
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination of azol(in)ium dithiocarboxylate ligands to Au(III): unexpected formation of a novel family of cyclometallated Au(III) complexes, DFT calculations and catalytic studies†
Paula
Pérez-Ramos
,
María A.
Mateo
,
David
Elorriaga
 *,
Daniel
García-Vivó
,
Raquel G.
Soengas
* and
Humberto
Rodríguez-Solla
*
*,
Daniel
García-Vivó
,
Raquel G.
Soengas
* and
Humberto
Rodríguez-Solla
*
Departamento de Química Orgánica e Inorgánica and Instituto Universitario de Química Organometálica “Enrique Moles”, Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: hrsolla@uniovi.es; rsoengas@uniovi.es; elorriagadavid@uniovi.es
First published on 14th May 2024
Abstract
A series of cyclometallated gold(III) complexes 21–27 of general formula [Au(dppta)(azdtc)Cl] (dppta = N,N-diisopropyl-P,P-diphenylphosphinothioic amide-κ2C,S; azdtc = azol(in)ium-2-dithiocarboxylate-κ1S) were prepared and characterized by spectroscopic and diffractometric techniques. Treatment of [Au(dppta)(azdtc)Cl] complexes with methanol led to their quantitative transformation into a novel family of (C^S, S^S)-cyclometallated gold(III) complexes of general formula [Au(dppta)(azmtd)] (azmdt = azol(in)ium-2-(methoxy)methanedithiol-κ2S,S) 28–34. All the [Au(dppta)(azdtc)Cl] complexes 21–27 catalyzed the alkylation of indoles, whereas [Au(dppta)(azmtd)] complexes 28–34 were inactive. Among the synthesized derivatives, complex 22 displayed the highest catalytic activity, leading to a series of functionalized indoles in excellent yields.
Introduction
Gold chemistry has attracted considerable attention in the past few years due to the potential applications in medicine, optoelectronics and catalysis.1 These studies have mostly focused on Au(I) complexes, with considerably less attention being paid to Au(III) complexes. However, recent studies have put Au(III) complexes on the spotlight as hot topic of research because of their potential interest as antitumor and antibacterial agents,2 their application in the development of luminescent materials3 and their ability to activate unsaturated groups in diverse catalytic processes.4,5 One of the mayor disadvantages of Au(III) complexes is their inherent instability, due to their tendency to undergo reduction.6 To avoid reduction, an interesting approach is the stabilization of the Au(III) centre by coordination to a cyclometallated ligand to render complexes of the general formula [AuX2(C–Y)], where C–Y represents a bis-coordinated ligand with a Au–C σ-bond and a coordinate bond through N, P or S donor atom (Fig. 1).7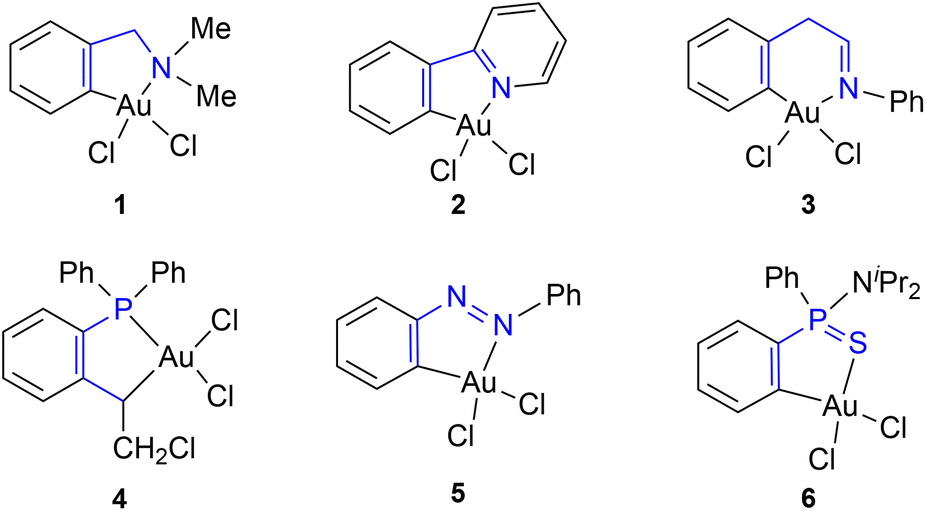 | ||
| Fig. 1 Some relevant cyclometallated gold(III) complexes. | ||
These Au(III) complexes have a square planar geometry with four coordination sites around the metal centre and through rational ligand design, the reactivity of the complexes can be modulated “à la carte”. For instance, the properties of Au(III) complexes can be tuned to achieve a right balance between stability and reactivity and obtain perfect candidates for their use as catalysts in various organic transformations.
In that sense, N-heterocyclic carbene (NHC) ligands are well known in gold chemistry due to their easy tuneability, electron-rich nature and steric bulk.2a,8 Related to NHC ligands, zwitterionic dithiocarboxylate azol(in)iums can coordinate to metals as monodentate, chelate bidentate or bridging bidentate (Fig. 2).9 Even though the potential of these ligands has been studied with a certain number of transition metals,10 the presence of coordination complexes of dithiocarboxylate azolium ligands and gold in the literature is very limited. The first report dates back to 1987, when Borer et al. described the formation of Au(I) complexes by reaction of dithiocarboxylate azolium ligands and NaAuCl4·2H2O.11 Later on, Wilton-Ely and co-workers reported an in-depth study of the coordination of these zwitterionic ligands to Au(I), as well as their suitability for an efficient stabilization of gold nanoparticles.12 In this same vein, these authors subsequently studied the chemisorption of dithiocarboxylate azolium ligands on solid gold substrates.13
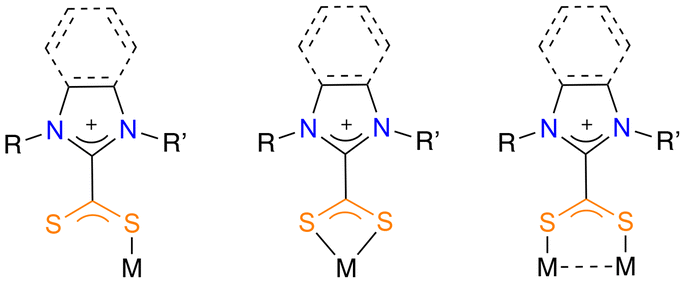 | ||
| Fig. 2 Bonding modes of azol(in)ium-2-dithiocarboxylate ligand to metal centres. | ||
Here we report the synthesis of a novel class of cyclometallated Au(III) compounds bearing azol(in)ium-2-dithiocarboxylate κ1S-donor ligands as auxiliary ligands. Moreover, their unprecedent transformation into κ2S,S-azol(in)ium-2-(methoxy)methanedithiolate cyclometallated gold(III) complexes was also achieved. Additionally, catalytic performance of all the synthesized complexes was studied for the alkylation of indoles.
Results and discussion
Synthesis of the azol(in)ium dithiocarboxylate ligands
The most convenient procedure to synthesize the azol(in)ium dithiocarboxylate zwitterionic ligands is by means of the deprotonation of azolium salts and subsequent addition of CS2. Thus, we treated commercially available benzimidazolium 7, imidazolinium 8, and imidazolium 9–13 salts with Cs2CO3 in acetonitrile for 30 minutes and then we added carbon disulfide. After stirring the resulting mixture at r.t. for 4 hours, compounds 14, 15 and 20 were obtained in almost quantitative yields (Scheme 1). However, this protocol was unsuccessful for N-alkyl substituted imidazolium salts, which need a stronger base for the deprotonation step. Therefore, treatment of imidazolium salts 9–12 with tBuOK in THF for 30 minutes and subsequent addition of carbon disulfide, afforded the corresponding azolium dithiocarboxylate zwitterionic ligands 16–19 as orange-red solids (Scheme 1). Ligands 14, 15 and 20 were previously reported in the literature,9d,f whereas ligands 16, 17, 18 and 19 have been prepared and characterized for the first time in this work. The 13C{1H} NMRs of all these ligands show a characteristic signal around 224 ppm which correspond to the carbon atom of the CS2 core. In general terms, the data obtained for ligands 14–20 fit with their zwitterionic nature and agree with the data previously reported in the literature.9d,f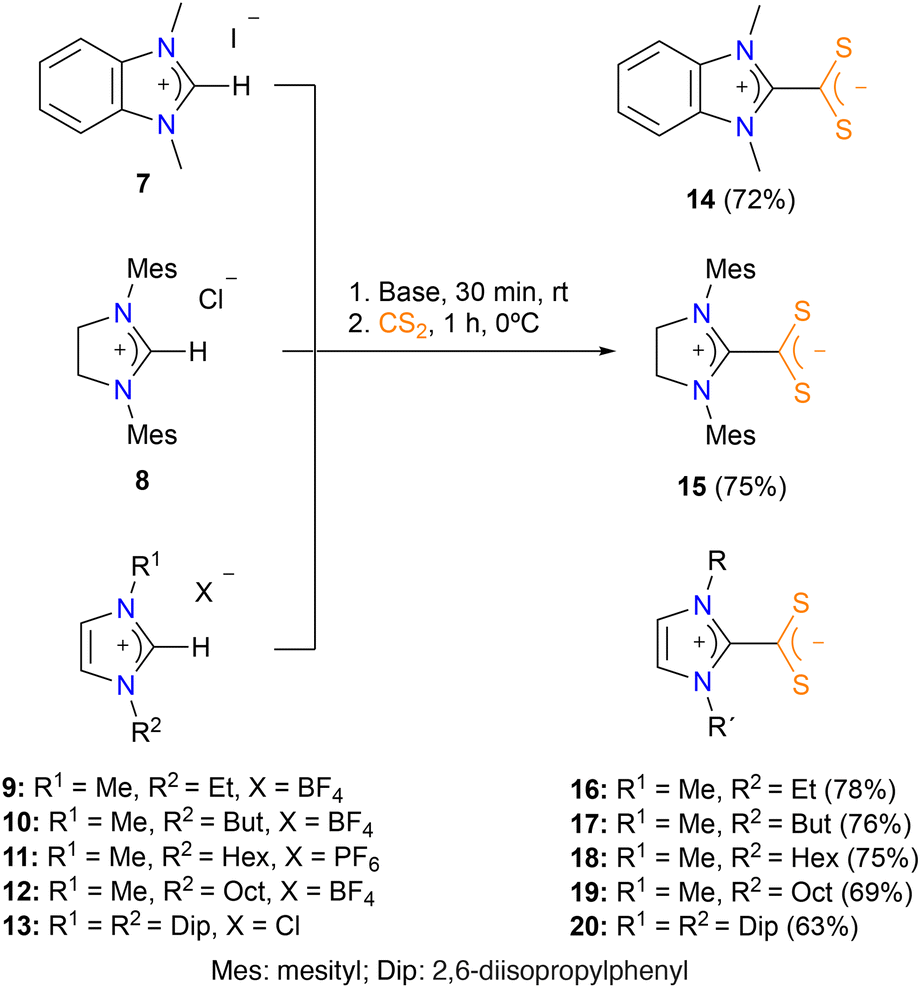 | ||
| Scheme 1 Synthesis of dithiocarboxylate azol(in)ium zwitterionic ligands 14–20. | ||
Formation of azol(in)ium dithiocarboxylate Au(III) complexes
Our recent studies on Au(III) complexes have focused on the synthesis of (S^C)-cycloaurated compounds for the evaluation of their chemical and biological properties.14 Taking into account our previous experience, we decided to investigate the complexation of (S^C)-cycloaurated dichloride complex 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7b to azol(in)ium dithiocarboxylate ligands.
7b to azol(in)ium dithiocarboxylate ligands.
Thus, reaction of stoichiometric amounts of the azol(in)ium dithiocarboxylate zwitterionic ligands 14–20 with the Au(III) precursor 6 in acetonitrile for 16 hours at room temperature and subsequent addition of aqueous KPF6 afforded the corresponding azol(in)ium dithiocarboxylate-κ1S complexes 21–27 as red solids in good yields (54–82%) (Scheme 2).
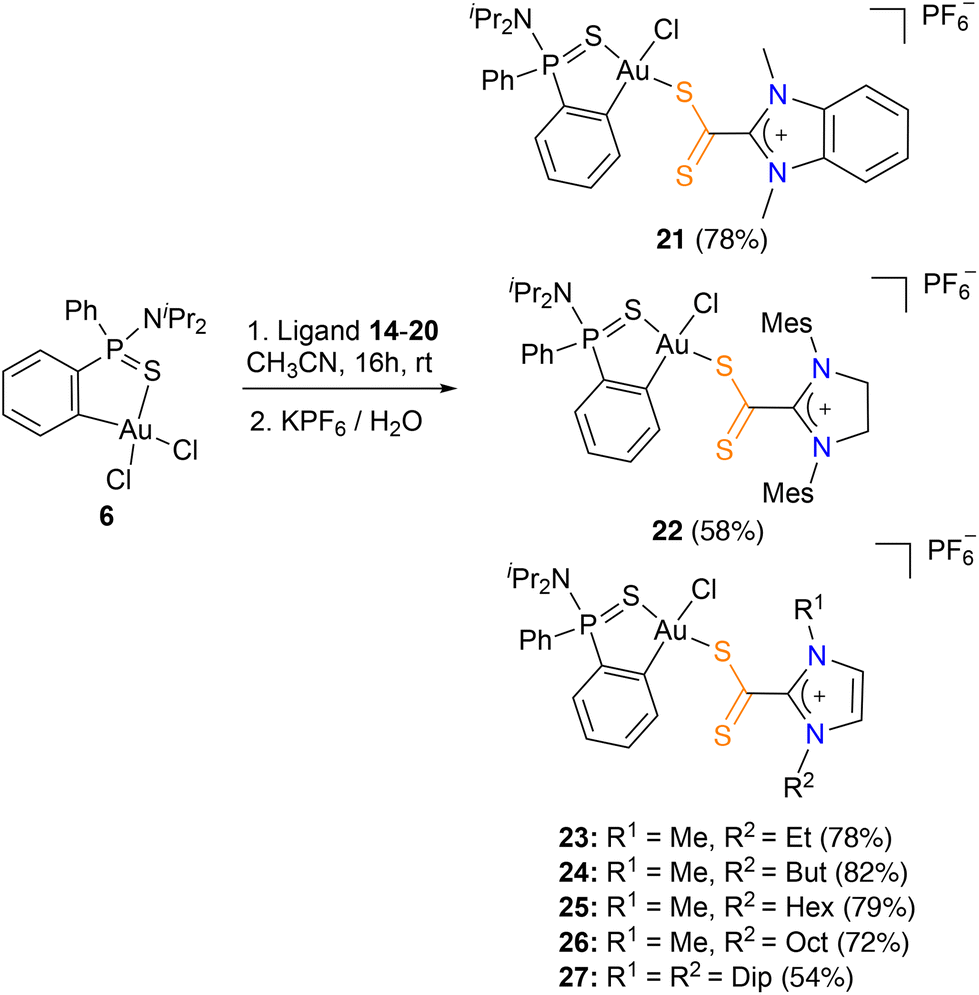 | ||
| Scheme 2 Synthesis of azol(in)ium-2-dithiocarboxylate complexes 21–27. | ||
The obtained dithiocarboxylate azol(in)ium complexes 21–27 were fully characterized by multinuclear NMR spectroscopy, MS, IR and, in several cases, X-ray diffraction studies. Two sets of proton and carbon signals were observed, indicating the formation of the two possible isomers, cis or trans. However, the isomers with the two S atoms in trans configuration are preferentially formed in all cases (de 73–94%). This is possibly due to the preferential mutual trans disposition of the ligand with the highest trans influence (C) and the hardest base (Cl) due to the antisymbiotic effect,15 a feature commonly observed in gold(III) complexes.16 The 1H NMR spectra of complexes 21–27 show that proton ortho to the gold ion (7.55–8.16 ppm) is shifted significantly upfield relative to the precursor complex [Au(dppta)Cl2] 6 (8.47 ppm). On the other hand, the 13C{1H}-NMR spectra showed a upfield displacement of the signal corresponding to the CS2 moiety (206.1–210.4 ppm) with respect to the free ligand (224.6–224.7 ppm), which is in accordance with the examples of azol(in)ium dithiocarboxylate metal complexes reported so far in the literature (205–225 ppm).17
Fortunately, X-ray quality single crystals of the trans-isomers of 22 and 27 were obtained from a saturated solution of the compound in THF layered with hexane. The molecular structures of 22 and 27 showing the atomic numbering scheme are depicted in Fig. 3. Diffraction studies revealed a mononuclear compound and confirmed the coordination of the dithiocarboxylate ligand in a κ1S-monodentate mode, so that both coordinated sulfur atoms are in a mutual trans-disposition. The S1–Au1 and S2–Au1 distances are very similar (∼2.31 Å and ∼2.33 Å, respectively). On the other hand, in contrast with the free ligand, the S3–C19 distance (∼1.63 Å) is slightly shorter than the S2–C19 distance (∼1.70 Å) which suggests a greater π-contribution over the S3–C19 bond. However, both bonds are in an intermediate situation between the values expected for single and double C–S bonds (approx. 1.61 Å for the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and approx. 1.75 Å for the S–C bonds).18 The metallacycle in complex 22 differ slightly from the planar geometry, with the phosphorus atom puckered out the plane and the plane S1–P1–C2 tilted 29.27° form the plane formed between S1–Au1–C1. This effect is even more evident in complex 27, for which this angle is even larger (46.6°), which seems to be due to the presence of an S–π interaction between S3 and the centroid of the aromatic ring facing it at a distance of 3.51 Å (Fig. 3c); this distance falls whithin the range described in the literature for this type of S–π interactions.19 All these effects translate into a geometry around the gold atom that deviates slightly from the ideal square planar geometry, as indicated by the structural parameter τ4, with values of 0.06 and 0.09 for 22 and 27, respectively.20
C bonds and approx. 1.75 Å for the S–C bonds).18 The metallacycle in complex 22 differ slightly from the planar geometry, with the phosphorus atom puckered out the plane and the plane S1–P1–C2 tilted 29.27° form the plane formed between S1–Au1–C1. This effect is even more evident in complex 27, for which this angle is even larger (46.6°), which seems to be due to the presence of an S–π interaction between S3 and the centroid of the aromatic ring facing it at a distance of 3.51 Å (Fig. 3c); this distance falls whithin the range described in the literature for this type of S–π interactions.19 All these effects translate into a geometry around the gold atom that deviates slightly from the ideal square planar geometry, as indicated by the structural parameter τ4, with values of 0.06 and 0.09 for 22 and 27, respectively.20
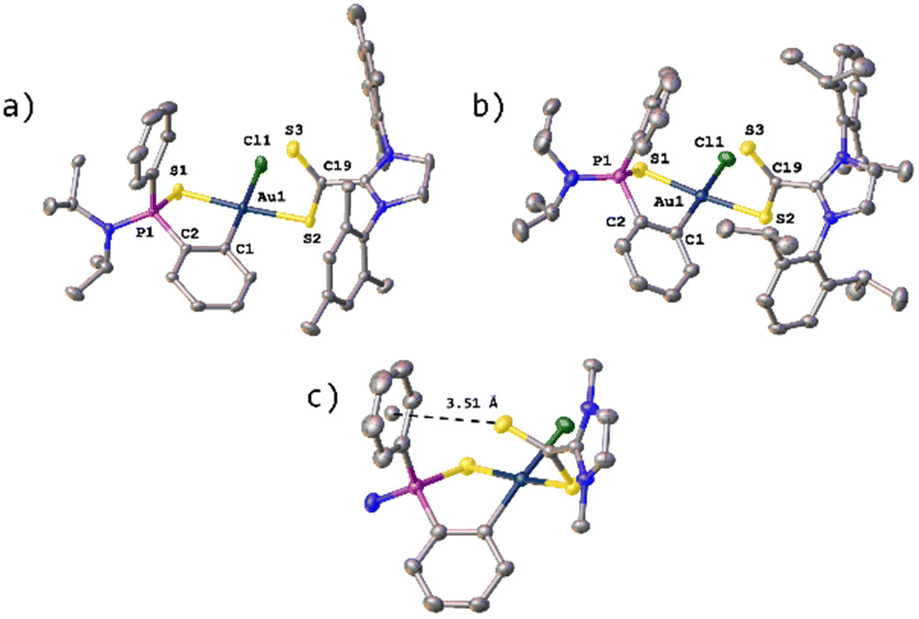 | ||
| Fig. 3 Molecular structure of compounds 22 (a) and 27 (b) with hydrogen atoms, solvent molecules and PF6− anions omitted for clarity. (c) S–Ph π interaction in complex 27. | ||
Surprisingly, the outcome of the reaction of the azol(in)ium dithiocarboxylate ligands 14–20 with the gold precursor 6 is strongly dependent on the solvent used. Thus, when the reaction is carried out using methanol as solvent, a new family of cyclometallated complexes 28–34 was obtained (Scheme 3). In these new complexes, a methoxy group bonds to the dithiocarboxyl carbon atom, forming a azol(in)ium-2-(methoxy)methanedithiolate which bonds to the Au(III) metal center in a κ2S,S coordination mode. The new complexes 28–34 were obtained as yellow solids in good yields and characterized by multinuclear NMR. Two chiral centers are present in the product of this reaction; thus, there are two pairs of enantiomers possible from this reaction. The 1H-NMR spectra of all complexes showed two identical sets of signals in a 1:1 ratio which correspond with the mixture of syn/anti-isomers with respect to mutual orientation of the nitrogen atom of the NiPr2 and the oxygen atom of the methoxide motif. The presence of two singlets around 3–3.5 ppm (one for each isomer) confirm the incorporation of the methoxy group. The 31P{1H}-NMR spectrum are also consistent with the formation of diastereomeric complexes, showing two distinct signals at 70–80 ppm and a septuplet at around −140 ppm which corresponds to the PF6 anion. Regarding the 13C{1H}-NMR spectra, the most salient feature is that the signal for the CS2 moiety is around 91.4–93.3 ppm, which is significantly shielded when compared to complexes 21–27. This shift to high field is in agreement with the change of hybridization of the CS2 carbon atom from sp2 to sp3 upon incorporation of the methoxy group.
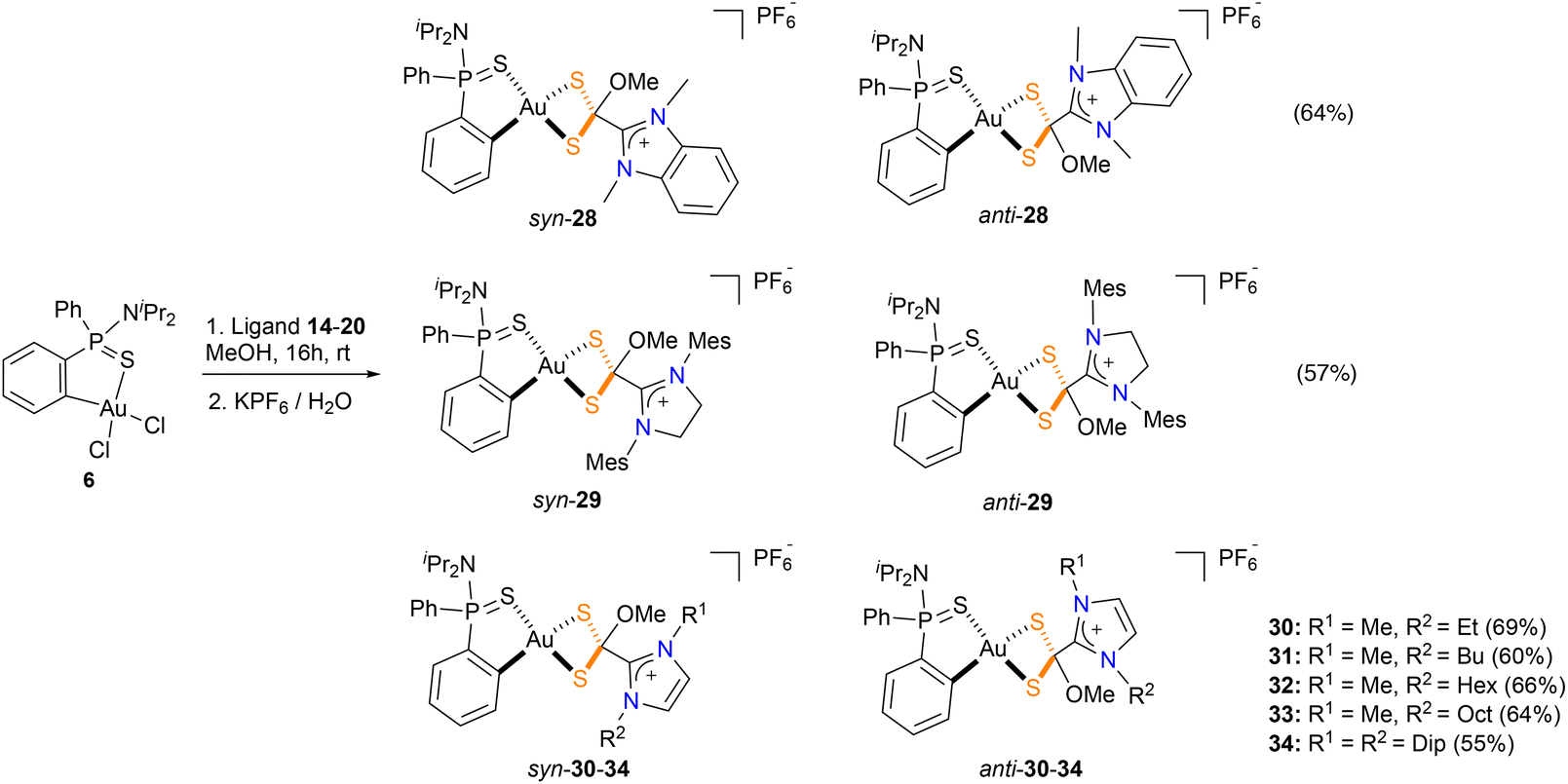 | ||
| Scheme 3 Synthesis of azol(in)ium-2-(methoxy)methanedithiol complexes 28–34. | ||
Single crystals of syn-34 suitable X-ray diffraction were obtained from a saturated solution of the compound in THF layered with hexane. The molecular structure displaying the atomic numbering scheme is shown in Fig. 4.
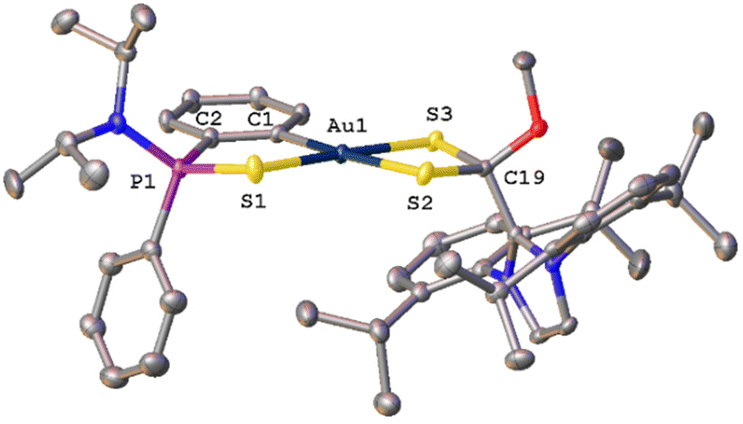 | ||
| Fig. 4 Molecular structure of compound syn-34 (H atoms, solvent molecules and PF6− anion are omitted for clarity). | ||
X-Ray diffraction studies revealed a mononuclear compound where the dithio ligand is now κ2-S,S′ coordinated to the gold centre; however, the involved distances S2–Au1 and S3–Au1 are 2.347 Å and 2.285 Å, respectively, with difference being attributed to the higher trans-influence of the C6H4 aromatic ring when compared to that of the SP moiety. Furthermore, the distance of the dative S1–Au1 bond is 2.347 Å, the same as the S2–Au1 distance. As suggested by the NMR data, the carbon atom of the CS2 core has changed hybridization from sp2 to sp3 during the reaction and has a methoxy group attached. Analogous to complexes 21–27, the metallacycle deviates slightly from the planar geometry, with the phenylphosphinothioic amide ligand tilted to give a dihedral angle of 15.03°. The value of structural parameter τ4 (0.10) suggest that the geometry around the gold atom is pseudo-square planar.19
To investigate the possible transformation of dithiocarboxylate into dithiolate complexes, compounds 21–27 were dissolved in methanol. After stirring the resulting mixture for a few minutes, the solution turned from red to yellow, suggesting the transformation of the starting azol(in)ium-2-dithiocarboxylate complexes 21–27 into the azol(in)ium-2-(methoxy)methanedithiolates 28–34. After 6 h (20 h for sterically hindered complexes 22 and 27), the corresponding dithiolate complexes 28–34 were isolated in almost quantitative yield (Scheme 4).
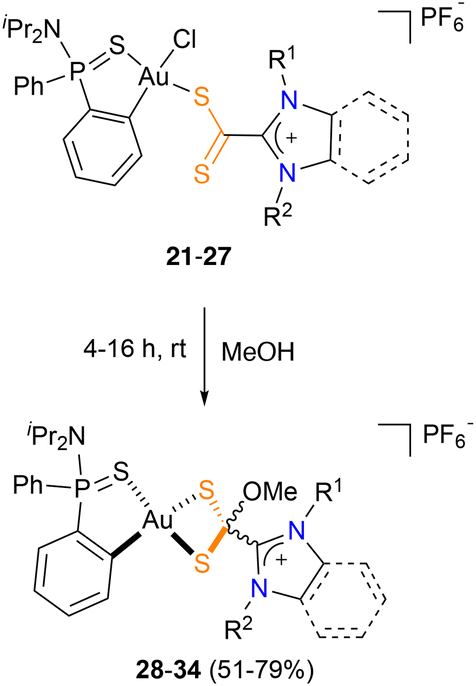 | ||
| Scheme 4 Transformation of azol(in)ium-2-dithiocarboxylate complexes 21–27 into azol(in)ium-2-(methoxy)methanedithiol complexes 28–34. | ||
In order to gain more insight on this unusual transformation, we monitored this transformation by 31P-NMR. Thus, an NMR tube was charged with 27 and deuterated methanol was added; 31P-NMR spectra were recorded at ten-minute intervals (Fig. S56 and S57, ESI†). Formation of significant amounts of 34 is evident after just 10 min of reaction and the complete disappearance of the starting dicarboxylate complex 27 takes 4.5 h. Interestingly, a new resonance appears after ca. 10 min of reaction in a intermediate position between the resonances of the parent chloride and those of the cyclometallated derivative 34 (δP = 69 ppm), and it completely disappears after ca. 5 hours. This new signal must be attributed to the formation of a long-lived reaction intermediate and can be tentatively assigned to a putative Au(III) complex derivative formed after metathesis of the chloride ligand by a methoxy (vide infra).
Aiming to shed some light into the transformation of 2-dithiocarboxylate complexes 21–27 into (methoxy)methanedithiolate complexes 28–34, we decided to explore the mechanism for such process by means of DFT calculations. To that end, we have used as model compound the 1,3-bis(2,6-diisopropylphenyl)-1H-imidazol-3-ium derivative 27. According to our calculations, the first step of the reaction would be a Cl to OMe ligand metathesis whereby a new gold methoxide complex (I1) is formed after formal HCl elimination (Fig. S58, ESI†). This substitution takes place through a transition state (TS1, Fig. 5) of moderate activation energy (ca. 10 kcal mol−1 above the starting chloride trans-27), and is preceded by the formation of a methanol adduct (trans-27·MeOH), in which the solvent molecule is bound by a strong MeOH⋯Cl hydrogen bond (dH–Cl = 2.38 Å), an interaction not altering significantly the coordination sphere of the gold atom. The gold centre in TS1 displays a five-coordinate distorted trigonal bipyramidal geometry which agrees with a classification of the overall process as an associative interchange mechanism (Ia), in which Au–Cl bond has been significantly elongated (ca. 0.6 Å) with concerted Au–O bond formation (dAu–O = 2.34 Å). Similar mechanisms have been proposed on the basis of computational studies for related chloride ligand substitution reactions at Au(III) complexes.21
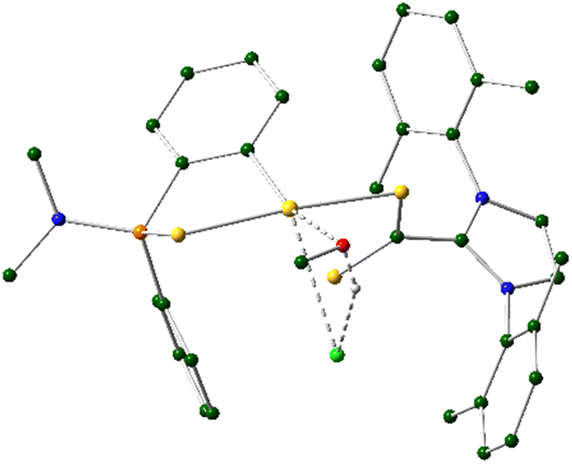 | ||
| Fig. 5 DFT computed structure of TS1 with most hydrogen atoms omitted for clarity. | ||
The gold methoxide intermediate I1 can be, tentatively, identified as the experimentally observed intermediate during the NMR monitoring experiments. Several potential pathways have been explored computationally to account for the transformation of this methoxide I1 into the final (methoxy)methanedithiolates 34. According to our calculations, the most likely mechanism would imply the participation of a second molecule of MeOH in the process in an essentially outer-sphere mechanism by which the gold atom is not directly involved in the process (Fig. 6). Thus, in first place the imidazoliumdithiolate group swings so that the sulfur atoms of the CS2 group exchange its positions to form intermediate I1′, a process with a small energy penalty (ca. 6 kcal mol−1) occurring through an energetically accessible TS2 located only +11 kcal mol−1 above I1. Then, addition of MeOH occurs in an exergonic manner (ca. −2 kcal mol−1) rendering the formation of I1′·MeOH adduct, in which the incoming solvent molecule binds to the Au bound methoxide group through a conventional strong hydrogen bond interaction (dO⋯H = 1.692 Å), while an almost perfect orientation of the oxygen atom of the methanol molecule to accomplish the nucleophilic attack to the carbon atom of the CS2 group is attained (dO⋯C = 3.031 Å). NBO analysis of the charges in this intermediate also provides additional support for the nucleophilic attack (qO = −0.682e, qC = +0.445e), which occurs through transition state TS3 which is located ca. 19 kcal mol−1 above the parent methoxide I1. Interestingly, the formation of the new C–O bond in TS3 (dCO = 2.029 Å) occurs in a concerted manner with a proton transfer from the former methanol to the gold bound methoxide, with the hydrogen atom being, in fact, more tightly bound to this latter group (dOH = 1.082 and 1.412 Å, respectively).
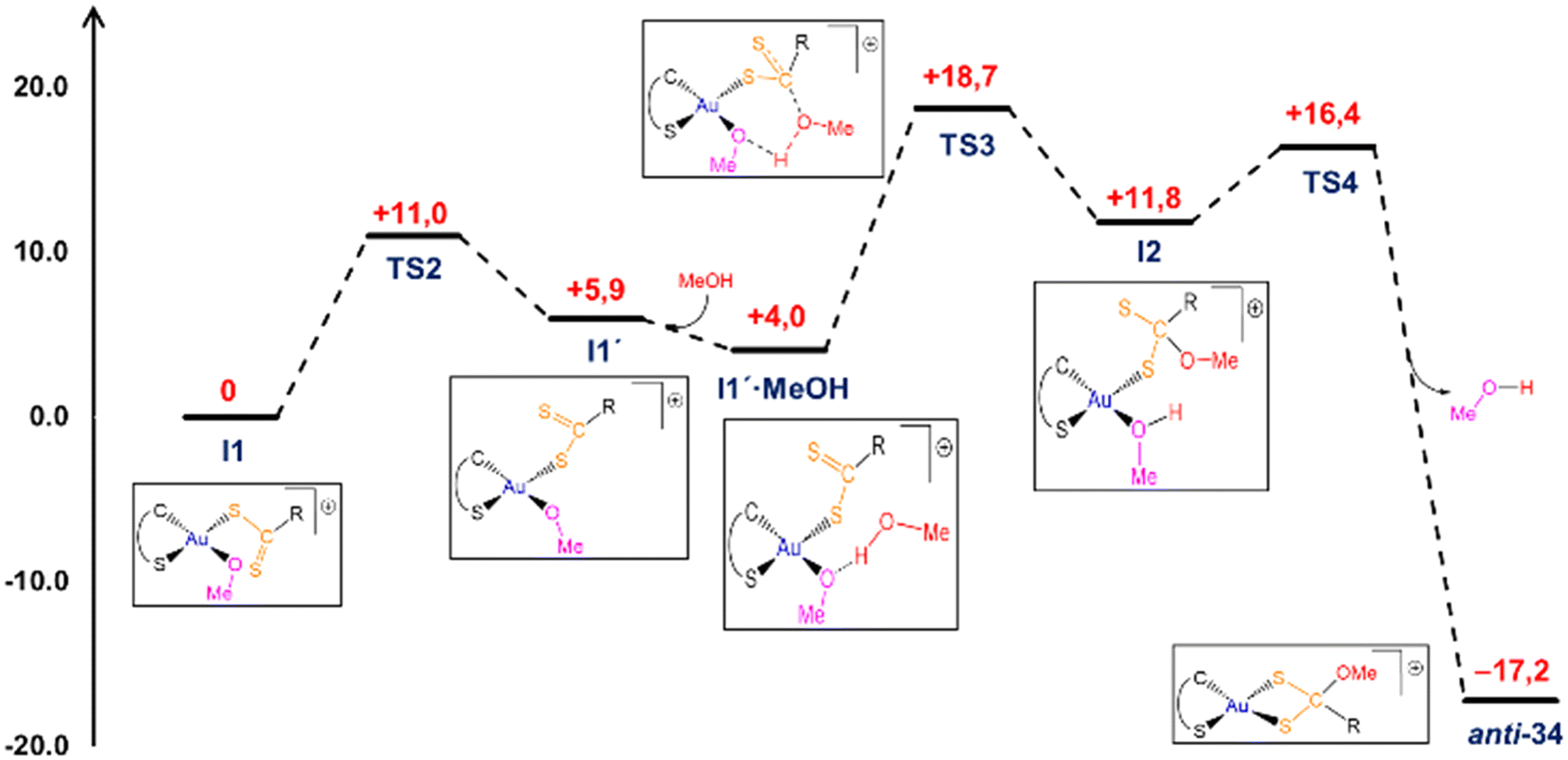 | ||
| Fig. 6 DFT computed reaction profile for the I1 → anti-34 transformation. | ||
The outcome of this step is the formation of a new intermediate (I2) placed ca. +12 kcal mol−1 above I1 and displaying a fully formed (methoxy)methanedithiolate group which is bound to the gold centre in a κ1S-monodentate mode, while the former methoxide group also remains coordinated to the metal now converted into a methanol molecule. From here, the system would evolve by methanol replacement by the non-coordinated sulfur atom of the dithiolate group, leading to the formation of the reaction product displaying an anti configuration (anti-34). This ligand substitution occurs through an energetically accessibly TS4 (ca. +16 kcal mol−1), and also follows an associative interchange mechanism, with concerted elimination of methanol (dAuO = 2.434 Å) and formation of Au–S bond (dAuS = 2.896 Å). Considering the complete mechanism, the rate determining step for the I1 to anti-34 transformation would correspond to the nucleophilic attack of the methanol oxygen atom to the carbon atom of the CS2 group with an overall reaction barrier of ca. 19 kcal mol−1, which is kinetically accessible at room temperature. In order to account for the formation of the corresponding syn-34 isomer, a similar mechanism has been computationally verified (Fig. S59, ESI†) with the main differences then being that the initial coordination of the methanol molecule would take place directly at the methoxide I1 and that the overall energetic barrier would be slightly higher (ΔΔG‡ = +1.6 kcal mol−1), yet remaining compatible with a room temperature transformation, and accounting for the experimental observation of formation of both syn- and anti-isomers of 34.
Catalytic activity of the synthesized Au(III) complexes
As mentioned before, one of the most relevant applications of Au(III) complexes is as catalysts in the activation of unsaturated bonds. To this end, it is of the uttermost importance to reach the right balance between stability and reactivity. In this regard, we hypothesized that cationic (S^C, S^S)-cyclometallated gold(III) complexes might be able to stabilize the cationic Au(III) center while maintaining catalytic activity. In order to evaluate the catalytic performance of κ1S-azol(in)ium-2-dithiocarboxylate complexes 21–27 and κ2S,S-azol(in)ium-2-(methoxy)methanedithiolate complexes 28–34, we focused on the Au(III)-catalyzed preparation of 3-substituted indoles,22 important structural motifs found in many natural products and pharmaceuticals.23With the aim of investigating the catalytic activities of κ1S-azol(in)ium-2-dithiocarboxylate complexes 21–27 and κ2S,S-azol(in)ium-2-(methoxy)methanedithiolate complexes 28–34 in the gold–silver cooperative dual catalytic alkylation of indoles, the reaction of alkynyl alcohol 35a with indole 36a was used as a model. The results of the catalytic studies are given in Table 1.
|
|
|||
|---|---|---|---|
| Entry | Au catalyst (mol%) | Ag catalyst (mol%) | Yielda (%) |
| a Determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. | |||
| 1 | 21 (2.5) | AgPF6 (5) | 94 |
| 2 | 22 (2.5) | AgPF6 (5) | 97 |
| 3 | 23 (2.5) | AgPF6 (5) | 78 |
| 4 | 24 (2.5) | AgPF6 (5) | 80 |
| 5 | 25 (2.5) | AgPF6 (5) | 85 |
| 6 | 26 (2.5) | AgPF6 (5) | 88 |
| 7 | 27 (2.5) | AgPF6 (5) | 84 |
| 8 | 28 (2.5) | AgPF6 (5) | 6 |
| 9 | 29 (2.5) | AgPF6 (5) | 8 |
| 10 | 22 (2.5) | — | 0 |
| 11 | — | AgPF6 (5) | 6 |
Using 2.5 mol% of azol(in)ium-2-dithiocarboxylate complexes 21–27 (2.5 mol%) with AgPF6 (5 mol%), alkylated indole 37a was obtained in good yields in all cases (entries 1–7).
In contrast, azol(in)ium-2-(methoxy)methanedithiolate complexes 28–34 displayed very poor catalytic activity (entries 8 and 9). This result is in agreement with the findings obtained by DFT calculations, which show a high stability of the Au(III) centre in the azol(in)ium-2-(methoxy)methanedithiolate complexes. Therefore, the monocyclometallated Au(III) center in the azol(in)ium-2-dithiocarboxylate complexes can effectively activate the alkyne moiety, whereas the cyclometallated Au(III) center in the azol(in)ium-2-(methoxy)methanedithiolate complexes is too stable to undergo any type of reaction. The best results were obtained with complex 22, affording alkylated indole 37a in an excellent 97% yield (entry 2). In general terms, the catalytic activity increases with increasing size of the side chain in the azol(in)ium moiety. This trend could be related to differences in the geometry of the complexes, since bulkier ligands somehow distort the square planar geometry of the metal center. In this regard, it has been reported that distorted square planar geometry in Au(III) complexes is associated with enhanced catalytic activity.22a In the control experiments (entries 10 and 11), only low yields were observed when a single metal catalyst was used.
Using the optimized conditions, we investigated the substrate scope of the dual metal catalysis for indole alkylation. Thus, a solution of alkynyl alcohol 35 and indole 36 in CH2Cl2 in the presence of a 2.5 mol% of complex 22 and a 5 mol% of AgPF6 was stirred at r.t. for 2 h.
According to the results compiled on Table 2, alkynyl alcohols of different chain lengths 35a–d and indoles with different substituents 36a–b afforded alkylated indoles 37a–e with good to excellent yields (71–97%) and total regioselectivity. The reactivity and regioselectivity observed for the indole alkylation reaction were consistent with those reported in the literature.22a,24
Conclusions
In conclusion, we have described the preparation and characterization of a novel family of Au(III) cyclometallated κ1S-azol(in)ium-2-dithiocarboxylatecomplexes. These derivatives undergo a fascinating rearrangement in the presence of methanol to afford the corresponding Au(III) cyclometallated κ2-S,S′-azol(in)ium-2-(methoxy)methanedithiolate complexes, a family of Au(III) compounds that have no precedent in the literature. In addition, this transformation was thoroughly investigated by the means of NMR studies and DFT calculations, allowing us to propose a plausible mechanism.Furthermore, the catalytic behavior of both families of Au(III) complexes in the alkylation of indoles has been explored. In this regard, κ1-S-azol(in)ium-2-dithiocarboxylate complexes display excellent performance, providing alkylated indoles in good to excellent yields and total regioselectivity. On contrary, κ2-S,S′-azol(in)ium-2-(methoxy)methanedithiolate complexes showed poor catalytic activity, consistent with their high stability, as predicted by the DFT calculations.
Author contributions
Conceptualization: R. G. S., H. R. S.; investigation: P. P. R., M. A. M., R. G. S., D. E., D. G. V.; validation: P. P. R., R. G. S.; supervision: R. G. S., H. R. S., D. E.; funding acquisition: R. G. S., H. R. S.; project administration: R. G. S., H. R. S.; writing – original draft: R. G. S., D. E.; writing – review & editing: R. G. S., H. R. S., D. E., D. G. V., P. P. R., M. A. M.Conflicts of interest
All authors declare that they have no conflicts of interest.Acknowledgements
This work has received financial support from the Instituto de Salud Carlos III (PI22/00148) and MINECO (PID2022-137893OB-I00 and PID2021-123964NB-I00). Partial financial support by Instituto de Salud Global de Barcelona (FUO-23-194) is gratefully acknowledged. We thank the SCBI of the Universidad de Málaga, Spain, for access to computing facilities. Authors would like to thank the use of RIAIDT-USC analytical facilities. P. P. R. thanks Programa Investigo for a predoctoral contract (AYUD/2022/9313, EU Next Generation).References
- (a) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS; (b) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed; (c) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS; (d) J. Ruiz, D. Sol, M. A. Mateo, M. Vivanco and R. Badía-Laiño, Dalton Trans., 2020, 49, 6561 RSC; (e) G. Moreno-Alcántar, P. Picchetti and A. Casini, Angew. Chem., Int. Ed., 2023, 62, 1433 CrossRef PubMed; (f) P. Crochet and V. Cadierno, Catalysts, 2023, 13, 436 CrossRef CAS.
- (a) B. D. Glišić and M. I. Djuran, Dalton Trans., 2014, 43, 5950 RSC; (b) D. Fontinha, S. A. Sousa, T. S. Morais, M. Prudencio, J. H. Leitao, Y. Le Gal, D. Lorcy, R. A. L. Silva, M. F. G. Velho, D. Belo, M. Almeida, J. F. Guerreiro, T. Pinheiro and F. Marques, Metallomics, 2020, 12, 974 CrossRef CAS PubMed.
- (a) V. W.-W. Yam and A. S-Y. Law, Coord. Chem. Rev., 2020, 414, 213298 CrossRef CAS; (b) V. W-W. Yam, K. M.-C. Wong, L.-L. Hung and N. Zhu, Angew. Chem., 2005, 117, 3167 CrossRef.
- (a) A. Arcadi and S. Di Giuseppe, Curr. Org. Chem., 2004, 8, 795 CrossRef CAS; (b) H. Schmidbaur and A. Schier, Arabian J. Sci. Eng., 2012, 37, 1187 CrossRef CAS.
- (a) L. Rocchigiani and M. Bochmann, Chem. Rev., 2021, 121, 8364 CrossRef CAS PubMed; (b) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248 CrossRef CAS PubMed; (c) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC; (d) P. Garcia, M. Malacria, C. Aubert, V. Gandon and L. Fensterbank, ChemCatChem, 2010, 2, 493 CrossRef CAS; (e) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236 CrossRef CAS PubMed.
- E. A. Pacheco, E. R. T. Tiekink and M. W. Whitehouse, Gold Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, vol. 283 Search PubMed.
- (a) B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209 RSC; (b) E. Belmonte-Sánchez, M. J. Iglesias, H. El Hajjouji, L. Roces, S. García-Granda, P. Villuendas and F. López-Ortiz, Organometallics, 2017, 36, 1962 CrossRef.
- (a) I. Özdemir, N. Tenelli, S. Günal and S. Demir, Molecules, 2010, 15, 2203 CrossRef PubMed; (b) S. Hussaini, R. A. Haque and M. R. Razali, J. Organomet. Chem., 2019, 882, 96 CrossRef CAS.
- (a) L. Delaude, A. Demonceau and J. Wouters, Eur. J. Inorg. Chem., 2009, 1882 CrossRef CAS; (b) O. Sereda, A. Blanrue and R. Wilhelm, Chem. Commun., 2009, 1040 RSC; (c) D. Konieczna, A. Blanrue and R. Wilhelm, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 2014, 69, 596 CrossRef CAS; (d) P. A. Gokturk, S. E. Donmez, B. Ulgut, Y. E. Türkmen and S. Suzer, New J. Chem., 2017, 41, 10299 RSC; (e) L. Di Marco, M. Hans, L. Delaude and J.-C. M. Monbaliu, Chem. – Eur. J., 2016, 22, 4508 CrossRef CAS PubMed; (f) F. Mazars, M. Hrubaru, N. Tumanov, J. Wouters and L. Delaude, Eur. J. Org. Chem., 2021, 2025 CrossRef CAS; (g) A. R. Katritzky, D. Jishkariani, R. Sakhuja, C. D. Hall and P. J. Steel, J. Org. Chem., 2011, 76, 4082 CrossRef CAS PubMed.
- (a) T. F. Beltrán, G. Zaragoza and L. Delaude, Dalton Trans., 2017, 46, 9036 RSC; (b) T. F. Beltrán and L. Delaude, J. Cluster Sci., 2017, 28, 667 CrossRef; (c) D. Elorriaga, B. Parra-Cadenas, P. Pérez-Ramos, R. G. Soengas, F. Carrillo-Hermosilla and H. Rodríguez-Solla, Crystals, 2023, 13, 1304 CrossRef CAS.
- L. L. Borer, J. V. Kong, P. A. Keihl and D. M. Forkey, Inorg. Chim. Acta, 1987, 129, 223 CrossRef CAS.
- S. Naeem, L. Delaude, A. J. P. White and J. D. E. T. Wilton-Ely, Inorg. Chem., 2010, 49, 1784 CrossRef CAS PubMed.
- U. Siemeling, H. Memczak, C. Bruhn, F. Vogel, F. Träger, J. E. Baio and T. Weidner, Dalton Trans., 2012, 41, 2986 RSC.
- (a) C. Ratia, V. Cepas, R. Soengas, Y. Navarro, M. Velasco-de Andrés, M. J. Iglesias, F. Lozano, F. López-Ortiz and S. M. Soto, Front. Microbiol., 2022, 13, 815622 CrossRef PubMed; (b) C. Ratia, S. Sueiro, R. G. Soengas, M. J. Iglesias, F. López-Ortiz and S. M. Soto, Antibiotics, 2022, 11, 1728 CrossRef CAS PubMed; (c) C. Ratia, V. Ballén, Y. Gabasa, R. Soengas, I. A. Velasco-de, M. J. Iglesias, Q. Cheng, F. Lozano, E. S. J. Arnér, F. López-Ortiz and S. M. Soto, Front. Microbiol., 2023, 14, 1198473 CrossRef PubMed.
- R. Navarro and E. P. Urriolabeitia, J. Chem. Soc., Dalton Trans., 1999, 4111 RSC.
- R. G. Pearson, Inorg. Chem., 1973, 12, 712 CrossRef CAS.
- (a) A. L. Thompson, L. Delaude and J. D. E. T. Wilton-Ely, Chem. – Eur. J., 2010, 16, 1097 Search PubMed; (b) T. F. Beltrán, G. Zaragoza and L. Delaude, Polyhedron, 2021, 197, 115055 CrossRef; (c) M. Z. Aldin, G. Zaragoza, W. Deschamps, J.-C. D. Tomani, J. Souopgui and L. Delaude, Inorg. Chem., 2021, 60, 16769 CrossRef PubMed.
- F. H. Allen, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, in International Tables for Crystallography, ed. E. Prince, Springer, Berlin, 2006, vol. C, pp. 790–811 Search PubMed.
- J. Hwang, P. Li, M. D. Smith, C. E. Warden, D. A. Sirianni, E. C. Vik, J. M. Maier, C. J. Yehl, C. D. Sherrill and K. D. Shimizu, J. Am. Chem. Soc., 2018, 140, 13301 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- (a) A. Djeković, B. Petrović, Ž. D. Bugarčić, R. Puchtab and R. van Eldik, Dalton Trans., 2012, 41, 3633 RSC; (b) H. F. Dos Santos, D. Paschoal and J. V. Burda, Chem. Phys. Lett., 2012, 548, 64 CrossRef CAS.
- (a) H.-M. Ko, K. K.-Y. Kung, J.-F. Cui and M.-K. Wong, Chem. Commun., 2013, 49, 8869 RSC; (b) J. Rodriguez, A. Zeineddine, E. D. Sosa Carrizo, K. Miqueu, N. Saffon-Merceron, A. Amgoune and D. Bourissou, Chem. Sci., 2019, 10, 7183 RSC.
- (a) N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159 CrossRef CAS PubMed; (b) A. Dorababu, RSC Med. Chem., 2020, 11, 1335 RSC; (c) D. Sarkar, A. Amin, T. Qadir and P. K. Sharma, Open Med. Chem. J., 2021, 15, 1 CrossRef; (d) N. G. Bajad, S. K. Singh, S. K. T. D. Singh and M. Singh, Curr. Res. Pharmacol. Drug Discov., 2022, 3, 100119 CrossRef PubMed; (e) P. A. Pacheco and M. M. Santos, Molecules, 2022, 27, 319 CrossRef CAS PubMed.
- S. Bhuvaneswari, M. Jeganmohan and C.-H. Cheng, Chem. – Eur. J., 2007, 13, 8285 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2346965–2346967. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01184h |
| This journal is © The Royal Society of Chemistry 2024 |