
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation and functionalisation of carbon dioxide by bis-tris(pyrazolyl)borate-supported divalent samarium and trivalent lanthanide silylamide complexes†
Tajrian
Chowdhury
 ,
Claire
Wilson
and
Joy H.
Farnaby
*
,
Claire
Wilson
and
Joy H.
Farnaby
*
School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Joy.Farnaby@glasgow.ac.uk
First published on 24th June 2024
Abstract
Synthesis and reactivity with carbon dioxide (CO2) of divalent samarium in the bis-tris(pyrazolyl)borate ligand environment has been reported. In addition, CO2 activation and functionalisation by lanthanide silylamides in the bis-tris(pyrazolyl)borate ligand environment was demonstrated. Reduction of the Sm(III) precursor [Sm(Tp)2(OTf)] (Tp = hydrotris(1-pyrazolyl)borate; OTf = triflate) with KC8 yielded the insoluble Sm(II) multi-metallic coordination polymer [{Sm(Tp)2}n] 1-Sm. Addition of 1,2-dimethoxyethane (DME) to 1-Sm enabled isolation of the monomeric complex [Sm(Tp)2(DME)] 1-Sm(DME). Complex 1-Sm(DME) reduced CO2 to yield the oxalate-bridged dimeric Sm(III) complex [{Sm(Tp)2}2(μ-η2:η2-O2CCO2)] 2-Sm. The reactions of heteroleptic Ln(III) silylamide complexes [Ln(Tp)2(N′′)] (Ln = Y, Sm; N′′ = N(SiMe3)2) with CO2 yielded monomeric Ln(III) silyloxides [Ln(Tp)2(OSiMe3)] 3-Ln and trimethylsilyl isocyanate (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNSiMe3). Complexes 3-Ln are the first crystallographically characterised examples of Ln(III)–OSiMe3 bonds accessed via CO2 activation and functionalisation. Full characterisation data are presented for all complexes, including solid-state molecular structure determination by single-crystal X-ray diffraction.
CNSiMe3). Complexes 3-Ln are the first crystallographically characterised examples of Ln(III)–OSiMe3 bonds accessed via CO2 activation and functionalisation. Full characterisation data are presented for all complexes, including solid-state molecular structure determination by single-crystal X-ray diffraction.
Introduction
Carbon dioxide (CO2) activation and functionalisation by complexes of transition metals (TM),1 rare-earth metals lanthanides (Ln, where Ln = Sc, Y, La–Lu) and actinides,2 and heterobimetallic systems,3 have attracted significant interest from scientific communities worldwide, since CO2 is the largest single source contributor to the greenhouse effect and climate change. CO2 is a renewable one-carbon (C1) building block and can bind to metal ions in various ways during activation,4 which upon functionalisation yields valuable synthetic products and commercially important chemicals.5Divalent Sm(II) ion, which is a powerful one electron reductant,6 can activate CO2 to yield oxos (O2−), oxalates (C2O42−), and carbonates (CO32−), the mechanism of which is well-modelled by computational methods (Fig. 1(a)).7 For Sm(II) in an organometallic ligand environment, the first step of CO2 activation at a Sm(II) metal centre involves the cooperative double reduction of CO2 to CO22− by two Sm(II) ions to form a reactive bimetallic intermediate [Sm]–(μ-η2:η1-CO2)–[Sm] ([Sm] = SmIII). This intermediate further reacts in two possible ways, where either (i) reaction with another molecule of CO2 induces a C–C coupling reaction to form an oxalate-bridged dimeric complex [Sm]–(μ-η2:η2-O2CCO2)–[Sm] (Fig. 1(a)(i)) or (ii) where it loses a CO molecule and the resultant bimetallic oxo complex [Sm]–(μ-O)–[Sm] reacts with a free CO2 molecule to form a bimetallic carbonate complex [Sm]–(μ-CO3)–[Sm] (Fig. 1(a)(ii)).7c In the activation of CO2 by [Sm(Cp*)2(THF)2] (Cp* = C5Me5), pathway (i) from Fig. 1(a) was shown to be the predominant pathway both experimentally and computationally,7c and any radical dimerization pathways have been deemed unlikely at the Sm(II) metal centre.7 Outcome of the reactivity of CO2 with low valent rare-earth metals is challenging to predict from first principles. The selectivity of CO2 activation reactions of Sm(II) to yield oxalates vs. carbonates, can sometimes be rationalised based on the steric influence of the ancillary ligands of the Sm(II) ion, for example the more sterically bulky organometallic cyclopentadienyl ligand derivatives and macrocyclic ligands result in formation of carbonates.7f,h For trivalent U(III) it was shown that tuning the steric bulk of the ancillary cyclopentadienyl ligand (more sterically bulky ancillary ligands favoured the formation of carbonates over oxalates)8 or where utilising different reaction temperatures led to the isolation of the thermodynamic (carbonate) vs. kinetic (oxalate) products.9
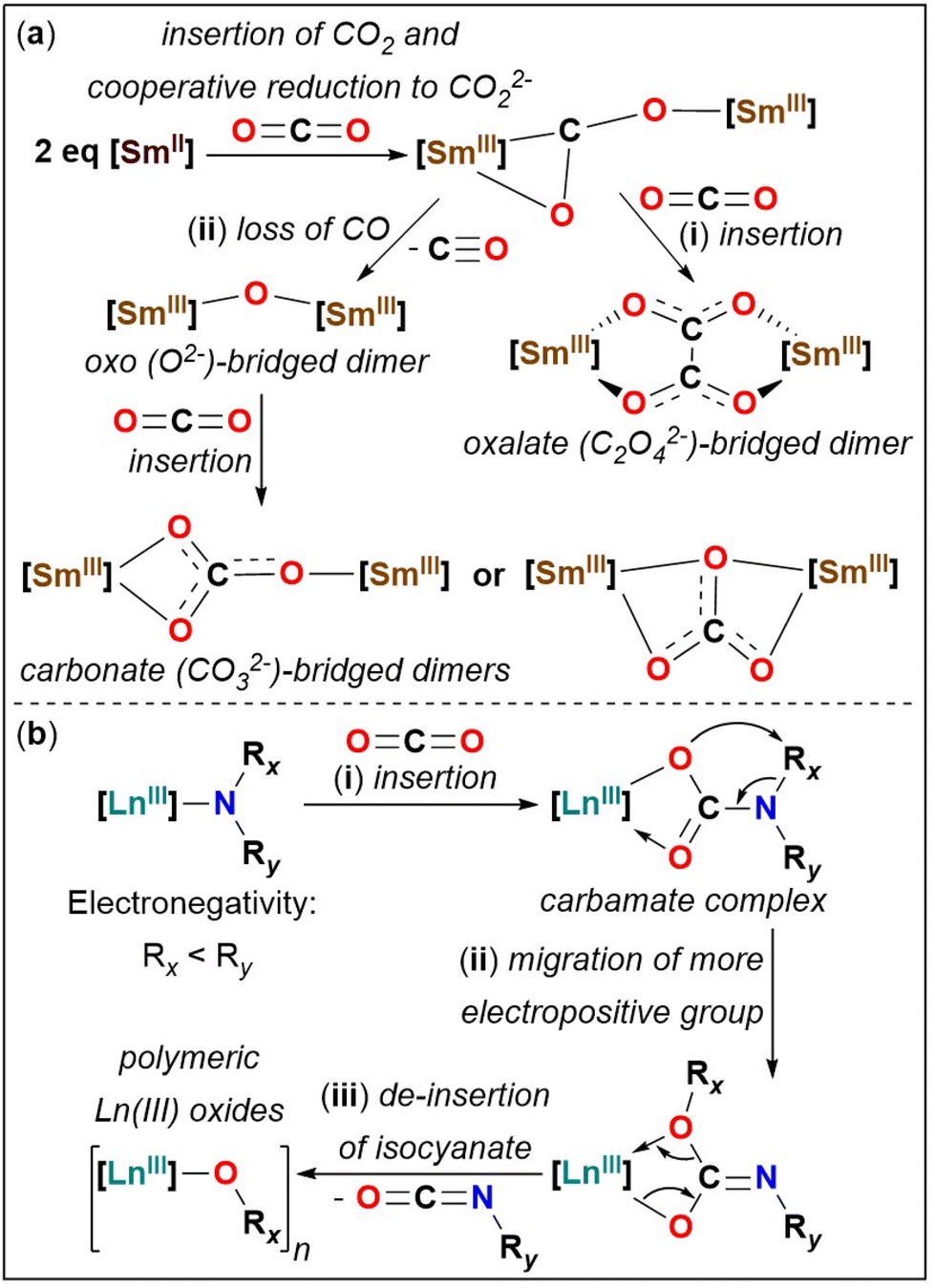 | ||
| Fig. 1 Mechanistic pathway of CO2 activation and functionalisation by (a) Sm(II)7c and (b) Ln(III)-amides.10b | ||
Lewis acidic lanthanides activate CO2 in various ways such as (a) the insertion of CO2 into Ln(III)–X σ-bonds (X = amide, alkyl, alkyl/aryloxy, hydride, chalcogenide, carbene)2a and (b) cooperative reduction of CO2 by Ln(II).7a,c,e Since Ln–N(amide) bonds exhibit remarkable substrate insertion scope10 and catalytic activity,11 the homoleptic Ln(III) silylamides [Ln(N′′)3] (N′′ = N(SiMe3)2)12 have been studied for small-molecule activation.2a,13 The activation and functionalisation of CO2 at the Ln(III) metal centre of a Ln–N(amide) bond proceeds via three steps. As shown in Fig. 1(b), these are: (i) insertion of CO2 into [Ln]–NR2 σ-bonds to form a carbamate complex [Ln]–(κ2-O,OCNR2) (Fig. 1(b)(i)), (ii) migration of the more electropositive amide substituent from the nitrogen atom to the oxygen atom within the carbamate complex (Fig. 1(b)(ii)), and (iii) de-insertion of isocyanate to yield Ln(III) oxides (Fig. 1(b)(iii)).2a,10b Isolation of carbamate complexes has proven challenging in the reactivity of CO2 with [Ln(N′′)3], owing to the facile thermodynamic migration of the SiMe3 group (as in (ii)), leading to the formation of insoluble polymeric Ln(III) silyloxides [{Ln(OSiMe3)3}n].2a,10b,14 Therefore, in absence of isolation of discrete Ln(III) silyloxides, CO2 activation by Ln–N′′ bonds have been exclusively studied by isolating the corresponding organic products: OCNSiMe3, (Me3Si)2O, Me3SiNCNSiMe3, analogous to main-group metal–N′′ mediated CO2 activation.2a,10b,15 Ln(III) silyloxides are potential precursors towards silicate materials and catalysts for organic transformations.14b Various synthetic routes to access Ln–OSiR3 bonds exist, however insertion of CO2 into Ln–N(amide) bonds to access molecular Ln(III) silyloxides remains a rare route.14b,16
Trofimenko's tridentate scorpionate N-donor hydrotris(1-pyrazolyl)borate (Tp) ligand and its derivatives (TpR) are robust ancillary ligands in Ln chemistry, which provide great structural diversity to Ln complexes and result in useful physical (optical, magnetic) and chemical (small-molecule activation, catalysis) properties.17 The Sm(II) complexes [Sm(TpR)2] display activation of a wide range of small-molecules (CO,18 NO,19 O2,20 S8,21 dichalcogenides,22 azobenzene,23 aldehyde,24 ketones,24,25 quinones,24,25 azines24). Reduction of heavy-metal (Hg(I), Tl(I)) salts with [Sm(Tp*)2] (Tp* = hydrotris(3,5-dimethyl-1-pyrazolyl)borate) was utilised to synthesise organometallic complexes: (a) the first Tp/cyclopentadienyl-mixed complex of a lanthanide26 and (b) an alkynide.27 Reactions of [Sm(Tp*)2] with TM carbonyl complexes enabled isolation of heterometallic TM/Sm complexes.28 The synthetic routes to [Ln(TpR)2] complexes, with one exception,29 have predominantly relied on Ln(II) precursors.17a,23,29,30 Reduction of Ln(III) [Ln(Tp)2(OTf)] (OTf = triflate) was shown as a viable route to access Ln(II) [Ln(Tp)2] (Ln = Eu, Yb) complexes.31 Utilising two small unsubstituted Tp ligands enabled isolation of the reactive Ln(III) amide complexes [Ln(Tp)2(N′′)],32 which were inaccessible in the bulky [Ln(Tp*)2]+ ligand environment.33
Here we report the reduction of [Sm(Tp)2(OTf)],32c to the Sm(II) complex [Sm(Tp)2(DME)] 1-Sm(DME). The reactivity of 1-Sm(DME) with small-molecules such as pnictogens and CO2 has been investigated. Furthermore, the activation and functionalisation of CO2 by the bis-Tp Ln(III) amide complexes [Ln(Tp)2(N′′)] (Ln = Y, Sm) has been reported.
Results and discussion
Synthesis of [Sm(Tp)2(DME)] 1-Sm(DME)
The formal reduction potential (E0) of Sm3+/Sm2+ (−1.55 V) in aqueous solution vs. the Normal Hydrogen Electrode (NHE) makes it the most accessible classical Ln(III) candidate for reduction after Ln = Eu (−0.35 V) and Yb (−1.15 V).34 Recently, the synthesis of soluble adduct-free Ln(II) [Ln(Tp)2] (Ln = Eu, Yb) complexes was reported by reduction of Ln(III) [Ln(Tp)2(OTf)] (OTf = triflate) with KC8 in toluene.31 However, in pursuit of non-classical Ln(II) ions,35 as investigated herein, the bis-Tp ligand environment has been shown to be unsuccessful in stabilising non-classical Ln(II) ions. For example, the reaction between Ln(III) [Ln(Tp)2(OTf)] (Ln = Y,32a Dy![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32b) and KC8 yielded complicated reaction mixtures containing [Ln(Tp)3]32b,36 and this was observed irrespective of reaction conditions and solvent choices (see ESI section A3† for NMR data for the reaction of [Dy(Tp)2(OTf)] with KC8 in toluene). Additionally for Ln = Y, fragmentation of the Tp ligand was observed resulting in formation of [Y(Tp)2(κ2-pz)] (pz = pyrazolyl). Such fragmentation of the Tp ligand by cleavage of a B–N bond, in the absence of a metal-based reduction, often leading to the isolation of pz-bound byproducts, has been observed in rare-earth Tp/TpR chemistry.33,36c,37 Purification of [Y(Tp)2(κ2-pz)] away from complicated reaction mixtures containing [Y(Tp)3] proved challenging, however, single-crystals were isolated on one occasion (see ESI section A1.3† for NMR data for [Y(Tp)2(κ2-pz)] and Fig. S65† for the molecular structure of [Y(Tp)2(κ2-pz)]).
32b) and KC8 yielded complicated reaction mixtures containing [Ln(Tp)3]32b,36 and this was observed irrespective of reaction conditions and solvent choices (see ESI section A3† for NMR data for the reaction of [Dy(Tp)2(OTf)] with KC8 in toluene). Additionally for Ln = Y, fragmentation of the Tp ligand was observed resulting in formation of [Y(Tp)2(κ2-pz)] (pz = pyrazolyl). Such fragmentation of the Tp ligand by cleavage of a B–N bond, in the absence of a metal-based reduction, often leading to the isolation of pz-bound byproducts, has been observed in rare-earth Tp/TpR chemistry.33,36c,37 Purification of [Y(Tp)2(κ2-pz)] away from complicated reaction mixtures containing [Y(Tp)3] proved challenging, however, single-crystals were isolated on one occasion (see ESI section A1.3† for NMR data for [Y(Tp)2(κ2-pz)] and Fig. S65† for the molecular structure of [Y(Tp)2(κ2-pz)]).
Reduction of the Sm(III) triflate complex [Sm(Tp)2(OTf)]32c in toluene yielded highly insoluble dark brown solids, precluding isolation of a molecular complex. However, the reduction of [Sm(Tp)2(OTf)] in THF under ambient conditions, resulted in a dark red-brown solution containing the Sm(II) complex [Sm(Tp)2(THF)2] 1-Sm(THF) (Scheme 1). The reaction mixture was filtered to exclude excess KC8 and graphite. THF was removed in vacuo and the dark red-brown solids were extracted into toluene. Filtration of the dark orange-brown suspension to exclude K(OTf) and removal of toluene in vacuo (10−2 mbar) from the extract led to desolvation of THF from 1-Sm(THF) (observed on the NMR-scale) and isolation of the insoluble dark-brown multi-metallic coordination polymer [{Sm(Tp)2}n] 1-Sm (91%, Scheme 1).
 | ||
| Scheme 1 Synthesis of Sm(II) [Sm(Tp)2(DME)] 1-Sm(DME)via [Sm(Tp)2(THF)2] 1-Sm(THF) by reduction of [Sm(Tp)2(OTf)] with KC8 in THF and DME complexation to [{Sm(Tp)2}n] 1-Sm. Note: The box shows numbering of the pyrazolyl carbon atoms of the Tp ligand. | ||
Since the solid-state molecular structure of [{Eu(Tp)2}2] exhibits two bridging μ-κ1:η5 Tp ligands,31 it is anticipated that 1-Sm can adopt similar extended bridging modes of the Tp ligand leading to insolubility of polymeric 1-Sm, owing to the slightly larger ionic radius of Sm(II) compared to Eu(II).38 In order to isolate a molecular complex of 1-Sm, after removal of toluene in the synthesis, excess DME (1,2-dimethoxyethane) was added resulting in the formation of the DME-adduct complex [Sm(Tp)2(DME)] 1-Sm(DME). Since 1-Sm(DME) does not undergo desolvation upon application of vacuum (10−2 mbar), drying in vacuo led to the isolation of 1-Sm(DME) (89%, Scheme 1). Complex 1-Sm(DME) has excellent solubilities in coordinating (THF) and non-coordinating (benzene, toluene) solvents, but poor solubilities in hydrocarbon solvents (hexane). Elemental analysis of 1-Sm(DME) is consistent with the [Sm(Tp)2(DME)] formulation. Desolvation of Lewis base adducts from 1-Sm(LB) (LB = THF, DME) is observed in solution of non-coordinating aromatic solvents slowly over time (LB = THF, 2 days; DME, 2 weeks) under ambient conditions or quickly with heating (1 hour in both cases), leading to liberation of free Lewis bases and formation of insoluble 1-Sm and [Sm(Tp)3].36b
Spectroscopy of [Sm(Tp)2(DME)] 1-Sm(DME)
By relative integration, in the 1H NMR spectrum of paramagnetic 1-Sm(DME) in d8-THF, the Tp-pyrazolyl protons in 1-Sm(DME) are observed at δ = 0.30, 4.14 and 11.49 ppm, the Tp-borohydride at δ = −1.20 ppm, and the DME ligand protons at δ = 3.29 and 3.42 ppm, in the expected 6:6:6:2:6:4 ratio. The proton and carbon resonances of the Tp ligands and DME of 1-Sm(DME) were assigned using DEPT-135 13C{1H} and 1H-13C HSQC NMR experiments (see ESI Fig. S10 and Fig. S11,† respectively). The 11B NMR spectrum of 1-Sm(DME) exhibits a resonance at δ = −25.36 ppm, corresponding to the Tp ligands, consistent with [Sm(TpR)2].29,30e The ATR-IR spectra of 1-Sm and 1-Sm(DME), both exhibit weak absorptions between 2350–2450 cm−1 assigned to the characteristic borohydride stretching frequencies (υBH) for the Tp ligands in Ln(II) [Ln(TpR)2]29,30f and [Ln(Tp)2] complexes.31 Additionally, weak absorptions are observed between 2820–2930 cm−1, which are assigned to the aliphatic sp3-carbon hydrogen bond stretching frequencies (υsp3-CH) of the DME ligand in 1-Sm(DME), consistent with literature.39
Crystallography of [Sm(Tp)2(LB)x] (LB = THF, x = 2; DME, x = 1) 1-Sm(LB)
Lath-shaped red single-crystals of 1-Sm(THF) suitable for X-ray diffraction were grown from a saturated THF solution at −35 °C overnight and shard-shaped red single-crystals of 1-Sm(DME) suitable for X-ray diffraction were grown from a saturated toluene solution with a hexane antisolvent at −35 °C over three weeks. The structures of 1-Sm(LB) are monomeric, containing an 8-coordinate Sm(II) ion bound to two κ3-coordinated Tp ligands and two oxygen atoms for two THF adducts (Fig. 2(a)) or one DME adduct (Fig. 2(b)), arranged around the Sm(II) ion in a distorted square antiprismatic geometry. Important structural metrics are tabulated in the ESI in section B,† with bond metrics and relevant structural data comparisons in Table 1 and crystallographic information in Table 2.† The Sm–N(Tp) bond distances in 1-Sm(LB) (LB = THF, 2.662(6)–2.744(7) Å; DME, 2.6805(16)–2.7180(16) Å) are indistinguishable to those in the Eu(II) complex [Eu(Tp)2(THF)2] and longer than those in the Yb(II) complexes [Yb(Tp)2(LB)] (LB = THF, DME) consistent with the size of the different Ln(II) ionic radii Sm(II) ∼ Eu(II) ≫ Yb(II).31,38 The Sm–N(κ3-Tp) bond distances in 1-Sm(LB) are consistent with adduct-free [Sm(TpR)2] (see ESI Table 1†).23,29,30e Since a range of Ln–N(κ3-Tp) bond distances is normally observed in literature [Ln(TpR)2] complexes (ESI Table 1†), therefore small structural differences e.g. neighbouring Ln(II) ionic radii or substitution of the Tp ligand (TpR) do not result in statistically significant Ln–N(κ3-Tp) distances. The increase in ionic radius upon reduction of Sm(III) to Sm(II) is reflected by the longer Sm–N(Tp) bond distances in 1-Sm(LB) when compared to the THF-adduct of the Sm(III) precursor [Sm(Tp)2(OTf)(THF)] (2.512(2)–2.583(2) Å).32c | ||
| Fig. 2 Molecular structures of [Sm(Tp)2(LB)x] 1-Sm(LB) LB = THF (a), DME (b). Hydrogen atoms and lattice solvent molecules omitted for clarity and carbon atoms of Tp pyrazolyl, THF, and DME displayed in wireframe. Displacement ellipsoids drawn at 50% probability level. Selected bond distances: (a) Sm–N(Tp) 2.662(6)–2.744(7) Å, Sm–O(THF) 2.692(4) Å, (b) Sm–N(Tp) 2.6805(16)–2.7180(16) Å, Sm–O(DME) 2.6696(14), 2.7044(14) Å. | ||
Reactivity of 1-Sm(DME) with molecular pnictogens
Owing to the diverse reactivity exhibited by [Sm(TpR)2],18–28 the scope of 1-Sm(DME) in small-molecule activation was investigated in collaboration with the Roesky group. Since the reactivity of Ln(II) [Ln(TpR)2] with pnictogens has not been reported to date, the reactivity of 1-Sm(DME) with white phosphorus (P4) was tested, targeting motifs such as [{(Cp*)2Sm}4(μ4-η2:η2:η2:η2-P8)].13a,40 Reactions of 1-Sm(DME) with freshly sublimated P4 or P4 vapour, resulted in formation of insoluble Sm(III) products, where [Sm(Tp)3]41 was crystallised on several occasions as the major product, irrespective of reaction conditions and solvents. Reactions of [Yb(Tp)2]31 with white P4, led to the formation of [Yb(Tp)3]36a and recovery of unreacted [Yb(Tp)2], consistent with the lower reducing strength of Yb(II) vs. Sm(II).34 Reactivity of 1-Sm(DME) with heavier pnictogens like elemental nanoparticles of arsenic (As0nano),42 were also unsuccessful.Reactivity of 1-Sm(DME) with carbon dioxide (CO2) in the synthesis of [{Sm(Tp)2}2(μ-η2:η2-O2CCO2)] 2-Sm
Activation and functionalisation of CO2 by Sm(II) in various ligand environments have been reported in literature to yield oxalate-bridged Sm(III) dimers (Fig. 1(a)).7a–e The synthesis of the oxalate-bridged dimeric Ln(III) complexes [{Ln(Tp)2}2(μ-η2:η2-O2CCO2)] (Ln = Y, Sm, Dy, Yb, Lu) were previously reported by salt metathesis between Ln(III) chlorides [LnCl3·(H2O)n], K(Tp), and Na2(C2O4) in a 2:4:1 ratio in water.43 Since the reactivity of Ln(II) [Ln(TpR)2] with CO2 has not been reported to date, the reaction of 1-Sm(DME) with CO2 was studied in d8-THF to yield the Sm(III) complex [{Sm(Tp)2}2(μ-η2:η2-O2CCO2)] 2-Sm (92%, Scheme 2). Complex 2-Sm has poor solubilities in all solvents. Elemental analysis of 2-Sm is consistent with the [{Sm(Tp)2}2(μ-η2:η2-O2CCO2)] formulation.
 | ||
| Scheme 2 Reaction of 1-Sm(DME) with CO2 to yield oxalate-bridged dimeric Sm(III) complex [{Sm(Tp)2}2(μ-η2:η2-O2CCO2)] 2-Sm. | ||
Spectroscopy of [{Sm(Tp)2}2(μ-η2:η2-O2CCO2)] 2-Sm
Due to the insolubility of 2-Sm post-work-up, the NMR data discussed here are from the NMR-scale reaction of 1-Sm(DME) with CO2 in d8-THF (see ESI section A2.1†). In the 1H NMR spectrum of 2-Sm in d8-THF, the Tp-pyrazolyl protons in 2-Sm are observed at δ = 4.87, 6.04 and 8.84 ppm, consistent with NMR of this complex reported elsewhere.43 The proton and carbon resonances of the pyrazolyl rings of the Tp ligands of 2-Sm were assigned as far as possible, by using 13C{1H} and 1H–13C HSQC NMR experiments (see ESI Fig. S45 and Fig. S46,† respectively). In the 13C{1H} NMR spectrum of 2-Sm, the resonance corresponding to the oxalate ligand carbon could not be identified. This observation is similar to the 13C{1H} NMR spectra of [{Sm(CpR)2}2(μ-η2:η2-O2CCO2)],7a,b where the oxalate ligand carbon atoms were not observed unless reactions were performed with 13CO2.7a The 11B NMR spectrum of 2-Sm exhibits a resonance at δ = 2.91 ppm, corresponding to the Tp ligands, consistent with other [Sm(Tp)2]+ examples.32c In the ATR-IR spectrum of 2-Sm, besides weak absorptions between 2350–2450 cm−1 assigned to the characteristic borohydride stretching frequencies (υBH) for the Tp ligands, a weak absorption is also observed at 1649 cm−1, which is assigned to the carbonyl stretching frequencies (υCO) of the oxalate ligand in 2-Sm, analogous to reported [{Ln(Tp)2}2(μ-η2:η2-O2CCO2)] (Ln = Y, La, Ce, Nd, Sm, Dy, Yb, Lu) (υCO = 1620–1660 cm−1).43
Crystallography of [{Sm(Tp)2}2(μ-η2:η2-O2CCO2)] 2-Sm
The solid-state structure of 2-Sm was not previously reported.43 Block-shaped colourless single-crystals of 2-Sm suitable for X-ray diffraction were grown from a saturated THF solution with a hexane antisolvent at −35 °C over four months. The structure of 2-Sm (Fig. 3) is dimeric, containing two 8-coordinate Sm(III) ions where each Sm(III) ion is bound to two κ3-coordinated Tp ligands and two oxygens of the μ-η2:η2-O2CCO22− oxalate ligand bridging the two Sm(III) ions, in a distorted square antiprismatic geometry. The Sm–N(κ3-Tp) bond distances of 2.480(6)–2.592(7) Å in 2-Sm are consistent with [Sm(Tp)2]+.32c The Sm–O(C2O4) bond distances of 2.423(6) and 2.425(5) Å in 2-Sm are consistent with the literature of oxalate-bridged dimeric Sm(III) complexes (see ESI Table 1†).7b,d The Sm–N(κ3-Tp) and Sm–O(C2O4) bond distances in 2-Sm are longer than those in [{Dy(Tp)2}2(μ-η2:η2-O2CCO2)] (see ESI Table 1†) and consistent with differences in size of the different Ln(III) ionic radii Sm(III) > Dy(III).38 | ||
| Fig. 3 Molecular structure of [{Sm(Tp)2}2(μ-η2:η2-O2CCO2)] 2-Sm. Hydrogen atoms and lattice solvent molecules omitted for clarity and carbon atoms of Tp pyrazolyl displayed in wireframe. Displacement ellipsoids drawn at 50% probability level. Selected bond distances: Sm–N(Tp) 2.480(6)–2.592(7) Å, Sm–O(C2O4) 2.423(6), 2.425(5) Å. | ||
Reactivity of [Ln(Tp)2(N′′)] (Ln = Y, Sm; N′′ = N(SiMe3)2 with CO2) in the synthesis of [Ln(Tp)2(OSiMe3)] 3-Ln
The heteroleptic Ln(III) amide complexes [Ln(Tp)2(N′′)] (Ln = Y, Yb, Dy) are reactive towards protonolysis with alcohols32a and primary amines.32b To aid investigation of CO2 activation by the paramagnetic [Sm(Tp)2(N′′)], the analogous diamagnetic reaction with Ln = Y was also studied. Reactions of d6-benzene solutions of [Ln(Tp)2(N′′)] (Ln = Y,32a Sm)32c with CO2 led to the complete consumption of [Ln(Tp)2(N′′)]. The reaction proceeds likely by insertion of CO2 to form the carbamate intermediate [Ln(Tp)2(κ2-O,OCN′′)] (see Fig. 1(b)(i)),2a,10b which is not observed, but the subsequent intermediate complex [Ln(Tp)2(κ2-O,O(SiMe3)CNSiMe3)] resulting from an intramolecular silyl-migration (see Fig. 1(b)(ii)), was observed on the NMR-scale (see ESI section A2.2† for Ln = Y and section A2.3 for Ln = Sm). Upon work up of the reaction mixtures by removing d6-benzene, excess CO2, and OCNSiMe3in vacuo, and washing the crude with hexane, the monomeric Ln(III) silyloxides [Ln(Tp)2(OSiMe3)] 3-Ln (Ln = Y, 59%, Sm 53%; Scheme 3) were isolated. Complexes 3-Ln have excellent solubilities in toluene and moderate solubilities in hexane.
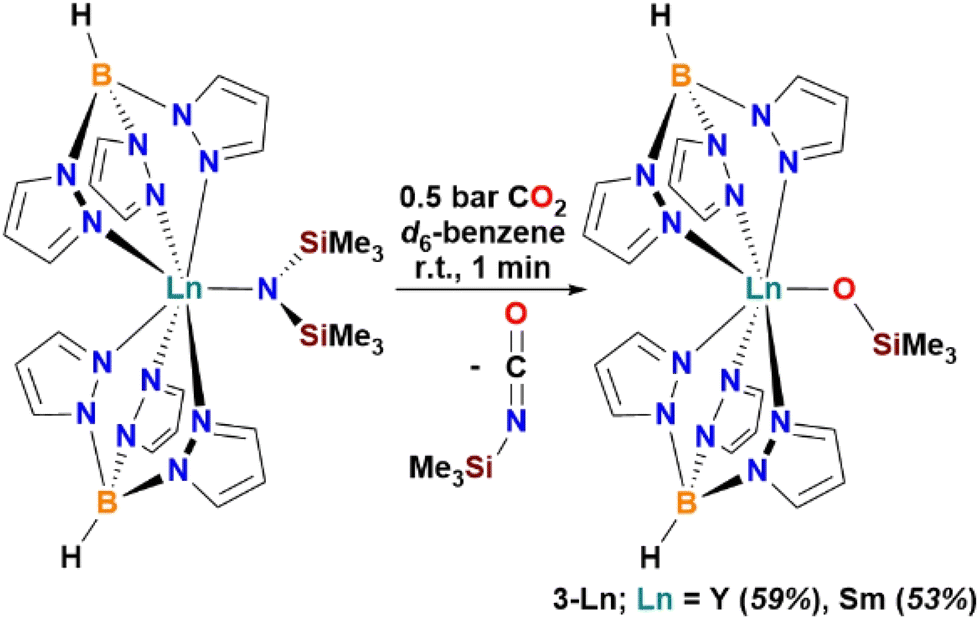 | ||
| Scheme 3 CO2 activation by Ln(III) complexes [Ln(Tp)2(N′′)] (Ln = Y, Sm; N′′ = N(SiMe3)2) to yield monomeric Ln(III) silyloxides [Ln(Tp)2(OSiMe3)] 3-Ln. | ||
The [Ln(Tp)2(N′′)] complexes are air/moisture-sensitive and adventitious moisture-mediated decomposition of [Y(Tp)2(N′′)] yields the hydroxide-bridged dimer [{Y(Tp)2(μ-OH)}2].32a On one occasion during exploration of the reactivity of [Sm(Tp)2(N′′)] with small-molecules, similar adventitious moisture-mediated decomposition of [Sm(Tp)2(N′′)] yielded a hydroxide-bridged cluster [Sm5(Tp)6(μ2-OH)6(μ3-OH)2(μ4-OH)] 4-Sm (see ESI section A1.6† for NMR data for 4-Sm and Fig. S69† for the connectivity-only molecular structure of 4-Sm). The formation of a hydroxide-bridged {Sm5} cluster instead of a hydroxide-bridged {Ln2} dimer as seen for Y(III) is consistent with the larger size of the Sm(III) ion when compared to the Y(III) ion.38
Spectroscopy of [Ln(Tp)2(OSiMe3)] 3-Ln (Ln = Y, Sm)
In the 1H NMR spectrum of 3-Y in d6-benzene, the Tp-pyrazolyl protons in 3-Y are observed at δ = 5.81, 7.26 and 7.49 ppm, the Tp-borohydride at δ = 4.82 ppm, and the trimethylsilyl protons at δ = −0.06 ppm, in the expected 6:6:6:2:9 ratio. In the 1H NMR spectrum of 3-Sm in d6-benzene, the Tp-pyrazolyl protons in 3-Sm are observed at δ = 2.61, 5.64 and 8.96 ppm, the Tp-borohydride at δ = 8.22 ppm, and the trimethylsilyl protons at δ = 2.56 ppm, in the expected 6:6:6:2:9 ratio. The OSiMe3 ligand resonance in 3-Y is comparable to that observed in [Y{2,2′-bis-((tBuMe2Si)N)-6,6′-Me2-biphenyl}(OSiMe3)(THF)2] (δ = 0.26 ppm).44 The proton and carbon resonances of the Tp ligands and the OSiMe3 ligand of 3-Ln were assigned by using 13C{1H} and 1H-13C HSQC NMR experiments. The 11B NMR spectra of 3-Ln exhibit a resonance (δ = −2.93 ppm 3-Y, δ = 5.43 ppm 3-Sm), corresponding to the Tp ligands, similar to the chemical shifts observed in the amide complexes [Ln(Tp)2(N′′)] (Ln = Y, δ = −2.96 ppm;32a Sm, δ = 6.25 ppm).32c In the ATR-IR spectrum of 3-Ln, besides weak absorptions between 2350–2470 cm−1 assigned to the characteristic borohydride stretching frequencies (υBH) for the Tp ligands, weak absorptions were also observed between 2890–2960 cm−1, which are assigned to the aliphatic sp3-carbon hydrogen bond stretching frequencies (υsp3-CH) of the OSiMe3 ligand.16,44
Crystallography of [Ln(Tp)2(OSiMe3)] 3-Ln (Ln = Y, Sm)
Rod-shaped colourless single-crystals of 3-Y suitable for X-ray diffraction were grown from a saturated d6-benzene solution with a hexane antisolvent at −35 °C overnight, and tablet-shaped colourless single-crystals of 3-Sm suitable for X-ray diffraction were grown from a saturated hexane solution at −35 °C over three months. The structures of 3-Ln are monomeric, containing a 7-coordinate Ln(III) ion bound to two κ3-coordinated Tp ligands and a monodentate OSiMe3 anion, arranged around the Ln(III) ion in a pentagonal bipyramidal geometry (Fig. 4). The [Ln(Tp)2(κ2-O,O(SiMe3)CNSiMe3)] intermediate in the formation of 3-Ln could not be isolated by crystallisation of the crude reaction mixtures, since 3-Ln was crystallised from all attempts. The Ln–N(Tp) bond distances (3-Sm: 2.538(3)–2.589(3) Å, 3-Y: 2.451(6)–2.567(8) Å) and Ln–O(OSiMe3) bond distances (3-Sm: 2.155(2) Å, 3-Y: 2.094(6) Å), are consistent with differences in size of the different Ln(III) ionic radii Sm(III) > Y(III) and the fact that a range of Ln–N(κ3-Tp) bond distances is normally observed in the literature [Ln(Tp)2]+ complexes.38 The Ln–N(κ3-Tp) bond distances in 3-Ln are consistent with [Ln(Tp)2(X)].32 The Ln–O(OSiMe3) bond distances in 3-Ln are consistent with heteroleptic monomeric Ln(III) silyloxides containing Ln–O(OSiMe3) (Ln = Y,44,45 Sm)46 bonds and homoleptic monomeric Ln(III) silyloxides [Ln(OSiR3)3(LB)x] (Ln = Y, R = OtBu,47 Ph;48 Sm, R = SiMe3,49 Ph)50 (see ESI Table 1† for detailed data comparisons). The Sm–N(Tp) bond distances in 3-Sm are shorter than those in 1-Sm(LB), consistent with the smaller ionic radius of Sm(III) vs. Sm(II).38
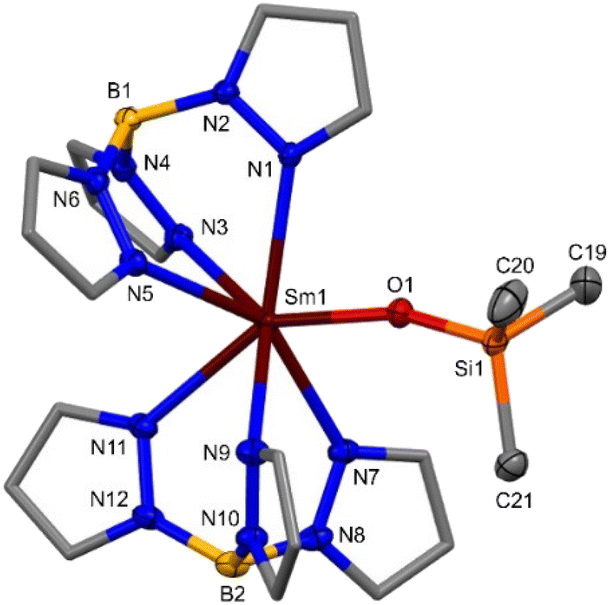 | ||
| Fig. 4 Molecular structure of 3-Sm (see ESI Fig. S67† for the molecular structure of 3-Y). Hydrogen atoms omitted for clarity and pyrazolyl carbon atoms of Tp displayed in wireframe. Displacement ellipsoids drawn at 50% probability level. Selected bond distances in 3-Sm: Sm–N(Tp) 2.538(3)–2.589(3) Å, Sm–O(OSiMe3) 2.155(2) Å; Selected bond distances in 3-Y: Y–N(Tp) 2.451(6)–2.567(8) Å, Y–O(OSiMe3) 2.094(6) Å. | ||
Conclusions
The reduction of heteroleptic Ln(III) triflate complexes [Ln(Tp)2(OTf)] (Tp = hydrotris(1-pyrazolyl)borate; OTf = triflate) was successfully extended to Sm(II). The reduction of [Sm(Tp)2(OTf)] with KC8 in THF yielded the insoluble Sm(II) [{Sm(Tp)2}n] 1-Sm multi-metallic coordination polymer, which was successfully isolated as the monomeric 1,2-dimethoxyethane (DME) adduct complex [Sm(Tp)2(DME)] 1-Sm(DME). The reactivity of 1-Sm(DME) was investigated with various small molecules such as pnictogens and CO2. The reactions of 1-Sm(DME) with molecular pnictogens (white P4, As0nano) yielded intractable Sm(III) products and [Sm(Tp)3]. The reaction of 1-Sm(DME) with CO2 yielded the oxalate-bridged dimeric Sm(III) homobimetallic complex [{Sm(Tp)2}2(μ-η2:η2-O2CCO2)] 2-Sm. Furthermore, CO2 activation by lanthanide silylamide complexes in the bis-Tp ligand environment was investigated. Reactions of the heteroleptic Ln(III) amides [Ln(Tp)2(N′′)] (Ln = Y, Sm; N′′ = N(SiMe3)2) with CO2, was proposed to proceed via formation a carbamate intermediate [Ln(Tp)2(κ2-O,OCN′′)], which was not observed, but the subsequent intermediate [Ln(Tp)2(κ2-O,O(SiMe3)CNSiMe3)] in this pathway, resulting from an intramolecular silyl-migration was observed on the NMR-scale. Upon workup of the reaction mixtures and removal of the CO2-functionalised product OCNSiMe3, the monomeric Ln(III) silyloxides [Ln(Tp)2(OSiMe3)] 3-Ln were isolated. The bis-Tp ligand environment on the Ln(III) ion enabled isolation of 3-Ln as discrete molecular silyloxides, which are the first crystallographically characterised examples of Ln(III)–OSiMe3 bonds accessed via CO2 activation and functionalisation.
Experimental
General experimental considerations
All air-sensitive manipulations were carried out in an MBraun glovebox (inert atmosphere of N2, O2 < 0.1 ppm, H2O < 1.5 ppm) or by using standard Schlenk techniques under N2. All glassware was dried at 130 °C overnight, in a Binder ED53 Drying Oven/Hot Air Steriliser, prior to use. An Innovative Technology Inc. Pure Solv 400-5-MD solvent purification system (activated alumina columns) was used to obtain anhydrous toluene and tetrahydrofuran (THF). Anhydrous hexane (95%) and 1,2-dimethoxyethane (DME, 99.5%) were purchased from Merck. Anhydrous solvents were degassed, sparged with N2, and stored in ampoules over activated 3.0 Å molecular sieves (25–35% weight by volume) under N2. Absence of water/levels of residual water in bulk solvents were confirmed by using a sodium benzophenone ketyl solution in THF after 24–48 hours. Deuterated benzene (d6-benzene) and deuterated THF (d8-THF) were purchased from Merck and dried by directly transferring from sealed glass ampoules onto activated 3.0 Å molecular sieves and stored in ampoules in a N2 atmosphere glovebox. Deuterated toluene (d8-toluene) was purchased from Merck and degassed by three freeze–pump–thaw degassing cycles and dried by storing in ampoules over activated 3.0 Å molecular sieves in a N2 atmosphere glovebox. Deuterated acetonitrile (d3-MeCN) was purchased from Merck and dried by refluxing over CaH2, degassed by three freeze–pump–thaw degassing cycles, filtered across a frit coated with Celite® into a Büchner flask, to exclude excess CaH2 and stored in ampoules over activated 3.0 Å molecular sieves in a N2 atmosphere glovebox. Absence of water in deuterated solvents was confirmed by 1H NMR after 48 hours. Carbon dioxide (CO2, 99.98%, vapour withdrawal) was purchased from BOC. Potassium hydrotris(1-pyrazolyl)borate K(Tp), K(N′′) (N′′ = N(SiMe3)2), Ln(OTf)3 (OTf = CF3SO3; Ln = Y, Sm, Dy), [Ln(Tp)2(OTf)] (Ln = Y, Sm, Dy), [Ln(Tp)2(N′′)] (Ln = Y, Sm), were synthesised according to published procedures.32Physical methods
NMR data were recorded on an AVIII 400 MHz spectrometer operating at frequencies 400.1 MHz (1H), 100.6 MHz (13C{1H}), 128.4 MHz (11B and 11B{1H}). The NMR data were referenced internally to the appropriate residual proteo-solvent and reported relative to tetramethylsilane (δ = 0 ppm) for 1H and 13C{1H} NMR. 11B and 11B{1H} NMR data were reported relative to 15% BF3·OEt2 in CDCl3 (δ = 0 ppm). All spectra were recorded at a constant temperature of 25 °C (298 K). Coupling constants (J) are reported in hertz (Hz). Standard abbreviations for multiplicity were used as follows: m = multiplet, t = triplet, d = doublet, s = singlet. For broad intensities, abbreviated as br, the full width at half-maximum intensity (FWHM) is provided in Hz. ATR-IR spectra were collected in air at ambient temperature using ThermoFisher Scientific Nicolet Summit LITE FTIR Spectrometer (containing a LiTaO3 detector) equipped with Everest ATR. Abbreviations for the intensity of stretching frequencies were used as follows: s = strong, m = medium, w = weak. Single-crystal X-ray diffraction data for 1-Sm(THF), 1-Sm(DME), Y-pz, 3-Y, and 3-Sm were collected at 150 K using Mo-Kα radiation (λ = 0.71073 Å) on a Bruker D8 VENTURE diffractometer equipped with a Photon II CPAD detector, with Oxford Cryosystems n-Helix device mounted on an IμS 3.0 (dual Cu and Mo) microfocus sealed tube generator. Single-crystal X-ray diffraction data for 2-Sm (on beamline I19 at the Diamond Light Source) and 4-Sm were collected by the EPSRC UK National Crystallography Service (University of Southampton). Deposition numbers 2347954–2347960† contain the supplementary crystallographic data for this paper. Elemental analyses were performed by Orla McCullough at the London Metropolitan University, using a Flash 2000 Organic Elemental Analyzer, Thermo Scientific analyser. The samples for the measurements were prepared using V2O5 (to ensure complete combustion of all complexes) in tin capsules inside an inert argon glovebox atmosphere.Synthesis of Sm(II) [{Sm(Tp)2}n] 1-Sm and [Sm(Tp)2(LB)x] (LB = THF, x = 2; DME, x = 1) 1-Sm(LB)
C), 1402 (s), 1384 (s), 1293 (s), 1209 (s), 1118 (s), 1045 (s), 973 (s), 925 (w), 876 (w), 803 (w), 752 (s), 722 (s), 668 (s), 620 (s) cm−1. The multinuclear NMR data below for 1-Sm(THF) was obtained by extracting product into d8-toluene from the dried filtrate in THF (after synthesis) and the data were collected within 12 hours. 1H NMR (d8-toluene): δ −3.64 (2H, very br m, FWHM = 289.4 Hz, Tp-BH) −1.56 (6H, s, FWHM = 25.0 Hz, Tp-CH), 0.18 (8H, s, FWHM = 19.9 Hz, THF-CH2), 2.27 (8H, s, FWHM = 27.2 Hz, THF-CH2), 4.02 (6H, s, FWHM = 26.3 Hz, Tp-CH), 15.10 (6H, very br s, FWHM = 106.4 Hz, Tp-CH) ppm; 13C{1H} NMR (d8-toluene): δ 25.3 (s, THF-C), 74.2 (s, Tp-C), 85.0 (very br s, THF-C), 112.7 (s, Tp-C), 204.7 (br s, Tp-C) ppm; 11B NMR (d8-toluene): δ −32.00 (d, 1JB–H = 82.4 Hz, Tp-B) ppm; 11B{1H} NMR (d8-toluene): δ −32.00 (s, Tp-B) ppm. Desolvation of THF-adducts from 1-Sm(THF) is observed in solution in 48 hours in d8-toluene. When heated up to 65 °C in d8-toluene, 1-Sm(THF) desolvates completely in an hour, liberating free THF, 1-Sm, and [Sm(Tp)3]. Lath-shaped red single-crystals of [Sm(Tp)2(THF)2·(THF)] 1-Sm(THF) suitable for X-ray diffraction were grown from a saturated THF solution at −35 °C overnight.
C), 1402 (s), 1386 (s), 1296 (s), 1211 (s), 1118 (s), 1040 (s), 973 (s), 923 (w), 887 (w), 854 (w), 805 (w), 752 (s), 720 (s), 664 (s), 620 (s) cm−1.
Reaction of [Dy(Tp)2(OTf)] with KC8 in toluene resulting in the observation of [Dy(Tp)3]
In the glovebox, a 20 mL scintillation vial was charged with a stirrer bar, and then the white powder [Dy(Tp)2(OTf)] (31.6 mg, 0.043 mmol, 1.0 eq.) was suspended in toluene (3 mL) and stirred. To this white suspension, the bronze powder KC8 (10.9 mg, 0.081 mmol, 1.9 eq.) was added by spatula in small portions over two minutes, with stirring at ambient temperature. The resultant dark-brown suspension was stirred at ambient temperature for 22.5 h, after which the resultant black-brown suspension was filtered across a frit into a Büchner flask, to exclude excess KC8, graphite, and K(OTf). Toluene (3 mL) was removed in vacuo to yield a dark orange-brown solid, which was dried in vacuo (10−2 mbar, 4.5 h). Subsequently, d6-benzene (0.5 mL) was added, and the resultant orange solution was analysed via multinuclear NMR to show the NMR resonances consistent with [Dy(Tp)3]36b as a major product amongst other unidentified Dy(III) products.Reaction of [Y(Tp)2(OTf)] with KC8 in THF resulting in isolation of [Y(Tp)2(κ2-pz)] Y-pz (pz = pyrazolyl)
In the glovebox, a 20 mL scintillation vial was charged with a stirrer bar, and then the white powder [Y(Tp)2(OTf)·(toluene)0.07] (42.2 mg, 0.063 mmol, 1.0 eq.) was dissolved in THF (3.5 mL) and stirred. To this colourless solution, the bronze powder KC8 (20.0 mg, 0.148 mmol, 2.4 eq.) was added by spatula in small portions over two minutes, with stirring at ambient temperature. The resultant dark-brown suspension was stirred at ambient temperature for 20 h, after which the suspension was filtered across a frit into a Büchner flask, to exclude excess KC8 and graphite. THF (3.5 mL) was removed in vacuo to yield a yellow-brown oil, which was dried in vacuo (10−2 mbar, 1.5 h). The yellow-brown oil was washed with hexane (3 × 1 mL) and the washings filtered through a pipette containing a Kimwipe. The pale-yellow filtrate was cooled down to −35 °C and the product [Y(Tp)2(κ2-pz)] Y-pz crystallised overnight as colourless plates, including crystals of [Y(Tp)3] (see ESI Fig. S17(b)† for overlay of 1H NMR data for Y-pz and [Y(Tp)3]32b). 1H NMR (d6-benzene): δ 4.77 (2H, very br m, FWHM = 269.9 Hz, Tp-BH), 5.74 (6H, t, 3JH–H = 2.1 Hz, Tp-C4H), 6.59 (1H, t, 3JH–H = 1.6 Hz, pz-C4H), 6.88 (6H, d, 3JH–H = 1.8 Hz, Tp-C3H), 7.42 (6H, dd, 3JH–H = 2.2 Hz, 4JH–H = 0.6 Hz, Tp-C5H), 7.70 (2H, d, 3JH–H = 1.6 Hz, pz-C3H and pz-C5H) ppm; 11B NMR (d6-benzene): δ −2.86 (d, 1JB–H = 84.9 Hz, Tp-B) ppm; 11B{1H} NMR (d6-benzene): δ −2.82 (s, Tp-B) ppm.Reaction of 1-Sm(DME) and [Ln(Tp)2(N′′)] (Ln = Y, Sm) with carbon dioxide (CO2) gas
O), 1505 (m, υCC), 1431 (w), 1424 (w), 1404 (m), 1383 (m), 1294 (s), 1213 (s), 1198 (m), 1186 (m), 1120 (s), 1062 (m), 1045 (s), 1007 (m), 974 (s), 924 (w), 901 (w), 878 (w), 854 (w), 839 (w), 805 (w), 778 (m), 768 (s), 750 (s), 740 (s), 724 (s), 669 (s), 620 (m), 549 (w) cm−1.
CNSiMe3 were removed in vacuo from the colourless solution to yield a colourless oil. Hexane (1 mL) was added to precipitate white solids, which were washed with hexane (1 mL) and the washing subsequently decanted away to remove impurities. All solvents were removed in vacuo and the resultant white powder was scraped with a spatula and dried in vacuo (10−2 mbar, 1 h) yielding [Y(Tp)2(OSiMe3)] 3-Y (11.3 mg, 0.019 mmol, 59%). Complex 3-Y has excellent solubility in toluene and moderate solubility in hexane. Rod-shaped colourless single-crystals of 3-Y suitable for X-ray diffraction were grown from a saturated d6-benzene solution with a hexane antisolvent at −35 °C overnight. 1H NMR (d6-benzene): δ −0.06 (9H, s, OSi(CH3)3), 4.82 (2H, very br m, FWHM = 228.1 Hz, Tp-BH), 5.81 (6H, t, 3JH–H = 2.1 Hz, Tp-C4H), 7.26 (6H, d, 3JH–H = 1.4 Hz, Tp-C3H), 7.49 (6H, d, 3JH–H = 2.1 Hz, Tp-C5H) ppm; 13C{1H} NMR (d6-benzene): δ 3.2 (s, OSi(CH3)3), 104.5 (s, Tp-C4), 135.2 (s, Tp-C5) 142.0 (s, Tp-C3) ppm; 11B NMR (d6-benzene): δ −2.93 (d, 1JB–H = 44.8 Hz, Tp-B) ppm; 11B{1H} NMR (d6-benzene): δ −2.72 (s, Tp-B) ppm. Anal. Calcd for C21H29B2N12OSiY: C, 41.75%; H, 4.84%; N, 27.82%. Found: C, 36.68%; H, 3.08%; N, 24.81%. Lower CHN values than calculated are the result of minor byproducts that could not be eliminated by recrystallisation of 3-Y. IR (ATR): 3616 (w), 3139 (w, υsp2-CH), 2947 (w, υsp3-CH), 2456 (w, υBH), 2416 (w, υBH), 2373 (w, υBH), 1720 (w), 1504 (m, υCC), 1404 (m), 1387 (m), 1299 (s), 1252 (w), 1240 (w), 1214 (s), 1200 (m), 1187 (m), 1120 (s), 1065 (w), 1046 (s), 975 (s), 950 (s), 924 (w), 887 (2), 824 (w), 781 (m), 737 (s), 722 (s), 670 (s), 621 (s) cm−1.
CNSiMe3 were removed in vacuo from the white suspension to yield a colourless oil. Hexane (1 mL) was added to precipitate white solids, which were washed with hexane (1 mL) and the washing subsequently decanted away to remove impurities. All solvents were removed in vacuo and the resultant white powder was scraped with a spatula and dried in vacuo (10−2 mbar, 1 h) yielding [Sm(Tp)2(OSiMe3)] 3-Sm (8.8 mg, 0.013 mmol, 53%). Complex 3-Sm has excellent solubility in toluene and moderate solubility in hexane. Tablet-shaped colourless single-crystals of 3-Sm suitable for X-ray diffraction were grown from a saturated hexane solution at −35 °C over three months. 1H NMR (d6-benzene): δ 2.56 (9H, s, overlapped with the resonance at δ 2.61 ppm integrating to 6H, FWHM = 16.1 Hz, OSi(CH3)3), 2.61 (6H, br s, overlapped with the resonance at δ 2.56 ppm integrating to 9H, FWHM = 26.3 Hz, Tp-C4H), 5.64 (6H, d, 3JH–H = 1.8 Hz, Tp-C3/5H), 8.22 (2H, very br m, FWHM = 283.8 Hz, Tp-BH), 8.96 (6H, d, 3JH–H = 1.9 Hz, Tp-C3/5H) ppm; 13C{1H} NMR (d6-benzene): δ 6.0 (s, OSi(CH3)3), 103.4 (s, Tp-C3/5), 136.3 (s, Tp-C3/5), 139.9 (s, Tp-C4) ppm; 11B NMR (d6-benzene): δ 5.43 (d, 1JB–H = 76.5 Hz, Tp-B) ppm; 11B{1H} NMR (d6-benzene): δ 5.43 (s, Tp-B) ppm. Anal. Calcd for C21H29B2N12OSiSm: C, 37.89%; H, 4.39%; N, 25.25%. Found: C, 39.00%; H, 3.83%; N, 23.65%. IR (ATR): 3626 (w), 3146 (w, υsp2-CH), 2949 (w, υsp3-CH), 2893 (w, υsp3-CH), 2452 (w, υBH), 2412 (w, υBH), 2367 (w, υBH), 1503 (m, υCC), 1403 (m), 1383 (m), 1297 (s), 1254 (w), 1240 (w), 1212 (s), 1198 (m), 1187 (m), 1118 (s), 1065 (m), 1044 (s), 974 (s), 956 (s), 924 (w), 825 (m), 805 (w), 760 (s), 738 (s), 721 (s), 670 (s), 621 (s) cm−1.
Isolation of cluster [Sm5(Tp)6(μ2-OH)6(μ3-OH)2(μ4-OH)] 4-Sm in the adventitious moisture-mediated decomposition of [Sm(Tp)2(N′′)]
On one occasion a toluene solution of [Sm(Tp)2(N′′)] reacted with adventitious moisture in the solvent yielding [Sm5(Tp)6(μ2-OH)6(μ3-OH)2(μ4-OH)] 4-Sm. Plate-shaped colourless single-crystals of 4-Sm suitable for X-ray diffraction were grown from a dilute hexane solution at −35 °C over a week. 1H NMR (d6-benzene): δ −1.47 (9H under integrating as 3H, br s, FWHM = 21.4 Hz, μ-OH), 5.14 (18H, very br s, overlapped with the resonance at δ 5.71 ppm integrating to 18H, FWHM = 282.0 Hz, Tp-CH), 5.71 (18H, br s, overlapped with the resonance at δ 5.14 ppm integrating to 18H, FWHM = 26.4 Hz, Tp-CH), 6.79 (6H, overlapped with the d6-benzene resonance, FWHM = 329.2 Hz, Tp-BH), 8.40 (18H, s, FWHM = 10.4 Hz, Tp-CH) ppm; 13C{1H} NMR (d6-benzene): δ 103.6 (s, Tp-C), 135.3 (s, Tp-C), 141.2 (br s, Tp-C) ppm; 11B NMR (d6-benzene): δ 1.55 (d, 1JB–H = 39.5 Hz, Tp-B) ppm; 11B{1H} NMR (d6-benzene): δ 1.36 (s, Tp-B) ppm. IR (ATR): 3146 (w, υsp2-CH), 3118 (w, υsp2-CH), 2441 (w, υBH), 2409 (w, υBH), 2374 (w, υBH), 1505 (m, υCC), 1431 (w), 1423 (w), 1402 (m), 1383 (m), 1295 (s), 1212 (s), 1198 (m), 1186 (m), 1119 (s), 1062 (m), 1045 (s), 974 (m), 923 (w), 900 (w), 879 (w), 806 (w), 778 (m), 768 (s), 750 (s), 739 (m), 723 (s), 669 (m), 620 (m) cm−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the University of Glasgow (UoG) for funding, and the award of a College of Science and Engineering (UoG) PhD Scholarship to T. C. We thank Dr Zeliha Ertekin from the research group of Professor Dr Mark D. Symes (UoG) for lending us a carbon dioxide gas cylinder. We are grateful to Professor Dr Peter W. Roesky at the Karlsruhe Institute of Technology (KIT, Germany) for valuable discussions on reactivity of Sm(II) and Mr David Frick for investigating the reactivity of Ln(II) 1-Sm(DME) and [Yb(Tp)2] with pnictogens (P4, As0nano) at KIT. We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data for 2-Sm and 4-Sm and additionally Diamond Light Source for an award of beamtime on I19 (CY31778) for 2-Sm.References
- (a) K. A. Grice, Coord. Chem. Rev., 2017, 336, 78–95 CrossRef CAS; (b) K. Blaziak, D. Tzeli, S. S. Xantheas and E. Uggerud, Phys. Chem. Chem. Phys., 2018, 20, 25495–25505 RSC.
- (a) U. Bayer and R. Anwander, Dalton Trans., 2020, 49, 17472–17493 RSC; (b) O. P. Lam and K. Meyer, Polyhedron, 2012, 32, 1–9 CrossRef CAS.
- M. Pérez-Jiménez, H. Corona, F. de la Cruz-Martínez and J. Campos, Chem. – Eur. J., 2023, 29, e202301428 CrossRef PubMed.
- (a) D. H. Gibson, Chem. Rev., 1996, 96, 2063–2096 CrossRef CAS PubMed; (b) A. Paparo and J. Okuda, Coord. Chem. Rev., 2017, 334, 136–149 CrossRef CAS.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- (a) J. C. Wedal and W. J. Evans, J. Am. Chem. Soc., 2021, 143, 18354–18367 CrossRef CAS PubMed; (b) M. Szostak and D. J. Procter, Angew. Chem., Int. Ed., 2012, 51, 9238–9256 CrossRef CAS PubMed.
- (a) W. J. Evans, C. A. Seibel and J. W. Ziller, Inorg. Chem., 1998, 37, 770–776 CrossRef CAS; (b) W. J. Evans, J. M. Perotti, J. C. Brady and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 5204–5212 CrossRef CAS PubMed; (c) L. Castro, S. Labouille, D. R. Kindra, J. W. Ziller, F. Nief, W. J. Evans and L. Maron, Chem. – Eur. J., 2012, 18, 7886–7895 CrossRef CAS PubMed; (d) L. Castro, D. P. Mills, C. Jones and L. Maron, Eur. J. Inorg. Chem., 2016, 2016, 792–796 CrossRef CAS; (e) H. M. Nicholas and D. P. Mills, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2017, 1–10. DOI:10.1002/9781119951438.eibc2453; (f) M. Xémard, V. Goudy, A. Braun, M. Tricoire, M. Cordier, L. Ricard, L. Castro, E. Louyriac, C. E. Kefalidis, C. Clavaguéra, L. Maron and G. Nocton, Organometallics, 2017, 36, 4660–4668 CrossRef; (g) M. Xémard, M. Cordier, E. Louyriac, L. Maron, C. Clavaguéra and G. Nocton, Dalton Trans., 2018, 47, 9226–9230 RSC; (h) N. W. Davies, A. S. P. Frey, M. G. Gardiner and J. Wang, Chem. Commun., 2006, 4853–4855, 10.1039/B611784H.
- N. Tsoureas, L. Castro, A. F. R. Kilpatrick, F. G. N. Cloke and L. Maron, Chem. Sci., 2014, 5, 3777–3788 RSC.
- C. J. Inman, A. S. P. Frey, A. F. R. Kilpatrick, F. G. N. Cloke and S. M. Roe, Organometallics, 2017, 36, 4539–4545 CrossRef CAS.
- (a) J. Zhang and X. Zhou, Dalton Trans., 2011, 40, 9637–9648 RSC; (b) H. Yin, P. J. Carroll and E. J. Schelter, Chem. Commun., 2016, 52, 9813–9816 RSC; (c) W. J. Evans, C. H. Fujimoto and J. W. Ziller, Organometallics, 2001, 20, 4529–4536 CrossRef CAS.
- (a) R. D. Dicken, A. Motta and T. J. Marks, ACS Catal., 2021, 11, 2715–2734 CrossRef CAS; (b) R. Anwander, in Organolanthoid Chemistry: Synthesis, Structure, Catalysis, Springer Berlin Heidelberg, Berlin, Heidelberg, 1996, pp. 33–112. DOI:10.1007/BFb0015595.
- (a) E. C. Alyea, D. C. Bradley and R. G. Copperthwaite, J. Chem. Soc., Dalton Trans., 1972, 1580–1584, 10.1039/dt9720001580; (b) D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 1021–1023, 10.1039/dt9730001021; (c) P. G. Eller, D. C. Bradley, M. B. Hursthouse and D. W. Meek, Coord. Chem. Rev., 1977, 24, 1–95 CrossRef CAS; (d) C. A. P. Goodwin and D. P. Mills, in Organometallic Chemistry, ed. I. Fairlamb, J. M. Lynam, N. J. Patmore and P. Elliott, The Royal Society of Chemistry, 2017, vol. 41 Search PubMed.
- (a) Z. R. Turner, Inorganics, 2015, 3, 597–635 CrossRef CAS; (b) M. G. Gardiner and D. N. Stringer, Materials, 2010, 3, 841–862 CrossRef CAS.
- (a) M. N. Bochkarev, E. A. Fedorova, Y. F. Radkov, S. Y. Khorshev, G. S. Kalinina and G. A. Razuvaev, J. Organomet. Chem., 1983, 258, C29–C33 CrossRef CAS; (b) C. Krempner and B. McNerney, in Encyclopedia of Inorganic and Bioinorganic Chemistry, 2012, DOI:10.1002/9781119951438.eibc2089.
- L. R. Sita, J. R. Babcock and R. Xi, J. Am. Chem. Soc., 1996, 118, 10912–10913 CrossRef CAS.
- T. J. Boyle and L. A. M. Ottley, Chem. Rev., 2008, 108, 1896–1917 CrossRef CAS PubMed.
- (a) J. Takats, J. Alloys Compd., 1997, 249, 52–55 CrossRef CAS; (b) A. C. Hillier, S. Y. Liu, A. Sella and M. R. J. Elsegood, J. Alloys Compd., 2000, 303–304, 83–93 CrossRef CAS; (c) N. Marques, A. Sella and J. Takats, Chem. Rev., 2002, 102, 2137–2160 CrossRef CAS PubMed; (d) C. Hossack, C. Cahill and C. Besson, Dalton Trans., 2023, 52, 17656–17665 RSC.
- A. C. Hillier, A. Sella and M. R. J. Elsegood, J. Organomet. Chem., 2002, 664, 298–305 CrossRef CAS.
- G. H. Maunder, M. R. Russo and A. Sella, Polyhedron, 2004, 23, 2709–2714 CrossRef CAS.
- X. Zhang, G. R. Loppnow, R. McDonald and J. Takats, J. Am. Chem. Soc., 1995, 117, 7828–7829 CrossRef CAS.
- M. Kühling, R. McDonald, P. Liebing, L. Hilfert, M. J. Ferguson, J. Takats and F. T. Edelmann, Dalton Trans., 2016, 45, 10118–10121 RSC.
- (a) A. C. Hillier, S.-Y. Liu, A. Sella and M. R. J. Elsegood, Inorg. Chem., 2000, 39, 2635–2644 CrossRef CAS PubMed; (b) I. Lopes, A. C. Hillier, S. Y. Liu, Â. Domingos, J. Ascenso, A. Galvão, A. Sella and N. Marques, Inorg. Chem., 2001, 40, 1116–1125 CrossRef CAS PubMed.
- J. Takats, X. W. Zhang, V. W. Day and T. A. Eberspacher, Organometallics, 1993, 12, 4286–4288 CrossRef CAS.
- I. Lopes, R. Dias, Â. Domingos and N. Marques, J. Alloys Compd., 2002, 344, 60–64 CrossRef CAS.
- Â. Domingos, I. Lopes, J. C. Waerenborgh, N. Marques, G. Y. Lin, X. W. Zhang, J. Takats, R. McDonald, A. C. Hillier, A. Sella, M. R. J. Elsegood and V. W. Day, Inorg. Chem., 2007, 46, 9415–9424 CrossRef PubMed.
- I. Lopes, G. Y. Lin, A. Domingos, R. McDonald, N. Marques and J. Takats, J. Am. Chem. Soc., 1999, 121, 8110–8111 CrossRef CAS.
- G. Lin, R. McDonald and J. Takats, Organometallics, 2000, 19, 1814–1816 CrossRef CAS.
- (a) A. C. Hillier, S. Y. Liu, A. Sella, O. Zekria and M. R. J. Elsegood, J. Organomet. Chem., 1997, 528, 209–215 CrossRef CAS; (b) A. C. Hillier, A. Sella and M. R. J. Elsegood, J. Organomet. Chem., 1999, 588, 200–204 CrossRef CAS.
- A. C. Hillier, X. W. Zhang, G. H. Maunder, S. Y. Liu, T. A. Eberspacher, M. V. Metz, R. McDonald, Â. Domingos, N. Marques, V. W. Day, A. Sella and J. Takats, Inorg. Chem., 2001, 40, 5106–5116 CrossRef CAS PubMed.
- (a) M. A. J. Moss, R. A. Kresinski, C. J. Jones and W. J. Evans, Polyhedron, 1993, 12, 1953–1955 CrossRef CAS; (b) Â. Domingos, J. Marçalo, N. Marques, A. P. D. Matos, A. Galvão, P. C. Isolani, G. Vicentini and K. Zinner, Polyhedron, 1995, 14, 3067–3076 CrossRef; (c) X. Zhang, R. McDonald and J. Takats, New J. Chem., 1995, 19, 573–585 CAS; (d) K. O. Saliu, J. Takats and M. J. Ferguson, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m643–m644 CrossRef CAS PubMed; (e) A. Momin, L. Carter, Y. Yang, R. McDonald, S. Essafi, F. Nief, I. Del Rosal, A. Sella, L. Maron and J. Takats, Inorg. Chem., 2014, 53, 12066–12075 CrossRef CAS PubMed; (f) M. Kühling, C. Wickleder, M. J. Ferguson, C. G. Hrib, R. McDonald, M. Suta, L. Hilfert, J. Takats and F. T. Edelmann, New J. Chem., 2015, 39, 7617–7625 RSC.
- T. Chowdhury, M. J. Evans, M. P. Coles, A. G. Bailey, W. J. Peveler, C. Wilson and J. H. Farnaby, Chem. Commun., 2023, 59, 2134–2137 RSC.
- (a) T. Chowdhury, S. J. Horsewill, C. Wilson and J. H. Farnaby, Aust. J. Chem., 2022, 75, 660–675 CrossRef CAS; (b) T. Chowdhury, C. Wilson, C. Maichle-Mössmer, R. Anwander and J. H. Farnaby, Eur. J. Inorg. Chem., 2024, 27, e202300731 CrossRef CAS; (c) T. Chowdhury, F. Murphy, A. R. Kennedy, C. Wilson, J. H. Farnaby and C. E. Weetman, Inorg. Chem., 2024, 63, 9390–9394 CrossRef CAS PubMed.
- J. L. Galler, S. Goodchild, J. Gould, R. McDonald and A. Sella, Polyhedron, 2004, 23, 253–262 CrossRef CAS.
- (a) L. J. Nugent, R. D. Baybarz, J. L. Burnett and J. L. Ryan, J. Phys. Chem., 1973, 77, 1528–1539 CrossRef CAS; (b) L. R. Morss, Chem. Rev., 1976, 76, 827–841 CrossRef CAS.
- (a) F. Nief, Dalton Trans., 2010, 39, 6589 RSC; (b) D. H. Woen and W. J. Evans, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2016, vol. 50, pp. 337–394 Search PubMed.
- (a) M. V. R. Stainer and J. Takats, J. Am. Chem. Soc., 1983, 105, 410–415 CrossRef CAS; (b) C. Apostolidis, J. Rebizant, B. Kanellakopulos, R. Von Ammon, E. Dornberger, J. Müller, B. Powietzka and B. Nuber, Polyhedron, 1997, 16, 1057–1068 CrossRef CAS; (c) I. Lopes, B. Monteiro, G. Lin, Â. Domingos, N. Marques and J. Takats, J. Organomet. Chem., 2001, 632, 119–125 CrossRef CAS.
- (a) J. H. Farnaby, F. G. N. Cloke, M. P. Coles, J. C. Green and G. Aitken, C. R. Chim., 2010, 13, 812–820 CrossRef CAS; (b) F. Ortu, H. Zhu, M.-E. Boulon and D. Mills, Inorganics, 2015, 3, 534–553 CrossRef CAS.
- (a) R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef; (b) R. E. Cramer, J. M. Rimsza and T. J. Boyle, Inorg. Chem., 2022, 61, 6120–6127 CrossRef CAS PubMed.
- (a) S. J. Swamy, J. Loebel, J. Pickardt and H. Schumann, J. Organomet. Chem., 1988, 353, 27–34 CrossRef CAS; (b) M. Håkansson, M. Vestergren, B. Gustafsson and G. Hilmersson, Angew. Chem., Int. Ed., 1999, 38, 2199–2201 CrossRef.
- S. N. Konchenko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, H. Schnöckel and P. W. Roesky, J. Am. Chem. Soc., 2009, 131, 5740–5741 CrossRef CAS PubMed.
- (a) H. Reddmann, C. Apostolidis, O. Walter, J. Rebizant and H.-D. Amberger, Z. Anorg. Allg. Chem., 2005, 631, 1487–1496 CrossRef CAS; (b) C. Apostolidis, A. Kovács, A. Morgenstern, J. Rebizant and O. Walter, Inorganics, 2021, 9, 44 CrossRef CAS.
- C. Schoo, S. Bestgen, A. Egeberg, J. Seibert, S. N. Konchenko, C. Feldmann and P. W. Roesky, Angew. Chem., Int. Ed., 2019, 58, 4386–4389 CrossRef CAS PubMed.
- M. A. J. Moss and C. J. Jones, Polyhedron, 1989, 8, 2367–2370 CrossRef CAS.
- T. I. Gountchev and T. D. Tilley, Organometallics, 1999, 18, 2896–2905 CrossRef CAS.
- Y. Ma, Y.-Q. Zhai, Y.-S. Ding, T. Han and Y.-Z. Zheng, Chem. Commun., 2020, 56, 3979–3982 RSC.
- M. Karl, G. Seybert, W. Massa, K. Harms, S. Agarwal, R. Maleika, W. Stelter, A. Greiner, W. H. B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 1999, 625, 1301–1309 CrossRef CAS.
- (a) A. Mortis, C. Maichle-Mössmer and R. Anwander, Dalton Trans., 2022, 51, 1070–1085 RSC; (b) G. Lapadula, A. Bourdolle, F. Allouche, M. P. Conley, I. del Rosal, L. Maron, W. W. Lukens, Y. Guyot, C. Andraud, S. Brasselet, C. Copéret, O. Maury and R. A. Andersen, Chem. Mater., 2014, 26, 1062–1073 CrossRef CAS.
- (a) M. J. McGeary, P. S. Coan, K. Folting, W. E. Streib and K. G. Caulton, Inorg. Chem., 1989, 28, 3283–3284 CrossRef CAS; (b) P. S. Gradeff, K. Yunlu, T. J. Deming, J. M. Olofson, R. J. Doedens and W. J. Evans, Inorg. Chem., 1990, 29, 420–424 CrossRef CAS; (c) M. J. McGeary, P. S. Coan, K. Folting, W. E. Streib and K. G. Caulton, Inorg. Chem., 1991, 30, 1723–1735 CrossRef CAS.
- T. J. Boyle, F. Guerrero, R. E. Cramer, P. C. Reuel, D. M. Boye and H. L. Brooks, Inorg. Chem., 2022, 61, 5048–5059 CrossRef CAS PubMed.
- Z. Xie, K. Chui, Q. Yang, T. C. W. Mak and J. Sun, Organometallics, 1998, 17, 3937–3944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2347954–2347960. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01382d |
| This journal is © The Royal Society of Chemistry 2024 |