
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of mono- and dinuclear thiosemicarbazone platinacycles in the proliferation of a colorectal carcinoma cell line†
Francisco
Reigosa-Chamorro‡
a,
Sandra
Cordeiro‡
bc,
M. Teresa
Pereira
a,
Beatriz
Filipe
 bc,
Pedro V.
Baptista
bc,
Alexandra R.
Fernandes
*bc and
José M.
Vila
*a
bc,
Pedro V.
Baptista
bc,
Alexandra R.
Fernandes
*bc and
José M.
Vila
*a
aDepartamento de Química Inorgánica, Universidade de Santiago de Compostela, Avenida das Ciencias s/n, 15782 Santiago de Compostela, Spain. E-mail: josemanuel.vila@usc.es
bAssociate Laboratory i4HB – Institute for Health and Bioeconomy, NOVA School of Science and Technology, NOVA University Lisbon, 2819-516 Caparica, Portugal. E-mail: ma.fernandes@fct.unl.pt
cUCIBIO, Departamento de Ciências da Vida, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Portugal
First published on 23rd August 2024
Abstract
Herein, we describe the synthesis and characterization of a series of thiosemicarbazone platinacycles. Their activity towards HCT116 and A2780 cancer cell lines as well as normal fibroblasts was explored and conclusions about the influence of their structures were drawn based on the results. Ligands L1–3, tetranuclear compounds [Pt(L1–3)]4, [Pt(L1–3)(PPh3)], and [Pt(L1–L3)2{Ph2P(CH2)4PPh2}], and phosphine derivatives, were deemed unpromising owing to their lack of activity. However, mono-coordinated diphosphine complexes [Pt(L1–L3)(Ph2PCH2PPh2-P)] showed high selectivity and low IC50 values, and their antiproliferative activity was further studied. The three studied derivatives 3a, 3b and 3c showed a fast internalization of HCT116 colorectal cancer cells with similar IC50 values, which induced a depolarization of mitochondrial membrane potential, with the subsequent triggering of apoptosis and autophagy in the case of 3c. In the case of compounds 3a and 3b, cell death mechanisms (extrinsic and intrinsic apoptosis, respectively) were triggered via the induction of reactive oxygen species (ROS). The three compounds were not toxic to a chicken embryo in vivo (after 48 h), and, importantly, showed an anti-angiogenic potential after exposure to the IC50 of compounds 3a, 3b and 3c.
Introduction
Since the seminal paper by Cope and Siekman1 describing cyclometallated compounds, i.e., metallacycles, achieving the activation of aromatic C–H bonds using transition metals was published, the chemistry of such species has attracted much attention. Although many transition metals can potentially be applied to achieve corresponding metallacycles, the primary ones include palladium and platinum. This is a result of the numerous applications of these two transition metals in reactivity,2–4 metallomesogens,5,6 and synthetic chemistry,7,8 where they are used in the functionalization of aromatic carbons through insertion reactions.9,10 Palladium and platinum have also found application in catalysis after the discovery of phosphine palladacycles by Herrmann et al.11–13 Furthermore, being of particular note are the Suzuki14–19 and Mizoroki–Heck cross-coupling reactions.19,20 One application that has expanded the most for obvious reasons is their usage as antineoplastic substances, where the functionality of metallacycles is mainly marked by palladium or platinum.21–26 The choice of the ligand in the synthesis of new compounds is of great importance, especially when bioactive applications are to be considered, since their properties will depend on the final structure of the compound. Thiosemicarbazone ligands are typical ligands in cyclometallation chemistry owing to the ease of their preparation via a simple condensation reaction, which is also compatible with a number of functional groups. Their selection is enhanced because they are bioactive on their own27 as well as when they are combined with the metals of coordination compounds and cyclometallated species.28 Their effects as antiplasmodic agents were studied for years, but research interests and new findings have shifted their application to other diseases such as cancer.29 We have reported that in thiosemicarbazone metallacycles, the organic ligand is able to bind to a metal centre as tridentate [C,N,S] in a tetranuclear30 structure through M–Schelating and M–Sbridging bonds. This favours the addition of an appropriate leaving group on the metal in the resulting mononuclear compound, holding fast the thiosemicarbazone/metal moiety. This safeguards metallacycle structural integrity during the transport in biological fluids until it reaches cancer cells, securing its biological/drug activity. This also allows to modify the characteristics of the compounds with diverse ancillary ligands, which has proved to be vital in the proficiency of the parent compound in catalytic reactions, spectroscopic and emissive chemiluminescence and antiproliferative effect. In this sense, phosphine ligands can be a great way to add variability to the resulting structures by using different ligands and changing the reaction conditions. Our previous experience with cyclometallated compounds tested the potential of mononuclear and dinuclear species as anticancer agents. The results allowed us to conclude that the inclusion of a second metal via palladacycle metallo-ligands bearing a monocoordinated diphosphine did not produce a noticeable improvement, and that the combination of the boronic acid function and dppm showed great potential. Nevertheless, the enhanced effect could not be attributed to one factor alone. Furthermore, it was unclear if all types of boronic acid derivatives, regardless of the ancillary ligands, could be as effective.26 The results presented here confirm the effectiveness of the platinum analogues, and point to the dppm ligand as a key structural moiety in the biological effect of these species. In the present work, we aimed to expand on the knowledge of the family of thiosemicarbazone platinacycles by adjustment of the imine groups and the ancillary ligands in a series of compounds derived from acetylphenylboronic acid to determine the effect of this tuning in their bioactivity as anticancer drugs.Experimental section
General procedures
All solvents were used without any previous purification. All chemicals were of reagent grade. The phosphines PPh3 (triphenylphosphine), PPh2(CH2)PPh2 [bis(diphenylphosphino)methane, dppm] and PPh2(CH2)4PPh2 [bis(diphenylphosphino)-butane, dppb] were purchased from Sigma-Aldrich. Elemental analyses were performed in a THERMO FINNIGAN, model FLASH 1112. IR spectra were acquired with a JASCO FT/IR-4600 spectrometer equipped with an ATR, model ATR-PRO ONE. The NMR spectra were acquired on Varian INOVA 400 or Bruker DPX-250 spectrometers, using the solvent signal (CDCl3, δ1H = 7.26, DMSO-d6, δ1H = 2.50; acetone-d6, δ1H = 2.05), or external H3PO4 (85%), as appropriate. Coupling constants are reported in Hz.Synthesis of the ligands
L1 (a) 4-Acetylphenylboronic acid (500 mg, 3.05 mmol) was added to 3-thiosemicarbazide (277.92 mg, 3.05 mmol), hydrochloric acid (35%, 0.65 cm3) and water (40 cm3), resulting in a clear solution that was stirred at room temperature (RT) for 3 h. The formed white solid powder was filtered off, washed with cold water, and dried in vacuo. White solid. Yield: 650.7 mg, 90%. Anal. Found: 45.8; H, 5.2; N, 17.7; S, 13.5%; C9H12BN3O2S (237.08 g mol−1) requires C, 45.6; H, 5.1; N, 17.7; S, 13.5%. IR cm−1ν(O–H) 3458; ν(N–H) 3272, 3330; ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) 1596; ν(B–O) 1352; ν(CS) 813. 1H NMR (400 MHz, DMSO-d6, δ): 10.24 (s, 1H, NNH), 8.31 (s, 1H, NH2), 8.14 (s, 2H, B(OH)2), 7.96 (s, 1H, NH2), 7.89 (vd, N = 7.8 Hz, 2H, H2/H6), 7.78 (v, N = 7.8 Hz, 2H, H3/H5), 2.29 (s, 3H, MeCN).
N) 1596; ν(B–O) 1352; ν(CS) 813. 1H NMR (400 MHz, DMSO-d6, δ): 10.24 (s, 1H, NNH), 8.31 (s, 1H, NH2), 8.14 (s, 2H, B(OH)2), 7.96 (s, 1H, NH2), 7.89 (vd, N = 7.8 Hz, 2H, H2/H6), 7.78 (v, N = 7.8 Hz, 2H, H3/H5), 2.29 (s, 3H, MeCN).
L2 (b) was prepared similarly from 3.05 mmol of acetylphenylboronic acid and methylthiosemicarbazone. White solid. Yield: 719.8 mg, 94%. Anal. Found: C, 47.6; H, 5.5; N, 16.8; S, 12.8%; C10H14BN3O2S (251.11 g mol−1) requires C, 47.8; H, 5.6; N, 16.7; S, 12.8%. IR cm−1ν(O–H) 3500; ν(N–H) 3314, 3341; ν(CN) 1595; ν(B–O) 1363; ν(CS) 831. 1H NMR (400 MHz, DMSO-d6, δ): 10.26 (s, 1H, NNH), 8.49 (br, 1H, NHMe), 8.17 (s, 2H, B(OH)2), 7.90 (d, N = 7.6 Hz, 2H, H2/H6), 7.80 (vd, N = 7.8 Hz, 2H, H3/H5), 3.03 (d, 3J = 4.0 Hz, 3H, NHMe), 2.29 (s, 3H, MeCN).
L3 (c) was made from 3.05 mmol of acetylphenylboronic acid and methylthiosemicarbazone following the same procedure. White solid. Yield: 768.1 mg, 95%. Anal. Found: C, 50.0; H, 6.1; N, 16.0; S, 12.0%; C11H16BN3O2S (265.14 g mol−1) requires C, 49.8; H, 6.1; N, 15.9; S, 12.1%. IR cm−1ν(O–H) 3450; ν(N–H) 3317, 3321; ν(CN) 1592; ν(B–O) 1357; ν(CS) 819. 1H NMR (400 MHz, DMSO-d6, δ): 10.16 (s, 1H, NNH), 8.55 (br, 1H, NHEt), 8.13 (s, 2H, B(OH)2), 7.88 (vd, N = 7.8 Hz, 2H, H2/H6), 7.80 (vd, N = 7.8 Hz, 2H, H3/H5), 3.62 (dq, 3J = 7.0 Hz, 2H, CH2), 2.28 (s, 3H, MeCN), 1.14 (t, 3J = 7.0 Hz, 3H, Me).
[PtL1]
4
(
1a
). L1 (50 mg, 0.21 mmol) was added to a suspension of potassium tetrachloroplatinate (0.18 mmol, 1 eq.) in a mixture of ethanol (20 mL) and water (0.5 mL). The mixture was stirred at 55 °C for 48 h. Ethanol was removed under reduced pressure and the resulting orange solid was washed with water, centrifuged, and dried in vacuo. Yield: 84.9 mg, 92%. Anal. Found: C, 24.8; H, 2.2; N, 9.6; S, 7.6%; (C9H10BN3O2PtS)4 (1720.6 g mol−1) requires C, 25.1; H, 2.3; N, 9.8; S, 7.5%. IR cm−1ν(O–H) 3448; ν(N–H) 3289, 3162; ν(CN) 1577; ν(B–O) 1317. 1H NMR (400 MHz, DMSO-d6, δ): 7.83 (s, 1H, H5), 7.16 (d, 3J = 7.6 Hz, 1H, H3), 6.60 (s, 2H, NH2), 6.41 (d, 3J = 7.6 Hz, 1H, H2), 1.88 (s, 3H, MeCN).
[PtL2]
4
(1b) was prepared from L2 (50 mg, 0.20 mmol) and potassium tetrachloroplatinate (75 mg, 0.18 mmol) following the same procedure. Yield: 72.5 mg, 82%. Anal. Found: C, 27.0; H, 2.5; N, 9.3; S, 7.5%; (C10H12BN3O2PtS)4 (1776.72 g mol−1) requires C, 27.1; H, 2.7; N, 9.5; S, 7.2%. IR cm−1ν(O–H) 3472; ν(N–H) 3305, 3210; ν(CN) 1582; ν(B–O) 1351. 1H NMR (400 MHz, DMSO-d6, δ): 7.93 (s, 1H, H5), 7.58 (s, 2H, B(OH)2), 7.36 (d, 3J = 7.6 Hz, 1H, H3), 6.70 (d, 3J = 7.6 Hz, 1H, H2), 6.49 (s, 1H, NHMe), 2.93 (d, 3J = 4.7 Hz, 3H, NHMe), 1.51 (s, 3H, MeCN).
[PtL3]
4
(1c) was prepared employing L3 (53 mg, 0.20 mmol) and potassium tetrachloroplatinate (75 mg, 0.18 mmol) as starting materials. Yield: 69.1 mg, 80%. Anal. Found: C, 28.9; H, 3.1; N, 9.4; S, 7.2%; (C11H14BN3O2PtS)4 (1832.84 g mol−1) requires C, 28.8; H, 3.1; N, 9.2; S, 7.0%. IR cm−1ν(O–H) 3419; ν(N–H) 3244, 2972; ν(CN) 1575; ν(B–O) 1320. 1H NMR (400 MHz, DMSO-d6, δ): 7.92 (s, 1H, H5), 7.35 (d, 3J = 7.6 Hz, 1H, H3), 6.68 (d, 3J = 7.6 Hz, 1H, H2), 6.29 (s, 1H, NHEt), 1.54 (s, 3H, MeCN), 1.11 (t, 3J = 7.2 Hz, 3H, Me).
[PtL1(PPh
3
)] (
2a
). Compound 1a (50 mg, 0.029 mmol) and triphenylphosphine (30.5 mg, 0.11 mmol, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) were added in a carrousel reaction flask fitted with a stirring rod, and vacuum/argon cycles were performed; then, deoxygenated acetone was added through a syringe. This mixture was stirred for 6 h to yield a clear solution. After solvent removal, the resulting solid was triturated with dichloromethane/n-hexane and centrifuged. The coral solid was then dried under vacuum. Yield: 63.6 mg, 79%. Anal. Found: 46.6; H, 3.6; N, 6.2; S, 4.8%; C27H25BN3O2PPtS (692.44 g mol−1) requires C, 46.8; H, 3.7; N, 6.1; S, 4.6%. IR cm−1ν(O–H) 3452; ν(N–H) 3049; ν(CN) 1574; ν(B–O) 1328. 1H NMR (400 MHz, acetone-d6, δ): δ 7.71–7.64 (m, 6H, o-PPh3), 7.49–7.39 (m, 11H, PPh3), 7.31 (dd, J = 7.5, 1.2 Hz, 1H, H3), 6.98 (d, J = 7.6 Hz, 1H, H2), 6.96 (q, J = 1.8 Hz, 1H, H5), 6.05 (s, 2H, NH2), 2.43 (s, 3H, MeCN). 31P–{1H} NMR (400 MHz, acetone-d6) δ 23.91, 1J(PtP) = 3892.86 Hz.
:4) were added in a carrousel reaction flask fitted with a stirring rod, and vacuum/argon cycles were performed; then, deoxygenated acetone was added through a syringe. This mixture was stirred for 6 h to yield a clear solution. After solvent removal, the resulting solid was triturated with dichloromethane/n-hexane and centrifuged. The coral solid was then dried under vacuum. Yield: 63.6 mg, 79%. Anal. Found: 46.6; H, 3.6; N, 6.2; S, 4.8%; C27H25BN3O2PPtS (692.44 g mol−1) requires C, 46.8; H, 3.7; N, 6.1; S, 4.6%. IR cm−1ν(O–H) 3452; ν(N–H) 3049; ν(CN) 1574; ν(B–O) 1328. 1H NMR (400 MHz, acetone-d6, δ): δ 7.71–7.64 (m, 6H, o-PPh3), 7.49–7.39 (m, 11H, PPh3), 7.31 (dd, J = 7.5, 1.2 Hz, 1H, H3), 6.98 (d, J = 7.6 Hz, 1H, H2), 6.96 (q, J = 1.8 Hz, 1H, H5), 6.05 (s, 2H, NH2), 2.43 (s, 3H, MeCN). 31P–{1H} NMR (400 MHz, acetone-d6) δ 23.91, 1J(PtP) = 3892.86 Hz.
The remaining phosphine and/or diphosphine derivatives were prepared similarly.
[PtL2(PPh
3
)] (2b) was prepared from 0.1 mmol of triphenylphosphine and 0.025 mmol of 1b. Yield: 56.5 mg, 71%. Anal. Found: C, 47.4; H, 3.8; N, 6.0; S, 4.4%; C28H27BN3O2PPtS (706.47 g mol−1) requires C, 47.6; H, 3.8; N, 5.9; S, 4.5%. IR cm−1ν(O–H) 3412; ν(N–H) 3324; ν(CN) 1574; ν(B–O) 1335. 1H NMR (400 MHz, acetone-d6, δ): 7.75–7.63 (m, 6H, PPh3), 7.47–7.35 (m, 9H, PPh3), 7.30 (d, 3J = 7.5 Hz, 1H, H3), 6.98 (d, 3J = 8.0 Hz, 1H, H2), 6.94 (d, 4J = 1.6 Hz, 1H, H5), 6.04 (s, 1H, NHMe), 2.96 (d, 3J = 4.5 Hz, 3H, NHMe), 2.42 (s, 3H, Me). 31P–{1H} NMR (400 MHz, acetone-d6) δ 23.91 1J(PtP) = 3888 Hz.
[PtL3(PPh
3
)] (2c) was prepared from 0.1 mmol of triphenylphosphine and 0.025 mmol of 1c. Yield: 62.9 mg, 80%. Anal. Found: C, 48.1; H, 4.0; N, 5.9, S, 4.6%; C29H29BN3O2PPtS (720.50 g mol−1) requires C, 48.3; H, 4.1; N, 5.8, S, 4.5%. IR cm−1ν(O–H) 3434; ν(N–H) 3052; ν(CN) 1574; ν(B–O) 1330. 1H NMR (400 MHz, acetone-d6, δ): 7.67 (dd, 3J(PH) = 11.3 Hz, 3J = 7.7 Hz, 6H, o-PPh3), 7.45 (dd, J = 8.2, 5.2 Hz, 9H), 7.31 (d, 3J = 7.6 Hz, 1H, H3), 6.99 (d, 3J = 8.3 Hz, 1H, H2), 6.96 (s, 1H, H5), 6.11 (s, 1H, NHEt), 3.40 (p, 3J = 6.9 Hz, 2H, CH2), 2.44 (s, 3H, Me), 1.17 (td, 3J = 7.1, 2.3 Hz, 3H, Me). 31P–{1H} NMR (400 MHz, acetone-d6) δ 23.83 1J(PtP) = 3912.3 Hz.
[PtL1(dppm-P)] (3a) was prepared from 0.2 mmol of dppm and 0.05 mmol of 1a. Yield: 101.3 mg, 57% Anal. Found: C, 49.8; H, 3.7; N, 5.1; S, 3.8%; C34H32BN3O2P2PtS (814.55 g mol−1) requires C, 50.1; H, 3.9; N, 5.2; S, 3.9. IR cm−1ν(O–H) 3444; ν(N–H) 3051; ν(N–H) 1575; ν(B–O) 1330. 1H NMR (400 MHz, acetone-d6, δ): 7.90–7.82 (m, 4H, PPh2), 7.42–7.33 (m, 6H, PPh2), 7.33–7.27 (m, 4H, PPh2), 7.21–7.13 (m, 5H, PPh2), 7.03 (d, J = 1.6 Hz, 1H, H5), 6.89 (d, 3J = 7.6 Hz, 1H, H2), 6.28 (s, 2H, B(OH)2), 6.08 (s, 2H, NH2), 3.72–3.51 (m, 2H, PCH2P), 2.37 (s, 3H, MeCN). 31P–{1H} NMR (400 MHz, acetone-d6) δ 12.54 (d, 2J(PP) = 78.9 Hz, 3J(PtP) = 3863.70 Hz), −23.51 (d, 2J(PP) = 79.0 Hz, 3J(PtP) = 77.76 Hz).
[PtL2(dppm-P)] (3b) was prepared from 0.2 mmol of dppm and 0.05 mmol of 1b. Yield: 88.8 mg, 62%. Anal. Found: C, 50.6; H, 4.1; N, 5.0; S, 3.8%; C35H34BN3O2P2PtS (828.58 g mol−1) requires C, 50.7; H, 4.1; N, 5.1; S, 3.9%. IR cm−1ν(O–H) 3391; ν(N–H) 3051; ν(CN) 1574; ν(B–O) 1331. 1H NMR (400 MHz, acetone-d6, δ): 7.86 (t, J = 9.7 Hz, 4H, PPh2), 7.33 (q, J = 7.3 Hz, 10H, PPh2), 7.18 (p, J = 7.7 Hz, 6H, PPh2), 7.03 (s, 1H, H5), 6.90 (d, 3J = 7.6 Hz, 1H, H2), 6.32 (s, 2H, B(OH)2), 6.12 (s, 1H, NHMe), 3.72–3.55 (m, 2H, PCH2P), 2.99 (t, 3J = 3.5 Hz, 3H, NHMe), 2.37 (s, 3H, MeCN). 31P–{1H} NMR (400 MHz, acetone-d6) δ 12.55 (d, 2J(P–P) = 79.5 Hz, 1J(Pt–P) = 3860.46 Hz), −23.59 (d, 2J(PP) = 79.0 Hz, 3J(PtP) = 38.6 Hz).
[PtL3(dppm-P)] (3c) was prepared from 0.2 mmol of dppm and 0.05 mmol of 1c. Yield: 89.4 mg, 63%. Anal. Found: C, 51.1; H, 4.2; N, 5.1; S, 3.9%; C36H36BN3O2P2PtS (842.60 g mol−1) requires C, 51.3; H, 4.3; N, 5.0; S, 3.8%. IR cm−1ν(O–H) 3391; ν(N–H) 3054; ν(CN) 1574; ν(B–O) 1334. 1H NMR (400 MHz, acetone-d6, δ): 7.86 (t, J = 9.8 Hz, 4H), 7.49 (s, 1H), 7.33 (q, J = 7.5 Hz, 10H), 7.18 (t, J = 8,4 Hz, 6H), 7.03 (s, 1H, H5), 6.89 (dd, 3J = 7.6, 2.5 Hz, 1H, H2), 6.31 (d, J = 2.5 Hz, 2H, B(OH)2), 6.12 (s, 1H, NHEt), 3.62 (dd, J = 17.9, 9.8 Hz, 2H, PCH2P), 3.43 (p, 3J = 7.2 Hz, 2H, PCH2P), 2.36 (d, J = 2.5 Hz, 3H, MeCN), 1.20 (td, 3J = 7.2, 2.3 Hz, 3H, Me). 31P–{1H} NMR (400 MHz, acetone-d6) δ 12.55 (d, 2J(P–P) = 78.9 Hz, 1J(Pt–P) = 3859.6 Hz), −23.53 (d, 2J(P–P) = 78.4 Hz, 3J(Pt–P) = 38.7 Hz).
[(PtL1)
2
(μ-dppb)] (4a) was prepared from 0.05 mmol of dppb and 0.025 mmol of 1a. Yield: 53.8 mg, 72%. Anal. Found: C, 42.6; H, 3.7; N, 6.7; S, 4.7%; C46H48B2N6O4P2Pt2S2 (1286.78 g mol−1) requires C, 43.0; H, 3.8; N, 6.5; S, 5.0%. IR cm−1ν(O–H) 3389; ν(N–H) 3050; ν(CN) 1574; ν(B–O) 1331. 1H NMR (400 MHz, acetone-d6) δ 7.84–7.71 (m, 8H, PPh2), 7.50 (q, J = 9.4, 8.1 Hz, 6H, PPh2), 7.40 (td, J = 5.2, 2.7 Hz, 6H, PPh2), 7.37–7.30 (m, 3H, PPh2), 7.05 (d, 3J = 7.5 Hz, 2H, H2), 6.94 (t, J = 6.6 Hz, 2H, H5), 6.43 (s, 4H, B(OH)2), 6.17 (s, 4H, NH2), 2.35 (d, J = 5.9 Hz, 6H, MeCN), 2.30–2.16 (m, 4H, PCH2), 1.92–1.85 (m, 4H, CH2). 31P–{1H} NMR (400 MHz, acetone-d6) δ 16.04, 15.06, 1J(PtP) = 3818.23 Hz.
[(PtL2)
2
(μ-dppb)] (4b) was prepared from 0.05 mmol of dppb and 0.025 mmol of 1b. Yield: 50.3 mg, 68%. Anal. Found: C, 44.3; H, 4.1; N, 6.2; S, 4.7%; C48H52B2N6O4P2Pt2S2 (1314.84 g mol−1) requires C, 43.9; H, 4.0; N, 6.4; S, 4.9%. IR cm−1ν(O–H) 3394; ν(N–H) 3054; ν(CN) 1576; ν(B–O) 1333. 1H NMR (400 MHz, acetone-d6) δ 7.82–7.75 (m, 6H, PPh2), 7.55–7.46 (m, 6H, PPh2), 7.39 (dq, J = 6.1, 2.5 Hz, 4H, PPh2), 7.36–7.32 (m, 4H, PPh2), 7.07 (t, J = 3.8 Hz, 2H, H5), 6.96 (d, 3J = 7.5 Hz, 2H, H2), 6.33 (s, 4H, B(OH)2), 6.10 (s, 2H, NHMe), 2.98 (dd, J = 8.6, 4.6 Hz, 6H, NHMe), 2.40 (d, J = 7.8 Hz, 6H, MeCN), 2.31–2.17 (m, 4H, PCH2), 1.92–1.76 (m, 4H, CH2). 31P–{1H} NMR (400 MHz, acetone-d6) δ 16.04, 15.06, 1J(PtP) = 3818.23 Hz.
[(PtL3)
2
(μ-dppb)] (
4c
) was prepared from 0.05 mmol of dppb and 0.025 mmol of 1c. Yield: 57.1 mg, 78%. Anal. Found: C, 44.9; H, 4.3; N, 6.1; S, 4.6%; C50H56B2N6O4P2Pt2S2 (1342.89 g mol−1) requires C, 44.7; H, 4.2; N, 6.3; S, 4.8%. IR cm−1ν(O–H) 3399; ν(N–H) 3046; ν(CN) 1572; ν(B–O) 1330. 1H NMR (400 MHz, acetone-d6) δ 7.82–7.75 (m, 6H, PPh2), 7.55–7.46 (m, 6H, PPh2), 7.39 (dt, J = 6.1, 2.1 Hz, 4H, PPh2), 7.36–7.32 (m, 4H, PPh2), 7.09–7.05 (m, 2H, H5), 6.96 (d, J = 7.5 Hz, 2H, H2), 6.31 (s, 4H, B(OH)2), 6.17 (s, 2H, NHEt), 3.43 (ddd, J = 9.2, 7.1, 5.1 Hz, 4H, CH2), 2.40 (d, J = 7.1 Hz, 6H, MeCN), 2.34–2.17 (m, 4H, PCH2), 1.94–1.73 (m, 4H, CH2), 1.19 (q, 3J = 7.1 Hz, 6H, Me). 31P–{1H} NMR (400 MHz, acetone-d6) δ 16.06, 15.01, 1J(PtP) = 3821.58 Hz.
The compound synthesis reactions are shown in Scheme 1.
 | ||
| Scheme 1 Reaction sequence for the synthesis of the reported compounds. | ||
Cell culture
The HCT116 colorectal cancer cell line and the primary human dermal fibroblasts were obtained from American Type Culture Collection (ATCC®, Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium (DMEM), while the A2780 carcinoma cell line was acquired from Sigma-Aldrich (Madrid, Spain) and cultured in Roswell Park Memorial Institute (RPMI) medium. Media were supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) of a penicillin/streptomycin solution. The media and supplements were obtained from Thermo Fischer Scientific (Waltham, Massachusetts, USA).Cells were cultured in 25 cm2 and/or 75 cm2 T-flasks in a CO2 incubator with a humidified atmosphere at 37 °C and 5% (v/v) CO2 (SANYO CO2 Incubator, Electric Biomedical Co., Osaka, Japan).
Cell viability assays
Cells were initially seeded in 96-well plates at a cell density of 0.75 × 105 cells per mL and incubated (37 °C, 5% (v/v) CO2) for 24 h. After this period, the media were replaced with fresh media containing the desired compounds (concentrations ranging from 0.1 µM to 50 µM), 0.1% (v/v) of DMSO or 0.4 µM doxorubicin (Dox). After 48 h of incubation under the same conditions, the CellTiter 96® aqueous one solution cell proliferation assay kit (Promega, Madison, USA) was used to evaluate the cell viability by measuring the absorbance at 490 nm in a Tecan Infinite M200 microplate reader (Tecan, Männedorf, Switzerland). Data were analysed with the GraphPad Prism 8 software, where viability-concentration response curves facilitated the calculation of the concentration of compounds that induced a 50% reduction in cell viability (IC50).Complex stability in biological media
The stability of the three platinum compounds (3a, 3b and 3c) was analysed via UV-Visible spectroscopy (Shimadzu Scientific Instruments) with a quartz cuvette (1 cm path length) over a wavelength range of 220–700 nm for three different incubation times, namely 0, 3, 24 and 48 h. Compounds were diluted in RPMI medium without phenol red and FBS after being first dissolved in 100% (v/v) DMSO. Compounds 3a and 3c were analysed at a final concentration of 50 μM, while 3b was analysed at a final concentration of 150 μM.ICP-AES (inductively coupled plasma-atomic emission spectrometry)
To evaluate the compound internalization in HCT116 cells, inductively coupled plasma-atomic emission spectroscopy (ICP-AES) was performed. HCT116 cells were seeded in 25 cm2 T-flasks at a density of 5 × 105 cells/T-flask and incubated for 24 h. The culture medium was then replaced with fresh medium with 10× IC50 concentrations of the studied compounds or 0.1% (v/v) DMSO, and cells were incubated for 3 h. After this period, the cell culture medium was recovered in a 15 mL Falcon tube and the cells were washed with PBS. After this washing step, the PBS washing solution was also recovered in the 15 mL Falcon tube. Cells were detached from the T-flask with 2 mL of TrypLE Express and centrifuged at 750g (Sigma 3-16K Sartorius, Germany) for 5 min. The supernatant was removed and added to the 15 mL Falcon tube, and the cell pellet was washed 2 more times with PBS. Aqua regia (3:1 HCl/HNO3) was prepared, and added to the 15 mL Falcon tubes containing the previous washing solutions and the supernatant (non-cellular fraction) or to the cell pellets (cellular fraction). Samples were incubated at room temperature (RT) for 24 h in a hood fume, and then delivered to Laboratório de Análises/LAQV to quantify the platinum levels by ICP-AES.
Analysis of apoptosis induction by flow cytometry
Apoptosis was evaluated using the Alexa Fluor® 488 Annexin V/dead cell apoptosis kit (Invitrogen, Thermo Fisher Scientific, MA, USA). HCT116 cells were seeded in 6-well plates (at a density of 2 × 105 cells per well) for 24 h, and later incubated for 48 h period with the IC50 concentrations of compounds 3a, 3b and 3c. Cells treated with 0.1% (v/v) DMSO were used as the vehicle control, while cells treated with 0.4 μM Dox were used as positive controls. After 48 h of incubation, cells were washed with PBS and detached from the wells with TrypLE Express (Invitrogen), and then washed again with PBS. Lastly, cells were incubated with the Alexa® Fluor 488-Annexin V solution and 100 μg mL−1 of propidium iodide (PI) at RT for 15 min.Samples were evaluated by the Attune® acoustic focusing flow cytometer (Life Technologies, Carlsbad, USA), and the results were analysed with the Attune® Cytometric software.
Quantification of BAX and BCL-2 protein levels by western blot
HCT116 cells were cultivated, incubated and collected, as described previously.32 After collection, cells were submitted to 5 ultrasound pulses on ice (2 min on ultrasound, followed by 30 s on ice; Elma D-78224; Singen/Htw, Germany) and centrifuged at 1000g for 5 min. The total amount of protein in the supernatant extracts was quantified with Pierce 660 nm Protein Assay Reagent (Thermo Fisher Scientific, Waltham, MA, USA). For SDS-PAGE, 20 μg of protein was loaded on 10% polyacrylamide gel and transferred to a 0.45 μm PVDF membrane (GE Healthcare Life Sciences, Germany) (BAX and BCL-2 proteins). The membrane was blocked for 2 h with 5% (w/v) non-fat milk in TBST (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% (v/v) Tween-20), and incubated at room temperature for 1 h under constant agitation with the respective primary antibody solution (anti-BAX, 1:5000, Abcam, United Kingdom; and anti BCL-2, 1:1000, Sigma, St Louis, MO, USA). After incubation, membranes were washed with TBST buffer three times, for a duration of 5 min each (procedure also repeated after the secondary antibody incubation) and exposed to the secondary antibody solution (1:3000, anti-mouse IgG, horseradish peroxidase HPR-linked antibody or 1:2000, anti-rabbit IgG, HPR-linked antibody; Cell Signalling Technology, USA). Membranes were treated with the WesternBright ECL substrate (Advansta, USA) for 5 min and later exposed to a film in a dark room. After that, to normalize the results, membranes were incubated two times with stripping buffer (0.1 M glycine, 20 mM magnesium acetate, 50 mM KCl, pH 2.0) for a duration of 10 and 20 min, respectively, and then incubated with anti-β actin (1:5000; Sigma, St Louis, USA). BAX and BCL-2 protein quantification (densitometry) was performed using the Image J software. BAX and BCL-2 protein levels were normalized to β actin levels.
Mitochondrial membrane potential (ΔΨm) analysis by flow cytometry
ΔΨm was analysed using the JC-1 mitochondrial membrane potential assay kit (Abnova Corporation, Walnut, CA, USA). HCT116 cells were seeded in 6-well plates with a density of 2 × 105 cells per well, incubated for 24 h, and later incubated for another 48 h period with the IC50 concentrations of compounds 3a, 3b and 3c or with 0.1% (v/v) DMSO. As positive controls, 0.4 μM Dox and 5 μM Cis were used. After that, cells were washed with PBS, detached with TrypLE Express, and washed with DMEM medium. Cells were then resuspended in DMEM medium without phenol red + 5% (v/v) FBS, and stained with the JC-1 staining solution for 20 min at 37 °C. Lastly, cells were resuspended in DMEM medium without phenol red + 5% (v/v) FBS, and analysed in the Attune® Acoustic Focusing Flow Cytometer (Life Technologies, Carlsbad, CA, USA).Caspase-8 activity
HCT116 cells were seeded in 25 cm2 T-flasks at a density of 2 × 106 cells/T-flasks. After 24 h incubation, under the same conditions as described previously, the medium was replaced with fresh medium containing DMSO 0.1% (v/v), 0.4 µM Dox, 5 µM Cis or IC50 concentrations of the compounds 3a, 3b and 3c, and incubated for 48 h. After that, cells were detached using cold PBS and a cell scraper, and centrifuged at 500g for 5 min. Then, the instructions provided in the caspase-8 assay kit (Abcam) procedure were followed. Caspase 8 activity was obtained by measuring the absorbance at 400 nm of each sample. Caspase 8 activities were normalized to the corresponding value in DMSO samples.Analysis of autophagy by flow cytometry
The autophagic potential of HCT116 cells was evaluated using the autophagy assay kit (ab139484) (Abcam, Cambridge, United Kingdom). HCT116 cells were seeded in 6-well plates with a density of 2 × 105 cells per well and incubated for 24 h. After the replacement of the culture medium with fresh medium containing the IC50 concentrations of compounds 3a, 3b and 3c, cells were incubated for another 48 h. 0.1% (v/v) DMSO, 0.4 μM Dox and 5 μM Cis were used as controls. In addition, 15 h before finishing the 48 h incubation time, 0.5 μM Rapamycin was added to the respective wells. After that, cells were washed with PBS, detached with TrypLE Express, washed with DMEM medium without phenol red + 5% (v/v) FBS, and then incubated with the green stain solution in DMEM medium without phenol red + 5% (v/v) FBS for 30 min at RT. After this period, cells were washed, resuspended in the assay buffer, and then analysed with the Attune® Acoustic Focusing Flow Cytometer (Life Technologies, Carlsbad, CA, USA). The results were analysed with the Attune® Cytometric software.Analysis of reactive oxygen species (ROS) production by flow cytometry
HCT116 cells were seeded in 6-well plates at a density of 2 × 105 cells per well and incubated for 24 h. The culture medium was replaced with fresh medium containing the IC50 concentrations of compounds 3a, 3b and 3c, 0.1% (v/v) DMSO, 0.4 μM Dox, 5 μM cisplatin or 30 μM TBHP (positive control as indicated by the manufacturer), and cells were incubated for another 48 h period. Cells were then washed with PBS, detached with TrypLE Express, washed with PBS, and later incubated with 10 μM of 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) (Thermo Fisher Scientific, Waltham, MA, USA) in PBS for 20 min. Cells were later analysed with the Attune® Acoustic Focusing Flow Cytometer (Life Technologies, Carlsbad, USA), and the results were treated with the Attune® Cytometric software.Ex ovo CAM assay
The pro-/anti-angiogenic potential of the compounds was evaluated with the ex ovo chorioallantoic membrane (CAM) assay, as previously described in (Reigosa-Chamorro et al., 202126).30,31 Chicken embryos were first incubated for 24 h and later exposed to the IC50 concentrations of compounds 3a, 3b and 3c dissolved in PBS (in the centre of O-rings) or 0.1% (v/v) DMSO. The compounds’ and DMSO's distribution in the embryos were always performed in a different order. Then, embryos were incubated for another 48 h at 37 °C and its images were captured after 0, 24 and 48 h with a digital USB Microscope Camera (Opti-Tekscope OT-V1) to manually count the newly formed blood vessels via ImageJ software. The ex ovo CAM assay fulfils the Directive 2010/63/EU of the European Parliament to protect the animal models for scientific purposes.
Statistical analysis
The presented data are referred to as mean ± SEM from at least three biological independent experiments, unless otherwise stated. One-way ANOVA or Student's t-test were performed to determine the statistical significance (p < 0.05) with the GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA).Results and discussion
The compounds and respective reactions are shown in Scheme 1 for simplification. The thiosemicarbazone ligands L1–L3, a, b and c were prepared by reaction of 4-acetylphenylboronic acid with the corresponding thiosemicarbazide as pure air-stable solids, as appropriate (see Experimental section). The NNH proton resonated ca. 10.2 ppm, whilst the NH2 protons, a, gave rise to two characteristic resonances in the 1H NMR spectrum which were attributed to the restricted rotation of the NH2 group about the C(S)NH2 bond axis. The NHR protons, b, c, showed a broad resonance at ca. δ 8.5 ppm. The characteristics of the ligand spectra included two virtual doublets stemming from the aromatic AA′XX′ spin system with an N value at ca. 8.0–7.8 ppm. From them, the new cyclometallated compounds were obtained as described in Scheme 1. The preparative details and characteristic microanalytical and spectroscopic data are given in the Experimental section. Reaction of a, b or c, as appropriate, with potassium tetrachloroplatinate in water/ethanol gave clear solutions. These solutions were used to isolate the tetranuclear compounds 1a–1c as pure air-stable solids, with the ligand in the E,Z configuration. Absence of the NH resonance agreed with deprotonation at the hydrazine group. The 1H NMR spectra showed the absence of the AA′XX′ spin system upon metalation of the para-substituted phenyl ring, and distinct resonances were accordingly assigned to the H2, H3 and H5 nuclei (see Experimental).Reaction of [Pt(L1–3)]41a–1c with tertiary phosphines PPh3, Ph2PCH2PPh2 (dppm), and Ph2P(CH2)4PPh2 (dppb), in 1:4 or 1:2 molar ratios, as appropriate, gave the compounds [Pt(L1–3)(PPh3)] 2a–2c, [Pt(L1–L3)(Ph2PCH2PPh2-P)] 3a–3c, and [Pt(L1–L3)2{μ-Ph2P(CH2)4PPh2}] 4a–4c as pure air-stable solids (Scheme 1). The microanalytical and spectroscopic data are presented in the Experimental section. The use of excess diphosphine only gave cleavage of the Pt–Sbridging. Therefore, in compounds with diphosphines, coordination to the metal was only through one phosphorus atom. Usage of excess phosphine did not cleave the Pt–Schelate bond. The 1H NMR spectra showed the high-field shift of the H5 resonance at ca. 1 ppm with respect to the corresponding tetranuclear compound, owing to the shielding effect of the phosphine phenyl rings. The 31P NMR spectra for the 3a–3c compounds showed two doublets assigned to the two non-equivalent phosphorus nuclei. The lower field resonance, at ca. 12.5 ppm, was assigned to the 31P nucleus bonded to platinum, in agreement with the J(Pt–P) values. Meanwhile, for 4a–4c, the 31P resonance was a singlet signal in accordance with the existence of equivalent phosphorus nuclei; the chemical shift values were consistent with a phosphorus to nitrogen trans geometry.32,33 The resonance for the ABXY spin system of the PCH2P protons appeared at ca. at δ 3.52. Attempts to produce mononuclear species analogous to 3a–3c with diphosphines other than Ph2PCH2PPh2 were unsuccessful.
Suitable crystals of 1b were grown by slowly evaporating an acetone solution of the complex. The ORTEP illustration of compound 1b is shown in Fig. 1. The tetranuclear compound crystallizes in the Pcan space group with two acetone molecules. Within each molecule, the metalated moieties are displayed as two sets of almost coplanar antiparallel pairs separated at ca. 3.5 Å. Each platinum atom of the internal Pt4S4 nucleus is bonded to a tridentate C,N,Schelating ligand and to the sulfur atom, Sbridging, of another metalated species. The longer Pt–Schelating bond lengths, as opposed to Pt–Schelating, reflect the differing trans influence of the phenyl carbon and nitrogen atoms of the ligands.
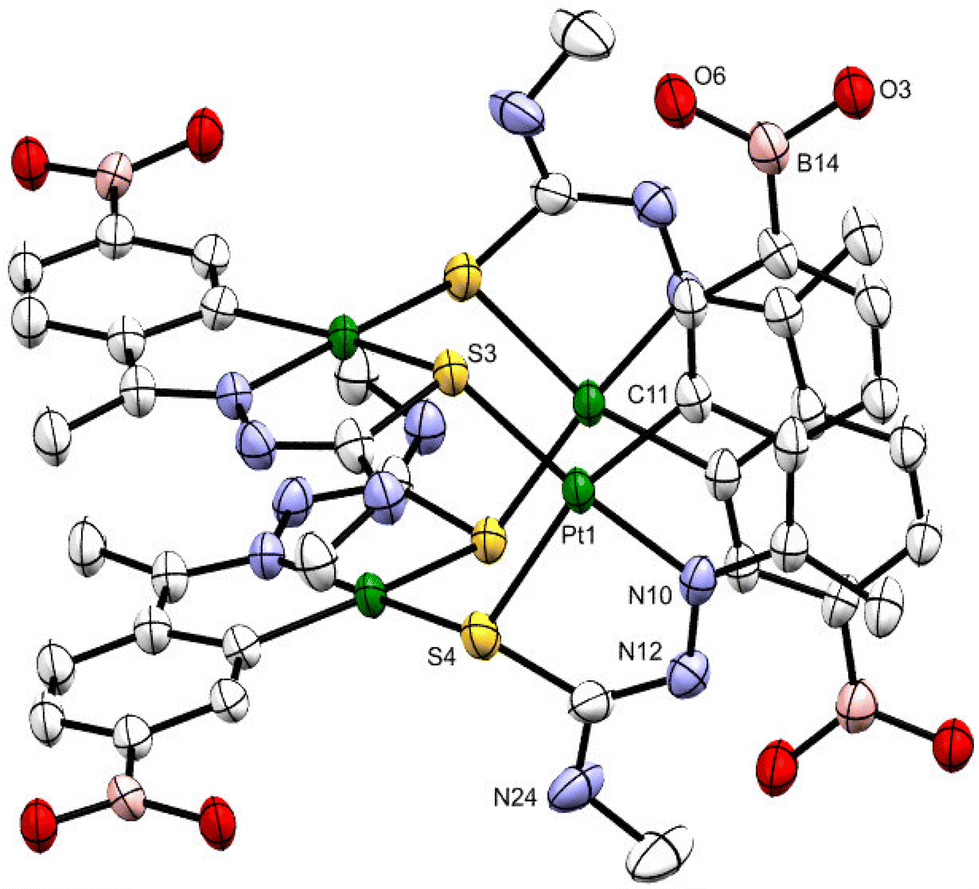 | ||
| Fig. 1 ORTEP drawing of the platinacycle 1b with thermal ellipsoid plot shown at the 50% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. Selected bond distances (Å) and angles (°) for 10: Pt(1)–C(11) 2.013(8), Pt(1)–N(10) 1.988(7), Pt(1)–S(3) 2.287(2), Pt(1)–S(4) 2.347(2), S(4)–Pt(1)–S(3) 100.76(8), N(10)–Pt(1)–S(3) 174.5(2), N(10)–Pt(1)–S(4) 84.1(2), C(11)–Pt(1)–S(3) 94.6(3), C(11)–Pt(1)–S(4) 163.9(3), and C(11)–Pt(1)–N(10) 80.8(3). | ||
Suitable crystals of 2c were grown by slowly evaporating a chloroform solution of the complex. The crystals were triclinic with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The ORTEP illustration is shown in Fig. 2. The structure of compound 2c comprises a molecule with the platinum(II) atom bonded in a slightly distorted square planar coordination to four different donor atoms, a tridentate thiosemicarbazone through the aryl C(6) carbon, the imine N(1) nitrogen, and the thioamide S(1) sulfur atom, and to a phosphorus atom P(1) of the triphenylphosphine. The bond distances and angles are within the expected values for analogous structures, as was provided by Mogul 2020.3 from the CCDC program package. The angles at platinum are close to 90° with allowance for the somewhat smaller C(6)–Pt(1)–N(1) and S(1)–Pt(1)–N(1) angles, 79.81° and 83.4°, respectively, and larger C(6)–Pt(1)–P(1) and S(1)–Pt(1)–P(1) angles, 99.80° and 96.97°, also respectively, consequent upon chelation. The crystal packing shows the molecules are arranged in sheets held together by means of hydrogen bonding interactions between the boronic acid function, the chlorine atom of the solvent molecule and the amide nitrogen, as well as by π–π stacking.
space group. The ORTEP illustration is shown in Fig. 2. The structure of compound 2c comprises a molecule with the platinum(II) atom bonded in a slightly distorted square planar coordination to four different donor atoms, a tridentate thiosemicarbazone through the aryl C(6) carbon, the imine N(1) nitrogen, and the thioamide S(1) sulfur atom, and to a phosphorus atom P(1) of the triphenylphosphine. The bond distances and angles are within the expected values for analogous structures, as was provided by Mogul 2020.3 from the CCDC program package. The angles at platinum are close to 90° with allowance for the somewhat smaller C(6)–Pt(1)–N(1) and S(1)–Pt(1)–N(1) angles, 79.81° and 83.4°, respectively, and larger C(6)–Pt(1)–P(1) and S(1)–Pt(1)–P(1) angles, 99.80° and 96.97°, also respectively, consequent upon chelation. The crystal packing shows the molecules are arranged in sheets held together by means of hydrogen bonding interactions between the boronic acid function, the chlorine atom of the solvent molecule and the amide nitrogen, as well as by π–π stacking.
 | ||
| Fig. 2 ORTEP drawing of the platinacycle 2c with thermal ellipsoid plot shown at the 50% probability level. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond distances (Å) and angles (°) for 10: Pt(1)–C(6) 2.038(6), Pt(1)–N(1) 2.032(5), Pt(1)–S(1) 2.3558(17), Pt(1)–P(1) 2.2234(18), S(1)–Pt(1)–P(1) 96.97(6), N(1)–Pt(1)–S(1) 83.44(16), N(1)–Pt(1)–P(1) 179.24(18), C(6)–Pt(1)–S(1) 162.95(19), C(6)–Pt(1)–P(1) 99.79(19), and C(6)–Pt(1)–N(1) 79.8(2). | ||
Antiproliferative activity
The in vitro antiproliferative potential of compounds 1a–c, 2a–c, 3a–c, 4a–c and the respective ligands a–c was assessed using the CellTiter 96®Aqueous non-radioactive cell proliferation assay (MTS assay), as described in the Experimental section.34 This antiproliferative activity was analysed by exposure of the ovarian carcinoma (A2780) and colorectal carcinoma (HCT116) cell lines and normal human primary dermal fibroblasts to 0.1–50 µM of all compounds for 48 h (ESI Fig. S1†). As shown in Fig. S1,† a reduction of the cell viability is observed upon increase of the compounds concentrations. Cell viability-concentration curves (GraphPad software) allowed us to calculate the relative IC50 values of each compound for each cell line (Table 1). Moreover, as positive controls, the antiproliferative activity of the Dox and Cis was evaluated through the exposure of A2780, HCT116 and Fibroblast cells to 0.1–50 µM of these antitumor drugs for 48 h (ESI Fig. S2†). The selectivity index (SI) of each compound – the ratio IC50 in fibroblasts/IC50 in the tumour cell line – was determined, and it is reported in Table 1 to evaluate the selectivity of the compound towards tumour cell lines. Higher SI values were correlated with higher selectivity of the compound for a particular tumour cell line compared to the normal cells.35| Complex | Cell line | IC50 (µM) | SI |
|---|---|---|---|
| > indicates that the IC50 is higher than the value indicated; – SI values not calculated. | |||
| 1a | HCT116 | >50 | — |
| A2780 | 10 < IC50 < 50 | — | |
| Fibroblasts | >50 | — | |
| 1b | HCT116 | >50 | — |
| A2780 | 35.7 | >1.4 | |
| Fibroblasts | >50 | — | |
| 1c | HCT116 | >50 | — |
| A2780 | 10 < IC50 < 50 | — | |
| Fibroblasts | >50 | — | |
| 2a | HCT116 | >50 | — |
| A2780 | >50 | — | |
| Fibroblasts | >50 | — | |
| 2b | HCT116 | >50 | — |
| A2780 | 31.9 | >1.6 | |
| Fibroblasts | >50 | — | |
| 2c | HCT116 | >50 | — |
| A2780 | >50 | — | |
| Fibroblasts | >50 | — | |
| 3a | HCT116 | 2.1 | >23.8 |
| A2780 | 5.0 | >10 | |
| Fibroblasts | >50 | — | |
| 3b | HCT116 | 1.9 | >26.3 |
| A2780 | 1.9 | >26.3 | |
| Fibroblasts | >50 | — | |
| 3c | HCT116 | 8.6 | >5.8 |
| A2780 | 8.0 | >6.3 | |
| Fibroblasts | >50 | — | |
| 4a | HCT116 | >50 | — |
| A2780 | 10 < IC50 < 50 | — | |
| Fibroblasts | >50 | — | |
| 4b | HCT116 | >50 | — |
| A2780 | 31.0 | >1.6 | |
| Fibroblasts | >50 | — | |
| 4c | HCT116 | >50 | — |
| A2780 | 10 < IC50 < 50 | — | |
| Fibroblasts | >50 | — | |
| L1 | HCT116 | >50 | — |
| A2780 | >50 | — | |
| Fibroblasts | >50 | — | |
| L2 | HCT116 | >50 | — |
| A2780 | >50 | — | |
| Fibroblasts | >50 | — | |
| L3 | HCT116 | >50 | — |
| A2780 | >50 | — | |
| Fibroblasts | >50 | — | |
| Doxorubicin (Dox) | HCT116 | 0.5 ± 0.10 | 24.2 |
| A2780 | 0.1 ± 0.04 | 121 | |
| Fibroblasts | 12.1 ± 0.20 | — | |
| Cisplatin (Cis) | HCT116 | 15.6 ± 5.30 | 0.6 |
| A2780 | 1.9 ± 0.20 | 4.6 | |
| Fibroblasts | 8.8 ± 2.90 | — | |
Comparing the IC50 values obtained for the HCT116 cancer cell line for all of the tested compounds (Table 1), compounds 3a–c presented the lowest IC50 values (2.1, 1.9 and 8.6 µM, respectively) with the following cytotoxicity order: 3b > 3a > 3c. Moreover, A2780 cells compounds 3a–c also presented the lowest IC50 values (5.0, 1.9, 8.0, respectively), maintaining the same cytotoxicity order of 3b > 3a > 3c (Table 1). Interestingly, in the HCT116 cells, these 3 compounds presented a higher cytotoxic potential when compared to the chemotherapeutic agent, cisplatin (IC50 value of 15.6 µM). All the other compounds do not demonstrate cytotoxicity in the HCT116 colorectal carcinoma cell line (IC50 > 50 µM). This clearly indicates that coordination with dppm – Ph2PCH2PPh2-P provides a higher cytotoxicity compared to the other phosphines (PPh3 or dppb) (Table 1).
In A2780 ovarian carcinoma cells, compounds 1a–c, 2b, and 4a–c show moderate cytotoxicity values (10 µM < IC50 < 50 µM) (Table 1). Among all compounds, it seems that compounds with substitution b (RMe) show more cytotoxic effects, although ligands 1, 2 and 3 did not present any cytotoxicity when tested separately (Table 1). We have yet to find a plausible explanation for the differing values for compounds 3a, 3b as compared to 3c. The modification of the –NHR group was not thought to have a significant effect on the modes of action of the compounds, but more in the fine-tuning of internalization and their properties. It is possible that R groups with longer carbon chains affect interactions with the biological membranes and/or targets. This puts forward the need to examine distinct options when designing a metallodrug.
One of the main reasons and interest to develop new platinum-based compounds is to circumvent the high cytotoxicity of approved platinum drugs, and to reduce the side effects in healthy tissues. Therefore, it is important that compounds 3a–c have much higher IC50 values in a normal human cell line when compared to the values obtained for the studied tumor cell lines. Fibroblasts were used as healthy cells due to their importance in the tumor microenvironment,36 and the IC50 values obtained are reported in Table 1.
Interestingly, when analysing the IC50 values obtained for fibroblasts (Table 1), all of the compounds showed low cytotoxic potential (IC50 values >50 µM), which is reflected in the high SI values. At the limit, if we consider the IC50 of compounds 3a–c in fibroblasts as equal to 50 µM, compounds 3a and 3b showed the highest SI values (23.8 and 26.3, respectively) in the HCT116 cancer cell line, meaning that those compounds are at least 23.8× more cytotoxic for HCT116 cancer cells than for fibroblasts. Considering the same approach, compounds 3a and 3b also show higher SI values in the A2780 cancer cell line (10 and 26.3, respectively). Compound 3c presented a SI value of 5.8 in the HCT116 cell line, and a slightly higher SI value (6.3) for the A2780 cell line. It is also interesting to note that all of the ligands (a, b and c) do not show a cytotoxic effect in the tested cell lines (IC50 > 50 µM in tumor and normal cells). Interestingly, our previous data37 show high cytotoxicity for the free PPh3 (a ligand that is present in compounds 2a, 2b and 2c) and for other free diphenylphosphines in normal fibroblasts. In the present work, we show a high cytotoxicity for compounds, particularly those harbouring dppm, in tumor cell lines and no cytotoxicity in fibroblasts. This means that this effect is probably attributed to the effects of the compounds per se, and not of the free ligands, even though free phosphines also have high cytotoxicity in these tumor cell lines.37 Thus, compounds 3a, 3b and 3c show an overall very good therapeutic window and potential in the HCT116 cell line and from now on, all of the remaining stability and biological analysis will be assessed for these 3 compounds in the HCT116 cell line.
Complex stability in biological media
After selection of the best compounds for pursuing additional biological characterization, the stability of the compound (3a, 3b and 3c) in biological medium was analyzed using UV-Visible spectroscopy over a wavelength range of 220 to 700 nm for different incubation times (−0, 3 h, as shown in Fig. 3; 0, 24, 48 h in ESI Fig. S5†).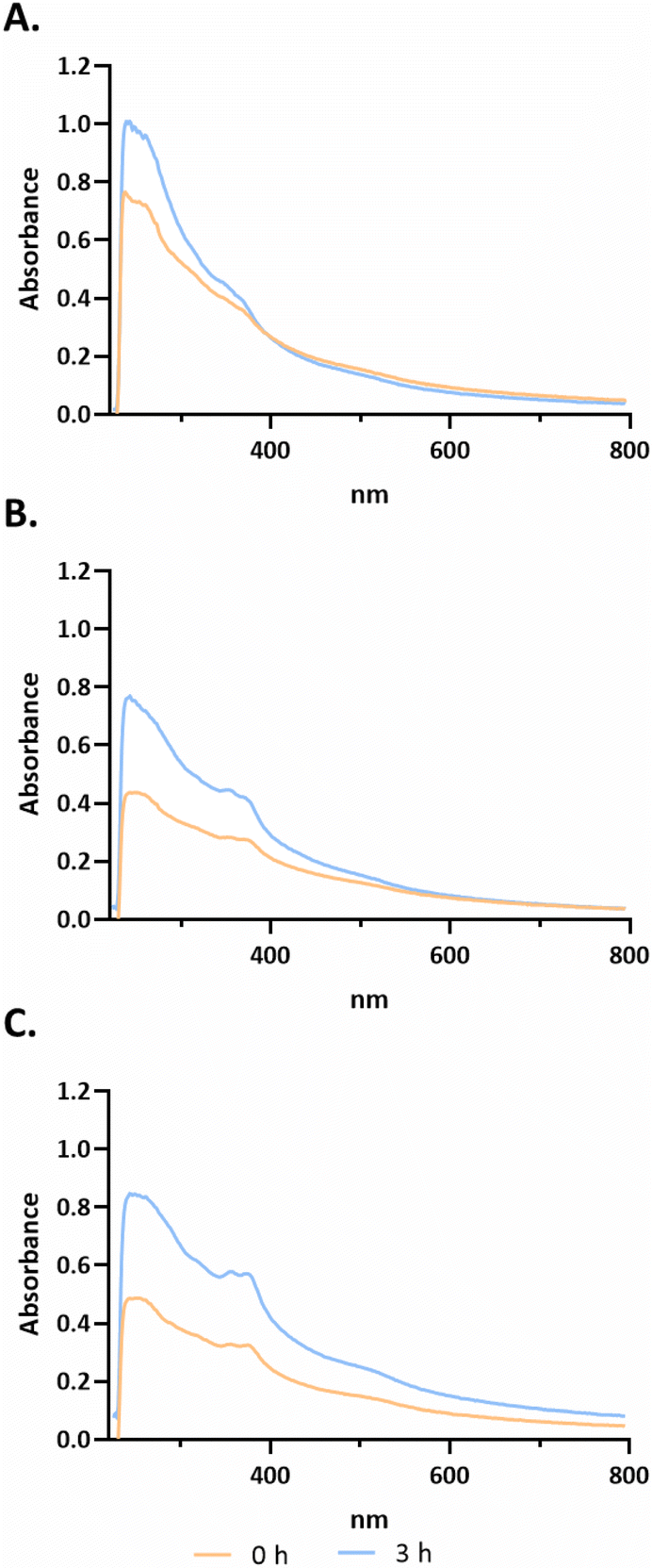 | ||
| Fig. 3 Evaluation of the stability of compounds 3a (A), 3b (B) and 3c (C) via UV-visible spectroscopy for 3 h. Absorbance spectra of 50 µM of compounds 3c and 3a and 150 µM 3b in an RPMI medium without phenol red and FBS at different incubation times: 0 h (orange) and 3 h (blue). | ||
Analysing the spectra of all compounds, high-energy absorption bands corresponding to π → π* and n → π* transitions with peaks in the 230–330 and 330–400 nm ranges, respectively, that are associated with the aromatic rings of terpyridines can be observed.38 According to Fig. 3, for compound 3a, bands are observed at approximately 235 nm, 265 nm and 360 nm at 0 h. For compound 3b, four characteristic bands can be identified at approximately 293, 325, 352 and 371 nm. Finally, compound 3c presents bands at approximately 235 nm, 265 nm, 327 nm, 365 nm, 380 nm and 530 nm. As observed in Fig. 3, from 0 h to 3 h, there is an increase of absorbance that is most probably due to a better solubilization of the complexes in the medium, maintaining their characteristic bands. However, analysing the data from ESI Fig. S5,† it is possible to observe a change in the peak at 265 nm to 272 nm from 0 h to 48 h for compound 3c. However, for 3a at 24 h, the band at 360 nm is no longer observed. Furthermore, at 48 h there is a peak at approximately 325 nm, which may indicate that the complex is not stable in solution (ESI Fig. S5†). Furthermore, there is not a large decrease in the absorbance values throughout the spectrum between the different acquisition times. Regarding compound 3b, the four characteristic bands remained at the same wavelength throughout the 48 h. However, between 0 h and 24 h, there is a slight decrease in all absorbances throughout the spectrum (ESI Fig. S5†).
Considering the stability of all compounds during the first three hours, we further accessed their internalization in HCT116 cells in this period. Nevertheless, for ensuring the best solubility and stability of the compounds in all biological assays, they were always performed with freshly prepared solutions of these compounds.
Compound cellular internalization by ICP-AES
The internalization of the platinum compounds 3a, 3b and 3c in HCT116 cells was evaluated by ICP-AES technique, which allows a quantitative measurement of the amount of metal present in each sample. This assay allows us to understand if the compounds under study are indeed internalized by HCT116 cells.39 To perform this technique, HCT116 cells were exposed to 10× the IC50 values of each platinum compounds for 3 h.The obtained results showed an internalization of almost 100% for all compounds after 3 h, with compound 3c being the one with a higher percentage of internalization (97.3 ± 0.2%), followed by compound 3b (96.5 ± 0.3%) and ultimately compound 3a (95.7 ± 0.1%). Considering this, the compounds are stable and soluble in biological medium by the time they are internalized in HCT116 cells (Fig. 3).
Cell death by apoptosis and/or necrosis
As the platinum compounds can induce a decrease in HCT116 cell viability, it is important to understand the type of cell death that is been triggered by their presence. Thus, the levels of apoptosis (a programmed cell death mechanism) and necrosis (a non-programmed cell death mechanism) after the exposure of HCT116 cells for 48 h to compounds 3a–c (at their IC50 concentrations) were measured through flow cytometry using Annexin V-Alexa fluor 488/PI double staining (Fig. 4). 0.1% DMSO (v/v) was used as the vehicle control (negative control) and Dox (0.4 μM) was used as the positive control. | ||
| Fig. 4 Apoptosis induction in HCT116 cells exposed to the IC50 of the compounds 3a–c for 48 h via flow cytometry using Annexin V/PI double staining. DMSO 0.1% (v/v) was used as a negative control (vehicle control) and Dox (0.4 µM) as a positive control. Data are expressed as mean ± SD of at least two independent assays. The symbols ****, *** and * represent p < 0.0001, p < 0.001 and p < 0.1, respectively. | ||
Annexin V has a high affinity for phosphatidylserine, which is present on the inner surface of the lipid bilayer of plasma membranes in viable cells. Once cells initiate the apoptotic process, phosphatidylserine is translocated to the outer layer of the membrane, favouring the interaction between annexin V and phosphatidylserine and the identification of cells in apoptosis by flow cytometry.39–41 Furthermore, PI (a fluorescent molecule that is impermeable to membranes in viable cells) allows for the detection of cells with a compromised membrane – cells in necrosis.40,42 Therefore, this method allows for distinguishing between cells in early apoptosis (labelled with annexin V-Alexa fluor 488), cells in late apoptosis (labelled with both annexin V-Alexa fluor 488 and PI), and cells in necrosis (labelled with PI) from normal viable cells (not labelled).40,42 The results shown in Fig. 4 indicate that the HCT116 cells exposed to compound 3c present the highest percentage of apoptosis (40.3%), while cells exposed to compounds 3a and 3b present about 30% apoptosis. On the contrary, cells exposed to DMSO presented 75.1% viable cells, 24.3% apoptosis and 0.6% necrosis. The exposure of the cells to Dox leads to cell values of 40% in apoptosis and 20.2% in necrosis. These results are in agreement with that previously observed in the literature.43
Comparing the results obtained for each complex to those obtained for DMSO, compound 3c shows a significant increase of 1.7× of the percentage of cells in apoptosis, while compounds 3a and 3b show an increase of 1.2× of the percentage of cells in apoptosis. Therefore, the platinum compounds induce apoptosis in HCT116 cells in levels that are relatively close to those exhibited in the presence of the positive control, Dox (Fig. 4). Fig. 4 also reveals that the percentage of necrotic cells was low under all analysed conditions, except for HCT116 cells exposed to Dox (∼20%), a result that correlates with the literature.44,45 This result is highly relevant as necrosis is a non-programmed cell death mechanism that is usually associated with the trigger of inflammation, a process that is not ideal in a tumor context.44,45 It is interesting to note that compound 3c has a slightly higher internalization by ICP-AES after 3 h, which correlates with its higher level of apoptosis (Fig. 4).
To evaluate whether the platinum compounds could induce apoptosis by the intrinsic pathway, the expression of BAX and BCL-2, a pro-apoptotic and an anti-apoptotic protein, respectively, was analysed in HCT116 cells after 48 h of exposure to the IC50 of each compound (Fig. 5). Usually, a BAX/BCL-2 ratio of >1 indicates that cell death by apoptosis is promoted. Conversely, a ratio lower than 1 results in cell survival. Thus, the balance of these two proteins is an important factor to determine the cell fate.46,47
 | ||
| Fig. 5 Relative expression of BAX (A) and BCL-2 (B) proteins in HCT116 cells after 48 h exposure to the IC50 of compounds 3a, 3b and 3c or 0.1% DMSO (vehicle control). (C) BAX/BCL-2 ratio. (D) Western blot bands used for quantification of BAX and BCL-2 proteins. Data were normalized to the DMSO vehicle control (value of 1 is represented with a dotted line) after an initial normalization with β-actin. Results are expressed as the mean ± SEM of two independent assays. The symbols **** and ** represent p < 0.0001 and p < 0.01, respectively. | ||
An increase of the expression of BAX protein when HCT116 cells are exposed to compounds 3b and 3c, compared to the levels shown by the control, is accompanied by a decrease of the expression of the BCL-2 protein (Fig. 5A and B). On the contrary, cells exposed to compound 3a have the same levels of expression of the BAX and BCL-2 proteins when compared to the DMSO control. The BAX/BCL-2 ratio (Fig. 5C) shows that compounds 3b and 3c have a 2.6-fold or 4-fold increase over the control, respectively, an indication that they can trigger the intrinsic/mitochondrial apoptotic pathway in the HCT116 cell line. On the contrary, compound 3a exhibits a BAX/BCL-2 ratio that is near 1, which suggests that this compound does not seem to induce an apoptotic pathway that is dependent on the expression of BAX. Nevertheless, as we have demonstrated, compound 3a induces apoptosis (Fig. 4) without other alternative pathways, such as the extrinsic apoptotic pathway or an intrinsic pathway that is dependent on BAK or other pro-apoptotic protein.
To further confirm that compounds 3b and 3c can induce the intrinsic apoptotic pathway, their effect in disrupting the mitochondrial membrane potential (ΔΨm) should also be analysed.
Evaluation of the mitochondrial membrane potential (ΔÑ°m)
The induction of the intrinsic/mitochondrial pathway of apoptosis correlates with changes in the mitochondrial membrane potential.48,49 In this regard, it is essential to study the effect of the compounds in the mitochondria and in its membrane potential. The cationic dye JC-1 has been widely used to evaluate the integrity/changes in the mitochondrial membrane potential.49,50 This dye naturally accumulates within the mitochondria due to its cationic properties, which enables the formation of aggregates that show a red fluorescence (from 532 to 590 nm).49,50 When the inner mitochondrial membrane is compromised and has a negative potential (high ΔΨm), the JC-1 dye will leave the mitochondria, accumulating in the cytosol in the monomeric form, which changes the dye's fluorescence to green (from 510 to 560 nm).49,50 Therefore, cells with depolarized mitochondria will display a lower red/green fluorescence ratio. The HCT116 cell line was exposed to the IC50 concentrations of each platinum compound for 48 h, and the red/green fluorescence ratios were obtained and are represented in Fig. 6.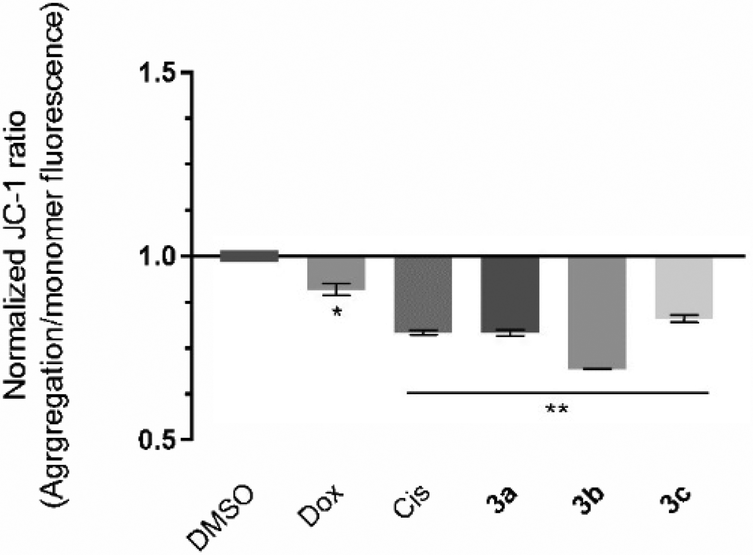 | ||
| Fig. 6 Evaluation of the mitochondrial membrane potential of HCT116 cells exposed for 48 h to the IC50 concentrations of the compounds 3a–cvia flow cytometry. 0.1% (v/v) DMSO was used as the vehicle control, and 5 μM Cis and 0.4 μM Dox were used as positive controls. Data are normalized to the control of DMSO and expressed as the mean ± SEM of at least two independent biological assays. The symbols ** and * represent p < 0.0001 and p < 0.01, respectively. | ||
The results presented in Fig. 6 show that all of the compounds promote significant changes in the mitochondrial membrane when compared to the DMSO control. Compound 3b affects the mitochondrial membrane the most, with a JC-1 ratio of 0.69. According to the previous results (Fig. 5), exposure of the cells to compounds 3b and 3c promote a loss of ΔÑ°m and permeability occurs, which leads to the release of mitochondria proteins (e.g., cytochrome C) into the cytosol, and activation of the intrinsic apoptosis pathway (Fig. 6). Compound 3a has previously been shown to induce apoptosis (Fig. 4) and a disruption of the mitochondrial membrane potential (Fig. 6), which is not in line with the results obtained for the expression of BAX and BCL-2 protein since their ratio was only slightly above 1 (1.27) (Fig. 5). This indicates that the depolarization of the mitochondria membrane potential is probably dependent on a different pro-apoptotic protein (e.g., BAK), or via an alternative extrinsic pathway involving t-BID.51,52 The extrinsic apoptotic pathway is usually activated through external signals recognized by death receptors at the cell membrane. This triggers activation of caspase-8 that may cleave BID into t-BID.52,53 Compound 3a may also directly affect the mitochondria, as described previously in the literature.53
Evaluation of caspase-8 activity
As explained above, external signals recognized by the death cellular receptors are able to trigger caspase 8, which can induce the cleavage of several molecules, such as t-BID, mitochondrial or nuclear proteins, or directly downstream caspases.52 The chromogenic substrate IEDT-pNA, which is constituted by the IETD (Ile-Glu-Thr-Asp) peptide conjugated with the chromophore p-nitroanilide (pNA), is usually used as a way to quantify the caspase 8 activity of the total protein extracts.54 When the substrate is cleaved by caspase 8, the pNA is released, which can be quantified by measuring the absorbance at 400 nm.55 To evaluate caspase 8 activity induced by the compounds, HCT116 cells were incubated for 48 h with the IC50 concentration of compound 3a (Fig. 7).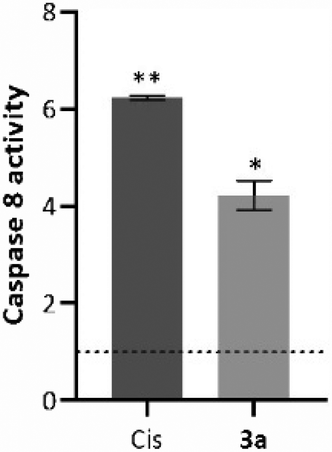 | ||
| Fig. 7 Caspase-8 activity in HCT116 cells exposed to the IC50 of compound 3a for 48 h. 0.1% (v/v) DMSO was used as the vehicle control and 5 μM Cis as positive control. Caspase 8 activity was quantified using the caspase 8 assay kit (Abcam). Date are normalized to the control of DMSO (value represented with a dotted line) and expressed as the mean ± SEM of at least two independent biological assays. The symbols ** and * represent p < 0.0001 and p < 0.001, respectively. | ||
Analysing the data presented in Fig. 7, exposure of the HCT116 cells to compound 3a or Cis leads to an increase of caspase-8 activity when compared to the DMSO control (4× and 6× higher, respectively). These data agree with previous results (Fig. 5 and 6), and confirm that compound 3a can induce cell death via the extrinsic apoptotic pathway.
Evaluation of autophagy induction
Besides apoptosis, there are other different types of programmed cell death, such as autophagy, that are commonly triggered by different metal compounds.56 Autophagy is associated with an increase in autophagosomes (which are intracellular double membrane vesicles) that can fuse with lysosomes, resulting in cellular material degradation.57 Therefore, the induction of autophagy in HCT116 cells after 48 h of exposure to IC50 concentrations of the compounds 3a–c was evaluated (Fig. 8). | ||
| Fig. 8 Induction of autophagy in HCT116 cells after 48 h of exposure to the IC50 of compounds 3a, 3b and 3c. 0.1% (v/v) DMSO was used as the vehicle control, while 5 μM Cis, 0.4 μM Dox and 500 nM Rapamycin were used as positive controls. Data are expressed as the mean ± SEM of at least two independent biological assays. The symbols ***, ** and * represent p < 0.0001, p < 0.01 and p < 0.1, respectively. | ||
Fig. 8 shows that compound 3c, Dox and Cis can induce the autophagic process in HCT116 cells (77.1%, 61.3% and 65.2% of cells in autophagy, respectively), demonstrating 1.6-, 1.3- and 1.4-fold increase of autophagic vesicles over the control, respectively. Compounds 3a and 3b do not seem to induce this type of cell death mechanism. These results indicate that compound 3c can activate both cell death mechanisms through activation of the apoptosis and autophagy in HCT116 cells. These results are supported by the literature, where it is described that platinum compounds can induce apoptosis and autophagy.56
To further understand the triggering process of those cell death processes and loss of viability by the presence of the platinum compounds, the levels of intracellular reactive oxygen species (ROS) were measured.58
Production of reactive oxygen species (ROS)
The level of intracellular ROS was quantified in HCT116 cells after their exposure to the platinum compounds (Fig. 9) using the probe 2′,7′-dichlorohydrofluorescein diacetate (H2DCF-DA). This probe can diffuse into the cells, and once inside, can be deacetylated by intracellular esterases and oxidized by ROS, originating from the fluorescent molecule 2′,7′-dichlorofluorescein (DCF), which can be detected by flow cytometry.58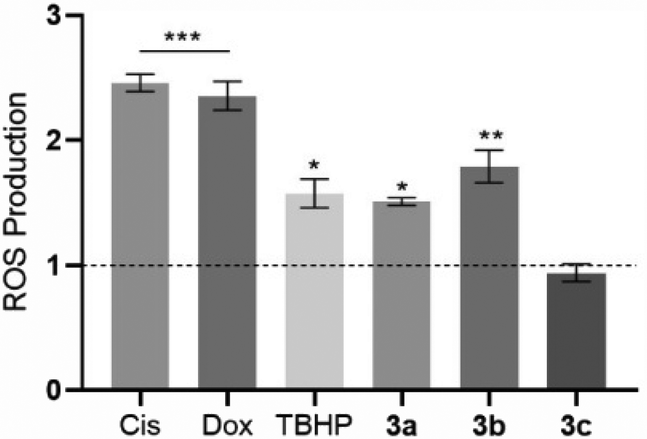 | ||
| Fig. 9 ROS produced by HCT116 cells after 48 h of exposure to the IC50 of compounds 3a–c. 0.1% (v/v) DMSO was used as the vehicle control, and 30 μM TBHP (tert-butyl hydroperoxide), 5 μM Cis and 0.4 μM Dox were used as positive controls. Data were normalized against the DMSO control (value represented as a dot line at y = 1) and expressed as the mean ± SEM of two independent assays. The symbols ***, ** and * represent p < 0.0001, p < 0.01 and p < 0.1, respectively. | ||
The results shown in Fig. 9 indicate that the compounds 3a and 3b can trigger ROS production in HCT116 cells, with a respective increase by 1.5-fold and 1.8-fold, over the DMSO control, with these values being close to the positive control TBHP (1.6× higher than the DMSO control). Intracellular ROS production by HCT116 cells in the presence of compounds 3a and 3b may lead to the activation of cell death by apoptosis.59,60 Induction of ROS production by the action of the platinum compounds has been described previously in the literature.61 Compound 3c does not seem to induce ROS production (Fig. 9).
Ex ovo chorioallantoic membrane (CAM) in vivo assay
The angiogenesis process, which consists of the formation of new blood vessels, has a determinant role in the tumor development, since it increases the nutrition of cancer cells and enables the invasion of adjacent tissues.62,63 Therefore, it is important to understand the role of new compounds in this process to achieve better cancer treatment.64The ex ovo chorioallantoic membrane (CAM) assay is a great in vivo model to evaluate the cytotoxicity of different compounds and their potentiality to modulate angiogenesis, being pro-angiogenic or anti-angiogenic. In this regards, CAMs of chicken embryos were treated with the IC50 concentrations of compounds 3a–c for 48 h. All newly formed vessels were counted at different time points (0 h (control), 24 h and 48 h) after exposure, and then compared with the formed vessels for DMSO 0.1% vehicle controls (Fig. 10).
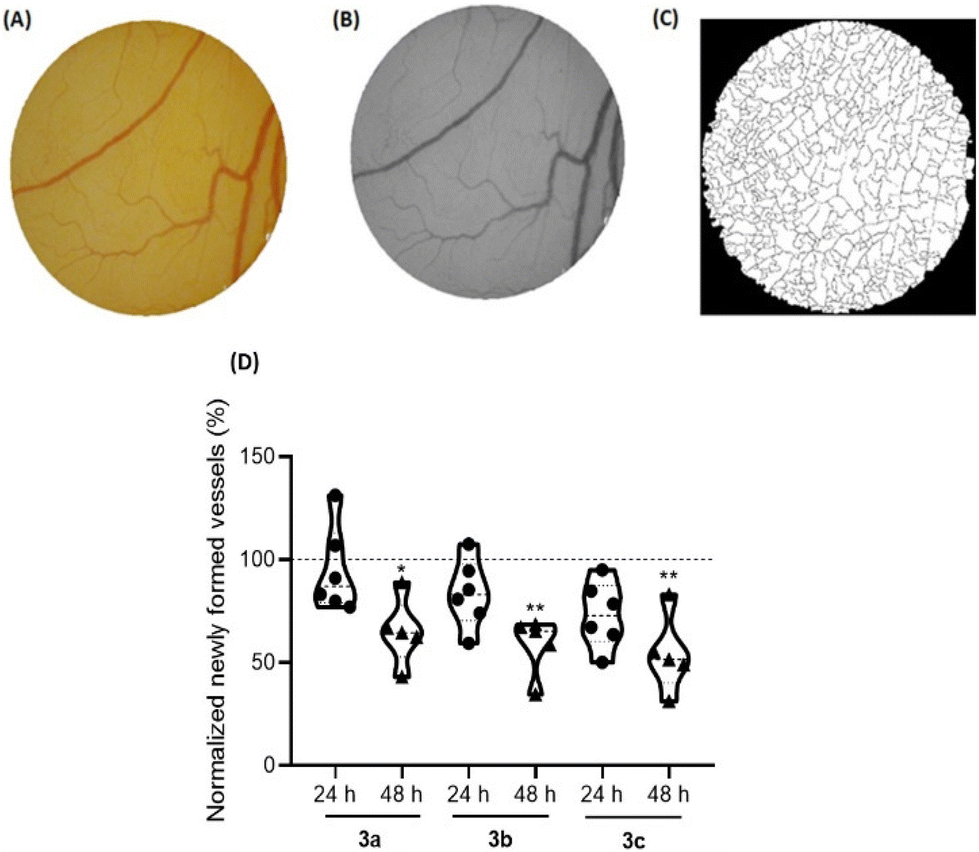 | ||
| Fig. 10 Newly formed blood vessels after exposure of chick embryos to the IC50 of compounds 3a–c for a duration of 48 h. (A) RGB image of the O-ring interior; (B) green channel of the same image used for counting the number of veins; (C) binary of the segmented image used to calculate the number of branches; (D) percentage of newly formed vessels in ex ovo YSMs after 24 h and 48 h exposure to IC50 concentrations of compounds 3a–c. At least 7 independent chicken embryos experiments were used for each condition. The values were normalized to the number of tertiary veins obtained after exposure to the control (0.1% DMSO) and the number of tertiary veins obtained in the corresponding o-ring at 0 hours of incubation in the same embryo. The 100% value refers to the 0.1% (v/v) DMSO sample in PBS. Symbols: dots – 24 hours, triangles – 48 h. The symbols ** and * represent p < 0.01 and p < 0.1, respectively. | ||
Based on Fig. 10, exposure to all compounds lead to a reduction of the newly formed blood vessels, indications that are in line with their anti-angiogenic potential at their IC50 concentrations.
Importantly, after 48 h of exposure to the IC50 concentration of these three platinum compounds, no in vivo toxicity to embryos were observed, considering their survival throughout the duration of the assay.
Conclusions
The study of the different structures synthesized in this work reveals a trend in the anticancer effect of the series. While the metallated compounds show differing levels of cytotoxicity, the tested ligands a, b and c do not show cytotoxicity in the tested tumor cell lines, disclosing that the observed cytotoxicity for the platinum compounds might not be attributed to them. However, the effect of the ancillary ligands was far greater than the substitution of the amine group. Among all studied compounds, the ones bearing dppm stood out for their high cytotoxicity in HCT116 and A2780 cancer cell lines. Moreover, compounds 3a, 3b and 3c do not show cytotoxicity in normal cells (fibroblasts), which is in line with their high selectivity index towards cancer cells when compared to normal cells (fibroblasts). All compounds were able to induce apoptosis via a depolarization of the mitochondria membrane potential. In the case of compound 3a, this is triggered via an extrinsic mechanism (probably associated with t-BIB). Conversely, compounds 3b and 3c can induce mitochondrial or intrinsic apoptosis via BAX. Moreover, compound 3c is able to induce cell death by autophagy. Compounds 3a and 3b are able to induce intracellular ROS, which might be associated with the loss of cell viability and cell death. It is also worth mentioning that these compounds are shown to be non-toxic in vivo in an ex ovo CAM model, although they show an anti-angiogenic potential after exposure to the IC50 of compounds 3a, 3b and 3c.Data availability
All the data are available within the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financed by national funds from FCT - Fundação para a Ciência e a Tecnologia, I.P., in the scope of the project DOI: https://doi.org/10.54499/UIDP/04378/2020 and DOI: https://doi.org/10.54499/UIDB/04378/2020 of the Research Unit on Applied Molecular Biosciences - UCIBIO and the project DOI: https://doi.org/10.54499/LA/P/0140/2020 of the Associate Laboratory Institute for Health and Bioeconomy - i4HB and doctoral grant 2021.08629.BD (S. Cordeiro). We thank Tatiana Fernandes for her help in performing the stability spectral measurements and western blot assay.This work was also made possible thanks to the financial support received from the Xunta de Galicia (Galicia, Spain) under the Grupos de Referencia Competitiva Programme (project GRC2019/14). F. R. thanks the Spanish Ministry of Education (grant FPU15/07145).
References
- A. C. Cope and R. W. Siekman, Formation of Covalent Bonds from Platinum or Palladium to Carbon by Direct Substitution, J. Am. Chem. Soc., 1965, 87(14), 3272–3273, DOI:10.1021/ja01092a063.
- R. Ujjval, M. Deepa, J. M. Thomas, C. Sivasankar and N. Thirupathi, Unusual [Pt{κ2(C,N)}]+ → [Pt{κ2(N,N)}]+ Coordination Mode Flip of the Guanidinate(1-) Ligand in Cationic N,N′,N″-Tris(3,5-Xylyl)Guanidinatoplatinum(II) Bis(Phosphine) Complexes. Syntheses, Structural and Theoretical Studies, Organometallics, 2020, 39(20), 3663–3678, DOI:10.1021/acs.organomet.0c00408.
- A. I. D'Aquino, H. F. Cheng, J. Barroso-Flores, Z. S. Kean, J. Mendez-Arroyo, C. M. McGuirk and C. A. Mirkin, An Allosterically Regulated, Four-State Macrocycle, Inorg. Chem., 2018, 57(7), 3568–3578, DOI:10.1021/acs.inorgchem.7b02745.
- M. E. Moustafa, P. D. Boyle and R. J. Puddephatt, Reactivity and Mechanism in Reactions of Methylene Halides with Cycloneophylplatinum(II) Complexes: Oxidative Addition and Methylene Insertion, J. Organomet. Chem., 2021, 941, 121083, DOI:10.1016/j.jorganchem.2021.121803.
- A. Ionescu, N. Godbert, A. Crispini, R. Termine, A. Golemme and M. Ghedini, Photoconductive Nile Red Cyclopalladated Metallomesogens, J. Mater. Chem., 2012, 22(44), 23617–23626, 10.1039/c2jm34946a.
- M. Iliş, M. Micutz and V. Cîrcu, Luminescent Palladium(II) Metallomesogens Based on Cyclometalated Schiff Bases and N-Benzoyl Thiourea Derivatives as Co-Ligands, J. Organomet. Chem., 2017, 836–837, 81–89, DOI:10.1016/j.jorganchem.2017.03.015.
- Á. Vivancos, D. Bautista and P. González-Herrero, Phosphorescent Tris-Cyclometalated Pt(IV) Complexes with Mesoionic N-Heterocyclic Carbene and 2-Arylpyridine Ligands, Inorg. Chem., 2022, 61(30), 12033–12042, DOI:10.1021/acs.inorgchem.2c02039.
- H. Torralvo, J. Albert, X. Ariza, M. Font-Bardia, J. Garcia, J. Granell and M. Martinez, Pyridine- A Nd Quinoline-Derived Imines as N, N-Bidentate Directing Groups in Palladium versus Platinum C-H Bond Activation Reactions, Organometallics, 2021, 40(2), 203–217, DOI:10.1021/acs.organomet.0c00703.
- H. Takahashi and J. Tsuji, Organic Syntheses by Means of Noble Metal Compounds: XXXIII. Carbonylation of Azobenzene-Palladium Chloride Complexes, J. Organomet. Chem., 1967, 10(3), 511–517 CrossRef CAS.
- J. Vicente, I. Saura-Llamas, J. Turpín, D. Bautista, C. R. De Arellano and P. G. Jones, Insertion of One, Two, and Three Molecules of Alkyne into the Pd-C Bond of Ortho-Palladated Primary and Secondary Arylalkylamines, Organometallics, 2009, 28(14), 4175–4195, DOI:10.1021/om9002895.
- W. A. Herrmann, C. Brossmer, K. Öfele, C.-P. Reisinger, T. Priermeier, M. Beller and H. Fischer, Palladacycles as Structurally Defined Catalysts for the Heck Olefination of Chloro– and Bromoarenes, Angew. Chem., Int. Ed. Engl., 1995, 34(17), 1844–1848, DOI:10.1002/anie.199518441.
- M. Beller, H. Fischer, W. A. Herrmann, K. Öfele and C. Brossmer, Palladacycles as Efficient Catalysts for Aryl Coupling Reactions, Angew. Chem., Int. Ed. Engl., 1995, 34(17), 1848–1849 CrossRef CAS.
- W. A. Herrmann, V. P. W. Böhm and C.-P. Reisinger, Application of Palladacycles in Heck Type Reactions, J. Organomet. Chem., 1999, 576(1), 23–41 CrossRef CAS.
- N. Miyaura and A. Suzuki, Stereoselective Synthesis of Arylated (E)-Alkenes by the Reaction of Alk-1-Enylboranes with Aryl Halides in the Presence of Palladium Catalyst, J. Chem. Soc., Chem. Commun., 1979, 19, 866–867 RSC.
- A. Suzuki, Carbon–Carbon Bonding Made Easy, Chem. Commun., 2005, 18, 4759–4763 RSC.
- F. Lucio-Martínez, L. A. Adrio, P. Polo-Ces, J. M. Ortigueira, J. J. Fernández, H. Adams, M. T. Pereira and J. M. Vila, Palladacycle Catalysis: An Innovation to the Suzuki-Miyaura Cross-Coupling Reaction, Dalton Trans., 2016, 45(44), 17598–17601, 10.1039/c6dt03542f.
- H. Qian, T. Zhang, L. Song, S. Yu, Q. Yuan, L. Sun, D. Zhang, Z. Yin and Y. Dai, Di- and Tetranuclear Palladium(II) Complexes Containing C,N-Bidentate Furoylhydrazone for Suzuki–Miyaura Reactions, Eur. J. Org. Chem., 2017, 10, 1337–1342, DOI:10.1002/ejoc.201700027.
- W. C. Wang, K. F. Peng, M. T. Chen and C. T. Chen, Palladacycles Bearing Tridentate CNS-Type Benzamidinate Ligands as Catalysts for Cross-Coupling Reactions, Dalton Trans., 2012, 41(10), 3022–3029, 10.1039/c2dt11097k.
- S. Ramírez-Rave, D. Morales-Morales and J. M. Grévy, Microwave Assisted Suzuki-Miyaura and Mizoroki-Heck Cross-Couplings Catalyzed by Non-Symmetric Pd(II) CNS Pincers Supported by Iminophosphorane Ligands, Inorg. Chim. Acta, 2017, 462, 249–255, DOI:10.1016/j.ica.2017.03.044.
- D. A. Alonso, C. Nájera and M. C. Pacheco, Oxime-Derived Palladium Complexes as Very Efficient Catalysts for the Heck-Mizoroki Reaction, Adv. Synth. Catal., 2002, 344(2), 172–183, DOI:10.1002/1615-4169(200202)344:2<172::AID-ADSC172>3.0.CO;2-9.
- C. Navarro-Ranninger, I. López-Solera, V. M. González, J. M. Pérez, A. Alvarez-Valdés, A. Martín, P. R. Raithby, J. R. Masaguer and C. Alonso, Cyclometalated Complexes of Platinum and Palladium with N-(4-Chlorophenyl)-α-Benzoylbenzylideneamine. In Vitro Cytostatic Activity, DNA Modification, and Interstrand Cross-Link Studies, Inorg. Chem., 1996, 35(18), 5181–5187, DOI:10.1021/ic960050y.
- N. Cutillas, G. S. Yellol, C. De Haro, C. Vicente, V. Rodríguez and J. Ruiz, Anticancer Cyclometalated Complexes of Platinum Group Metals and Gold, Coord. Chem. Rev., 2013, 257(19–20), 2784–2797, DOI:10.1016/j.ccr.2013.03.024.
- M. D. Aseman, S. Aryamanesh, Z. Shojaeifard, B. Hemmateenejad and S. M. Nabavizadeh, Cycloplatinated(II) Derivatives of Mercaptopurine Capable of Binding Interactions with HSA/DNA, Inorg. Chem., 2019, 58(23), 16154–16170, DOI:10.1021/acs.inorgchem.9b02696.
- M. Clemente, I. H. Polat, J. Albert, R. Bosque, M. Crespo, J. Granell, C. López, M. Martínez, J. Quirante, R. Messeguer, C. Calvis, J. Badía, L. Baldomà, M. Font-Bardia and M. Cascante, Platinacycles Containing a Primary Amine Platinum(II) Compounds for Treating Cisplatin-Resistant Cancers by Oxidant Therapy, Organometallics, 2018, 37(20), 3502–3514, DOI:10.1021/acs.organomet.8b00206.
- D. V. Aleksanyan, S. G. Churusova, Z. S. Klemenkova, R. R. Aysin, E. Y. Rybalkina, Y. V. Nelyubina, O. I. Artyushin, A. S. Peregudov and V. A. Kozlov, Extending the Application Scope of Organophosphorus(V) Compounds in Palladium(II) Pincer Chemistry, Organometallics, 2019, 38(5), 1062–1080, DOI:10.1021/acs.organomet.8b00867.
- F. Reigosa-Chamorro, L. R. Raposo, P. Munín-Cruz, M. T. Pereira, C. Roma-Rodrigues, P. V. Baptista, A. R. Fernandes and J. M. Vila, In Vitro and in Vivo Effect of Palladacycles: Targeting A2780 Ovarian Carcinoma Cells and Modulation of Angiogenesis, Inorg. Chem., 2021, 60(6), 3939–3951, DOI:10.1021/acs.inorgchem.0c03763.
- R. Matsa, P. Makam, M. Kaushik, S. L. Hoti and T. Kannan, Thiosemicarbazone Derivatives: Design, Synthesis and in Vitro Antimalarial Activity Studies, Eur. J. Pharm. Sci., 2019, 137, 104986, DOI:10.1016/j.ejps.2019.104986.
- G. Kalaiarasi, S. R. J. Rajkumar, S. Dharani, F. R. Fronczek, M. S. A. Muthukumar Nadar and R. Prabhakaran, Cyclometallated Ruthenium(II) Complexes with 3-Acetyl-2[H]-Chromene-2-One Derived CNS Chelating Ligand Systems: Synthesis, X-Ray Characterization and Biological Evaluation, New J. Chem., 2018, 42(1), 336–354, 10.1039/c7nj02877f.
- G. Kalaiarasi, S. R. Jeya Rajkumar, S. Dharani, F. R. Fronczek and R. Prabhakaran, Biological Evaluation of New Organoruthenium(II) Metallates Containing 3-Acetyl-8-Methoxy-2H-Chromen-2-One Appended CNS Donor Schiff Bases, J. Organomet. Chem., 2018, 866, 223–242, DOI:10.1016/j.jorganchem.2018.04.030.
- J. M. Vila, M. T. Pereira, J. M. Ortigueira, M. Graña, D. Lata, A. Suárez, J. J. Fernández, A. Fernández, M. López-Torres and H. Adams, Formation, Characterization, and Structural Studies of Novel Thiosemicarbazone Palladium(II) complexes. Crystal structures of [{Pd[C6H4C(Et)=NN=C(S)NH2]}4], [Pd{C6H4C(Et)=NN= C(S)NH2}(PMePh2)] and [{Pd[C6H4C(Et)=NN=C(S)NH2]}2(m-Ph2PCH2PPh2)], J. Chem. Soc., Dalton Trans., 1999, 23, 4193–4201, 10.1039/A906276I.
- D. Sequeira, P. V. Baptista, R. Valente, M. F. M. Piedade, M. H. Garcia, T. S. Morais and A. R. Fernande, Cu(I) complexes as new antiproliferative agents against sensitive and doxorubicin resistant colorectal cancer cells: syntehis, characterization, and mechanisms of action, J. Chem. Soc., Dalton Trans., 2021, 1845–1865, 10.1039/d0dt03566a.
- P. S. Pregosin and R. W. Kuntz, in 31P and 13C NMR of Transition Metal Phosphine Complexes, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer, 1979 Search PubMed.
- J. Albert, J. Granell, J. Sales, M. Font-Bardía and X. Solans, Optically Active Exocyclic Cyclopalladated Derivativesof Benzylidene-(R)-(1-phenylethy1)amines: Syntheses and X-ray Molecular Structures of [Pd(2-{(E)-(R)-CHMeN=CH-2′,6′-Cl2C6H3}C6H4)Cl(PPh3)] and [Pd(2-{(Z)-(R)-CHMeN=CH-2′,6′-F2C6H3}C6H4)I(PPh3)], Organometallics, 1995, 14, 1393–1404 CrossRef CAS.
- Promega Corporation. CellTiter 96 ® AQueous One Solution Cell Proliferation Assay. [accessed 03-01-2023] https://pl.promega.com/resources/protocols-/technical-bulletins/0/celltiter-96-aqueous-one-solution-cell-proliferation-assay-system-protocol/2012.
- G. Indrayanto, G. S. Putra and F. Suhud, Validation of in vitro bioassay methods: Application in herbal drug research. Profiles of Drug Substances, Excipients and Related Methodology, Elsevier Inc., 2021, vol. 46 Search PubMed.
- C. Roma-Rodrigues, R. Mendes, P. V. Baptista and A. R. Fernandes, Targeting tumor microenvironment for cancer therapy, Int. J. Mol. Sci., 2019, 20, 840, DOI:10.3390/ijms20040840.
- N. Svahn, A. J. Moro, C. Roma-Rodrigues, R. Puttreddy, K. Rissanen, P. V. Baptista, A. R. Fernandes, J. C. Lima and L. Rodríguez, The Important Role of the Nuclearity, Rigidity, and Solubility of Phosphane Ligands in the Biological Activity of Gold(I) Complexes, Chem. – Eur. J., 2018, 24(55), 14654–14667, DOI:10.1002/chem.201802547.
- K. Choroba, B. Machura, A. Szlapa-Kula, J. G. Malecki, L. Raposo, C. Roma-Rodrigues, S. Cordeiro, P. V. Baptista and A. R. Fernandes, Square Planar Au(III), Pt(II) and Cu(II) Complexes with Quinoline-Substituted 2,2′:6′,2″-Terpyridine Ligands: From in Vitro to in Vivo Biological Properties, Eur. J. Med. Chem., 2021, 218, 113404 CrossRef CAS PubMed.
- P. Nicholas, Cheremisinoff. Elemental and Structural Characterization Tests, in Polymer Characterization: Laboratory Techniques and Analysis, 1996, pp. 43–81 Search PubMed.
- M. Van Engeland, L. J. W. Nieland, F. C. S. Ramaekers, B. Schutte and C. P. M. Reutelingsperger, Annexin V–Affinity assay: A review on an apoptosis detection system based on Phosphatidylserine Exposure, Cytometry, 1998, 9, 1–9 CrossRef.
- K. Emoto, N. Toyama-Sorimachi, H. Karasuyama, K. Inoue and M. Umeda, Exposure of phosphatidylethanolamine on the surface of apoptotic cells, Exp. Cell Res., 1997, 232, 430–434 CrossRef CAS PubMed.
- Z. Darzynkiewicz, G. Juan, X. Li, W. Gorczyca, T. Murakami and F. Traganos, Cytometry in cell necrobiology: Analysis of apoptosis and accidental cell death (necrosis), Cytometry, 1997, 27, 1–20 CrossRef CAS PubMed.
- Q.-P. Qin, S.-L. Wang, M.-X. Tan, Y.-C. Liu, T. Meng, B.-Q. Zou and H. Liang, Synthesis of two platinum(II) complexes with 2-methyl-8-quinolinol derivatives as ligands and study of their antitumor activities, Eur. J. Med. Chem., 2019, 161, 334–342 CrossRef CAS PubMed.
- F. Mihlon, C. E. Ray and W. Messersmith, Chemotherapy agents: A primer for the interventional radiologist, Semin. Intervent. Radiol., 2010, 27, 384–390 CrossRef PubMed.
- A. M. Meredith and C. R. Dass, Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism, J. Pharm. Pharmacol., 2016, 68, 729–741 CrossRef CAS PubMed.
- E. Khodapasand, N. Jafarzadeh, F. Farrokhi, B. Kamalidehghan and M. Houshmand, Is Bax/Bcl-2 ratio considered as a prognostic marker with age and tumor location in colorectal cancer?, Iran. Biomed. J., 2015, 19, 69–75 Search PubMed.
- B. Kulsoom, T. S. Shamsi, N. A. Afsar, Z. Menom, N. Ahmed and S. N. Hasnain, Bax, Bcl-2, and Bax/Bcl-2 as prognostic markers in acute myeloid leukemia: Are we ready for bcl-2-directed therapy?, Cancer Manage. Res., 2018, 10, 403–416 CrossRef CAS PubMed.
- C. M. Pfeffer and A. T. K. Singh, Apoptosis: A target for anticancer therapy, Int. J. Mol. Sci., 2018, 19 CAS.
- J. D. Ly, D. R. Grubb and A. Lawen, The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update, Apoptosis, 2003, 8, 115–128 CrossRef CAS PubMed.
- A. Perelman, C. Wachtel, M. Cohen, S. Haupt, H. Shapiro and A. Tzur, JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry, Cell Death Dis., 2012, 3, 1–7 Search PubMed.
- K. F. Cooper, Till death do us part: The marriage of autophagy and apoptosis, Oxid. Med. Cell. Longevity, 2018, 2018, 4701275, DOI:10.1155/2018/4701275.
- M. S. D'Arcy, Cell death: a review of the major forms of apoptosis, necrosis and autophagy, Cell Biol. Int., 2019, 43, 582–592 CrossRef PubMed.
- S. Zaman, R. Wang and V. Gandhi, Targeting the apoptosis pathway in hematologic malignancies, Leuk. Lymphoma, 2014, 55, 1980–1992 CrossRef CAS PubMed.
- K. Suntharalingam, J. J. Wilson, W. Lin and S. J. Lippard, A dual-targeting, p53-independent, apoptosis-inducing platinum(II) anticancer complex, [Pt(BDIQQ)]Cl, Metallomics, 2014, 6(3), 437–443 CrossRef CAS PubMed.
- C. Roma-Rodrigues, G. Malta, D. Peixoto, L. M. Ferreira, P. V. Baptista, A. R. Fernandes and P. S. Branco, Synthesis of new hetero-arylidene-9(10H)-anthrone derivatives and their biological evaluation, Bioorg. Chem., 2020, 99, 103849 CrossRef CAS PubMed.
- T. Ichimiya, T. Yamakawa, T. Hirano, Y. Yokoyama, Y. Hayashi, D. Hirayama, K. Wagatsuma, T. Itoi and H. Nakase, Autophagy and autophagy–related diseases: A review, Int. J. Mol. Sci., 2020, 21, 1–21 Search PubMed.
- F. Yang, K. Xu, Y.-G. Zhou and T. Ren, Insight into autophagy in platinum resistance of cancer, Int. J. Clin. Oncol., 2023, 28, 354–362 CrossRef CAS PubMed.
- I. Liguori, G. Russo, F. Curcio, G. Bulli, L. Aran, D. Della-Morte, G. Gargiulo, G. Testa, F. Cacciatore, D. Bonaduce and P. Abete, Oxidative stress, aging, and diseases, Clin. Interventions Aging, 2018, 13, 757–772 CrossRef CAS PubMed.
- Abcam. DCFDA/H2DCFDA - Cellular ROS Assay Kit (ab113851). Oxidative Stress [accessed 04-01-2023] https://www.abcam.com/dcfda-h2dcfda-cellular-ros-assay-kit-ab113851.html.
- D. Kashyap, A. Sharma, V. Garg, H. S. Tuli, G. Kumar, M. Kumar and T. Mukherjee, Reactive Oxygen Species (ROS): an Activator of Apoptosis and Autophagy in Cancer, J. Biol. Chem. Sci., 2016, 3, 256–264 Search PubMed.
- Q. Xie, G. Lan, Y. Zhou, J. Huang, Y. Liang, W. Zheng, X. Fu, C. Fan and T. Chen, Strategy to enhance the anticancer efficacy of X-ray radiotherapy in melanoma cells by platinum complexes, the role of ROS-mediated signaling pathways, Cancer Lett., 2014, 354, 58–67 CrossRef CAS PubMed.
- F. Finetti and L. Trabalzini, Bidimentional In Vitro Angiogenic Assays to Study CCM Pathogenesis: Endothelial Cell Proliferation and Migration, in Methods in Molecular Biology, 2020, vol. 2152, pp. 377–385 Search PubMed.
- A. K. M. N. Hossian and G. Mattheolabakis, Cellular Migration Assay: An In Vitro Technique to Simulate the Wound Repair Mechanism, in Methods in Molecular Biology, 2021, vol. 2193, pp. 77–83 Search PubMed.
- D. Kobelt, W. Walther and U. S. Stein, Real-Time Cell Migration Monitoring to Analyze Drug Synergism in the Scratch Assay Using the IncuCyte System, in Methods in Molecular Biology, 2021, vol. 2294, pp. 133–142 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2309646 (1b) and 2309120 (2c). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01490a |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |