
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis and bonding of 4-ferrocenyl-1,2,4-triazol-5-ylidene complexes†
Michal
Franc
 ,
Jiří
Schulz
and
Petr
Štěpnička
*
,
Jiří
Schulz
and
Petr
Štěpnička
*
Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 40 Prague, Czech Republic. E-mail: stepnic@natur.cuni.cz
First published on 18th June 2024
Abstract
Ferrocene-substituted carbenes have emerged as attractive, redox-active ligands. However, among the compounds studied to date, ferrocenylated 1,2,4-triazol-5-ylidenes, which are closely related to the archetypal imidazol-2-ylidenes, are still unknown. Here, we demonstrate that the triazolium salt [CHN(Me)NCHN(Fc)]I (2; Fc = ferrocenyl), obtained by alkylation of 4-ferrocenyl-4H-1,2,4-triazole (1) with MeI, reacts selectively with metal alkoxide/hydroxide precursors [(cod)Rh(OMe)]2 and [(IPr)Au(OH)] (cod = cycloocta-1,5-diene, IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) to produce the ferrocene-substituted 1,2,4-triazol-5-ylidene complexes [(cod)RhI{CN(Me)NCHN(Fc)}] and [(IPr)Au{CN(Me)NCHN(Fc)}]I in good yields. The complexes were characterised by NMR and IR spectroscopy, mass spectrometry, cyclic voltammetry, and single-crystal X-ray diffraction analysis. Density function theory (DFT) calculations were used to rationalise the electrochemical behaviour of the carbene complexes and to elucidate the bonding situation in these compounds. An analysis using intrinsic bond orbitals (IBOs) revealed that the 1,2,4-triazol-5-ylidene ligand exerted a strong trans influence and showed a synergistic stabilisation by the negative inductive and positive π-donor effects of the nitrogen atoms adjacent to the carbene carbon atom; these effects were enhanced by conjugation with the CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond at the exterior, similar to that in imidazol-2-ylidenes.
N bond at the exterior, similar to that in imidazol-2-ylidenes.
Introduction
Pioneering experiments by Wanzlick1 and Öfele2 in the 1960s and subsequent isolation of the first3 N-heterocyclic carbene (NHC) three decades later4 opened a new, exciting area of chemistry. The discovered NHCs were rapidly recognised as attractive ligands for transition metals and metal-mediated reactions and as organocatalysts, which further accelerated the research focused on these compounds.5 Soon, the family of NHCs, which were originally derived from imidazoles, was expanded to include compounds based on other heterocyclic scaffolds, such as pyrrolidine, thiazole, and triazoles.5In this regard, carbenes resulting from readily accessible6 1,2,3-triazoles (i.e., 1,2,3-triazol-5-ylidenes), reported first in 2008,7 have a distinct position as examples of mesoionic carbenes, whose structures cannot be shown without adding charges to some of the ring atoms (Scheme 1). In contrast, isomeric compounds derived from 1,2,4-triazoles (i.e., 1,2,4-triazol-5-ylidenes), which can be regarded as imidazol-2-ylidene analogues with an additional nitrogen atom inserted in the ring, are less explored8,9 even though they are equally structurally versatile and widely applicable. Thus far, 1,2,4-triazolylidenes have been used as auxiliary ligands in the design of transition metal catalysts,10 new luminescent materials11 and biologically active compounds,12 and further utilised as organocatalysts.13
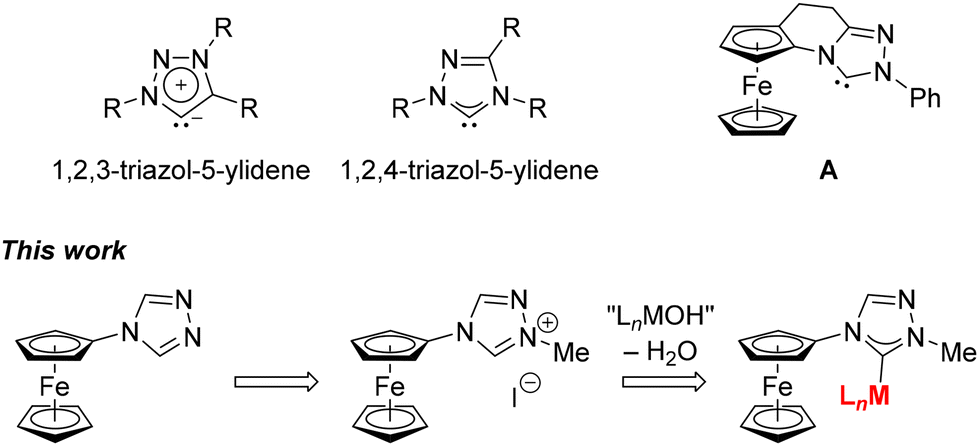 | ||
| Scheme 1 (top) General structures of 1,2,3- and 1,2,4-triazol-5-ylidenes and the ferrocene-based 1,2,4-triazol-5-ylidene A, and (bottom) the aim of this work. | ||
Despite their wide applications, 1,2,4-triazol-5-ylidenes have not yet established themselves among the ferrocene carbenes,14 unlike their 1,2,3-isomers, which have been studied as redox-active and redox-switchable ligands.15 To the best of our knowledge, the sole exception is the planar chiral carbene A (Scheme 1), which was trapped and structurally authenticated as a CuCl complex.16,17 Based on this, we decided to synthesise and study the 1,2,4-triazol-5-ylidene complexes derived from 4-ferrocenyl-4H-1,2,4-triazole (1),18 which bears the ferrocenyl substituent directly at the triazole ring. The synthesis was accomplished by using simple reactions of the corresponding triazolium salt with suitable metal hydroxide and alkoxide complexes19 that act as both the base and the metal source.
Results and discussion
The parent azole, 4-ferrocenyl-4H-1,2,4-triazole (1),18 was prepared by acid-catalysed condensation of aminoferrocene20 with N,N′-bis(dimethylaminomethylene)hydrazine21 in refluxing toluene (Scheme 2) and isolated in a 70% yield after chromatography (see the ESI†).22 Subsequent alkylation with methyl iodide produced triazolium salt 223 (88% yield). Attempts to obtain a doubly methylated derivative24 (as an entry to biscarbene complexes25) by reacting 1 with an excess of MeI or [Me3O][BF4] or, alternatively, by reacting 2 with [Me3O][BF4] were unsuccessful.26 | ||
| Scheme 2 Preparation and alkylation of 4-ferrocenyl-4H-1,2,4-triazole (1) (TsOH = 4-toluenesulfonic acid). | ||
The 1H and 13C{1H} NMR spectra of 1 showed the resonances due to the ferrocenyl substituent, with the signal of Cipso–N characteristically shifted to a low field (δC 90.5; cf. δC 105.6 for aminoferrocene27). The signals of the triazole ring (CHN) were detected at δH 8.36 and δC 143.0 (in CDCl3). Upon alkylation, the 1H and 13C{1H} NMR signals of the ferrocene CH groups shifted to a lower field, while the Cipso–N signal moved slightly upfield (δC 89.3 in CD2Cl2/CD3OD). Methylation also differentiated the triazole CH groups, particularly in the 1H NMR spectrum (δH 8.50, 11.6; δC ≈ 142.3 for both carbons); the signal of the methyl substituent was observed at δH 4.36 and δC 42.0.
Compound 2 cleanly reacted with [Rh(μ-OMe)(cod)]2 (cod = 1,5-cycloocta-1,5-diene) in dry dichloromethane to produce carbene complex 3 (Scheme 3),28 which was obtained in 73% yield after column chromatography. Compound 3 was stable under ambient conditions and dissolved well in common organic solvents, which made its crystallisation difficult. Crystals suitable for structure determination were eventually grown by diffusion of water vapour into a solution of the complex in acetone, which further illustrated the high chemical stability of this compound. In contrast, the complex decomposed when treated with MeI or [Me3O][BF4].
 | ||
| Scheme 3 Synthesis of carbene complexes 3 and 4. | ||
Similarly, the triazolium salt underwent a smooth reaction with the Au(I) hydroxide [(IPr)Au(OH)]29 (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene-κC2), generating bis-carbene complex 4 (Scheme 3),30 which was isolated by flash chromatography in a 90% yield. Even in this case, further methylation with MeI or Meerwein salt was not successful.
The formation of carbene complexes was indicated by their distinct NMR features. In particular, the 13C{1H} NMR spectrum of 3 displayed a low-field signal attributable to the carbene carbon (δC 185.9), split into a doublet by 103Rh (1JC–Rh = 49 Hz). Because of hindered molecular mobility, the CH groups in the ferrocene C5H4 ring were diastereotopic and produced four separate signals in the 1H and 13C{1H} NMR spectra, and similar features were observed for the CH and CH2 signals of the diene ligand. The 13C NMR signals of the η2-coordinated double bonds were observed at δC 71.9/72.5 (d, 1JC–Rh = 14 Hz) and 96.3/96.9 (d, 1JC–Rh = 7 Hz), being markedly distinguished by ligands in the trans positions.
For 4, two carbene 13C NMR signals were observed at δC 183.5 (triazolylidene) and 185.5 (IPr). Unlike 3, however, the signals of the ferrocene unit in 4 were degenerate; specifically, only two signals were found for the CH groups at the C5H4 ring, and even the resonances of the isopropyl substituents at the IPr ligand were undifferentiated despite the overall steric bulk. The electrospray ionisation (ESI) mass spectra showed ions due to [M − I]+ resulting from ligand loss (3) and simple dissociation (4).
Structure determination of 3 (Fig. 1) revealed square planar coordination around the Rh(I) centre with η2-coordinated double bonds oriented nearly perpendicular to the plane defined by the rhodium and the remaining donor atoms {Rh1, I1, C11} (angles 82.3(1)° and 81.0(1)° for the C21C22 and C25C26 bonds, respectively). The individual Rh–C distances (Rh1–C21 2.118(2), Rh1–C22 2.124(2), Rh1–C25 2.209(2), and Rh1–C26 2.220(2) Å) varied, depending on the trans influence of the remaining ligands (carbene > I),31,32 in agreement with the NMR data; the CC bond lengths were inversely affected (C21–C22 1.410(3) Å, C25–C26 1.373(3) Å).
 | ||
| Fig. 1 Molecular structure of 3. | ||
The Rh1–C11 distance in 3 (2.016(2) Å) did not significantly differ from those in the analogous complexes featuring ring-fused triazolylidenes33 and triazolium-ylidene ligands,34 while the Rh1–I1 bond (2.6849(4) Å) had a similar length as that in [RhI(cod)L] (L = 1,3-dimethylimidazolin-2-ylidene).28b The ferrocene unit in 3 showed similar Fe–C distances (2.032(2)–2.052(2) Å) and only minor tilting35 of the cyclopentadienyl rings (4.0(1)°).
The structure of 4·CHCl3 (Fig. 2) comprised symmetrically and linearly coordinated Au(I) ion (Au1–C11 2.020(4), Au1–C21 2.016(4) Å; C11–Au1–C21 178.5(2)°); the Au–C distances were unexceptional considering those reported for the symmetrical complexes [Au(IPr)2][BF4]36 and [AuL2][AuI2] (L = 1,4-dimethyl-1,2,4-triazol-5-ylidene).37 The carbene moieties in 4 were mutually twisted by 20.4(3)°, and the triazolylidene ring was rotated by 21.6(3)° from the plane of the cyclopentadienyl ring C(1–5). The ferrocene unit had its usual geometry with parallel cyclopentadienyl rings (Fe–C 2.022(4)–2.054(5) Å, tilt: 0.8(3)°).
 | ||
| Fig. 2 View of the complex cation in the structure of 4·CHCl3. | ||
While the structure of 3 was essentially molecular, the cooperative, soft hydrogen bonds between the polarised CH groups at the carbene ligands and the halogen atoms (the iodide anion and chlorine from the solvent molecule) were detected in the crystal assembly of ionic 4·CHCl3 (see the ESI, Fig. S3†).
The complexes were studied by cyclic voltammetry at a glassy carbon electrode in dichloromethane containing [Bu4N][PF6] (0.1 M) (data for triazole 1 are presented in the ESI†). The redox behaviour of Rh(I) complex 3 was relatively complicated (Fig. 3): the compound underwent one-electron oxidation at 0.19 V vs. the ferrocene/ferrocenium reference,38 which was followed by two irreversible redox steps at approximately 0.68 and 0.95 V (the anodic peak potentials at a scan range of 100 mV s−1 are given).39 The peak currents of the two irreversible oxidations were lower than that of the first reversible oxidation step, and the shape of the last oxidation indicated the possible involvement of adsorption phenomena. After traversing the second oxidative step, the cathodic branch of the first wave gained a “shoulder” as a result of convolution with a minor reductive wave due to an electrochemically generated species.
 | ||
| Fig. 3 Cyclic voltammograms of 3 (glassy carbon disc electrode, dichloromethane, 0.1 M Bu4N[PF6], scan rate: 100 mV s−1). The scan direction is indicated with an arrow, and the voltammograms recorded over different potential ranges are distinguished by colour; the second scan is shown as a dashed line. | ||
In contrast, complex 4 displayed single reversible oxidation at 0.32 V, which was preceded by a relatively weaker, broad peak, likely due to adsorbed species (Fig. 4). The oxidation occurred at a more positive potential than that of 3, in line with the cationic nature of complex 4 and with the absence of the purely donor ligands in the structure (the carbenes are π-acceptors), both making electron removal more difficult.
 | ||
| Fig. 4 Cyclic voltammogram of 4 (glassy carbon disc electrode, dichloromethane, 0.1 M Bu4N[PF6], scan rate: 100 mV s−1). The scan direction is indicated with an arrow, and the second scan is shown as a dashed line. | ||
An analysis of the frontier molecular orbitals (FMOs) using the natural atomic orbital (NAO) approach40 revealed that both HOMO and LUMO of complex 3 were localised mainly at the Rh(cod)I fragment, while for 4, the HOMO was confined to the ferrocene unit, and the LUMO was evenly distributed between the two Au-bound carbene ligands (for details, see ESI†). However, to rationalise the electrochemical behaviour, we investigated electron density changes associated with electron removal.41 Indeed, the calculated electron density change between the parent species and its oxidized form with the same geometry, ρ(3) − ρ(3+) and ρ(4) − ρ(4+), indicated that the initial oxidation indeed occurs at the ferrocene moiety (Fig. 5).
 | ||
| Fig. 5 Electron difference maps for ρ(3) − ρ(3+) (left), and ρ(4) − ρ(4+) (right; only the cation is shown for clarity) mapped at the geometry of the native species 3 and 4 (isosurfaces at ±0.02 a.u.). | ||
The bonding of the carbene complexes was investigated using intrinsic bond orbitals (IBOs).42 For complex 3, the bonding situation could be effectively described by the traditional Dewar–Chatt–Duncanson model:43 the diene ligand transfers four electrons from the bonding π orbitals of the two CC bonds to a metal-centred vacant orbital (Fig. 6), while the metal donates electrons back to the antibonding π*(CC) orbitals. However, a closer inspection of the IBOs describing these interactions showed that the coordination of the diene to the rhodium was asymmetrical, with the asymmetry reflecting the different trans influence of the other ligands:44 the coordination of the double bond in the trans position to the carbene ligand [0.84(C25):0.88(C26)/0.24(Rh)] was weakened compared to that of the double bond located trans to the iodide ligand [0.77(C21):0.84(C22)/0.36(Rh)]. A lower π donation diminished Rh→CC π-back donation [1.73(Rh)/0.12(C25):0.11(C26)]. Both effects were reflected in the different Rh–C bond lengths, as determined experimentally (vide supra) and calculated (Table 1), and in the different bond orders of the non-equivalent double bonds. The calculated Mayer bond order of the CC bond trans to the carbene ligand was greater (MBO 1.42), reflecting a lower back-donation to the antibonding π*(CC) orbitals, while the increased Rh→CC interaction [1.59(Rh)/0.21(C21):0.17(C22)] weakened the CC bond in the trans position with respect to the iodide ligand (MBO 1.23).
 | ||
| Fig. 6 Selected intrinsic bond orbitals (IBOs) of 3. The values in parentheses indicate the fraction of the bonding electrons assigned to the individual atoms. Atom labelling follows that in the crystal structure (see Fig. 1). The IBO corresponding to the standard I→Rh dative bond with the following parameters is not shown [1.64(I)/0.29(Rh)]. | ||
| Bond | Distance [Å] | MBO | WBI | |
|---|---|---|---|---|
| Exp. | Calc. | |||
| a Theoretical values were estimated at the PBE0(d3)/def2-TZVP:sdd(Rh,I) level of theory. | ||||
| Rh–C11 | 2.016(2) | 2.007 | 0.69 | 1.04 |
| Rh–I | 2.6849(4) | 2.721 | 0.82 | 1.54 |
| Rh–C21 | 2.118(2) | 2.101 | 0.66 | 0.84 |
| Rh–C22 | 2.124(2) | 2.124 | 0.68 | 0.80 |
| Rh–C25 | 2.209(2) | 2.209 | 0.59 | 0.66 |
| Rh–C26 | 2.220(2) | 2.220 | 0.54 | 0.63 |
| C21–C22 | 1.410(3) | 1.410 | 1.23 | 1.50 |
| C25–C26 | 1.373(3) | 1.373 | 1.42 | 1.64 |
The IBOs describing the Rh–carbene [1.54(C)/0.40(Rh)] and Rh–I bonds [1.64(C)/0.29(Rh)] showed values typical of L-type,45 dative σ interactions. For the carbene, the σ interaction was supported by Rh→Ctriaz π-back bonding [1.89(Rh)/0.07(C)], albeit less than for the cod ligand.
IBO analysis of complex 4 focused mainly on the comparison between the two carbene ligands. Both carbenes coordinated as L-type ligands, with the σ-bonding component dominating over π-back bonding (Fig. 7).46 The donor properties of both ligands were rather similar, although the IPr ligand appeared to be a better σ donor (based on the comparison of charge distribution in the respective IBOs: [1.54(CIPr)/0.40(Au)] vs. [1.56(Ctriaz)/0.38(Au)]), whereas the triazolylidene ligand behaved as a better acceptor ([1.91(Au)/0.03(CIPr)/0.04(Ctriaz)]).
 | ||
| Fig. 7 Selected intrinsic bond orbitals (IBOs) of 4. The values in parentheses indicate the fraction of bonding electrons assigned to the individual atoms. Atom labelling follows that in the crystal structure (see Fig. 2). | ||
Further analysis, focused on the bonding within the carbene ligands (see the ESI, Fig. S6 and S7†), revealed inductive and conjugative stabilisation of the carbene centres, in agreement with the generally accepted bonding scheme for NHCs.47 The IBOs corresponding to the C–N σ-bonds were polarised towards the nitrogen atoms, whereas the nitrogen lone electron pairs were delocalised into the vacant 2p(C) orbital. In the IPr ligands, these two bonding components were symmetrical, but in the triazolylidene, the lone electron pair of the ferrocenyl-bound nitrogen atom contributed less than the nitrogen from the N–N bond. Thus, the better π-acceptor properties of the triazole ligand potentially resulted from the lower charge accumulated on the carbene carbon atom by conjugation (∑(π) = 0.83 in the triazolylidene vs. ∑(π) = 0.87 in the IPr ligand). The lower π electron density at the carbene carbon in the triazolylidene thus reflected the replacement of one carbon in the conjugated double bond at the exterior for the more electronegative nitrogen, which reduced the net charge transferred to the carbene 2p(C) orbital from the adjacent nitrogen atoms.
Correspondingly, the IBO analysis of the hypothetical species [Au(SIPr){C(NMe)NCHN(Fc)}]+ (Fc = ferrocenyl) featuring the analogous, ring-saturated carbene ligand (SIPr = 1,3-bis(2,6-diisopropylphenyl)imidazolidin-2-ylidene) that lacks carbene stabilisation by the π electron density of the CC or CN bond showed that SIPr was indeed more electrophilic than the triazolylidene ligand. The cumulative charge contributed by the carbene π-system, ∑(π), was 0.71. The overall backdonation from Au to the carbene carbons remained the same as that in 4 but was reversed in favour of the SIPr ligand [1.90(Au)/0.03(Ctriaz)/0.04(CSIPr)].
Conclusion
In this study, we describe the synthesis of 1,2,4-triazol-5-ylidene complexes bearing a redox-active ferrocenyl substituent directly at the heterocyclic carbene ligand from the readily accessible 4-ferrocenyl-4H-1,2,4-triazole. The reactions between the N-alkylated triazole and metal hydroxide/alkoxide precursors proceed cleanly, rapidly, and with a high atom economy, producing the targeted carbene complexes in good yields. The collected characterisation data, supported by the DFT calculations, corroborate the high trans influence of the triazolylidene ligand and the mode of stabilisation of the carbene moiety typical for NHCs based on the synergy between the inductive effect of the electronegative nitrogen atoms in the adjacent positions that reduces the σ electron density at the carbon atom and the conjugation between the nitrogen lone pairs and the carbene 2p orbital. Compared to IPr as a prototypical carbene ligand, the triazolylidene appears to be a slightly weaker σ donor and stronger π acceptor.Experimental
Materials and methods
All syntheses were performed under a nitrogen atmosphere using standard Schlenk techniques and oven-dried glassware. Aminoferrocene48 and [(IPr)Au(OH)]49 were prepared by following the literature methods; the synthesis of 1 and 2 is described in the ESI.† Other chemicals were purchased from commercial suppliers (Sigma-Aldrich, TCI) and used as received. Toluene was dried over sodium metal and distilled under nitrogen. Dry dichloromethane was obtained from a PureSolv MD5 solvent purification system (Innovative Technology). Solvents used for chromatography and crystallisations were employed without any purification (Lach-Ner, analytical grade).The NMR spectra were recorded on a Varian UNITY Inova 400 spectrometer at 25 °C. Chemical shifts (δ/ppm) are expressed relative to internal SiMe4. FTIR spectra were recorded with a Nicolet iS50 instrument (Thermo Fisher Scientific) over the 400–4000 cm−1 range. ESI MS spectra were recorded with a Compact QTOF-MS spectrometer (Bruker Daltonics). The samples were dissolved in HPLC-grade methanol. Elemental analyses were performed with a PE 2400 Series II CHNS/O Elemental Analyser (PerkinElmer). The amount of residual solvent was corroborated by NMR analysis.
Electrochemical measurements were carried out at room temperature using an μAUTOLAB III instrument (Eco Chemie) and a three-electrode cell equipped with a glassy carbon disc (2 mm diameter) working electrode, a platinum auxiliary electrode, and an Ag/AgCl (3 M KCl) reference electrode. The samples were dissolved in dry dichloromethane to produce a solution containing approximately 1 mM of the analysed compound and 0.1 M [Bu4N][PF6] as the supporting electrolyte (Sigma-Aldrich, puriss. for electrochemistry). The solutions were deaerated by bubbling with argon before the measurement and then maintained under an argon flow. Decamethylferrocene (Alfa-Aesar) was used as an internal standard during the last scans, and the potentials were converted into the ferrocene/ferrocenium scale by subtracting 0.548 V.50
Details on the structure determination and DFT calculations are available in the ESI.†
Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The first yellow band was collected and evaporated, leaving complex 3 as a yellow-orange solid. Yield: 28.9 mg (73%). The single-crystal used for the structural determination was grown by dissolving the compound in acetone in a small vial and slowly diffusing water vapour in a large test tube.
:1). The first yellow band was collected and evaporated, leaving complex 3 as a yellow-orange solid. Yield: 28.9 mg (73%). The single-crystal used for the structural determination was grown by dissolving the compound in acetone in a small vial and slowly diffusing water vapour in a large test tube.
1H NMR (400 MHz, CDCl3): δ 1.50–1.65 (m, 1H, CH2 of cod), 1.67–1.79 (m, 1H, CH2 of cod), 1.82–1.99 (m, 3H, CH2 of cod), 2.12–2.39 (m, 3H, CH2 of cod), 2.98–3.08 (m, 1H, CH of cod), 3.32–3.46 (m, 1H, CH of cod), 4.20 (s, 3H, CH3), 4.24 (td, J = 2.6, 1.4 Hz, 1H, C5H4), 4.28 (s, 5H, C5H5), 4.35 (td, J = 2.6, 1.4 Hz, 1H, C5H4), 4.44 (td, J = 2.6, 1.4 Hz, 1H, C5H4), 5.25–5.33 (m, 2H, CH of cod), 6.36 (td, J = 2.6, 1.4 Hz, 1H, C5H4), 8.27 (s, 1H, NCH). 13C{1H} NMR (151 MHz, CDCl3): δ 29.52 (s, CH2 of cod), 29.57 (s, CH2 of cod), 31.57 (s, CH2 of cod), 32.40 (s, CH2 of cod), 40.74 (s, CH3), 62.03 (s, CH of C5H4), 66.16 (s, CH of C5H4), 66.52 (s, CH of C5H4), 67.62 (s, CH of C5H4), 70.04 (s, C5H5), 71.90 (d, 1JC–Rh = 14 Hz, CH of cod), 72.53 (d, 1JC–Rh = 14 Hz, CH of cod), 92.95 (s, Cipso–N C5H4), 96.32 (d, 1JC–Rh = 7 Hz, CH of cod), 96.90 (d, 1JC–Rh = 7 Hz, CH of cod), 142.27 (s, NCH), 185.94 (d, 1JC–Rh = 49 Hz, Cipso–Rh). ESI+ MS: m/z 478 ([M − I]+). IR (DRIFTS, KBr): νmax 3921 w, 3110 s, 3096 m, 3075 m, 3055 w, 2998 w, 2982 s, 2966 m, 2939 s, 2910 m, 2874 s, 2828 m, 1793 w, 1732 w, 1618 w, 1533 s, 1517 m, 1492 s, 1474 m, 1460 m, 1426 s, 1409 m, 1367 m, 1350 m, 1335 m, 1304 s, 1252 w, 1227 m, 1216 w, 1198 m, 1174 w, 1153 w, 1130 m, 1105 m, 1075 m, 1062 w, 1047 m, 1030 m, 1000 s, 993 m, 963 s, 959 s, 888 m, 877 s, 859 m, 843 m, 829 s, 824 s, 794 w, 784 m, 763 w, 720 m, 700 m, 681 w, 649 m, 637 w, 523 m, 506 s, 491 m, 471 s, 442 w, 430 w cm−1. Anal. calc. for C21H25FeIN3Rh (605.10): C 41.68, H 4.16, N 6.94%. Found: C 41.94, H 4.08, N 6.91%.
:1). Evaporation of a single yellow band produced complex 4 as a yellow-orange solid. The yield of 4·1/2CH2Cl2 was 92.0 mg (90%). The crystal used for structure determination was obtained from a dichloromethane solution layered with methyl tert-butyl ether.
1H NMR (400 MHz, CDCl3): δ 1.22 (d, 3JHH = 6.9 Hz, 12H, CHMe2), 1.27 (d, 3JHH = 6.9 Hz, 12H, CHMe2), 2.52 (sept, 3JHH = 6.9 Hz, 4H, CHMe2), 3.28 (s, 3H, CH3), 3.96 (t, J = 2.0 Hz, 2H, C5H4), 4.16 (s, 5H, C5H5), 4.40 (t, J = 2.0 Hz, 2H, C5H4), 7.36 (d, J = 7.8 Hz, 4H, Ph), 7.52 (s, 2H, CHCH of IPr), 7.61 (t, J = 7.8 Hz, 2H, Ph), 8.99 (s, 1H, NCH of triazole). 13C{1H} NMR (150 MHz, CDCl3): δ 24.03 (s, CHMe2), 24.85 (s, CHMe2), 28.87 (s, CHMe2), 39.36 (s, CH3), 63.56 (s, CH of C5H4), 67.14 (s, CH of C5H4), 70.24 (s, C5H5), 91.49 (s, Cipso–N of C5H4), 124.38 (s, CH of Ph), 124.78 (s, CHCH of IPr), 131.05 (s, CH of Ph), 133.59 (s, Cipso–N of Ph), 144.06 (s, NCH of triazole), 145.88 (s, Cipso–CHMe2 of IPr), 183.46 (s, Cipso–Au of triazolylidene), 185.46 (s, Cipso–Au of IPr). ESI+ MS: m/z 852 ([M − I]+). IR (DRIFTS, KBr): νmax 3568 w, 3357 w, 3110 m, 3071 m, 3024 m, 2960 s, 2926 s, 2868 m, 2711 w, 1725 w, 1621 w, 1592 w, 1533 m, 1492 s, 1471 s, 1459 s, 1423 m, 1400 w, 1386 m, 1364 m, 1351 m, 1329 m, 1311 m, 1274 s, 1250 s, 1241 s, 1220 s, 1182 m, 1146 m, 1126 w, 1107 m, 1061 m, 1017 s, 978 m, 949 w, 938 w, 874 m, 820 s, 811 s, 804 s, 759 s, 727 s, 719 s, 712 m, 674 w, 655 m, 640 w, 608 m, 576 m, 549 w, 534 w, 493 s, 474 m, 452 m, 431 w cm−1. Anal. calc. for C40H49AuFeIN5·1/2CH2Cl2 (1022.03): C 47.59, H 4.93, N 6.85%. Found: C 47.52, H 4.88, N 6.87%.
Data availability
The data will be available upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (project no. 23-06718S). Theoretical calculations were performed using the resources provided by the e-INFRA CZ project (ID: 90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic. The authors also thank Dr Ivana Císařová from the Department of Inorganic Chemistry, Faculty of Science, Charles University, for collecting the X-ray diffraction data.Notes and references
- (a) H.-W. Wanzlick and E. Schikora, Angew. Chem., 1960, 72, 49 CrossRef; (b) H.-W. Wanzlick and E. Schikora, Chem. Ber., 1961, 64, 2389 CrossRef; (c) H.-W. Wanzlick, Angew. Chem., Int. Ed. Engl., 1962, 1, 75 CrossRef.
- (a) K. Öfele, J. Organomet. Chem., 1968, 12, P42 CrossRef. See also: (b) D. J. Cardin, B. Cetinkaya and M. F. Lappert, Chem. Rev., 1972, 72, 545 CrossRef CAS; (c) M. F. Lappert, J. Organomet. Chem., 1988, 358, 185 CrossRef CAS.
- The very first isolable carbene, R2PCSiMe3, was reported even earlier in: A. Igau, H. Grützmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463 CrossRef CAS.
- (a) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS; (b) A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530 CrossRef CAS; (c) A. J. Arduengo, J. R. Goerlich and W. J. Marshall, J. Am. Chem. Soc., 1995, 117, 11027 CrossRef CAS.
- (a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (b) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862 CrossRef; (c) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC; (d) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (e) H. V. Huynh, Chem. Rev., 2018, 118, 9457 CrossRef CAS PubMed; (f) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711 CrossRef CAS PubMed.
- 1,2,3-Triazoles are accessible by Cu-catalysed Huisgen cycloaddition from azides and alkynes: (a) M. Meldal and C. Wenzel Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (b) M. Breugst and H.-U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293 CrossRef CAS PubMed.
- (a) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534 CrossRef CAS PubMed. For reviews, see: (b) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445 CrossRef CAS PubMed; (c) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755 CrossRef CAS; (d) G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236 CrossRef CAS PubMed; (e) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493 CrossRef PubMed; (f) R. Maity and B. Sarkar, JACS Au, 2022, 2, 22 CrossRef CAS PubMed.
- D. Enders, K. Breuer, G. Raabe, J. Runsink, J. H. Teles, J.-P. Melder, K. Ebel and S. Brode, Angew. Chem., Int. Ed. Engl., 1995, 34, 1021 CrossRef CAS.
- 1,2,4-Triazole-5-ylidenes remained overlooked; the carbene character of the long known analytical reagent nitron has been recognized only relatively recently: C. Färber, M. Leibold, C. Bruhn, M. Maurer and U. Siemeling, Chem. Commun., 2012, 48, 227 RSC.
- Representative examples: (a) F. Simal, D. Jan, L. Delaude, A. Demonceau, M.-R. Spirlet and A. F. Noels, Can. J. Chem., 2001, 79, 529 CrossRef CAS; (b) A. Zanardi, R. Corberán, J. A. Mata and E. Peris, Organometallics, 2008, 27, 3570 CrossRef CAS; (c) J. Iglesias-Sigüenza, A. Ros, E. Díez, M. Alcarazo, E. Álvarez, R. Fernández and J. M. Lassaletta, Dalton Trans., 2009, 7113 RSC; (d) C. Dash, M. M. Shaikh and P. Ghosh, Eur. J. Inorg. Chem., 2009, 1608 CrossRef CAS; (e) S. K. U. Riederer, P. Gigler, M. P. Högerl, E. Herdtweck, B. Bechlars, W. A. Herrmann and F. E. Kühn, Organometallics, 2010, 29, 5681 CrossRef CAS; (f) C. Dash, M. M. Shaikh, R. J. Butcher and P. Ghosh, Dalton Trans., 2010, 39, 2515 RSC; (g) S. K. U. Riederer, B. Bechlars, W. A. Herrmann and F. E. Kühn, Dalton Trans., 2011, 40, 41 RSC; (h) S. C. Holm, F. Rominger and B. F. Straub, J. Organomet. Chem., 2012, 719, 54 CrossRef CAS; (i) J. Turek, I. Panov, M. Semler, P. Štěpnička, F. De Proft, Z. Padělková and A. Růžička, Organometallics, 2014, 33, 3108 CrossRef CAS; (j) D. Yuan and H. V. Huynh, Organometallics, 2014, 33, 6033 CrossRef CAS; (k) S. K. Gupta, S. K. Sahoo and J. Choudhury, Organometallics, 2016, 35, 2462 CrossRef CAS; (l) V. H. Nguyen, M. B. Ibrahim, W. W. Mansour, B. M. El Ali and H. V. Huynh, Organometallics, 2017, 36, 2345 CrossRef CAS; (m) V. H. Nguyen, B. M. El Ali and H. V. Huynh, Organometallics, 2018, 37, 2358 CrossRef CAS; (n) V. H. Nguyen, H. H. Nguyen and H. H. Do, Inorg. Chem. Commun., 2020, 121, 108173 CrossRef CAS; (o) M. T. Lee, M. B. Goodstein and G. Lalic, J. Am. Chem. Soc., 2019, 141, 17086 CrossRef CAS PubMed; (p) V. H. Nguyen, T. T. Dang, H. H. Nguyen and H. V. Huynh, Organometallics, 2020, 39, 2309 CrossRef CAS; (q) S. Bauri, A. Mallik and A. Rit, Organometallics, 2020, 39, 3362 CrossRef CAS; (r) P. J. Quinlivan, A. Loo, D. G. Shlian, J. Martinez and G. Parkin, Organometallics, 2021, 40, 166 CrossRef CAS; (s) H.-Y. Hung, Y.-H. Hsu, H.-C. Pi, C.-H. Hu and H. M. Lee, Dalton Trans., 2022, 51, 18264 RSC.
- (a) K. M. Lee, H. M. J. Wang and I. J. B. Lin, J. Chem. Soc., Dalton Trans., 2002, 2852 RSC; (b) Y. Unger, D. Meyer and T. Strassner, Dalton Trans., 2010, 39, 4295 RSC; (c) M. Tenne, S. Metz, I. Muenster, G. Wagenblast and T. Strassner, Organometallics, 2013, 32, 6257 CrossRef CAS; (d) M. Tenne, S. Metz, G. Wagenblast, I. Muenster and T. Strassner, Dalton Trans., 2015, 44, 8444 RSC; (e) S. Aghazada, I. Zimmermann, V. Scutelnic and M. K. Nazeeruddin, Organometallics, 2017, 36, 2397 CrossRef CAS; (f) J. Soellner and T. Strassner, Organometallics, 2018, 37, 1821 CrossRef CAS; (g) B.-S. Yun, S.-Y. Kim, J.-H. Kim, S. Choi, S. Lee, H.-J. Son and S. O. Kang, ACS Appl. Electron. Mater., 2022, 4, 2699 CrossRef CAS.
- (a) A. Kumar, A. Naaz, A. P. Prakasham, M. K. Gangwar, R. J. Butcher, D. Panda and P. Ghosh, ACS Omega, 2017, 2, 4632 CrossRef CAS PubMed; (b) Y. Gothe, I. Romero-Canelón, T. Marzo, P. J. Sadler, L. Messori and N. Metzler-Nolte, Eur. J. Inorg. Chem., 2018, 2461 CrossRef CAS; (c) J. C. Mather, J. A. Wyllie, A. Hamilton, T. P. Soares da Costa and P. J. Barnard, Dalton Trans., 2022, 51, 12056 RSC; (d) H. T. T. Phung, H.-M. Vu, M. Q. H. Ly, H. H. Nguyen, T. H. Nguyen, H. T. T. Luong and V. H. Nguyen, Inorg. Chem. Commun., 2023, 154, 110898 CrossRef CAS.
- (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (b) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef CAS PubMed.
- (a) U. Siemeling, Eur. J. Inorg. Chem., 2012, 3523 CrossRef CAS; (b) P. Štěpnička, Dalton Trans., 2022, 51, 8085 RSC.
- (a) L. Hettmanczyk, S. Manck, C. Hoyer, S. Hohloch and B. Sarkar, Chem. Commun., 2015, 51, 10949 RSC; (b) L. Hettmanczyk, L. Suntrup, S. Klenk, C. Hoyer and B. Sarkar, Chem. – Eur. J., 2017, 23, 576 CrossRef CAS PubMed; (c) D. Aucamp, T. Witteler, F. Dielmann, S. Siangwata, D. C. Liles, G. S. Smith and D. I. Bezuidenhout, Eur. J. Inorg. Chem., 2017, 1227 CrossRef CAS; (d) S. Klenk, S. Rupf, L. Suntrup, M. van der Meer and B. Sarkar, Organometallics, 2017, 36, 2026 CrossRef CAS; (e) D. Aucamp, S. V. Kumar, D. C. Liles, M. A. Fernandes, L. Harmse and D. I. Bezuidenhout, Dalton Trans., 2018, 47, 16072 RSC; (f) R. Haraguchi, S. Hoshino, T. Yamazaki and S. Fukuzawa, Chem. Commun., 2018, 54, 2110 RSC; (g) R. Haraguchi, T. Yamazaki, K. Torita, T. Ito and S. Fukuzawa, Dalton Trans., 2020, 49, 17578 RSC; (h) K. Škoch, P. Vosáhlo, I. Císařová and P. Štěpnička, Dalton Trans., 2020, 49, 1011 RSC; (i) C. Hoyer, P. Schwerk, L. Suntrup, J. Beerhues, M. Nössler, U. Albold, J. Dernedde, K. Tedin and B. Sarkar, Eur. J. Inorg. Chem., 2021, 1373 CrossRef CAS.
- G. Forcher, A. Silvanus, P. de Frémont, B. Jacques, M. S. M. Pearson-Long, F. Boeda and P. Bertus, J. Organomet. Chem., 2015, 797, 1 CrossRef CAS.
- Some 1,2,4-triazoles with ferrocenyl substituents have been studied as N-donor ligands. For examples, see: (a) G. Gasser, J. D. Carr, S. J. Coles, S. J. Green, M. B. Hursthouse, S. M. Cafferkey, H. Stoeckli-Evans and J. H. R. Tucker, J. Organomet. Chem., 2010, 695, 249 CrossRef CAS; (b) H. S. Scott, A. Nafady, J. D. Cashion, A. M. Bond, B. Moubaraki, K. S. Murray and S. M. Neville, Dalton Trans., 2013, 42, 10326 RSC.
- T. Mochida, H. Shimizu, S. Suzuki and T. Akasaka, J. Organomet. Chem., 2006, 691, 4882 CrossRef CAS.
- (a) H. E. Bryndza and W. Tam, Chem. Rev., 1988, 88, 1163 CrossRef CAS; (b) D. J. Nelson and S. P. Nolan, Coord. Chem. Rev., 2017, 353, 278 CrossRef CAS.
- S. Sethi, P. Kumar Das and N. Behera, J. Organomet. Chem., 2016, 824, 140 CrossRef CAS.
- R. K. Bartlett and I. R. Humphrey, J. Chem. Soc. C, 1967, 1664 RSC.
- The yield significantly decreased when undried solvent or noninert conditions were used.
- Y. Miura, F. Shimizu and T. Mochida, Inorg. Chem., 2010, 49, 10032 CrossRef CAS PubMed.
- T. J. Curphey and K. D. Prasad, J. Org. Chem., 1972, 37, 2259 CrossRef CAS.
- (a) A. Zanardi, J. A. Mata and E. Peris, J. Am. Chem. Soc., 2009, 131, 14531 CrossRef CAS PubMed; (b) A. Zanardi, J. A. Mata and E. Peris, Organometallics, 2009, 28, 4335 CrossRef CAS; (c) A. Zanardi, J. A. Mata and E. Peris, Chem. – Eur. J., 2010, 16, 13109 CrossRef CAS PubMed; (d) S. Sabater, J. A. Mata and E. Peris, Chem. – Eur. J., 2012, 18, 6380 CrossRef CAS PubMed; (e) S. Guo and H. V. Huynh, Organometallics, 2012, 31, 4565 CrossRef CAS; (f) S. Guo and H. V. Huynh, Organometallics, 2014, 33, 2004 CrossRef CAS , and ref. 10b.
- Recently, Hölzel and Ganter achieved two-fold methylation of a 1,2,4-triazole upon reacting 5-(dimethylamino)-1,4-dimethyl-4H-1,2,4-triazol-1-ium iodide with an excess of methyl triflate: T. Hölzel and C. Ganter, J. Organomet. Chem., 2020, 915, 121234 CrossRef.
- D. C. D. Butler and C. J. Richards, Organometallics, 2002, 21, 5433 CrossRef CAS.
- (a) C. Köcher and W. A. Herrmann, J. Organomet. Chem., 1997, 532, 261 CrossRef; (b) M. V. Baker, S. K. Brayshaw, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2004, 357, 2841 CrossRef CAS.
- (a) S. P. Nolan, Acc. Chem. Res., 2011, 44, 91 CrossRef CAS PubMed; (b) T. Scattolin, G. Tonon, E. Botter, S. G. Guillet, N. V. Tzouras and S. P. Nolan, Chem. – Eur. J., 2023, 29, e202301961 CrossRef CAS PubMed.
- For an analogous reaction with a triazolium salt, see: S. Gaillard, P. Nun, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 5402 CrossRef CAS.
- (a) T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335 CrossRef CAS; (b) F. R. Hartley, Chem. Soc. Rev., 1973, 2, 163 RSC.
- (a) S. Fuertes, A. J. Chueca and V. Sicilia, Inorg. Chem., 2015, 54, 9885 CrossRef CAS PubMed; (b) F. Horký, J. Soellner, J. Schulz, I. Císařová, T. Strassner and P. Štěpnička, New J. Chem., 2023, 47, 18442 RSC.
- J. Iglesias-Sigüenza, A. Ros, E. Díez, M. Alcarazo, E. Álvarez, R. Fernández and J. M. Lassaletta, Dalton Trans., 2009, 7113 RSC.
- T. Hölzel, M. Otto, H. Buhl and C. Ganter, Organometallics, 2017, 36, 4443 CrossRef.
- J. C. Green, Chem. Soc. Rev., 1998, 27, 263 RSC.
- S. Gaillard, J. Bosson, R. S. Ramón, P. Nun, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2010, 16, 13729 CrossRef CAS PubMed.
- Q. Liu, M. Xie, X. Chang, S. Cao, C. Zou, W.-F. Fu, C.-M. Che, Y. Chen and W. Lu, Angew. Chem., Int. Ed., 2018, 57, 6279 CrossRef CAS PubMed.
- G. Gritzner and J. Kůta, Pure Appl. Chem., 1984, 56, 461 CrossRef.
- S. Wolf and H. Plenio, J. Organomet. Chem., 2009, 694, 1487 CrossRef CAS.
- T. Lu and F. Chen, Acta Chim. Sin., 2011, 69, 2393 CAS.
- (a) J. Schulz, F. Uhlík, J. M. Speck, I. Císařová, H. Lang and P. Štěpnička, Organometallics, 2014, 33, 5020 CrossRef CAS; (b) K. Škoch, I. Císařová, F. Uhlík and P. Štěpnička, Dalton Trans., 2018, 47, 16082 RSC; (c) K. Škoch, J. Schulz, I. Císařová and P. Štěpnička, Organometallics, 2019, 38, 3060 CrossRef; (d) P. Vosáhlo, J. Schulz, I. Císařová and P. Štěpnička, Dalton Trans., 2021, 50, 6232 RSC.
- (a) G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834 CrossRef CAS PubMed; (b) G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518 CrossRef CAS PubMed. For applications in the chemistry of carbene ligands, see: (c) L. Nunes dos Santos Comprido, J. E. M. N. Klein, G. Knizia, J. Kästner and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 10336 CrossRef CAS PubMed; (d) F. F. Mulks, A. S. K. Hashmi and S. Faraji, Organometallics, 2020, 39, 1814 CrossRef CAS; (e) L. Nunes dos Santos Comprido, J. E. M. N. Klein, G. Knizia, J. Kästner and A. S. K. Hashmi, Chem. – Eur. J., 2016, 22, 2892 CrossRef CAS PubMed; (f) D. Sorbelli, L. Nunes dos Santos Comprido, G. Knizia, A. S. K. Hashmi, L. Belpassi, P. Belanzoni and J. E. M. N. Klein, ChemPhysChem, 2019, 20, 1671 CrossRef CAS PubMed.
- (a) M. J. S. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C71 Search PubMed; (b) J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939 RSC.
- (a) R. G. Pearson, Inorg. Chem., 1973, 12, 712 CrossRef CAS , and ref. 32.
- M. L. H. Green, J. Organomet. Chem., 1995, 500, 127 CrossRef CAS.
- (a) X. Hu, I. Castro-Rodriguez, K. Olsen and K. Meyer, Organometallics, 2004, 23, 755 CrossRef CAS; (b) D. M. Khramov, V. M. Lynch and C. W. Bielawski, Organometallics, 2007, 26, 6042 CrossRef CAS; (c) D. Nemcsok, K. Wichmann and G. Frenking, Organometallics, 2004, 23, 3640 CrossRef CAS; (d) S. V. C. Vummaleti, D. J. Nelson, A. Poater, A. Gómez-Suárez, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and L. Cavallo, Chem. Sci., 2015, 6, 1895 RSC; (e) G. Bistoni, L. Belpassi and F. Tarantelli, Angew. Chem., Int. Ed., 2013, 52, 11599 CrossRef CAS PubMed.
- (a) C. Heinemann, T. Müller, Y. Apeloig and H. Schwarz, J. Am. Chem. Soc., 1996, 118, 2023 CrossRef CAS; (b) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162 CrossRef CAS; (c) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed.
- A. Leonidova, T. Joshi, D. Nipkow, A. Frei, J.-E. Penner, S. Konatschnig, M. Patra and G. Gasser, Organometallics, 2013, 32, 2037 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742 RSC.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 1 and 2, details on structure determination and additional structure diagrams, computational details and further results from DFT calculations, copies of the NMR spectra (PDF file), and Cartesian coordinates of the DFT-optimised structures (XYZ file). CCDC 2345618 and 2345619. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01433b |
| This journal is © The Royal Society of Chemistry 2024 |