu, X. Gao and Q. Yao, Theranostics, 2022, 12, 2115–2132 CrossRef CAS PubMed.
- S. Rottenberg, C. Disler and P. Perego, Nat. Rev. Cancer, 2021, 21, 37–50 CrossRef CAS PubMed.
- K. Peng, B. B. Liang, W. Liu and Z. W. Mao, Coord. Chem. Rev., 2021, 449, 214210 CrossRef CAS.
- H. Shi, C. Imberti and P. J. Sadler, Inorg. Chem. Front., 2019, 6, 1623–1638 RSC.
- H. Shi and P. J. Sadler, Adv. Inorg. Chem., 2022, 80, 95–127 CrossRef CAS.
- L. Qi, Q. Luo, Y. Zhang, F. Jia, Y. Zhao and F. Wang, Chem. Res. Toxicol., 2019, 32, 1469–1486 Search PubMed.
- Z. Xu, Z. Wang, Z. Deng and G. Zhu, Coord. Chem. Rev., 2021, 442, 213991 CrossRef CAS.
- Z. Deng and G. Zhu, Curr. Opin. Chem. Biol., 2023, 74, 102303 CrossRef CAS PubMed.
- J. Gurruchaga-Pereda, Á. Martínez, A. Terenzi and L. Salassa, Inorg. Chim. Acta, 2019, 495, 118981 CrossRef CAS.
- J. Huang, W. Ding, X. Zhu, B. Li, F. Zeng, K. Wu, X. Wu and F. Wang, Front. Chem., 2022, 10, 876410 CrossRef CAS PubMed.
- M. Imran, W. Ayub, I. S. Butler and Zia-ur-Rehman, Coord. Chem. Rev., 2018, 376, 405–429 CrossRef CAS.
- D. Spector, K. Pavlov, E. Beloglazkina and O. Krasnovskaya, Int. J. Mol. Sci., 2022, 23, 14511 CrossRef CAS PubMed.
- P. Müller, B. Schröder, J. A. Parkinson, N. A. Kratochwil, R. A. Coxall, A. Parkin, S. Parsons and P. J. Sadler, Angew. Chem., Int. Ed., 2003, 42, 335–339 CrossRef PubMed.
- F. S. Mackay, J. A. Woods, H. Moseley, J. Ferguson, A. Dawson, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2006, 12, 3155–3161 CrossRef CAS PubMed.
- L. Ronconi and P. J. Sadler, Dalton Trans., 2011, 40, 262–268 RSC.
- L. Ronconi, A. M. Pizarro, R. J. McQuitty and P. J. Sadler, Chem. – Eur. J., 2011, 17, 12051–12058 CrossRef CAS PubMed.
- F. S. Mackay, J. A. Woods, P. Heringova, J. Kašpárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. MacKay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef CAS PubMed.
- R. R. Vernooij, T. Joshi, M. D. Horbury, B. Graham, E. I. Izgorodina, V. G. Stavros, P. J. Sadler, L. Spiccia and B. R. Wood, Chem. – Eur. J., 2018, 24, 5790–5803 CrossRef CAS PubMed.
- N. S. Ha, M. de Raad, L. Z. Han, A. Golini, C. J. Petzold and T. R. Northen, RSC Chem. Biol., 2021, 2, 1331–1351 RSC.
- M. Kandiah and P. L. Urban, Chem. Soc. Rev., 2013, 42, 5299–5322 RSC.
- X. Huang, H. Liu, D. Lu, Y. Lin, J. Liu, Q. Liu, Z. Nie and G. Jiang, Chem. Soc. Rev., 2021, 50, 5243–5280 RSC.
- J. Feldmann, A. Raab and E. M. Krupp, Anal. Bioanal. Chem., 2018, 410, 661–667 CrossRef CAS PubMed.
- S. C. Wilschefski and M. R. Baxter, Clin. Biochem. Rev., 2019, 40, 115–133 Search PubMed.
- A. R. Timerbaev, J. Anal. At. Spectrom., 2021, 36, 254–266 RSC.
- M. Patriarca, N. Barlow, A. Cross, S. Hill, A. Robson, A. Taylor and J. Tyson, J. Anal. At. Spectrom., 2022, 37, 410–473 RSC.
- J. G. Dorsey and K. A. Dill, Chem. Rev., 1989, 89, 331–346 CrossRef CAS.
- P. Žuvela, M. Skoczylas, J. J. Liu, T. Bączek, R. Kaliszan, M. W. Wong and B. Buszewski, Chem. Rev., 2019, 119, 3674–3729 CrossRef PubMed.
- H. R. Hansen and S. A. Pergantis, J. Anal. At. Spectrom., 2006, 21, 1240–1248 RSC.
- X. Yan, Y. Zhou, H. Li, G. Jiang and H. Sun, Ref. Modul. Chem. Mol. Sci. Chem. Eng., 2023, 53–76 Search PubMed.
- Y. Zhou, H. Li and H. Sun, Annu. Rev. Biochem., 2022, 91, 449–473 CrossRef CAS PubMed.
- M. Montes-Bayón, K. DeNicola and J. A. Caruso, J. Chromatogr. A, 2003, 1000, 457–476 CrossRef PubMed.
- D. P. Bishop, D. J. Hare, D. Clases and P. A. Doble, Trends Anal. Chem., 2018, 104, 11–21 CrossRef CAS.
- A. R. Timerbaev, J. Anal. At. Spectrom., 2014, 29, 1058–1072 RSC.
- L. H. Møller, C. S. Jensen, T. T. T. N. Nguyen, S. Stürup and B. Gammelgaard, J. Anal. At. Spectrom., 2015, 30, 277–284 RSC.
- G. Hermann, P. Heffeter, T. Falta, W. Berger, S. Hann and G. Koellensperger, Metallomics, 2013, 5, 636–647 CrossRef CAS PubMed.
- Z. Yao, B. Li, C. Li, B. Wang, M. Zhao and Z. Ma, J. Anal. At. Spectrom., 2022, 37, 1652–1657 RSC.
- J. Wang, J. Tao, S. Jia, M. Wang, H. Jiang and Z. Du, Pharmaceuticals, 2021, 14, 104 CrossRef CAS PubMed.
- G. A. Zachariadis and O. E. Misopoulou, Anal. Lett., 2018, 51, 1060–1070 CrossRef CAS.
- L. Galvez, M. Rusz, M. A. Jakupec and G. Koellensperger, J. Anal. At. Spectrom., 2019, 34, 1279–1286 RSC.
- R. Larios, M. E. Del Castillo Busto, D. Garcia-Sar, C. Ward-Deitrich and H. Goenaga-Infante, J. Anal. At. Spectrom., 2019, 34, 729–740 RSC.
- S. Cuello-Nuñez, R. Larios, C. Deitrich, T. Lekishvili, V. Nischwitz, B. L. Sharp and H. Goenaga-Infante, J. Anal. At. Spectrom., 2017, 32, 1320–1330 RSC.
- A. Martinčič, M. Cemazar, G. Sersa, V. Kovač, R. Milačič and J. Ščančar, Talanta, 2013, 116, 141–148 CrossRef PubMed.
- B. Michalke, J. Trace Elem. Med. Biol., 2010, 24, 69–77 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- D. Escudero, E. Heuser, R. J. Meier, M. Schäferling, W. Thiel and E. Holder, Chem. – Eur. J., 2013, 19, 15639–15644 CrossRef CAS PubMed.
- D. Escudero and W. Thiel, J. Chem. Phys., 2014, 140, 194105 CrossRef PubMed.
- Y. Zhang, Z. Guo and X. Z. You, J. Am. Chem. Soc., 2001, 123, 9378–9387 CrossRef CAS PubMed.
- A. Kastner, I. Poetsch, J. Mayr, J. V. Burda, A. Roller, P. Heffeter, B. K. Keppler and C. R. Kowol, Angew. Chem., Int. Ed., 2019, 58, 7464–7469 CrossRef CAS PubMed.
- I. Ritacco, G. Mazzone, N. Russo and E. Sicilia, Inorg. Chem., 2016, 55, 1580–1586 CrossRef CAS PubMed.
- J. Zhao, Z. Xu, J. Lin and S. Gou, Inorg. Chem., 2017, 56, 9851–9859 CrossRef CAS PubMed.
- G. I. Zhdankin, V. P. Grivin, V. F. Plyusnin, P. A. Tkachenko, D. B. Vasilchenko and E. M. Glebov, Mendeleev Commun., 2023, 33, 61–63 CrossRef CAS.
- E. M. Bolitho, C. Sanchez-Cano, H. Shi, P. D. Quinn, M. Harkiolaki, C. Imberti and P. J. Sadler, J. Am. Chem. Soc., 2021, 143, 20224–20240 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01587h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |