
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zinc and cadmium thioamidate complexes: rational design of single-source precursors for the AACVD of ZnS†
Max E.
Robson
ab and
Andrew L.
Johnson
 *a
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: a.l.johnson@bath.ac.uk
bCentre of Doctoral Training in Aerosol Science, University of Bristol, School of Chemistry, Cantock's Close, BS8 1TS, UK
First published on 18th June 2024
Abstract
A series of zinc(II) thioamidate complexes [Zn{SC(iPr)NR}2]n for R = iPr (n = 2) (2), tBu (3) (n = 1), Ph (4) (n = 2) and Cy (5) (n = 2) and one cadmium(II) thioamidate complex [Cd{SC(iPr)NtBu}2]3, (6), were designed and synthesised as single-source precursors for AACVD ZnS and CdS. Solid-state structures of all four zinc(II) compounds revealed distorted tetrahedral or trigonal bipyramidal geometries, with varying tendencies for dimeric association, mediated by {Zn–S} bridging bonds. The thermogravimetric analysis identified the {tBu} derivertive, 3, as the most promising precursor based on its low decomposition onset (118 °C) and clean conversion to ZnS. This was attributed to the greater availability of β-hydrogen atoms promoting the pyrolysis mechanism. The corresponding cadmium thioamide 6 was found to crystallise as a trimetallic molecule which lacked the thermal stability to be considered viable for AACVD. Hence, 3 was used to deposit ZnS thin films by AACVD at 200–300 °C. Powder X-ray diffraction confirmed phase-pure growth of hexagonal wurtzite ZnS, with approximate crystallite sizes of 15–20 nm. Scanning electron microscopy revealed densely packed spherical nanoclusters. The morphology and crystallinity were most consistent for depositions between 250–300 °C. Energy dispersive X-ray spectroscopy indicated slightly sulfur-deficient stoichiometries.
Introduction
Zinc sulfide (ZnS) and cadmium sulfide (CdS) are two examples of direct bandgap semiconductor materials with polar surfaces, excellent transport properties, good thermal stability and high electronic mobility.1–6 These physical properties not only result in rich morphologies at the nanoscale, but make ZnS one of the most important semiconductors in the electronics industry.7–10 Similarly, CdS has attracted considerable attention,11–14 however toxicity concerns have limited the development of Cd-based materials recently.15Given that ZnS and CdS commonly exist as two possible polymorphs – cubic zinc blende (ZB-ZnS) and hexagonal wurtzite (W-ZnS) – each with disparate band structures,16 selective and reproducible fabrication methods are sought.17 CdS films are often deposited using chemical bath deposition.18–20 In contrast chemical bath deposition rates for ZnS are substantially lower than for CdS.21,22 While formulation of ZnS has also been achieved using sputtering,23,24 chemical vapour deposition,25,26 atomic layer deposition27–29 and spray pyrolysis,30,31 aerosol-assisted chemical vapour deposition (AACVD) offers a versatile and scalable technique proven to yield high quality inorganic films.32 Gas phase transfer of precursors is simplified by atomisation, circumventing the need for volatile precursors. Instead, chemical precursors with the appropriate solubility can be delivered to a heated deposition chamber at room temperature without the need for low pressures.33,34 Deposition quality invariably depends on precursor design. In this context, employing single-source precursors containing pre-existing bonds between target elements (Zn and S) can offer all-important control.
Despite this, the deposition of ZnS thin films via AACVD stands relatively underexplored and, to date, only a handful of single-source precursors have been studied. The first AACVD ZnS precursor reported was [Zn{SOCCH3}2(tmeda)] (tmeda = N,N,N,N-tetramethylethylenediamine), which formed mixed crystallographic phase films (ZB-ZnS and W-ZnS) at temperatures as low as 125–250 °C, setting the benchmark for low temperature deposition.35 While zinc(II) dithiocarbamates [Zn{S2CNR2}2] (R = nBu, Cy, Me/Ph)36,37 were later reported to deposit ZnS at medium temperatures (375–450 °C), these suffered from co-deposition of both cubic and hexagonal phases, preferentially forming ZB-ZnS at lower temperatures and W-ZnS above 400 °C.36 Interestingly, this temperature dependence was reversed where zinc(II) dithiobiuret [Zn{N(SCNMe2)2}2] was used over a similar temperature range.38 Zinc(II) thiosemicarbazones have been reported to form hexagonal W-ZnS films at 450 °C.39 Sullivan et al. previously reported the deposition of ZnS thin films using thioureide precursor [Zn{SCN(iPr)(NMe2)}2] at temperatures as low as 150 °C, and crystalline W–Zn deposited at temperatures above 250 °C.40 This has prevailed as the only precursor to deposit phase-pure ZnS at low temperatures and, consequently, a variation of this design has been used elsewhere for the deposition of Zn(O,S) via AACVD at 400 °C.41
Unsurprisingly, investigations into CdS single-source precursors often overlap with those of ZnS, including the use of compounds featuring thiourea,42–44 thiobiurets,38,45 thiocarboxylates35 and thiocarbamates.46–52 Additionally, dithioactylacetonates,53 xanthates54–57 and dialkyldithiophosphates58–61 have been studied, often for deployment in solvothermal processes. Despite the interest in CdS buffer layers, only a handful of single-source precursor designs have been used in AACVD.51,53,55,61
Various divalent metal sulfides have been deposited with alternative precursor designs,34,62,63 often extrapolated from the successful deposition of their corresponding, isoelectronic metal oxides.64 Most notably, building on the successful deposition of zirconium oxide from zirconium amidate precursors,65 Catherall et al. reported the first use of the thioamidate ligand framework towards tin(II) sulfide formulation.66 Deposition was conducted from 200 °C, the success of which was attributed primarily to the decomposition pathway presented in Scheme 1. Such well-defined pyrolysis mechanisms are seldom reported for single-source precursors, and this presents an otherwise untapped ligand design with great potential for deposition of metal sulfides.
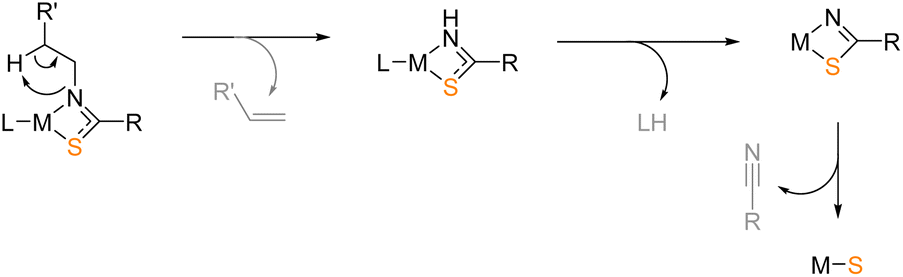 | ||
| Scheme 1 Suggested mechanism of metal thioamidate pyrolysis. | ||
Informed by this, a series of zinc thioamidate precursor complexes were sought and are presented here. Their synthesis and characterisation are reported, in addition to their solution-state dynamics and thermal profiling. These data informed the selection of a viable single-source precursor for AACVD of ZnS, a discussion of which is also presented. Informed by this, the corresponding cadmium thioamidate was also sought as a simple means to formulate CdS in an analogous manner.
Results and discussion
Pro-ligand synthesis
Given the requirement for volatile decomposition by-products for AACVD precursors, the zinc thioamidates targeted in this work contained an iso-propyl {C-iPr} backbone, which would form the isobutyronitrile by-product according to the decomposition pathway suggested by Catherall et al. (see Scheme 1). A series of N-substituted thioamide pro-ligands were synthesised, according to procedures reported in the literature.67 The insertion mechanism of an isothiocyanate into a metal alkyl species readily forms metal thioamidates. The appropriate Grignard reagents were used in THF solution and magnesium thioamidate intermediates formed (Scheme 2).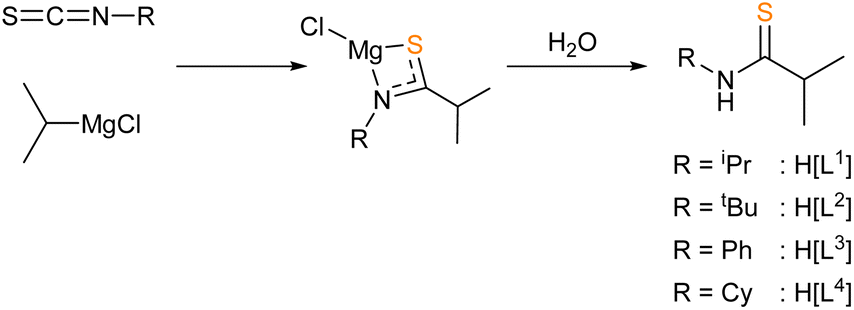 | ||
| Scheme 2 Synthesis of thioamides H[L1]–H[L4] via the insertion into a metal alkyl Grignard and aqueous workup. | ||
Aqueous work-up in situ formed the free thioamide. Both insertion and hydrolysis steps proceeded exothermically. After extraction with ethyl acetate, drying over sodium sulphate and in vacuo removal of volatiles, all pro-ligands H[L1]–H[L4] were isolated in high yields (73–77%) as microcrystalline off-white powders, of characteristic, sulfurous malodour.
All four pro-ligands were characterised by 1H and 13C{H} NMR spectroscopy. In benzene-d6, a resonance between, δH: 6.13–7.77 ppm, indicated the formation of the {N–H} moiety, in all four cases. 13C{H} spectra clearly showed the presence of a new carbon environment at the {C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S} position, with resonance shifts typical of thioamides (ranging δC: 209.8–210.5 ppm).
S} position, with resonance shifts typical of thioamides (ranging δC: 209.8–210.5 ppm).
Synthesis of zinc and cadmium bis-thioamidate complexes
The design of the metal thioimidate precursors was informed by the proposed pyrolysis mechanism of divalent metal sulfide precursors (Scheme 1), the second step of which sees the migration of a proton such to liberate a free spectator ligand. Considering this, the heteroleptic methyl zinc thioamide complex, 1, was initially targeted. It was hypothesised that this gas evolution (CH4), coupled with precursor asymmetry, would facilitate low-temperature decomposition and formation of ZnS. This was in a similar vein to our previous investigation of a family of zinc thioureide precursors of the general form [MeZn{SCN(R)(NMe2)}], [{(Me3Si)2N}Zn{SCN(R)(NMe2)}2] and [Zn{SCN(R)(NMe2)}2]2 using ZnMe2 and [Zn{N(SiMe3)2}2] ({N(SiMe3)2}![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) (HMDS)).40
(HMDS)).40
In an attempt to form the heteroleptic complex [[L1]ZnMe] (1), a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of thioamide H[L1] and dimethyl zinc (Scheme 3) was conducted. Toluene solutions of H[L1] and ZnMe2 were combined resulting in no visual change. It was noted that a small vacuum applied to the reaction vessel promoted the liberation of a gaseous by-product. After 1 hour of stirring, the solution was reduced in vacuo. On standing at −28 °C, colourless crystals formed and isolated by filtration. 1H NMR spectroscopy studies were conducted on the isolated crystals. No broad singlet resonance corresponding to the {N–H} proton of the pro-ligand was visible, and all iso-propyl proton resonances had shifted downfield, maintaining splitting. Interestingly, no resonance associated with the {Zn–CH3} moiety was observed. Single crystal X-ray diffraction analysis revealed the crystals to be the homoleptic species [Zn{L1}]2, rather that the targeted heteroleptic methyl-zinc complex, 1.
:1 reaction of thioamide H[L1] and dimethyl zinc (Scheme 3) was conducted. Toluene solutions of H[L1] and ZnMe2 were combined resulting in no visual change. It was noted that a small vacuum applied to the reaction vessel promoted the liberation of a gaseous by-product. After 1 hour of stirring, the solution was reduced in vacuo. On standing at −28 °C, colourless crystals formed and isolated by filtration. 1H NMR spectroscopy studies were conducted on the isolated crystals. No broad singlet resonance corresponding to the {N–H} proton of the pro-ligand was visible, and all iso-propyl proton resonances had shifted downfield, maintaining splitting. Interestingly, no resonance associated with the {Zn–CH3} moiety was observed. Single crystal X-ray diffraction analysis revealed the crystals to be the homoleptic species [Zn{L1}]2, rather that the targeted heteroleptic methyl-zinc complex, 1.
 | ||
| Scheme 3 Synthesis of targeted heteroleptic (1) and homoleptic (2–5) zinc thioamidate precursors. | ||
Although the formation of {Me–Zn} containing species under similar conditions has previously been reported,68,69 in our studies, no 1H NMR resonances attributable to the formation of a heteroleptic complexes could be observed. No further exploration of heteroleptic zinc-methyl compounds was pursued.
Comparable reaction of the pro-ligands H[L1]–H[L4] with [Zn{N(SiMe3)2}2] in a 2:1 ratio in toluene (Scheme 3) results in the formation of the bis-thioamidate complexes, 2–5, as white microcrystalline powders. Recrystallization from toluene yielded colourless crystals isolatable in moderate yields (23–52%). Higher yields (65–82%) could be readily obtained from successive concentration of reaction mixtures followed by storage at low temperatures and isolation of crystalline materials. The product formed from reactions between H[L1] and [Zn{N(SiMe3)2}2] and in 1:1 dimethyl zinc gave identical 1H NMR spectra, confirming the formation of Zn[L1]2 species, 2. The latter reaction may be the result of a Schlenk like equilibrium with ZnMe2 (Scheme 3).
As for 3–5, produced from reaction of pro-ligands with [Zn{N(SiMe3)2}2], 1H NMR spectra were devoid of resonances associated with {N(SiMe3)2} units (ca. 0.31–0.48 ppm) or broad resonances corresponding to the {N–H} moiety, consistent with the formation of each bis-thioamidate complexes (Scheme 3).
The 1H/13C{1H} NMR spectra of 2–5 were unexceptional, containing resonances solely assignable to the thioamide ligands, including 1H NMR resonances at δ = 2.73–3.07 ppm (heptet) and δ = 1.22–1.38 ppm (doublet) representative of the moiety. Characteristic 13C{1H} signals at ca. ∼204–207 ppm due to the quaternary {CS} moieties were also observed. The appearance of only one set of resonances in 1H and 13C{1H} NMR spectra suggested the ligands in each zinc bis-thioamidate system occupied identical environments on the NMR time scale. Elemental analysis of 2–5 is consistent with the formation of the bis-thioamidate complexes.
As part of our study the complex [Cd(SC(iPr)NtBu)2] (6) was identified as a possible precursor to CdS (infra vide). At room temperature, reaction between THF solutions of dimethyl cadmium, CdMe2, and of thioamide H[L2] (Scheme 4) was associated with mild effervescence and a colour change from colourless to pale yellow. After 1 hour stirring, the solution was concentrated in vacuo, from which pale-yellow crystalline solid could be isolated at −35 °C by filtration, in low yield (23%).
 | ||
| Scheme 4 Synthesis of the cadmium thioamidate 6. | ||
Interestingly, the 1H NMR spectrum corresponding to a solution of 6 in C6D6 contained six major resonance peaks: two heptets (2.96 and 2.52 ppm), two doublets (1.32 and 1.24 ppm) and two singlets (1.44 and 1.22 ppm). The relative intensities of these resonances indicated that two thioamidate environments were detected, in 2:1 ratio, potentially a result of two different ligand binding modes – commensurate with X-ray diffraction data discussed below.
Solid-state molecular structures
Crystalline samples of 2–6 sufficient for X-ray crystallographic analysis were obtained from cooled toluene (2–5) or THF (6) solutions.The molecular structures of 2, 4 and 5 are shown in Fig. 1. 2 and 4 crystalize in the monoclinic space groups P21/c and P21/n respectively, such that the asymmetric unit cell contains one whole [Zn{L}2]2 unit. With the second half of the dimer generated by specific symmetry operators. Complex 5, crystalises in the orthorhombic space group Pbcn, with one whole dimeric [Zn{L}2]2 unit.
 | ||
| Fig. 1 ORTEP diagrams of the solid-state molecular structure of the bis-thioureides 2 (a), 4 (b) and 5 (c). Thermal ellipsoids are shown at 50% probability and hydrogen atoms are omitted. Atoms with ‘A’ labels relate to those in the asymmetric unit cell generated by the symmetry operators. Symmetry transformations used to generate equivalent atoms: 2; −x + 1, −y + 1, −z + 1 and 4; −x, 1 − y, 1 − z. | ||
Table 1 shows selected bond lengths and angles for 2, 4 and 5. Each feature a zinc metal centre possessing a highly distorted 5-coordinate geometry, comprising of the sulfur and nitrogen atoms of the two chelating thioamidate ligands. The fifth, and final, coordinating atom is found to be a sulfur atom belonging to an adjacent chelating thioureide ligand, bridging two zinc atoms and resulting in the formation of three, fused four membered rings: {ZnSCN}/{ZnSZnS}/{ZnSCN}.
| 2 | 4 | 5 | |
|---|---|---|---|
| Selected bond lengths (Å) | |||
| Zn(1)–S(1) | 2.4822(7) | 2.3407(5) | 2.4801(17) |
| Zn(1)–S(2) | 2.865 | 2.892 | 2.8254(16) |
| Zn(1)–S(2A) | 2.3464(8) | 2.3489(5) | — |
| Zn(2)–S(3) | — | 2.4756(17) | |
| Zn(2)–S(4) | — | 2.8192(17) | |
| Zn(1)–N(1) | 2.027(2) | 2.1557(14) | 2.046(5) |
| Zn(1)–N(2) | 2.035(2) | 2.0266(13) | 2.056(5) |
| Zn(2)–N(3) | — | — | 2.064(5) |
| Zn(2)–N(4) | — | — | 2.050(5) |
| Zn(1)–S(4) | — | — | 2.3465(17) |
| Zn(2)–S(2) | — | — | 2.3650(18) |
| Selected bond angles (°) | |||
| S(1)–Zn(1)–N(1) | 68.81(6) | 69.82(4) | 68.76(14) |
| S(2)–Zn(1)–N(2) | 61.37(6) | 60.87(4) | 62.01(14) |
| S(3)–Zn(2)–N(3) | — | — | 68.59(14) |
| S(4)–Zn(2)–N(4) | — | — | 62.40(13) |
| S(1)–Zn(1)–S(2A) | 168.15(6) | 128.16(4) | 162.90(6) |
| N(1)–Zn(1)–N(2) | 121.76(9) | 108.71(5) | 127.5(2) |
| S(3)–Zn(2)–S(4) | — | — | 161.30(7) |
| N(3)–Zn(2)–N(4) | — | — | 124.1(2) |
The structures resemble the dimeric zinc units found in thioureide,40 diethyldithiocarbamates,70 diethyldiselenocarbamates,71 and selenocarbamate systems.72 The Zn–N bonds and Zn–S bond distances and angles are similar to those reported in related Zn-thioureide and Zn-dithiocarbamate systems.40
As can be seen in Fig. 2 the S-atoms engaged in both chelating/bridging interactions (3-coordinate), show significantly longer Zn–S contacts than their 2-coordinate counterparts resulting in asymmetric Zn–S interactions within the {Zn2S2} core. As with the previously reported zinc bis-thioureide complexes, the alkyl groups (iPr and Cy) in 2 and 5 chelate in transoidal fashion within each [Zn{L3}2] unit, resulting in trans–cis [Zn{L}2]2 dimer formation.40 In contrast, the phenyl derivative 4 possesses a cisoidal arrangement of ligands about the central zinc atom in the [Zn{L3}2] unit. Upon dimer formation the complex presents a relative cis–trans {[L3]2Zn}2 dimer configuration (Fig. 2). In all cases the C–S and C–N distances within the ligands are consistent with delocalisation across the “SCN” fragment of the metallacycle [2: C–Save = 1.751 Å; C–Nave = 1.291 Å, 4: C–Save = 1.751 Å; C–Nave = 1.297 Å, 5: C–Save = 1.750 Å; C–Nave = 1.292 Å], rather than a localised CS [1.67 Å] and C–N [1.33 Å] bonds.73,74
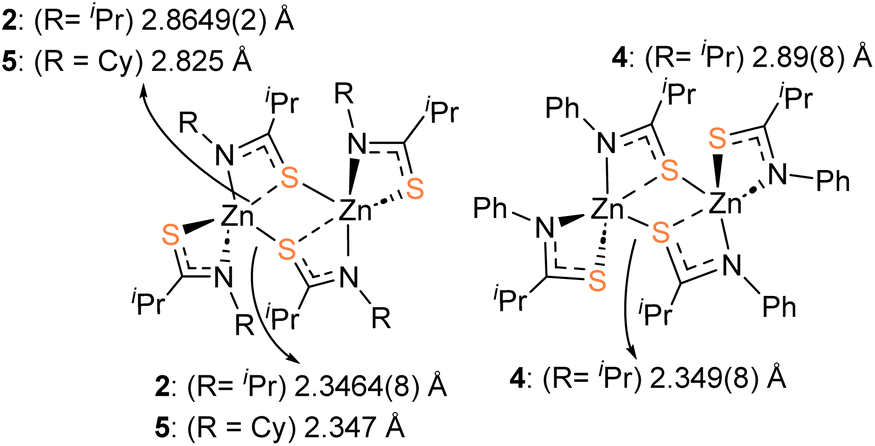 | ||
| Fig. 2 Diagram showing the asymmetry of the Zn–S bonding within the central {Zn2S2} core of the dimers 2, 4 and 5, as well as the relative orientation of ligands across the planar {Zn2S2} core. | ||
3 was found to be uniquely monomeric, in the solid-state (Fig. 3; Selected bond-lengths and bond-angles are provided in the figure caption.). The monometallic [Zn{L2}2]2 complex crystalises in the orthorhombic space group Pbcn, the unit cell containing half of the molecule, with the zinc atom occupying a special crystallographic position, such that symmetry operators generate the second half molecule. As can clearly be seen from Fig. 3, the metal coordination sphere consists of two κ2-N,S-thioamidiate ligands in a flattened tetrahedral geometry (τ4 = 0.64).75 Consequently, the angles at the metal are in the range 71.45(4)–138.31(4)°, the smallest of which being a consequence of ligand bite angle. The four membered {ZnNCS} metallacycles are near planar, with an angle of 84.50° between the two “ZnNCS” least-squares planes. Much like the dimeric systems above, C–S and C–N distances suggest delocalisation across the “SCN” fragment of the metallacycle [3: C–S = 1.755(2) Å; C–N = 1.292(2) Å].
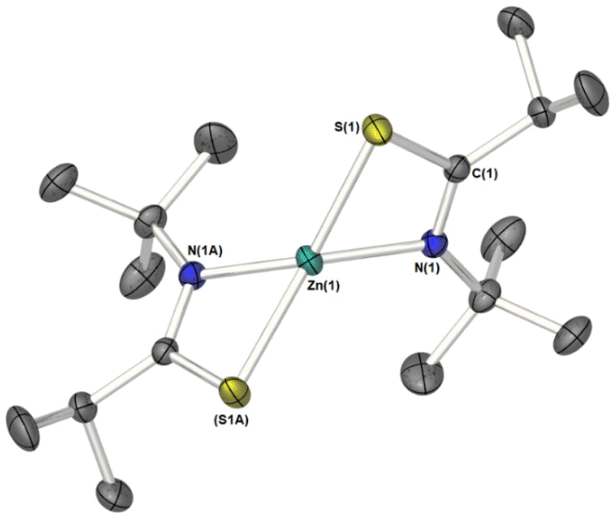 | ||
| Fig. 3 ORTOP diagram of solid-state molecular structure of 3. Thermal ellipsoids are shown at 50% probability. Atoms with ‘A’ labels relate to those in the asymmetric unit cell by the symmetry operator −x + 1, y, −z + 3/2. Selected bond lengths (Å): Zn(1)–S(1) 2.346(2), Zn(1)–N(1) 2.031(2), S(1)–C(1) 1.755(2), N(1)–C(1) 1.292(2); Selected bond angles (°): N(1)–Zn(1)–S(1) 71.45(4), N(1)–Zn(1)–S(1A) 130.85(4), S(1)–Zn(1)–S(1A) 138.31(3), N(1)–Zn(1)–N(1A) 123.66(8). | ||
As part of this study, attempts were made to synthesise the mixed ligand system [Zn{SC(iPr)NtBu}{OC(iPr)NtBu}]2 as a potential Zn(O,S) single source precursor.36 Despite repeated attempts the heteroleptic system could not isolated. However, crystal of the homoleptic amidate system [Zn{OC(iPr)NtBu}2]2 were isolated. The ESI† contains details of the synthesis characterisation and TGA for the resulting complex.
Crystals suitable for single crystal X-ray diffraction of 6 were isolated from a saturated THF solution, as pale-yellow crystals. 6 was found to crystallise in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, with the resulting structure presenting a trimetallic species with the molecular formula [Cd3{L2}6] (Fig. 4). Selected bond-lengths and bond-angles for complex 6 can be found in Table 2.
space group, with the resulting structure presenting a trimetallic species with the molecular formula [Cd3{L2}6] (Fig. 4). Selected bond-lengths and bond-angles for complex 6 can be found in Table 2.
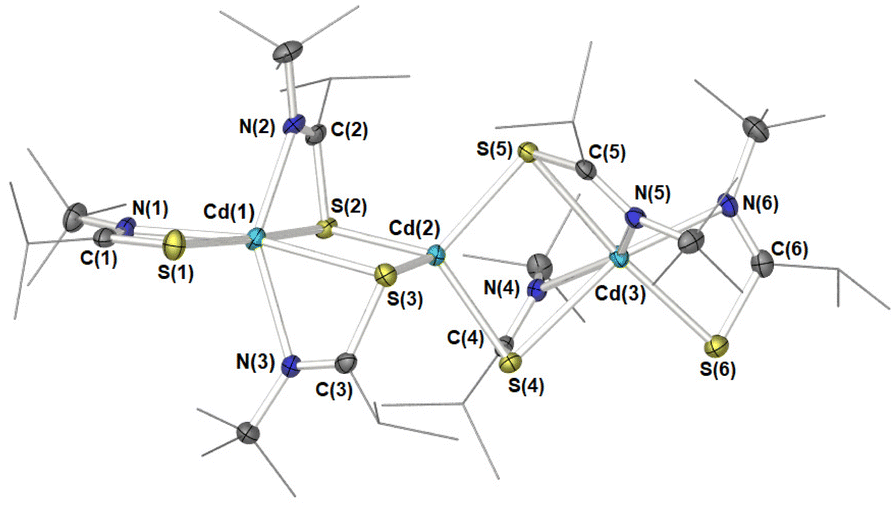 | ||
| Fig. 4 ORTEP diagram of the solid-state molecular structure of 6. Selected atoms have been labelled. Ancillary iso-propyl and tert-butyl groups are presented skeletally for clarity. All ellipsoids are at 50% probability with hydrogen atoms and one molecule of solvent of crystallisation (THF) has been omitted for clarity. | ||
| Bond lengths (Å) | Bond angles (°) | ||
|---|---|---|---|
| Cd(1)–S(1) | 2.5707(8) | S(1)–Cd(1)–N(1) | 62.88(6) |
| Cd(1)–S(2) | 2.8296(7) | S(2)–Cd(1)–N(2) | 59.54(6) |
| Cd(1)–S(3) | 2.8035(7) | S(3)–Cd(1)–N(3) | 59.39(5) |
| Cd(1)–N(1) | 2.409(2) | S(2)–Cd(2)–S(3) | 105.97(2) |
| Cd(1)–N(2) | 2.353(2) | S(3)–Cd(2)–S(4) | 111.74(2) |
| Cd(1)–N(3) | 2.442(2) | S(2)–Cd(2)–S(4) | 113.72(2) |
| Cd(2)–S(2) | 2.5543(7) | S(2)–Cd(2)–S(5) | 110.36(2) |
| Cd(2)–S(3) | 2.5316(7) | S(3)–Cd(2)–S(5) | 113.71(2) |
| Cd(2)–S(4) | 2.5234(7) | S(4)–Cd(2)–S(5) | 101.55(2) |
| Cd(2)–S(5) | 2.5480(7) | S(4)–Cd(3)–N(4) | 62.95(6) |
| Cd(3)–S(4) | 2.8257(7) | S(5)–Cd(3)–N(5) | 60.36(5) |
| Cd(3)–S(5) | 2.7532(7) | S(6)–Cd(3)–N(6) | 62.95(6) |
| Cd(3)–S(6) | 2.6055(7) | ||
| Cd(3)–N(4) | 2.413(2) | S(1)–Cd(1)–S(2) | 171.89(2) |
| Cd(3)–N(5) | 2.388(2) | N(1)–Cd(1)–S(3) | 157.37(6) |
| Cd(3)–N(6) | 2.360(2) | N(2)–Cd(1)–N(3) | 143.78(7) |
| S(5)–Cd(3)–S(6) | 174.00(2) | ||
| N(6)–Cd(3)–S(4) | 158.12(6) | ||
| N(4)–Cd(3)–N(5) | 148.41(7) | ||
As can be seen from Fig. 4 three cadmium atoms were arranged somewhat linearly [Cd(1)–Cd(2)–Cd(3) = 174.8(2)°], bridged by the sulfur atoms of two μ-S,κ2-S,N thioamidate ligands, to form two four-membered {Cd2S2} rings fused at the central Cd(2) atom. Cadmium atoms Cd(1) and Cd(3) each had one terminal κ2-S,N-bonded thioamidate ligand. The sum of valence bonds around all nitrogen atoms and carbon atoms C(1)–C(6) were close to 360°, indicating sp2 hybridisation and electron delocalisation within each {S–N–C} moiety. Interestingly, the terminal cadmium atoms Cd(1) and Cd(3) were ostensibly surrounded by three chelating ligands, thus each adopting a six-coordinate geometry, whereas the central Cd atom, Cd(2), bonded exclusively to sulfur atoms in a four-coordinate geometry. The bite angles of bridging ligands [{S–Cd–N}: 59.36(5)°–60.36(5)°] were smaller than those in terminal positions [S–Cd–N: 62.88(6)° and 62.95(6)°]. Presumably, the latter are not subject to the same degree of steric restraint, allowing them closer proximity to the metal centre. These acute angles made for two distorted octahedral geometries around Cd(1) and Cd(3). In contrast, the central Cd(2) atom adopts a geometry close to that of a perfect tetrahedron (τ4 = 0.94, τ′4 = 0.99)75,76 with all [S–Cd(2)–S’] angles falling between 101.55(2)° and 113.72(2)°.
The Cd–S bond distances varied according to ligand binding mode. The shortest were observed between Cd(2) and its four surrounding μ-S atoms [between 2.5234(7) Å–2.5543(7) Å]. These were significantly shorter than distances between terminal cadmium [Cd(1) and Cd(3)] and bridging sulfur atoms [2.7532(7) Å–2.8296(7) Å]. Bonding with sulfur atoms of the two terminal thioamidates fell between the two other modes [2.5707(8) Å and 2.6055(7) Å].
The relative arrangements of the thioimidate ligands in [Cd3{L2}6] 6 (four bridging and two terminally arranged) is consistent with the observed thioamidate environments (2:1) observed in 1H NMR studies, suggesting disparate exchange rates between terminal and bridging thioimidate ligands in the solution state.
Compared with the tin(II)66 and zinc(II) thioamidate compounds characterised a priori, 6 is uniquely trimetallic. While several oligomeric Cd–S-containing materials are known in the literature,49,77–86 only two definitively trimetallic {Cd–S} compounds have been reported previously. These included a trimer of the cadmium bis-dithiocarbamate [Cd(S2CN{CH2Tol}{CH2C4H3O}2)]3 which featured three CdL2 units fused by two {Cd2S2} rings.49,84 and the compound [Cd3(4,6-dimethylpyrimidine-2-thione)4(2,2′-bipyridine)2I2] was also found to crystallise as a trimetallic species.81
While crystallographic analysis verified the identity of 6 and revealed a trimetallic arrangement in the solid-state at 150 K, oligomerisation does not preclude solubility in conventional organic solvents (THF, CDCl3, and C6D6), suggesting 6 could be a potential single-source precursor for AACVD of CdS.
Solution state studies
As part of our study, all four Zn compounds (2–5) were investigated using 1H DOSY experiments. While crystallographic studies show a clear propensity for close molecular association of the zinc thioamidates 2, 4 and 5 in the solid-state, these complexes were designed for application in the solution-based process AACVD.321H DOSY provides an experimentally simple method to calculate diffusion coefficient, Dr, of an NMR-active chemical in solution, providing an indication of solution-state speciation. Using the Stokes–Einstein relationship between diffusion coefficient and radius, Evans et al. developed an approach accurately to relate these data to the molecular mass of diffusing compounds in different solvents; the method used here.87,88 We have also previously reported using 1H DOSY to calculate the estimated molecular weights of zinc phosphate complexes.89Previous AACVD studies have shown that organic solvents capable of metal-coordination (e.g., THF or diethyl ether) as well as those non-coordinating (e.g., toluene) can significantly affect the deposition process as well as thin film morphology.30,90,91
Therefore, 1H DOSY studies were carried out on these compounds in representative solvents, CDCl3 and THF-d8. The resulting diffusion coefficients are presented in Table 3. Initial 1H DOSY experiments were conducted in CDCl3, from which all four diffusion coefficients were found to correspond to dimeric species (2: 0.86 × 10−9 m2 s−1, 3: 0.83 × 10−9 m2 s−1, 4: 0.60 × 10−9 m2 s−1 and 5: 0.71 × 10−9 m2 s−1). 4 and 5 appeared to remain in the dimeric state, irrespective of solvent, as similar coefficients were found in THF-d8 solutions [0.68 × 10−9 m2 s−1 and 0.78 × 10−9 m2 s−1, respectively]. Contrastingly, in THF 2 (1.10 × 10−9 m2 s−1) and 3 (1.07 × 10−9 m2 s−1) exhibited significantly larger diffusion coefficients. These results suggested that THF is a sufficiently strong coordinating solvent to perturb dimer formation in solution, resulting in what we assume to be a THF coordination complex in solution.
| MWCal (g mol−1) | Solvent | Obs Dr (×10−9 m2 s−1) | Est Dr (×10−9 m2 s−1) | MWerra (%) | |
|---|---|---|---|---|---|
| a MWerr = [100 × (MWcal–MWdet)/MWcal].89 | |||||
| 2 | 353.89 | CDCl3 | 0.86 | 0.77 (d) | 10 |
| THF-d8 | 1.10 | 1.06 (m) | 4 | ||
| 3 | 381.95 | CDCl3 | 0.83 | 0.75 (d) | 10 |
| THF-d8 | 1.07 | 1.06 (m) | 1 | ||
| 4 | 421.93 | CDCl3 | 0.60 | 0.71 (d) | 15 |
| THF-d8 | 0.68 | 0.71 (d) | 4 | ||
| 5 | 434.02 | CDCl3 | 0.71 | 0.70 (d) | 1 |
| THF-d8 | 0.78 | 0.70 (d) | 11 | ||
Attempts to perform similar experiments on the Cd complex, 6, were limited by its instability, as determined via thermal profiling (infra vide).
Thermal studies
The zinc (2–5) and cadmium (6) bis-thioamidate compounds were designed as potential AACVD precursor candidates for ZnS and CdS formulation, for which ideal precursors would cleanly pyrolyze to form ZnS and CdS respectively. Thermogravimetric analysis (TGA) was used to survey their thermal profiles, the plots of which are presented in Fig. 5. | ||
| Fig. 5 (a) TGA data profiles corresponding to the pyrolysis of 2, 3, 4 and 5. (b) presents the first order differential of all TGA profiles. It is noted that the purple plot corresponding to 4 completely masks that of 2, which was near-identical. | ||
The TGA profile of 2 displayed a single thermal event (onset 142 °C) ending with a residual mass (17%) lower than that expected for zinc sulfide formation (27%). While this residual mass is consonant with the formation of metallic zinc (18.6% residual mass), the reductive elimination of two thioamidate ligands is deemed unlikely. We therefore assume that the observed thermal event was a combination of decomposition and volatilisation. Attempts to analyse the residual white solid by PXRD were unsuccessful.
The thermal activities of 4 and 5 each followed broadly the same profile. Each displayed two indiscrete thermal events, culminating in residual masses closely aligned with the target mass of ZnS (see Table 4). Decomposition onset temperatures are like that of 2 at 151 °C (5) and 171 °C (4). Interestingly, the first thermal event appeared to finish at percentage masses of 40% (4) and 39% (5). Secondary thermal events plateaued at ca. 400 °C, indicating no further thermal activity. In contrast, complex 3 decomposes at 118 °C, as can be seen in Fig. 5 and Table 4. As with 2, 4 and 5, complex 3 has an initial mass loss of ca. 56% with a residue of 44%.
Fig. 5b represents the first order differential of Fig. 5a, illustrating the rate of mass loss as a function of temperature. Fig. 5b clearly highlights the propensity of 3 to decompose more readily at lower temperatures, indicated by a peak rate of ca. 9% mass loss per degree Celsius at 140 °C. The qualitative observation that the peak of decomposition rate for 3 also alludes to a cleaner decomposition mechanism. It is noted that for both zinc(II) (here) and tin(II) thioamidates,66 containing a tert-butyl moiety at the N-position underwent pyrolysis at temperatures lower than any other derivative. Given the mechanistic evidence collected heretofore, the availability of β-hydrogen atoms clearly bears an influence on metal thioamidate pyrolysis.
Using the experimental apparatus described elsewhere,65 a sample of 3 was heated to 250 °C in vacuo, for 20 minutes. Volatile pyrolysis products were captured in a J Young Wildman NMR tube at −78 °C. 1H NMR spectroscopy was conducted separately on both the captured volatile components and a sample of non-volatile oil remaining from pyrolysis. Isobutylene (1H δ: 4.75 ppm, 1.60 ppm) and isobutyronitrile (1H δ: 1.75 and 0.60 ppm) resonances could clearly be observed as the major gaseous products,65 while all resonances observed for that of the solid residue could be assigned to the free thioamide pro-ligand (1H δ: 6.32, 2.11, 1.33 and 1.14 ppm). These results are consistent with the suggested decomposition mechanism (Scheme 1) and sufficiently accounted for all by-product elements required for clean ZnS formation. Given the thermal properties of all four zinc thioamidates studies here (2–5), it was clear that all were viable precursors for AACVD of zinc sulphide. There was also good evidence to suggest that 3 would deposit at the lowest temperatures, whilst undergoing clean decomposition to form ZnS.
In contrast, the thermal profile of 6 (Fig. 6) revealed major mass loss initiating at the low temperature of 77 °C and culminating at ca. 190 °C. The residual mass (48%) was notably higher than that calculated for the formation of CdS (34%). A subtle inflexion point was discernible at ca. 173 °C, bearing similarity to the two-stage decomposition of 3.
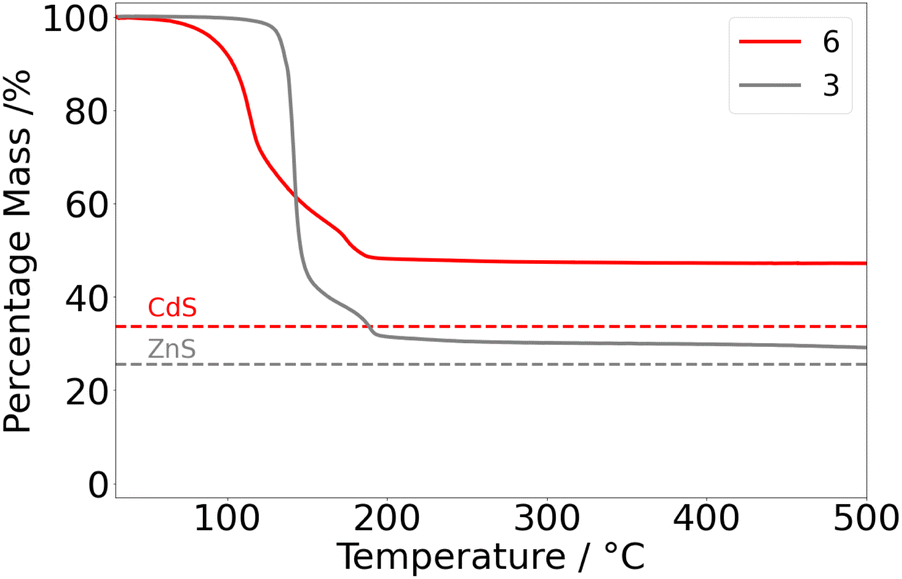 | ||
| Fig. 6 Thermogravimetric profile of 6 (red). The thermogravimetric profile of 3 (grey) is presented for comparison. The target percentage masses corresponding to CdS and ZnS are presented as dotted lines and labelled. | ||
Unfortunately, the low-temperature onset of decomposition of 6 is also indicative of ambient thermal instability, consistent with the observed solution state instability (vide supra), with samples of 6 showing signs of decomposition upon storage at ambient temperatures. Attempts to dissolve 6 in THF afford cloudy yellow solutions, analysis of which indicated the formation of free ligand as determined by 1H NMR studies. As such AACVD experiments and assessment of 6 as a precursor for CdS was discontinued.
Thin film deposition studies
3 was identified as the most promising precursor candidate for the deposition of zinc sulfide via AACVD and was, thus, carried forward for deposition trials. As a preliminary study, a 0.1M THF solution of 3 was deposited for 45 min, in a hot wall reactor (as described elsewhere55) onto fluorine-doped tin oxide (FTO), at a range of temperatures. In all cases, an argon flow rate of 1.8–2 L min−1 was used, resulting in 30–40 mL of the 50 mL solution being consumed. Deposition conditions are presented in the ESI (Table S1†).Depositions at 175 °C (A) were unsuccessful with no visible change to the FTO substrate. However, successful deposition was obtained at temperatures greater than 200 °C (B–E) with films appearing to the eye as cloudy white. There was no appearance of high carbon (black) or sulfur (yellow) contamination. At each temperature, areas appearing to have the most material deposited were selected for thin film analysis.
Powder X-ray diffraction (PXRD) was performed on samples (B–E). Representative PXRD patterns are presented in Fig. 7. The largest reflection intensities were indexed to the FTO surface of the substrates used. From the remaining reflection intensities, one major peak could be readily indexed to the (002) reflection of ZB-ZnS, for all samples – with films grown at 300 °C (D) exhibiting the highest qualifiable crystallinity. Most notably, films C and D appeared to be highly orientated with only the (002) ZB-ZnS reflection intensity observed. Films grown at 350 °C (E) also presented an intensity indexable to the (110) reflection of wurtzite ZnS. A more quantitative assessment of the (002) reflections was conducted to elucidate more structural information. Using the Scherrer formula, the minimum size of crystalline domains was estimated to lie between 15–20 nm.92
 | ||
| Fig. 7 PXRD patterns of films B–E resulting from AACVD of 3. Intensities from the FTO substrate are labeled. | ||
Morphological information was obtained using a field emission scanning electron microscope (FESEM) for samples B–E. Fig. 8 presents top-view images of all samples at the same magnification (x30000). The appearance of film B was markedly different from films deposited at higher temperatures; in this case, globular structures appeared to pack densely to form a high-coverage film. Conversely, films C, D and E appeared to grow as densely packed, approximately spherical nanoclusters. The largest of these were observed for film C.
 | ||
| Fig. 8 FESEM images captured at ×30000 magnification of films B, C, D and E. | ||
Encouragingly, the morphological consistencies observed between films deposited above 250 °C indicated independence from deposition temperature and consequential reproducibility.
Energy dispersive X-ray spectroscopy (EDX) was conducted on all (B–E) films. The resultant Zn:S ratios are presented in Table 5. All films were found to be deficient in sulfur – particularly B and E. Films C and D were found to obtain the best Zn:S ratios – 0.8 and 0.9, respectively. It is possible that deposition at temperatures 250–300 °C were conducted in an optimum temperature window, outside of which films may suffer from incomplete pyrolysis or sulfur leaching.
| Film | Zn:S ratio |
|---|---|
| B | ZnS0.5 |
| C | ZnS0.8 |
| D | ZnS0.9 |
| E | ZnS0.6 |
Conclusions
A series of four zinc bis-thioamidate precursors (2–5) and one cadmium bis-thioimidate complex (6) have been investigated. Their solid-state structure and solution-state dynamics were probed before thermal profiling to determine their viability as precursors for AACVD. The thioamidate ligand system was found to decompose most readily by the availability of β-hydrogen atoms on the substituted nitrogen. Consequently, 3 [Zn(SC(iPr)NtBu)2], was deemed most appropriate for low-temperature deposition of ZnS films, which was conducted successfully over a temperature range of 200–300 °C. Exclusive deposition and growth of hexagonal wurtzite ZnS was identified via PXRD of all films, in what appeared to be a phase-selective deposition process. The morphology of films grown at and above 250 °C was notably consistent, however, EDX spectroscopy analysis found each to suffer sulfur deficiencies.Complementary to these results, a cadmium(II) thioamidate [Cd{SC(iPr)NtBu}2] (6) was sought. In the solid-state, this crystallised as a trimetallic species [Cd{SC(iPr)NtBu}2]3 with three linearly arranged cadmium atoms surrounded by six thioamidate ligands. Thermal analysis found this compound to decompose at temperatures lower than 100 °C and lacked the thermal stability required of a viable AACVD precursor.
This work complements our previous work on thioamidate precursors65,66 and ZnS film formulation.40 In contrast to other work, we report phase-pure formulation of ZnS, excellent film coverage at temperatures as low as 200 °C and reproducible crystallinity at 250 °C. Work is ongoing to optimise deposition parameters towards ZnS film formulation, in addition to the development of metal thioamidate precursor chemistries for AACVD of other functional metal sulfides.
Experimental
General synthesis procedures
All reactions were carried out under an inert atmosphere using standard Schlenk techniques. Solvents were dried over activated alumina columns using an Innovative Technology solvent purification system and degassed under an argon atmosphere. [Zn{N(SiMe3)2}2] and ZnMe2 (used as a 1.0 M solution in hexane) were used as supplied (Sigma-Aldrich). All other reagents were purchased from commercial suppliers were used as received.Elemental analyses were performed externally by London Metropolitan University Elemental Analysis Service, UK. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 or 500 MHz FT-NMR spectrometer, as appropriate, in deuterated benzene-d6, at room temperature. Chemical shifts are given in ppm with respect to Me4Si (1H, 13C). High resolution mass spectrometry (HRMS) results were acquired on an externally calibrated Agilent QTOF UHR-ToF mass spectrometer coupled to an electrospray source (ESI-QToF). Molecular ions were detected as their protonated, or metalated (Li or Na) counterparts.
Synthesis of thioamide pro-ligands
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 4.72 (hep, 3JHH = 6.6 Hz, 1H, NCMe2), 2.17 (hep, 3JHH = 6.7 Hz, 1H, CMe2), 1.17 (d, 3JHH = 6.7 Hz, 6H, NCH
), 4.72 (hep, 3JHH = 6.6 Hz, 1H, NCMe2), 2.17 (hep, 3JHH = 6.7 Hz, 1H, CMe2), 1.17 (d, 3JHH = 6.7 Hz, 6H, NCH![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) 2), 0.86 (d, 3JHH = 6.5 Hz, 6H, CH2). 13C{1H} NMR (126 MHz, C6D6) δC: 209.9 (N
2), 0.86 (d, 3JHH = 6.5 Hz, 6H, CH2). 13C{1H} NMR (126 MHz, C6D6) δC: 209.9 (N![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) S), 46.3 (NHMe2), 44.4 (CHMe2), 22.9 (NCH2), 21.0 (CH2). HRMS (ESI+): calculated for [M + H]+ C7H16NS, 146.10; found, 146.10.
), 2.11 (hep, 3JHH = 6.9 Hz, 1H, CCMe2), 1.33 (s, 9H, NC3), 1.14 (d, 3JHH = 6.6 Hz, 6H, CH2). 13C{1H} NMR (126 MHz, C6D6) δC: 210.2 (NS), 54.8 (NMe3), 46.5 (HMe2), 27.6 (C3), 23.1 (CH2). HRMS (ESI+): calculated for [M + H]+ C8H18NS, 160.12; found, 160.12.
), 7.57 (d, 3JHH = 7.9 Hz, 2H, o-Ph), 7.08 (t, 3JHH = 7.9 Hz, 2H, m-Ph), 6.94 (t, 3JHH = 7.5 Hz, 1H, p-Ph), 2.27 (m, 1H, CMe2), 1.17 (d, 3JHH = 6.7 Hz, 6H, CH2). 13C{1H} NMR (101 MHz, C6D6) δC: 210.5 (NS), 139.4 (NMe3), 128.8 (m-Ph), 126.4 (p-Ph), 124.0 (o-Ph), 45.8 (HMe2), 23.0 (CH2). HRMS (ESI+): calculated for [M + H]+ C10H14NS, 180.08; found, 180.08.
), 4.56–4.48 (m, 1H, NC), 2.15 (hep, 3JHH = 6.7 Hz, 1H, CMe2), 1.92–1.84 (m, 2H, C2), 1.48–1.39 (m, 2H, C2), 1.41–1.33 (m, 1H, C2), 1.19 (d, 3JHH = 6.7 Hz, 6H, CH2), 1.18–1.06 (m, 2H, C2), 0.96–0.86 (m, 1H, C2), 0.80–0.72 (m, 2H, C2). 13C{1H} NMR (101 MHz, C6D6) δC: 209.8 (NS), 53.2 (NH), 44.6 (HMe2), 31.6 (H2), 25.8 (H2), 24.9 (H2), 23.04 (CH2). HRMS (ESI+): calculated for [M + H]+ C10H20NS, 186.13; found, 186.13.
S), 46.3 (NHMe2), 44.4 (CHMe2), 22.9 (NCH2), 21.0 (CH2). HRMS (ESI+): calculated for [M + H]+ C7H16NS, 146.10; found, 146.10.
), 2.11 (hep, 3JHH = 6.9 Hz, 1H, CCMe2), 1.33 (s, 9H, NC3), 1.14 (d, 3JHH = 6.6 Hz, 6H, CH2). 13C{1H} NMR (126 MHz, C6D6) δC: 210.2 (NS), 54.8 (NMe3), 46.5 (HMe2), 27.6 (C3), 23.1 (CH2). HRMS (ESI+): calculated for [M + H]+ C8H18NS, 160.12; found, 160.12.
), 7.57 (d, 3JHH = 7.9 Hz, 2H, o-Ph), 7.08 (t, 3JHH = 7.9 Hz, 2H, m-Ph), 6.94 (t, 3JHH = 7.5 Hz, 1H, p-Ph), 2.27 (m, 1H, CMe2), 1.17 (d, 3JHH = 6.7 Hz, 6H, CH2). 13C{1H} NMR (101 MHz, C6D6) δC: 210.5 (NS), 139.4 (NMe3), 128.8 (m-Ph), 126.4 (p-Ph), 124.0 (o-Ph), 45.8 (HMe2), 23.0 (CH2). HRMS (ESI+): calculated for [M + H]+ C10H14NS, 180.08; found, 180.08.
), 4.56–4.48 (m, 1H, NC), 2.15 (hep, 3JHH = 6.7 Hz, 1H, CMe2), 1.92–1.84 (m, 2H, C2), 1.48–1.39 (m, 2H, C2), 1.41–1.33 (m, 1H, C2), 1.19 (d, 3JHH = 6.7 Hz, 6H, CH2), 1.18–1.06 (m, 2H, C2), 0.96–0.86 (m, 1H, C2), 0.80–0.72 (m, 2H, C2). 13C{1H} NMR (101 MHz, C6D6) δC: 209.8 (NS), 53.2 (NH), 44.6 (HMe2), 31.6 (H2), 25.8 (H2), 24.9 (H2), 23.04 (CH2). HRMS (ESI+): calculated for [M + H]+ C10H20NS, 186.13; found, 186.13.
Synthesis of metal thioamide complexes
From ZnMe2. A 1.2 M solution of ZnMe2 in toluene (0.83 ml, 1 mmol) was diluted with THF (10 mL). This was combined with a solution of pro-ligand H[L1] in THF (10 mL) and left to stir at room temperature for 1 h. From the pale-yellow solution, colourless block crystals formed at −28 °C. 0.04 g (22%).
From [Zn{N(SiMe3)2}2]. [Zn{N(SiMe3)2}2] (1.16 g, 3 mmol) was solvated in toluene (30 mL), to which thioamide H[L1] (0.87 g, 6 mmol) was slowly added via syringe. After 1 h stirring at room temperature, a colour-less precipitate had dropped out, which was heated back into solution, allowing purification by crystallisation at 5 °C. 0.36 g (34%). 1H NMR (400 MHz, CD2Cl2) δH: 3.85 (hep, 3JHH = 6.3 Hz, 1H, NC
Me2), 2.99 (hep, 3JHH = 6.7 Hz, 1H, NCMe2), 1.22 (d, 3JHH = 6.7 Hz, 6H, NCH2), 1.13 (d, 3JHH = 6.3 Hz, 6H, CH2). 13C{1H} NMR (125.7 MHz, CD2Cl2) δC: 204.1 (NS), 50.2 (NHMe2), 33.4 (CHMe2), 24.2 (NCH2), 22.1 (CH2). Elemental analysis for C14H28N2S2Zn (expected): C 47.36% (47.52%), H 8.01% (7.98%), N 7.66% (7.92%).
Synthesis of [Zn{SC(iPr)NtBu}2] (3). [Zn{N(SiMe3)2}2] (0.77 g, 2 mmol) was solvated in toluene (15 mL). A toluene solution (15 mL) of thioamide H[L2] (0.63 g, 4 mmol) was then added and stirred at room temperature for 1 hour. Crystallisation at 5 °C yielded colourless block crystals. 0.40 g (52%). 1H NMR (400 MHz, CD2Cl2) δH: 2.86 (hep, 3JHH = 6.5 Hz, 2H, C
Me2), 1.24 (d, 3JHH = 6.5 Hz, 12H, CH2), 1.16 (s, 18H, Nt![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif)
![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) ). 13C{1H} NMR (125.7 MHz, CD2Cl2) δC: 207.2 (C
). 13C{1H} NMR (125.7 MHz, CD2Cl2) δC: 207.2 (C![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) N), 55.8 (NMe3), 36.0 (HMe2), 30.2 (NCMe3), 22.2 (CH2). Elemental analysis for C16H32N2S2Zn (expected): C 50.20% (50.31%), H 8.43% (8.45%), N 6.97% (7.33%).
N), 55.8 (NMe3), 36.0 (HMe2), 30.2 (NCMe3), 22.2 (CH2). Elemental analysis for C16H32N2S2Zn (expected): C 50.20% (50.31%), H 8.43% (8.45%), N 6.97% (7.33%).
Synthesis of [Zn{SC(iPr)NPh}2]2 (4). [Zn{N(SiMe3)2}2] (0.77 g, 2 mmol) was solvated in toluene (15 mL). A toluene solution (15 mL) of thioamide H[L3] (0.72 g, 4 mmol) was then added and stirred at room temperature for 1 hour. Crystallisation at 5 °C yielded colourless block crystals. 0.36 g (34%). 1H NMR (400 MHz, C6D6) δH: 7.12 (t, 3JHH = 7.7 Hz, 2H, m-Ph), 7.05–6.99 (m, 1H, o-Ph), 6.99–6.91 (m, 2H, p-Ph), 3.07 (hept, 3JHH = 6.6 Hz, 1H, NC
Me2), 1.38 (d, 3JHH = 6.6 Hz, 6H, CH2). 13C{1H} NMR (101 MHz, C6D6) δC: 203.9 (NS), 147.1(N–Cipso), 129.2 (m-Ph), 125.1 (o-Ph), 122.8 (p-Ph), 35.3 (HMe2), 22.3 (CH2). Elemental analysis for C20H24N2S2Zn (expected): C 57.56% (56.93%), H 5.58% (8.73%), N 6.50% (6.64%).
Synthesis of [Zn{SC(iPr)NCy}2]2 (5). [Zn{N(SiMe3)2}2] (0.77 g, 2 mmol) was solvated in toluene (15 mL). A toluene solution (15 mL) of thioamide H[L4] (0.74 g, 4 mmol) was then added. After 1 h stirring at room temperature, a colourless precipitate had formed, which was redissolved to recrystallise at 5 °C, affording colourless block crystals. 0.25 g (29%). 1H NMR (500 MHz, C6C6) δH: 3.28 (hept, 3JHH = 6.2 Hz, 1H, NC
), 2.73 (hept, 3JHH = 6.6 Hz, 1H, CH2), 1.69 (m, 2H, C2), 1.63–1.55 (m, 2H, C2), 1.54–1.37 (m, 3H, C2), 1.22 (d, 3JHH = 6.6 Hz, 6H, CH2), 1.13–0.97 (m, 3H, C2). 13C{1H} NMR (126 MHz, C6D6) δC: 203.9 (NS), 57.9 (NH), 34.7 (H2), 33.5 (HMe2), 25.5 (H2), 25.0 (H2), 22.4 (CH2). Elemental analysis for C20H24N2S2Zn (expected): C 54.81% (55.35%), H 8.23% (8.36%), N 6.17% (6.45%).
Synthesis of [Cd{SC(iPr)N(tBu)}2]3 (6). CdMe2, (0.58 g, 4.1 mmol) was solvated in THF (10 mL). A THF solution (10 mL) of thioamide H[L2] (1.31 g, 8.2 mmol) was then added and stirred at room temperature for 1 hour. The solution was concentrated in vacuo and crystallisation at −35 °C yielded yellow block crystals which were isolated via filtration. 0.40 g (23%).1H NMR (400 MHz, 298K, C6D6) δH: 2.96 (hept, J = 6.5 Hz, 1H, NC
Me2), 2.52 (hept, J = 6.6 Hz, 2H, NCMe2), 1.44 (br-s, 18H, C3), 1.32 (d, J = 6.5 Hz, 6H, CH2), 1.24 (d, J = 6.6 Hz, 12H, CH2), 1.22 (s, 9H, CMe3).13C NMR (101 MHz, 298K, C6D6) δC: 173.4 (NS), 158.9 (NS), 56.5 (N(CH3)3), 54.9 (N(CH3)3), 43.6 (H(CH3)2), 36.8 (H(CH3)2), 31.4 (C(H3)3), 29.7 (C(H3)3), 23.0 (CH(H3)2), 21.7 (CH(H3)2). Elemental analysis for C16H32N2S2Cd (expected): C 45.60% (44.80%), H 7.11% (7.52%), N 6.01% (6.53%).
Single crystal X-ray diffraction studies
Experimental details relating to the single-crystal X-ray crystallographic studies for 2–6 are summarized in Table S1 (ESI†). Single crystal X-ray crystallography data were collected at 150 K on RIGAKU SuperNova Dual wavelength diffractometer equipped with an Oxford Cryostream, featuring a micro source with MoKα radiation (λ = 0.71073 Å) and Cu Kα radiation (λ = 1.5418 Å). Crystals were isolated from an argon filled Schlenk flask and immersed under oil before being mounted onto the diffractometer. Structures were solved by direct methods throughout and refined on F2 data using the SHELXL-2014 suite of programs93 or OLEX2.94 All hydrogen atoms were included in idealized positions and refined using the riding model. Refinements were straightforward with no additional points that merit note. CCDC 2351130–2351135 contain the supplementary crystallographic data for this paper.AACVD experiments
Thin films were deposited using a horizontal hot wall reactor. The aerosol was generated with a TSI 3076 Constant Output Atomiser using N2 at 20 psi to generate the aerosol and act as the carrier gas. Films were grown on either 2.5 cm × 15 cm FTO glass (Pilkington NSG Ltd.), which were cleaned with isopropanol, water, and acetone and then dried under a flow of nitrogen gas, or commercial crystalline silicon. For each deposition a 0.1 M THF solution of 3 was made up in a glovebox and transferred into a bubbler under an inert atmosphere. Depositions were carried out at 175, 200, 250, 300 and 350 °C for 45 min using N2 as a carrier gas.The precursor solution was prepared within a glovebox under an atmosphere of argon and all solvents were dried and degassed prior to use. The precursor bubbler was kept under an atmosphere of argon, sealed, and attached onto to the AACVD apparatus. Once all substrates were prepared and mounted into the deposition chamber, nitrogen gas was allowed to flow through the system, bypassing the precursor holder, for 20 min to purge the system with nitrogen. Then with continuing gas flow the hot-wall furnace is switched on and allowed to reach the target deposition temperature and equilibrate for 20 min. Once this is achieved the gas flow is diverted to flow via the precursor solution which draws the solution into the TSI 3076 Constant Output Atomiser and out into the deposition chamber where the deposition commences, and the timer is started. Gas flow is monitored via bubbler and gas pressure (10 bar) until it reaches the atomizer, as described previously.40,55,57
Thermogravimetric analysis (TGA)
TGA was collected using a TGA 4000 PerkinElmer system. Samples were prepared air sensitively using a crimped aluminium sample pan. TGA was performed under a flow of N2 at 20 mL min−1 and heated from 30 to 600 °C at a ramp rate of 5 °C min−1 and results were analysed using Excel.Powder X-ray diffractometry (PXRD)
PXRD data was collected on a STOE STADI-P. The X-ray diffraction spectra were collected for the thin films using the flat plate mode from 5 to 70 2θ at 2° per minute. X-rays were generated from a Cu source at wavelengths of 1.54 Å.Field emission scanning electron microscopy (FESEM)
SEM was performed to visualize the morphology of the films as both cross sections (using a Field Emission Scanning Electron Microscope 6301F) and top down (JEOL 6480 Low Vacuum large stage SEM platform) images. The films were prepared by mounting onto steel SEM mounts with conductive carbon tape attached to the bottom and top surface of the films, to maximize conductivity of electrons and prevent charge accumulation. Samples were desiccated at 35 °C for 24 h prior to analysis.Energy dispersive X-ray spectroscopy (EDX)
EDX was performed using a JOEL JSM-IT300 scanning electron microscope (WD10 mm and 15 kV) with an Oxford Instruments X-max 80 (EDX) spectrometer and processed on INCA Wave software. All spectra were standardized and calibrated against a standard silicon wafer sample. The magnification, working distance, and beam energy (10 keV) were kept consistent between spectral analyses.Author contributions
ALJ devised and supervised the project, the main conceptual ideas and proof outline. MER performed all the synthesis, characterisation, and deposition work, including that of the thin films. ALJ collected all single crystal X-ray diffraction data. Both ALJ and MER, as authors, contributed to the final version of the manuscript: MER wrote and developed an outline manuscript, which was subsequently amended and worked on by ALJ.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. E. R. acknowledges financial support provided by the EPSRC Centre for Doctoral Training in Aerosol Science grant ref. (EP/S023593/1). The authors wish to acknowledge technical support from John Lowe, Gabriele Kociok-Köhn, Philip Fletcher and Diana Lednitzkythe at the Material and Chemical Characterisation facility (MC2), University of Bath, Claverton Down, Bath, BA2 7AY, U.K.References
- C. J. Barrelet, Y. Wu, D. C. Bell and C. M. Lieber, J. Am. Chem. Soc., 2003, 125, 11498–11499 CrossRef CAS PubMed.
- X. Fang, L. Wu and L. Hu, Adv. Mater., 2010, 23, 585–598 CrossRef PubMed.
- X. Fang, T. Zhai, U. K. Gautam, L. Li, L. Wu, Y. Bando and D. Golberg, Prog. Mater. Sci., 2011, 56, 175–287 CrossRef CAS.
- S. Ummartyotin and Y. Infahsaeng, Renewable Sustainable Energy Rev., 2016, 55, 17–24 CrossRef CAS.
- M. A. Shakil, S. Das, M. A. Rahman, U. S. Akther, M. K. Hassan and M. K. Rahman, Mater. Sci. Appl., 2018, 09, 751–778 CAS.
- V. S. Ganesha Krishna and M. G. Mahesha, Phys. Rev. B, 2022, 628, 413624 CrossRef.
- X. Wang, H. Huang, B. Liang, Z. Liu, D. Chen and G. Shen, Crit. Rev. Solid State Mater. Sci., 2013, 38, 57–90 CrossRef CAS.
- T. Lange, S. Reichenberger, S. Ristig, M. Rohe, J. Strunk, S. Barcikowski and R. Schlögl, Prog. Mater. Sci., 2022, 124, 100856 CrossRef.
- C. Feng, X. Jiang, Q. Zhou, T. Li, Y. Zhao, Z. Niu, Y. Wu, H. Zhou, M. Wang, X. Zhang, M. Chen, L. Ni, G. Diao and Y. Wei, J. Mater. Chem. A, 2023, 11, 18029–18045 RSC.
- A. Chauhan, R. Sharma, M. Singh and R. Sharma, Adv. Opt. Technol., 2024, 13, 1390474 CrossRef.
- G. Schmid, M. Bäumle, M. Geerkens, I. Heim, C. Osemann and T. Sawitowski, Chem. Soc. Rev., 1999, 28, 179–185 RSC.
- H. Li, X. Wang, J. Xu, Q. Zhang, Y. Bando, D. Golberg, Y. Ma and T. Zhai, Adv. Mater., 2013, 25, 3017–3037 CrossRef CAS PubMed.
- K. Deng and L. Li, Adv. Mater., 2014, 26, 2619–2635 CrossRef CAS PubMed.
- L. Cheng, Q. Xiang, Y. Liao and H. Zhang, Energy Environ. Sci., 2018, 11, 1362–1391 RSC.
- N. Chen, Y. He, Y. Su, X. Li, Q. Huang, H. Wang, X. Zhang, R. Tai and C. Fan, Biomaterials, 2012, 33, 1238–1244 CrossRef CAS PubMed.
- C.-Y. Yeh, Z. W. Lu, S. Froyen and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 10086–10097 CrossRef CAS PubMed.
- B. T. Diroll, B. Guzelturk, H. Po, C. Dabard, N. Fu, L. Makke, E. Lhuillier and S. Ithurria, Chem. Rev., 2023, 123, 3543–3624 CrossRef CAS PubMed.
- Y. Hashimoto, N. Kohara, T. Negami, N. Nishitani and T. Wada, Sol. Energy Mater. Sol. Cells, 1998, 50, 71–77 CrossRef CAS.
- C. Guillén, M. A. Martínez, C. Maffiotte and J. Herrero, J. Electrochem. Soc., 2001, 148, G602 CrossRef.
- M. Kemell, M. Ritala and M. Leskelä, Crit. Rev. Solid State Mater. Sci., 2005, 30, 1–31 CrossRef CAS.
- M. Montoya-Gómez, S. Paetel, W. Hempel, A. Kanevce, T. Magorian Friedlmeier and D. Hariskos, Thin Solid Films, 2023, 765, 139636 CrossRef.
- A. Z. Arsad, A. W. M. Zuhdi, S. F. Abdullah, C. F. Chau, A. Ghazali, I. Ahmad and W. S. W. Abdullah, Molecules, 2023, 28, 2780 CrossRef CAS PubMed.
- D. H. Hwang, J. H. Ahn, K. N. Hui, K. S. Hui and Y. G. Son, Nanoscale Res. Lett., 2012, 7, 26 CrossRef PubMed.
- D. Yoo, M. S. Choi, S. C. Heo, C. Chung, D. Kim and C. Choi, Met. Mater. Int., 2013, 19, 1309–1316 CrossRef CAS.
- D. M. Frigo, O. F. Z. Khan and P. O'Brien, J. Cryst. Growth, 1989, 96, 989–992 CrossRef CAS.
- K. Uno, Y. Yamasaki and I. Tanaka, Appl. Phys. Express, 2016, 10, 015502 CrossRef.
- A. Short, L. Jewell, A. Bielecki, T. Keiber, F. Bridges, S. Carter and G. Alers, J. Vac. Sci. Technol., A, 2014, 32, 01A125 CrossRef.
- V. V. Antipov, S. A. Kukushkin and A. V. Osipov, Phys. Solid State, 2016, 58, 629–632 CrossRef CAS.
- J. Kuhs, T. Dobbelaere, Z. Hens and C. Detavernier, J. Vac. Sci. Technol., A, 2017, 35, 01B111 CrossRef.
- A. Aadenan, N. A. Arzaee, M. F. M. Noh, M. N. M. Daud, I. N. N. Mumthas, D. H. B. Hisham, M. A. M. Anuar, N. A. Mohamed, M. A. Ibrahim, N. A. Ludin and M. A. M. Teridi, IOP Conf. Ser.: Mater. Sci. Eng., 2023, 1278, 012006 CrossRef.
- B. Elidrissi, M. Addou, M. Regragui, A. Bougrine, A. Kachouane and J. C. Bernède, Mater. Chem. Phys., 2001, 68, 175–179 CrossRef CAS.
- P. Marchand, I. A. Hassan, I. P. Parkin and C. J. Carmalt, Dalton Trans., 2013, 42, 9406–9422 RSC.
- X. Hou and K. L. Choy, Chem. Vap. Deposition, 2006, 12, 583–596 CrossRef CAS.
- C. E. Knapp and C. J. Carmalt, Chem. Soc. Rev., 2016, 45, 1036–1064 RSC.
- M. D. Nyman, M. J. Hampden-Smith and E. N. Duesler, Inorg. Chem., 1997, 36, 2218–2224 CrossRef CAS PubMed.
- M. A. Ehsan, T. A. N. Peiris, K. G. U. Wijayantha, H. Khaledi, H. N. Ming, M. Misran, Z. Arifin and M. Mazhar, Thin Solid Films, 2013, 540, 1–9 CrossRef CAS.
- A. A. Memon, M. Dilshad, N. Revaprasadu, M. A. Malik, J. Raftery and J. Akhtar, Turk. J. Chem., 2015, 39, 169–178 CrossRef CAS.
- K. Ramasamy, M. A. Malik, M. Helliwell, J. Raftery and P. O'Brien, Chem. Mater., 2011, 23, 1471–1481 CrossRef CAS.
- A. M. Palve, Front. Mater., 2019, 6, 46 CrossRef.
- H. S. I. Sullivan, J. D. Parish, P. Thongchai, G. Kociok-Köhn, M. S. Hill and A. L. Johnson, Inorg. Chem., 2019, 58, 2784–2797 CrossRef CAS PubMed.
- M. A. Bhide, K. L. Mears, C. J. Carmalt and C. E. Knapp, Chem. Sci., 2021, 12, 8822–8831 RSC.
- T. Mandal, V. Stavila, I. Rusakova, S. Ghosh and K. H. Whitmire, Chem. Mater., 2009, 21, 5617–5626 CrossRef CAS.
- M. J. Moloto, N. Revaprasadu, G. A. Kolawole, P. O'Brien, M. A. Malik and M. Motevalli, Eur. J. Chem., 2010, 7, 1148–1155 CAS.
- C. M. Siphamandla, S. Mlowe, A. S. Pawar, S. S. Garje and N. Revaprasadu, Russ. J. Inorg. Chem., 2019, 64, 1063–1071 CrossRef.
- K. Ramasamy, M. A. Malik, P. O'Brien and J. Raftery, Dalton Trans., 2010, 39, 1460–1463 RSC.
- P. O'Brien, J. R. Walsh, I. M. Watson, M. Motevalli and L. Henriksen, J. Chem. Soc., Dalton Trans., 1996, 2491–2496 RSC.
- P. O'Brien, J. R. Walsh, I. M. Watson, L. Hart and S. R. P. Silva, J. Cryst. Growth, 1996, 167, 133–142 CrossRef.
- M. Chunggaze, M. A. Malik and P. O'Brien, Adv. Mater. Opt. Electron., 1997, 7, 311–316 CrossRef CAS.
- V. Kumar, V. Singh, A. N. Gupta, K. K. Manar, M. G. B. Drew and N. Singh, CrystEngComm, 2014, 16, 6765–6774 RSC.
- A. Mietlarek-Kropidłowska, J. Chojnacki, M. Strankowski, A. Fahmi, M. Gazda and B. Becker, J. Therm. Anal. Calorim., 2014, 118, 993–1001 CrossRef.
- S. Mlowe, L. D. Nyamen, P. T. Ndifon, M. A. Malik, J. Raftery, P. O'Brien and N. Revaprasadu, Inorg. Chim. Acta, 2015, 434, 181–187 CrossRef CAS.
- S. A. Alderhami, R. Ahumada-Lazo, M. A. Buckingham, D. J. Binks, P. O'Brien, D. Collison and D. J. Lewis, Dalton Trans., 2023, 52, 3072–3084 RSC.
- K. Ramasamy, M. A. Malik, P. O'Brien and J. Raftery, Dalton Trans., 2009, 2196–2200 RSC.
- A. S. R. Chesman, N. W. Duffy, A. Martucci, L. De Oliveira Tozi, T. B. Singh and J. J. Jasieniak, J. Mater. Chem. C, 2014, 2, 3247–3253 RSC.
- M. A. Buckingham, A. L. Catherall, M. S. Hill, A. L. Johnson and J. D. Parish, Cryst. Growth Des., 2017, 17, 907–912 CrossRef CAS.
- A. A. K. Bakly, D. Collison, R. Ahumada-Lazo, D. J. Binks, M. Smith, J. Raftery, G. F. S. Whitehead, P. O'Brien and D. J. Lewis, Inorg. Chem., 2021, 60, 7573–7583 CrossRef CAS PubMed.
- M. A. Buckingham, K. Norton, P. D. McNaughter, G. Whitehead, I. Vitorica-Yrezabal, F. Alam, K. Laws and D. J. Lewis, Inorg. Chem., 2022, 61, 8206–8216 CrossRef CAS PubMed.
- Y. Takahashi, R. Yuki, M. Sugiura, S. Motojima and K. Sugiyama, J. Cryst. Growth, 1980, 50, 491–497 CrossRef CAS.
- C. Byrom, M. A. Malik, P. O'Brien, A. J. P. White and D. J. Williams, Polyhedron, 2000, 19, 211–215 CrossRef CAS.
- W. Lou, M. Chen, X. Wang and W. Liu, Mater. Lett., 2007, 61, 3612–3615 CrossRef CAS.
- V. Flores-Romero, O. L. García-Guzmán, A. Aguirre-Bautista, I. D. Rojas-Montoya, V. García-Montalvo, M. Rivera, O. Jiménez-Sandoval, M.-Á. Muñoz-Hernández and S. Hernández-Ortega, New J. Chem., 2020, 44, 10367–10379 RSC.
- D. Zeng, M. J. Hampden-Smith and E. N. Duesler, Inorg. Chem., 2002, 33, 5376–5377 CrossRef.
- J. M. Clark, G. Kociok-Köhn, N. J. Harnett, M. S. Hill, R. Hill, K. C. Molloy, H. Saponia, D. Stanton and A. Sudlow, Dalton Trans., 2011, 40, 6893–6900 RSC.
- T. Wildsmith, M. S. Hill, A. L. Johnson, A. J. Kingsley and K. C. Molloy, Chem. Commun., 2013, 49, 8773–8775 RSC.
- A. L. Catherall, M. S. Hill, A. L. Johnson, G. Kociok-Köhn and M. F. Mahon, J. Mater. Chem. C, 2016, 4, 10731–10739 RSC.
- A. L. Catherall, S. Harris, M. S. Hill, A. L. Johnson and M. F. Mahon, Cryst. Growth Des., 2017, 17, 5544–5551 CrossRef CAS.
- V. Pace, L. Castoldi, S. Monticelli, S. Safranek, A. Roller, T. Langer and W. Holzer, Chem. – Eur. J., 2015, 21, 18966–18970 CrossRef CAS PubMed.
- A. L. Johnson, N. Hollingsworth, G. Kociok-Köhn and K. C. Molloy, Inorg. Chem., 2008, 47, 12040–12048 CrossRef CAS PubMed.
- S. Chang, R. D. Sommer, A. L. Rheingold and D. P. Goldberg, Chem. Commun., 2001, 2396–2397 RSC.
- M. Motevalli, P. O'Brien, J. R. Walsh and I. M. Watson, Polyhedron, 1996, 15, 2801–2808 CrossRef CAS.
- M. B. Hursthouse, M. A. Malik, M. Motevalli and P. O'Brien, J. Mater. Chem., 1992, 2, 949–955 RSC.
- M. A. Malik and P. O'Brien, Chem. Mater., 2002, 3, 999–1000 CrossRef.
- A. R. Katritzky, N. M. Khashab, D. N. Haase, M. Yoshioka, I. Ghiviriga and P. J. Steel, J. Org. Chem., 2007, 72, 6742–6748 CrossRef CAS PubMed.
- C. Kulkarni, J. A. Berrocal, M. Lutz, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6302–6309 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- M. A. Romero-Molina, M. D. Gutierrez-Valero, R. Lopez-Garzon, J. M. Salas-Peregrin, M. I. Arriortua and F. J. Zuñiga, Inorg. Chim. Acta, 1987, 136, 87–92 CrossRef CAS.
- M. B. Hursthouse, O. F. Z. Khan, M. Mazid, M. Motevalli and P. O'Brien, Polyhedron, 1990, 9, 541–544 CrossRef CAS.
- I. G. Dance, R. G. Garbutt and M. L. Scudder, Inorg. Chem., 2002, 29, 1571–1575 CrossRef.
- E. S. Lang, G. M. de Oliveira, G. A. Casagrande and E. M. Vázquez-López, Inorg. Chem. Commun., 2003, 6, 1297–1301 CrossRef CAS.
- E. S. Lang, R. Stieler and G. M. de Oliveira, Polyhedron, 2009, 28, 3844–3848 CrossRef.
- J. Chaturvedi, S. Singh, S. Bhattacharya and H. Nöth, Inorg. Chem., 2011, 50, 10056–10069 CrossRef CAS PubMed.
- I. P. Ferreira, G. M. de Lima, E. B. Paniago, C. B. Pinheiro, J. L. Wardell and S. M. S. V. Wardell, Inorg. Chim. Acta, 2016, 441, 137–145 CrossRef CAS.
- K. K. Manar, M. K. Yadav, Anamika, M. G. B. Drew and N. Singh, Polyhedron, 2016, 117, 592–599 CrossRef CAS.
- C. E. Morrison, F. Wang, N. P. Rath, B. M. Wieliczka, R. A. Loomis and W. E. Buhro, Inorg. Chem., 2017, 56, 12920–12929 CrossRef CAS PubMed.
- Y. Wang, X. Xu, W. Lu, Y. Huo and L. Bian, Dalton Trans., 2018, 47, 4219–4227 RSC.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 3199–3202 CrossRef CAS PubMed.
- R. Evans, G. Dal Poggetto, M. Nilsson and G. A. Morris, Anal. Chem., 2018, 90, 3987–3994 CrossRef CAS PubMed.
- A. J. Straiton, J. D. Parish, J. J. Smith, J. P. Lowe and A. L. Johnson, Inorg. Chem., 2023, 62, 4770–4785 CrossRef CAS PubMed.
- C. Edusi, G. Sankar and I. P. Parkin, Chem. Vap. Deposition, 2012, 18, 126–132 CrossRef CAS.
- D. B. Potter, I. P. Parkin and C. J. Carmalt, RSC Adv., 2018, 8, 33164–33173 RSC.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534–534 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details for the synthesis, NMR spectra and TGA plots of [Zn{OC(iPr)NtBu}2]2 are included alongside experimental details for X-ray crystallographic studies of 2–6, 1H DOSY experimental data for complexes 2–5 as well as 1H NMR spectra of byproducts from pyrolysis of 3. EDAX data for thin films deposited from 3. CCDC 2351130–2351135. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01278j |
| This journal is © The Royal Society of Chemistry 2024 |