
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic routes and chemical structural analysis for guiding the strategies on new Pt(II) metallodrug design
Francisco
Aguilar Rico†
 a,
Maryam
Derogar†
a,
Leticia
Cubo
a and
Adoracion G.
Quiroga
*ab
a,
Maryam
Derogar†
a,
Leticia
Cubo
a and
Adoracion G.
Quiroga
*ab
aInorganic Chemistry Department, C/Francisco Tomás y Valiente, 7. Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: adoracion.gomez@uam.es
bIadChem, Institute for Advance Research in Chemistry, Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 6th August 2024
Abstract
Metals in medicine is a distinct and mature field of investigation. Its progress in recent times cannot be denied, as it provides opportunities to advance our knowledge of the properties, speciation, reactivity and biological effects of metals in a medicinal context. The development of novel Pt(II) compounds to combat cancer continues to make valuable contributions but it has not yet achieved a complete cure. The chemistry of this field is basic for drug design improvements and our analysis of the chemical procedures is a practical tool for achieving effective Pt(II) anticancer drugs. We present chemical approaches in a manner that can be used to strategically plot new synthetic routes choosing right pathways. Clarifying the chemical challenge will help the scientific community to be aware of the ease and/or difficulty of the procedure before and after further studies, such as speciation, reactivity and biological action which are also very arduous and costly. The work provides information to tackle many challenges in chemistry, combining the knowledge on the Pt(II) reagent preparation together with the reactivity of the biological units used in the Pt(II) drug design. We discuss and include the description of the chemical reactions, the importance of multiple steps and the right order of such reactions to achieve the final drugs, analyzing the coordination principles as well as the organic and organometallic basis. This thorough study of the routes helps to detect the simpler or more complicated reactivity and will serve to improve the synthesis performance with possible post-modifications.
Introduction
Multidisciplinary research leads us towards a more complete understanding of the many challenges in science. Metals in medicine is one of the best examples of interdisciplinary topics which benefit from the fields of chemistry, biology, physics and medicine.1 These branches of knowledge, all together, help to understand the mechanism of metabolites and metallodrugs at both the molecular level and living systems. Finding new metallodrugs implies performing difficult procedures from those diverse fields that are not common and their access is sometimes not straightforward in the long list of methods and publications.2On many occasions, the chemistry of metallodrugs is enshrouded in the numerous publications dealing with their mechanism of action. The chemistry of this field is basic for the improvement of drug designs using the structure–activity relationships of drug libraries. The analysis of the chemical procedures is then a practical tool for achieving new metallodrug designs, as we can use it to strategically plot new routes choosing right pathways. For example, performing the metal coordination to the ligand before or after the final synthesis can be the critical step in achieving a more efficient procedure.
Cisplatin and its second-generation analogues are a landmark of metals in medicine, as they are still the most used anticancer drugs in clinics. The chemical variations in these classic metallodrug structures are still ongoing, driving a new impulse in the last few years with the use of innovative designs and therapies.3 Many examples of Pt(II) antitumor drugs have been reported over these years, from nonconventional structures to those using combined therapies and targeting strategies.
Objectives and progress of this perspective
In this work, we aimed to revise the synthetic approaches used in the preparation of Pt(II) drugs with ligands bearing Biological Units (BUs). Chemical procedures vary dramatically when using organic compounds to when using organometallic or small metal ion sources. In 2016, Lippard et al. reviewed the structure–activity relationship for Pt(II) complexes, selecting those works where the mechanism studies were more advanced and completed which indicates clinical progress. These authors included modern formulations of Pt(II) agents such as targeted agents, nanoparticle-mediated drug delivery and Pt(IV) prodrugs.4 Instead, this review summarizes and analyzes by strategies the chemical routes used in these examples and updates those newly released.We define Biological Units, BUs, as those groups with a defined role in the biological system, such as intercalators, steroids or peptides which potentially can help to target DNA or its synthesis pathways. The reported synthetic routes of the metallodrugs are arranged into two subsections considering if the Pt(II) ion is integrated into the BU ligand from a classical starting material (route 1) or from a known active core (route 2).
We analyze the experimental data and methods to help the reader understand the progress of the synthetic procedures and the importance of their correct description for others to reproduce the work. Furthermore, we discuss the results and based on this analysis, the reader could foresee how easy or difficult any new possible functionalization would be.
Biological Units (BUs) being intercalating agents
Intercalating agents are those compounds which can bind non-covalently to DNA bases and consequently induce cell death through different mechanisms. Most of these examples’ objective is to synthesize effective drugs based on a possible synergism of the Pt(II) drugs and the intercalating agents.The most common Pt(II) classical precursors are iodido complexes, K2PtCl4 and cis-[PtCl2(DMSO)2]. In 2013, Wilson and Lippard reviewed the preparation of Pt(II) complexes describing these starting materials. They outlined the formation of cis-[PtI2L2] complexes through [PtCl3L]− or tetraiodido intermediates, defining the preparation of cis complexes as well like cis-[PtI2LL′] using iodido-bridged dimers and HClO4 treatment, where L stands for a N-donor ligand.5 Iodido complexes are intermediates for the synthesis of cisplatin, carboplatin and oxaliplatin, due to their higher reactivity. They were long rejected as antitumoral drugs until some research groups profited from the higher reactivity of the Pt–I bond and started to reevaluate their biological action. Many of these results manifest significant differences in their mechanism of action and since then a plethora of articles has been published. This myriad has been organized into a solid review article,6 where the complexes are classified according to the oxidation stage, the formula and the donor atoms of the ligand. We considered only hypoxanthine7 and benzimidazole derivatives8 as BUs, because they prompted biological response, and their synthesis is just a direct substitution on [PtI42−] by the chosen ligand or dinuclear formation of an iodido bridge. The cleavage of these dimers affords the desired complex with retention of the configuration. The most singular chemistry was the access of bimetallic Pt carbenes with polyamine linkers through oxidation of NHC-Pt0 species stabilized by divinyldisiloxane with I2, but no BUs were used in these models9 (NHC being a N-heterocyclic carbene).
The examples of this section are depicted in Fig. 1 and are organized by entries which authors are named in the discussion. The first entry is the work performed by Murray et al. in which they studied the DNA binding properties of four 9-aminoacridinecarboxamide-Pt(II) complexes.10 The authors synthesized the linkers by protecting diethylenetriamine (dien) first with one equivalent of ethyl trifluoroacetate to allow the protection of one NH2 group and the remaining amino groups are protected with an excess of Boc2O. Then, the trifluoroacetyl moiety is orthogonally cleaved employing ammonia yielding an amino group which renders an amide [Fig. 1 entry 1(i)] upon reaction with the acyl chloride derivative of 9-chloroacridine. The chloride bound to acridine is substituted through SNAr (aromatic nucleophilic substitution) using a nitrogen nucleophile (ammonia or ethanolamine), and the Boc group is removed with an acid. Finally, K2PtCl4 reacts in stoichiometric amounts with the final ligands to form a complex [Fig. 1 entry 1(ii)].
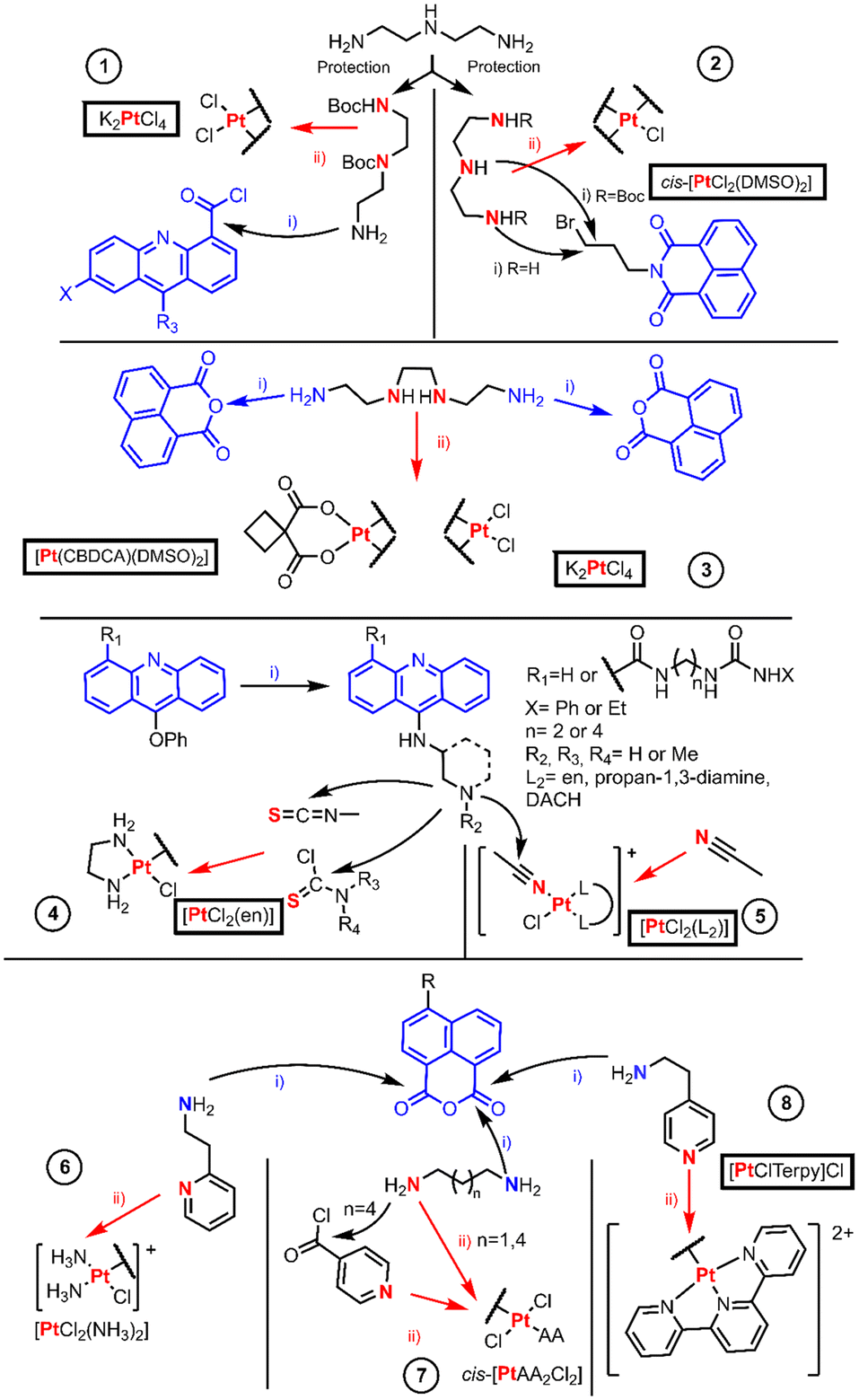 | ||
| Fig. 1 Synthetic routes used in the BUs being intercalating agents section. Pt(II) coordination reaction arrows and their donor atoms are shown in red and intercalator BU fragments are shown in blue. Pt(II) starting materials are boxed. | ||
Davey et al. designed two novel intercalator-Pt(II) compounds, in which only one chloride leaving group is present (Fig. 1 entry 2).11 The authors used N-(3-bromopropyl)naphthalimide with dien for the linear compound or dien diprotected with Boc at the terminal amino groups, so that the central N is the most prone to be alkylated. When the triamine linker is obtained, ligand exchange substitution takes place using cis-[PtCl2(DMSO)2].
Most authors have reported cis-[PtCl2(DMSO)2] synthesis as a “straightforward” method from the K2PtCl4 reaction with an excess of DMSO.12 However, other authors are more aware of the fact that a cis–trans isomerization can take place not only along its synthesis method but also when using it as a precursor in other reactions.13 Thus, its chemical behavior, its equilibria and potential speciation should be kept in mind before designing any synthetic route. For example, an alternative pathway to avoid the speciation is the use of silver salt reagents, Wilson's review describes methods in which both chloride leaving groups of cis-[PtCl2(DMSO)2] are substituted by a bidentate oxygen ligand, generating [Pt(DMSO)2(O2-Chel)] as a more reliable precursor for substitution reactions.5
The classical starting material, K2PtCl4, reactivity usually proceeds by coordination at the most favorable donor atom of the ligand. This binding is preferential on the N of the amine. When more than one amine is present, the non-selected position should be properly protected to avoid mixtures. This reactivity trend can also be observed in some examples where the reagent is an active Pt(II) core that also affords quick coordination.
The following examples lay in the frontier of the subsections appointed in this review article. The authors used, in the same publication, the classical platinum reagents and active Pt(II) fragments. At this hybrid point, cis-[Pt(CBDCA)(DMSO)2], where CBDCA is 1,1-cyclo-butane-dicarboxylate, is a good example, because although not reported as active its structure resembles carboplatin.
In 1999, Navarro-Ranninger et al. reported two novel Pt-bis-(naphthalimide) complexes different from each other in terms of leaving groups, being 1,1-cyclobutane dicarboxylate (CBDCA) or chloride, respectively [Fig. 1 entry 3]. The complexes were obtained by direct reaction of Pt(II) reagents including K2PtCl4 and cis-[Pt(CBDCA)(DMSO)2] with the selected bis(naphthalimide), where the Pt(II) ion binds to the two central N-donor atoms of the alkylamino linker.14
Bierbach et al. are well known for their efforts to develop mono-ionic Pt(II) acridine derivatives. Their early work was compiled by Lippard, but here we summarized their synthetic procedures, expanding information on the synthetic approaches in their most recent work (Fig. 1 entry 4). On their first publication, they synthesized {[Pt(ACRAMTU)Cl(en)](NO3)2}, where ACRAMTU is 1-[2-(ACRidin-9-ylAMino)ethyl]-1,3-dimethylThioUrea. The author used [PtCl2(en)] (en = ethane-1,2-diamine) as the starting material in a substitution reaction of one of the chloride leaving groups by the thiourea sulfur atom of the ACRAMTU.15 Later, they changed systematically the Pt-ACRAMTU structure by varying the acridine moiety16 or the thiourea linker.17 Such variations were performed always before the coordination to Pt(II) through key SNAr on the acridine employing amines. The formation of the thiourea is then straightforward by reacting with an isothiocyanate. As no advantage over these results was obtained, they replaced the thiourea connector by an amidine group.18 The key step for the obtention of this connection was the nucleophilic addition of an amine to a nitrile; the electrophilic character of this nitrile is enhanced when it is coordinated to Pt(II).
Bierbach's most recent work presents a new family to their monovalent Pt(II) library (Fig. 1, entry 5).19 They obtained these compounds by nucleophilic addition of aminoacridines to a Pt(II) coordinated acetonitrile, which was obtained after the chlorido replacement with AgNO3. Murray et al., discussed in the previous section,10 employed a similar method to Bierbach's strategy which involved the use of a diamine linker to achieve these functionalized acridine compounds. The selective functionalization of an amine group relies on the protection of the remaining reactive sites. These linkers were en and two sets of enantiopure 3-aminopiperidine and 3-aminopyrrolidine.
Yan et al. obtained the first example in this section, and this approach uses cisplatin with a stoichiometric amount of AgNO3 for the release of one of the chlorides affording AgCl as the secondary product. The formation of the cisplatin–DMF intermediate finally renders a cationic monofunctional Pt(II) complex, cis-[PtCl(NH3)2(NP)]NO3, and the ligand (NP, N-(2-ethylpyridine)-1,8-naphthalimide) was synthesized by a condensation reaction, Fig. 1 entry 6.20 Many authors have used this metathesis procedure, as it is a very common strategy to replace a halogen with silver salts.
The former common belief regarding Pt(II) drugs was that trans isomers could not exhibit antitumoral action as cisplatin because of the lack of activity of transplatin.21 However, in the eighties, several trans-Pt(II) complex examples were found to be cytotoxic towards cisplatin-resistant cell lines.5,22,23 The common observation was that upon controlling the aquation rate of the chlorido ligands by employing bulkier non-leaving ligands, the complexes became more active.24
Quiroga et al. reported specific intercalating naphthalimide compounds with different linkers using as an active core a trans-Pt(II) complex with aliphatic amines (AA) such as dimethylamine (dma) or isopropylamine (ipa) (Fig. 1 entry 7). The linkers were either alkylamines of different lengths (hexane-1,6-diamine and propane-1,3-diamine) that underwent condensation with naphthalic anhydride to render alkylamine naphthalimides (step ii) or a pyridine introduced to N-3-aminoproyl-naphthalimide by acylation with isonicotyl chloride (step ii). The ligands reacted with cis-[Pt(AA)2Cl2] to form a cationic tetramine species that after HCl treatment results in a trans-configured isomer.25
The last entry (8) from Fig. 1, a monofunctional Pt(II) complex, was synthesized by Banerjee et al. using a silver-aided substitution reaction between the active Pt(II) core [PtCl(terpy)]Cl and 4-N,N-dimethylamino-1,8-naphthalimide synthesized through condensation. This synthetic procedure rendered a water-soluble cationic complex with nitrate as a counterion.26
Steroidal ligands as targeting BU units
Traditional drug design focuses on biological targets where specific receptors or biomarkers are overexpressed by cancer cells, for example steroid hormones. Estrogens or progesterone are known to induce HMG1 overexpression and sensitize breast cancer cells to cisplatin and carboplatin.26 Hormone-dependent cancers share the same feature which is the overexpression of androgen, estrogen and progesterone receptors.26 Steroids are widely used in research as anticancer agents alone or combined with actual clinical drugs to potentiate them. Here we present the synthesis of the complexes obtained either from Pt(II) precursors or Pt(II) active cores.Bérubé et al. synthesized a series of compounds based on a chelated aminoethylpyridine Pt-estradiol. These series are designed to link the estradiol (16 position R in Fig. 2 entry 1) to one aminoethylpyridine through an alkyl or PEG (polyethyleneglycol) chain (Y). In addition, the series includes an example with a methylenehydroxy group (–CH2OH) at the 16-position. The syntheses started with an initial protection of the phenolic alcohol of estrone with either benzyl bromide (Pg is Bn) or tetrahydropyran (Pg is THP) followed by the introduction (step i) of methoxycarbonyl at the 16-position with NaH and dimethylcarbonate (step ii). This generates an acidic 1,3-ketoester which reacts upon basic treatment (step iii) with the diorganohalide when the linker chain is CH2OH or with the halide of the THP-protected alcohol when R = H.
 | ||
| Fig. 2 BU steroid targeted ligands and complex structures and their functionalization. The groups that participated in Sonogashira couplings are highlighted in blue. Coordination reaction arrows and their donor atoms are shown in red. Pt(II) starting materials are boxed. | ||
When X is OTHP, an aqueous treatment with LiCl (step iv) prompts the decarboxylation and the deprotection of the alcohol, which after an Appel reaction (with CBr4, PPh3) afforded the organohalide (step v). Step vi consists of a reduction reaction using a hydride donor (LiBH4 or LiAlH4), followed by the deprotection of the estradiol phenol (step vii, including X and Y), achieved either by an acidic treatment in ethanol (Pg is THP) or by Pd/C catalyzed hydrogenolysis (Pg is Bn). Finally, a nucleophilic substitution of the estradiol derivative was carried out employing 2-aminoethylpyridine (step viii), whereupon coordination to Pt takes place using K2PtCl4 (step ix).28
Osella et al. used a Sonogashira reaction in which an aryl halide bearing protected ethylenediamine group was linked to 17α-ethynylestradiol (Fig. 2 entry 2). The platination of the alkyl amine tail took place in the presence of K2PtCl4.29
Hannon et al. developed a variety of metallo-estrogen drugs based on 17α-ethynylestradiol (Fig. 2, entry 3). Of all the metal centers used in their work, we only focus on the synthetic route using Pt(II) starting materials, [PtCl2(PhCN)2] and [PtCl2(cod)] (cod = 1,5-cyclooctadiene). The authors prepared three different tridentate chelating ligands using a Sonogashira coupling reaction between ethynylestradiol and the linkers L1, L2 and L3. The reaction of the resulting ligand L1 with [PtCl2(PhCN)2] afforded a cationic [PtCl(L1)]Cl complex, whereas ligand L2 using the same method gave a mixture of products. However, the authors could achieve [PtCl(L2)]Cl by changing the solvent. Ligand L3 synthesis turned out to be more complicated, and the optimal procedure implied the base hydrolysis of the diethyl ester, whereas the other routes required tedious purifications. All the ligands coordinate to the Pt(II) to form a tridentate chelator with one chloride leaving group; the L3 derivative was discarded due to its poor solubility.30
Ghedini et al. designed a series of steroid-Pt(II)-o-catecholato complexes (Fig. 2 entry 4) from derivatives of estrone, estradiol and testosterone either functionalized in 17α or 17β.31 The Pt(II) reagent used bis-phosphine-Pt(II)-o-catecholato bearing an amine or a carboxylic group, which was obtained after the reaction of cis-[PtCl2(PPh3)2] with the corresponding catechol in a KOH medium. The carboxylic group either in the Pt(II) fragment or the steroid structure was activated employing NHS and DCC; nonetheless, they obtained very low yields of the final Pt(II) derivatives. The authors tested the use of isobutyl chloroformate as an activating reagent which did not improve the yield due to the formation of side products, such as the carbamate of the amine. They found that the most challenging activation was that of the α-aryl carboxylic group, which led to a mixture of compounds. The major product was the mixed anhydride. The authors made many attempts using harsher conditions, but the outcome of the reaction did not improve. However, they succeeded in avoiding the coordination of bromide to the Pt(II) core and the displacement of the catecholate, with a commercial alkylamino hydrobromide previously treated with a base.
Bérubé et al. investigated the biological activity of carboplatin and oxaliplatin analogs using an aminoethylpyridine chelated Pt-estradiol from a previous entry (Fig. 2, entry 1).32 To achieve the final analogs, the silver salts of CBDA and oxalic acid were used to prompt the replacement of the chlorido ligands by the corresponding carboxylate chelates.
Hannon et al. also reported an example using an active Pt core. They studied the testosterone conjugated to a monovalent diammino N-heteroaryl Pt(II) core (cis and trans isomers) (Fig. 2, entry 5).33,34 Sonogashira couplings were used to link ethisterone and N-heteroarenes, and to achieve these Pt(II) cores, they employed either cis or trans-platin with a stoichiometric amount of AgNO3 to displace only one chlorido ligand that renders the final testosterone conjugated ligand.
BUs based on therapeutic peptides for targeting
Introducing therapeutic peptides and their varying methods of action against tumor cells is a formidable example of targeting tumors.35 The so-called active targeting peptides are recognized by some receptors on the surface of tumor cells that are usually overexpressed.36 The selective metalation of peptide derivatives is a very challenging matter, as they possess multiple and diverse donor atoms suitable for the binding of Pt(II) fragments. Many examples use chelators like diamines or dicarboxylates as a proficient platform for the selective metalation of peptide derivatives.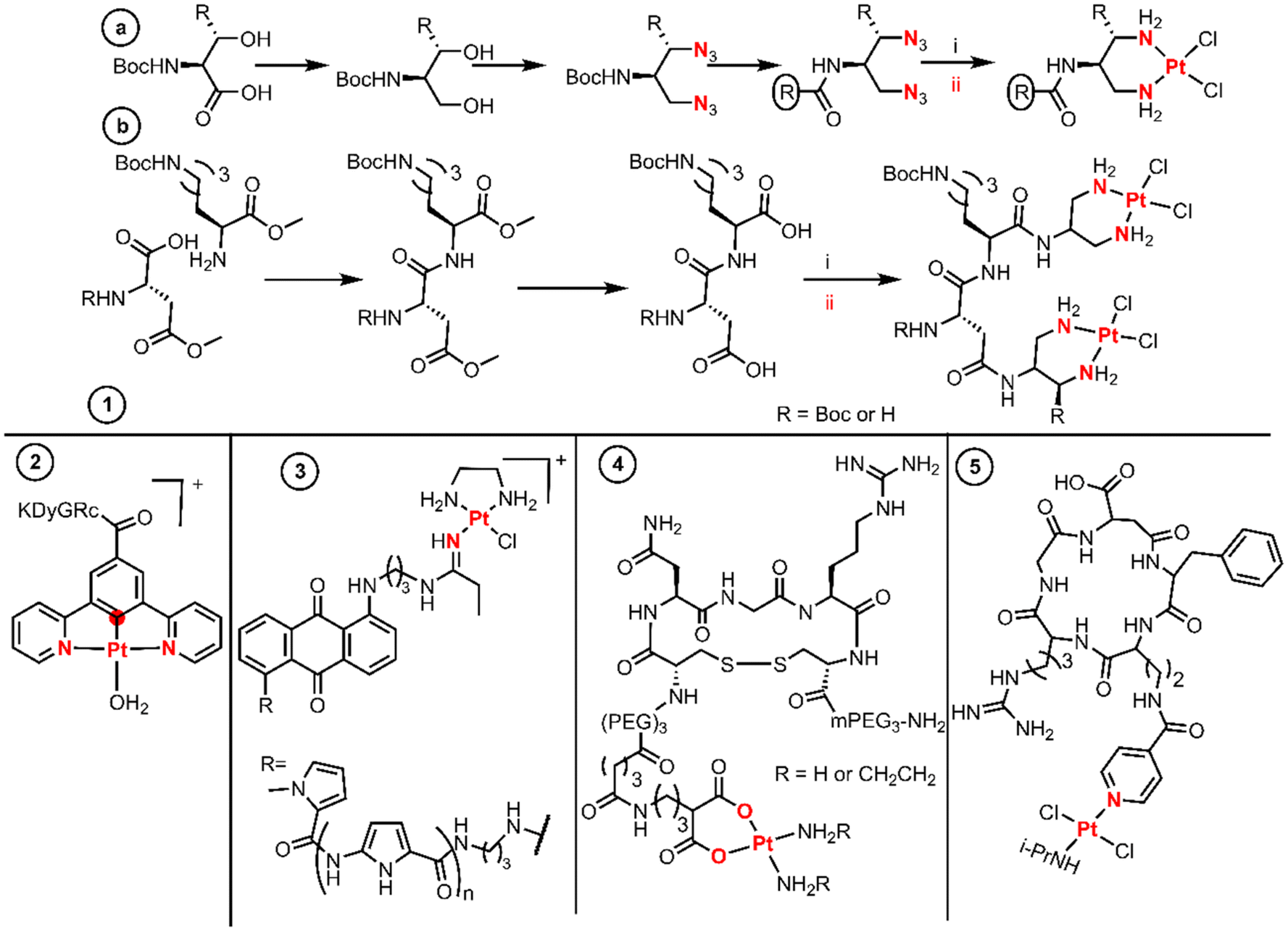 | ||
| Fig. 3 Synthetic routes and structures of BUs as peptides. Both coordination reaction steps and donor atoms are indicated in red. | ||
The diazide–peptide derivatives are finally reduced by hydrogenolysis, and the diamines react with K2PtCl4 leading to substitution by direct displacement and thereby generating the final divalent complexes. The authors used an extension of this methodology to achieve dinuclear Pt(II) complexes linked by a peptide sequence (Fig. 3 entry 1b). First, they performed peptide coupling between two protected amino acids. Then a LiOH solution hydrolyses the methyl esters on the C- and N-terminal residues affording the diacid species which condense twice with the same amine. The dinuclear Pt(II) complexes are finally achieved with a stoichiometric amount of K2PtCl4 by a substitution reaction.
Sarli et al. conjugated a monofunctional [N,C,N-Pt(II)] complex with the cyclic RGDyK peptide through an amide (Fig. 3, entry 2).38 The objective of this design was to develop a system for monitoring drug release and promising anticancer activity for photodynamic therapeutic applications. The synthetic route started with the Pd-catalysed borylation of methyl 3,5-dibromobenzoate with HBPin followed by a Suzuki–Muyaura coupling with chloropyridine catalyzed by Pd(OAc)2, PPh3 and Na2CO3, yielding methyl 3,5-di(pyridine-2-yl)benzoate. Subsequently, a substitution by direct displacement and cycloplatination was performed using K2PtCl4 followed by methyl ester hydrolysis. The Pt(II)–acid complex coupled with the lysine amino group of the c(RGDyK) peptide through EDC and NHS activation (Fig. 3, entry 2).
Hambley et al. prepared monofunctional Pt(II) complexes aiming to incorporate an anthraquinone intercalator and a minor groove pyrrole peptide binder into a Pt(II) complex (Fig. 3, entry 3).39 To achieve this design, the substituted anthraquinones were first coupled to pyrrole lexitropsins (polyamides) through a peptide bond, after which nucleophilic addition of the aminoalkyl anthraquinone to the coordinate nitrile ligand of the precursor [PtCl(en)(NCEt)]Cl was performed using the same procedure used by Bierbach (called the “click” reaction by the authors). The syntheses of the initial polyamides were performed using a solution phase peptide method in which the protected groups were Boc or methyl esters. The peptides were deprotected in basic media while the free amines were achieved using standard acidic media.
The last example of this section proves that the chelate platform is not essential for the conjugation of Pt(II) cores to peptides. Quiroga et al. isolated trans-Pt(II) compounds with isopropylamine trans to a picolinic acid and further functionalized it with the cyclic peptide cRGDfK (Fig. 3, entry 5).41
Including intercalating BUs within heterometallic structures
The designs with BUs aim to reduce side effects, overcome cisplatin resistance and improve the efficacy of Pt(II) drugs. Some authors have gone beyond a single metal center, introducing heterometallic complexes which potentially unchain dual mechanisms, combining the properties of both metallic cores. The chemistry to achieve heterometallic-based drugs is subject not only to the functionality of the organic moieties but to the reactivity of the metallic cores too. There are many examples of heterometallic with Pt(II) strategies, nicely compiled in a recent review.42 For our purpose, we have selected only the metallointercalator approaches.Ir, Rh and Ru octahedral complexes are the more commonly found intercalating derivatives bound to Pt(II). Fig. 4 encompasses the articles in which the chemistry has been proficiently reported, where the first example (entry 1) is an octahedral Ru(II)-bpp complex bound to Pt(II) where bpp is 2,3-di(pyridin-2-yl)pyrazine.43 The bpp ligand is introduced by chlorido substitution onto [RuCl2(phen)2] (phen = 1,10-phenanthroline) and further platination with cis-[PtCl2(DMSO)2].
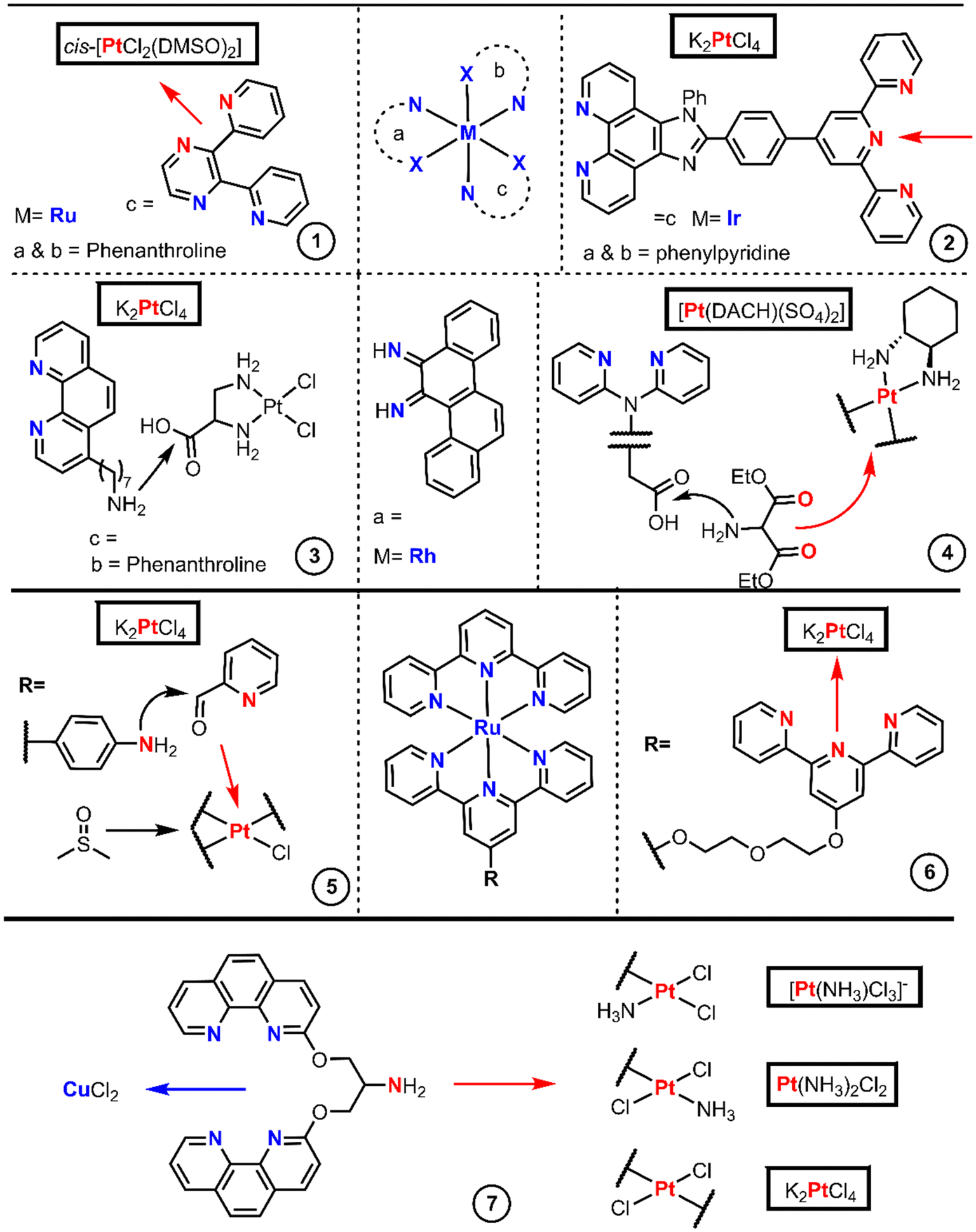 | ||
| Fig. 4 Synthetic routes and structures of heterometallic complexes bearing intercalator motifs. Coordination reaction arrows to Pt and their donor atoms are shown in red and to the other metal ions are shown in blue. Pt(II) starting materials are boxed. | ||
The use of N-donor connectors is not exclusive to octahedral Ru-based intercalators, Chao's group employed a similar strategy.44 They used a bridge splitting reaction with the [Ir2(μ-Cl)2(ppy)4] reagent, which implies the replacement of the two chlorides by a terpyridine-containing phenanthroline (terpy) ligand (Fig. 4 entry 2). The final octahedral Ir(I) complex was also platinated with cis-[PtCl2(DMSO)2].
Chrysi, chrysene-5,6-diimine, is an intercalator ligand used by Barton et al.45 to afford mismatch-specific metallointercalators and is presented in the center of Fig. 4. These authors first synthesized the Rh(III) complex core [Rh(chrysi)(NH3)2(phen)]Cl3, and later replaced the ammonia by a phenanthroline ligand which bears a heptylamino group. Then, they utilized the amino group to couple the Rh(III) complex to the Pt(II) core through EDC-mediated amide formation with [PtCl2(en-COOH)], generated by the direct chlorido substitution of K2PtCl4 (Fig. 4 entry 3). Barton et al. modulated their previous structure, in which the Rh(III)-Chrysi couple remains and the phenanthroline-type ligands are replaced by di(pyridin-2-yl)amine, one of which contains an acetic-2-yl acid bound to the amino-N (Fig. 4 entry 4).46 This heteroleptic Rh(III) core was synthesized analogously to their former example, in which they replace the amino ligands in [Rh(chrysi)(NH3)2(phen)](TFA)3 with di(pyridin-2-yl)glycine. The carboxylic acid acts as a linker, and it is then functionalized with diethyl aminomalonate. The removal of the ethyl chains with aqueous basic treatment renders the dicarboxylic acid providing a binding site for Pt(II). Ba(OH)2 treatment gave the Ba carboxylate, which afforded the final DACH-Pt-carboxylate complex upon reaction with a solution of [PtDACH(H2O)2]SO4.
The Reedijk group obtained a similar example to entry 2 by the initial formation of a Ru(terpy)Cl3 core from RuCl3. Reedijk's group utilized one terpyridyl47 as a connector and spaced with a polyethyleneglycol chain from other terpyridyl groups, and later platinated with [PtCl2(cod)], reaching a Ru terpyridine moiety linked to a monofunctional Pt(II) core (Fig. 4 entry 6) followed by silver-promoted substitution of the chlorido ligands by another tridentate ligand. Smythe's group, on the other hand, utilized an aminoterpyridine,48 which enabled the reductive amination of picolinaldehyde creating a new bidentate domain which can be platinated with K2PtCl4 and later treated with DMSO yielding a monofunctional Pt complex (Fig. 4 entry 5).
Several Pt(II)–Cu(II) complexes based on 3-Clip-Phen [1-(1,10-phenanthrolin-3-yloxy)-3-(1,10-phenanthrolin-8-yloxy)propan-2-amine] were prepared by Reedijk's group.49 The authors started with (NBu4)[PtCl3(NH3)] and 3-Clip-Phen, resulting in a cis isomer, whereas the trans compound synthesis requires the initial activation of cisplatin by removing one chloride aided by AgNO3 followed by reaction with 3-Clip-Phen in the excess of HCl. A third complex, also trans-configured, which bears two 3-Clip-Phen units and two chloride ligands, was obtained by treating K2PtCl4 with AgNO3 and reacted with an excess of 3-Clip-Phen to give the final complex (Fig. 4 entry 7).
In summary, we have observed that the incorporation of a second metal center into Pt(II) complexes generally proceeds by reactions of robust fragments on both metal ion reagents. Pt(II) active cores (cisplatin like) give good intermediates through metathesis with AgNO3, but in the heterobimetallic syntheses, [PtCl2(cod)] and cis-[PtCl2(DMSO)2] are more widely used because they trigger substitution reactions more quickly and efficiently.
Modern chemistry, using click chemistry with Pt(II) and biological units
Modern chemistry approaches are fundamental for achieving biological selectivity among the Pt(II) drugs. Click chemistry has gained the attention of the field whether for its use for functionalizing the Pt(II) coordinated ligands or for gaining information about these complexes’ mechanism of action in cells.Farrer and Griffith published a current opinion article with examples up to 2020.50 These authors published the first example to develop a steroid linked ligand using click chemistry and then coordinating Pt(II) using cis-[PtCl2(DMSO)2] and cis-[Pt(CBDCA)(DMSO)2]. However, it is more common to find examples in the literature where the click chemistry takes place after the coordination to Pt(II) (Fig. 5) which are discussed as follows.
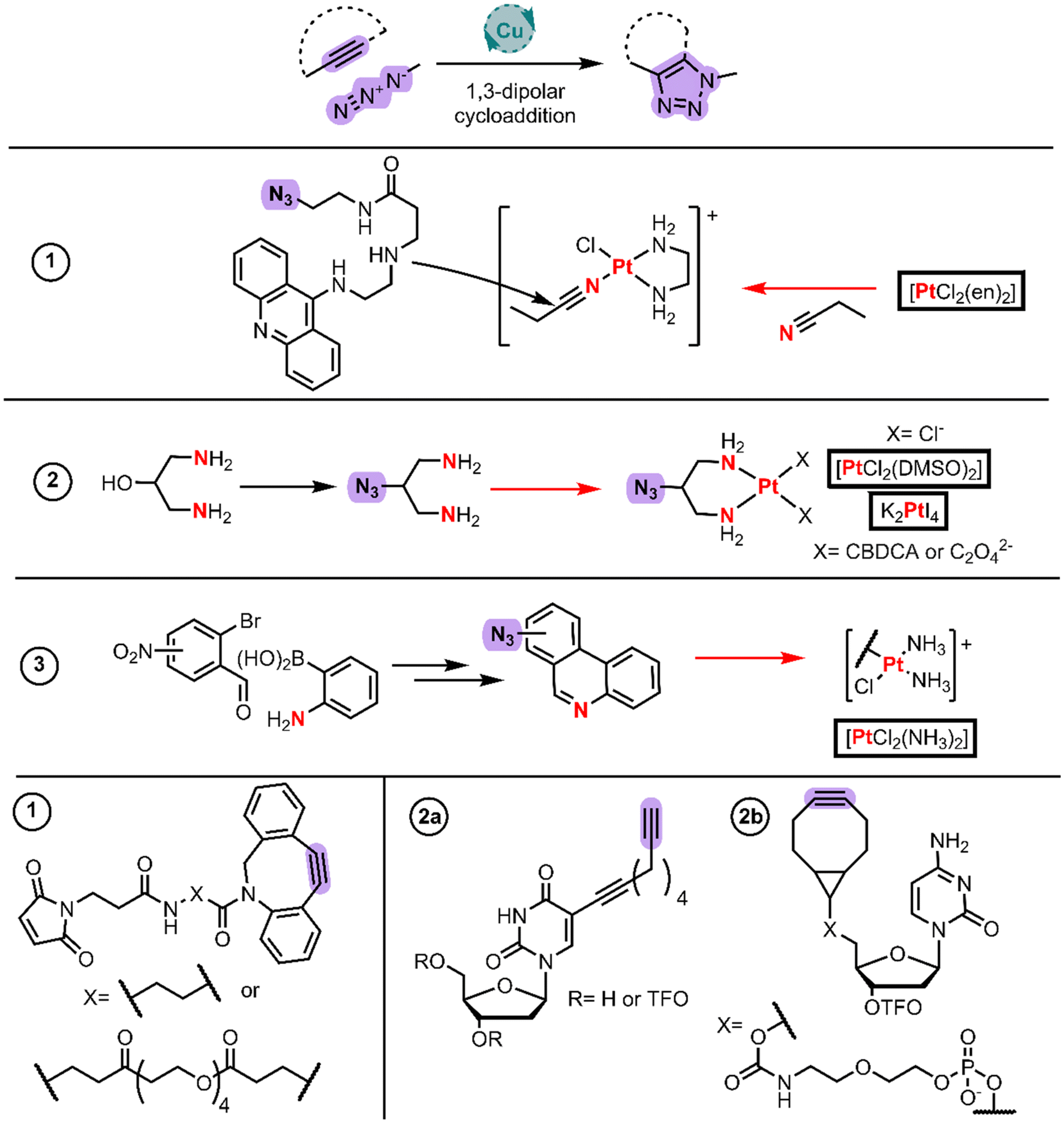 | ||
| Fig. 5 Synthetic routes and structures of click chemistry substrates in which participant groups are highlighted in purple. Coordination reaction arrows to Pt and their donor atoms are shown in red. Pt(II) starting materials are boxed. | ||
Bierbach et al. applied click chemistry to link cytotoxic Pt(II)–acridine complexes with a Michael acceptor to conjugate them with carrier proteins.51 They employed the same strategy described in the intercalator BU section (Fig. 1, entry 5), in which they initially synthesized the acridine-amine moiety that undergoes nucleophilic addition to the acetonitrile ligand bound to Pt(II). The selected amine was 3-((2-aminoethyl)amino)propanoic acid, which reacted through the terminal amino group with phenoxyacridine. The key step in this work was introducing a terminal azide group to the amine linker by CDI-assisted amide coupling to 2-azidoethan-1-amine, rendering the protected amino-azide-acridine derivative. After it was deprotected, the free amine derivative reacted with the electrophilic Pt–nitrile complex to form a cationic-Pt(II)-amidine-azide. This azide group enabled the authors to perform Cu-free cycloaddition chemistry with the alkyne (Fig. 5 alkyne 1) of the commercial maleimide-cyclooctyne derivatives.
Kellett et al. in 2022 took advantage of the click chemistry between a Pt(II) core functionalized with an azide and an alkyne bearing a triplex forming oligonucleotides (TFOs) (Fig. 5, entry 2).52 To reach the azide counterpart, they protected 1,3-diamino-2-propanol with Boc2O whereupon they mesylated the hydroxy group with MsCl in the presence of Et3N. This initial protection is crucial given the higher nucleophilic character of amines when compared to alcohols. The resulting mesylate reacted with sodium azide rendering the Boc-protected diamino chelate which, after the deprotection, rendered the hydrochloride salt of the ligand. They synthesized the cisplatin-like structure using cis-[PtCl2(DMSO)2] in DMF, whereas the carboplatin and oxaliplatin analogs were obtained by the reaction of the ligand with PtI42− (like cisplatin synthesis) through intermediates and AgNO3 metathesis explained with details in Wilson's review.5 On the other hand, to reach the alkyne counterpart, they synthesized the TFO with a nucleoside bearing a terminal alkyne or cyclooctyne (Fig. 5 alkyne 2a and 2b). The final step is dipolar cycloaddition affording the conjugated compounds.
More research seems to be still ongoing, the proof of which is the last work from ref. 53 with phenanthriplatin analogues functionalized with an azide group (Fig. 5, entry 3). They synthesized a phenanthroline functionalized with a nitro group by an initial tandem reaction. The tandem reaction consists of both a Schiff base condensation reaction and a Suzuki coupling reaction between 2-aminophenylboronic acid and 3-, 4- or 5-nitro-2-bromobenzaldehydes. These nitrophenanthrolines are then reduced to amines using Fe(0) and NH4Cl in aqueous MeOH. Nitrous acid treatment followed by the addition of sodium azide renders the phenanthroline azide through SNAr. These phenanthroline derivatives react with cisplatin by chlorido substitution to isolate the monofunctional complex with the aid of AgNO3. They have generated a very attractive example to further develop new libraries of compounds with click chemistry.
Conclusions
In conclusion, we would like to highlight the importance of chemical procedures in the design of metallodrugs. The chemical strategy of the metallodrug synthesis can be a vital step for its development and the optimization can make the process sustainable. Such optimization can benefit from the comparison of the similarities in synthetic parameters such as metal sources, solvents, substituents, protecting groups and linkers, which are common in the many steps within the reactions reported in the literature. We have found a broad variety of routes utilized in these syntheses, from amide formations to nucleophilic aliphatic substitutions, alkaline addition, hydrolysis of amides (protecting groups), reactions of acyl chlorides of alcohols and/or derivatives of amines, decarboxylation and a long list that includes metal assisted coupling reactions. The exploration of these reactivities brought to light two important observations:(a) When a classical Pt(II) reactant is used, the performance of the reactions is improved by selecting the platination position of the BU, protecting the rest of the coordinating sites that can be cleaved after the coordination takes place.
(b) When using an active core fragment, besides strategic protection, silver salts facilitate quick and orderly substitutions. The more modern chemistry takes place with a better yield using the organic substrate BU, but in the last few years organometallic chemistry (i.e. Sonogashira or Suzuki coupling), click chemistry and advanced peptide synthesis (among others) have proven that this methodology can be carried out in the presence of metal ion centers. Particularly, click chemistry goes beyond glassware experiments to conduct research inside the cell itself.
Data availability
The data supporting this article have been provided by Universidad Autonoma de Madrid licenses with access to scientific journals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from Spanish Agencia Estatal de investigacion: PID2019-106220RB100 and PID2022-137373OB-I00. F. A. R. is thankful for his PhD grant from Comunidad de Madrid: PIPF-2022/SAL-GL-25290.References
- K. J. Franz and N. Metzler-Nolte, Chem. Rev., 2019, 119, 727–729 CrossRef CAS PubMed.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- S. Rottenberg, C. Disler and P. Perego, Nat. Rev. Cancer, 2021, 21, 37–50 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- J. J. Wilson and S. J. Lippard, Chem. Rev., 2014, 114, 4470–4495 CrossRef CAS PubMed.
- P. Štarha, J. Vančo and Z. Trávníček, Coord. Chem. Rev., 2019, 380, 103–135 CrossRef.
- J. Vančo, Z. Trávníček, R. Křikavová, J. Gáliková, Z. Dvořák and M. Chalupová, J. Photochem. Photobiol., B, 2017, 173, 423–433 CrossRef.
- P. Štarha, J. Vančo, Z. Trávníček, J. Hošek, J. Klusáková and Z. Dvořák, PLoS One, 2016, 11, e0165062 CrossRef PubMed.
- M. Skander, P. Retailleau, B. Bourrié, L. Schio, P. Mailliet and A. Marinetti, J. Med. Chem., 2010, 53, 2146–2154 CrossRef CAS PubMed.
- H. W. Kava, W. Y. Leung, A. M. Galea and V. Murray, Bioorg. Med. Chem., 2021, 40, 116191 CrossRef CAS PubMed.
- E. Y. D. Chua, G. E. Davey, C. F. Chin, P. Dröge, W. H. Ang and C. A. Davey, Nucleic Acids Res., 2015, 43, 5284–5296 CrossRef CAS PubMed.
- J. H. Price, A. N. Williamson, R. F. Schramm and B. B. Wayland, Inorg. Chem., 1972, 11, 1280–1284 CrossRef CAS.
- J. H. Price, J. P. Birk and B. B. Wayland, Inorg. Chem., 1978, 17, 2245–2250 CrossRef CAS.
- J. M. Pérez, I. López-Solera, E. I. Montero, M. F. Braña, C. Alonso, S. P. Robinson and C. Navarro-Ranninger, J. Med. Chem., 1999, 42, 5482–5486 CrossRef.
- E. T. Martins, H. Baruah, J. Kramarczyk, G. Saluta, C. S. Day, G. L. Kucera and U. Bierbach, J. Med. Chem., 2001, 44, 4492–4496 CrossRef CAS PubMed.
- R. Guddneppanavar, G. Saluta, G. L. Kucera and U. Bierbach, J. Med. Chem., 2006, 49, 3204–3214 CrossRef CAS PubMed.
- M. C. Ackley, C. G. Barry, A. M. Mounce, M. C. Farmer, B.-E. Springer, C. S. Day, M. W. Wright, S. J. Berners-Price, S. M. Hess and U. Bierbach, JBIC, 2004, 9, 453–461 CrossRef CAS PubMed.
- Z. Ma, J. R. Choudhury, M. W. Wright, C. S. Day, G. Saluta, G. L. Kucera and U. Bierbach, J. Med. Chem., 2008, 51, 7574–7580 CrossRef CAS PubMed.
- S. Zhang, X. Yao, N. H. Watkins, P. K. Rose, S. R. Caruso, C. S. Day and U. Bierbach, Angew. Chem., Int. Ed., 2020, 59, 21965–21970 CrossRef CAS PubMed.
- Y. Guo, Y. He, S. Wu, S. Zhang, D. Song, Z. Zhu, Z. Guo and X. Wang, Inorg. Chem., 2019, 58, 13150–13160 CrossRef CAS PubMed.
- T. A. Connors, M. J. Cleare and K. R. Harrap, Cancer Treat. Rep., 1979, 1499–1502 CAS.
- L. R. Kelland, F. J. Barnard, I. G. Evans, B. A. Murrer, B. R. C. Theobald, S. B. Wyer, P. M. Goddard, M. Jones and M. Valenti, J. Med. Chem., 1995, 38, 3016–3024 CrossRef CAS PubMed.
- N. Farrell, T. T. B. Ha, J. P. Souchard, F. L. Wimmer, S. Cros and N. P. Johnson, J. Med. Chem., 1989, 32, 2240–2241 CrossRef CAS PubMed.
- A. G. Quiroga, J. Inorg. Biochem., 2012, 114, 106–112 CrossRef CAS PubMed.
- J. M. Herrera, F. Mendes, S. Gama, I. Santos, C. Navarro Ranninger, S. Cabrera and A. G. Quiroga, Inorg. Chem., 2014, 53, 12627–12634 CrossRef CAS PubMed.
- S. Banerjee, J. A. Kitchen, S. A. Bright, J. E. O'Brien, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Commun., 2013, 49, 8522–8524 RSC.
- J. Provencher-Mandeville, C. Descôteaux, S. K. Mandal, V. Leblanc, É. Asselin and G. Bérubé, Bioorg. Med. Chem. Lett., 2008, 18, 2282–2287 CrossRef CAS PubMed.
- J. Provencher-Mandeville, C. Debnath, S. K. Mandal, V. Leblanc, S. Parent, É. Asselin and G. Bérubé, Steroids, 2011, 76, 94–103 CrossRef CAS PubMed.
- E. Gabano, C. Cassino, S. Bonetti, C. Prandi, D. Colangelo, A. Ghiglia and D. Osella, Org. Biomol. Chem., 2005, 3, 3531–3539 RSC.
- A. Jackson, J. Davis, R. J. Pither, A. Rodger and M. J. Hannon, Inorg. Chem., 2001, 40, 3964–3973 CrossRef CAS.
- O. Gandolfi, M. Cais, G. Dolcetti, M. Ghedini and A. Modiano, Inorg. Chim. Acta, 1981, 56, 127–133 CrossRef CAS.
- P. Saha, C. Descôteaux, K. Brasseur, S. Fortin, V. Leblanc, S. Parent, É. Asselin and G. Bérubé, Eur. J. Med. Chem., 2012, 48, 385–390 CrossRef CAS PubMed.
- M. Huxley, C. Sanchez-Cano, M. J. Browning, C. Navarro-Ranninger, A. G. Quiroga, A. Rodger and M. J. Hannon, Dalton Trans., 2010, 39, 11353–11364 RSC.
- C. Sanchez-Cano, M. Huxley, C. Ducani, A. E. Hamad, M. J. Browning, C. Navarro-Ranninger, A. G. Quiroga, A. Rodger and M. J. Hannon, Dalton Trans., 2010, 39, 11365–11374 RSC.
- M. Xie, D. Liu and Y. Yang, Open Biol., 2020, 10, 200004 CrossRef CAS.
- M. Karami Fath, K. Babakhaniyan, M. Zokaei, A. Yaghoubian, S. Akbari, M. Khorsandi, A. Soofi, M. Nabi-Afjadi, H. Zalpoor, F. Jalalifar, A. Azargoonjahromi, Z. Payandeh and A. Alagheband Bahrami, Cell. Mol. Biol. Lett., 2022, 27, 33 CrossRef CAS.
- S. Kumbhakonam, K. Vellaisamy, S. Saroj, N. Venkatesan, K. D. and M. Kannoth Manheri, New J. Chem., 2018, 42, 2450–2458 RSC.
- T. Chatzisideri, S. Thysiadis, S. Katsamakas, P. Dalezis, I. Sigala, T. Lazarides, E. Nikolakaki, D. Trafalis, O. A. Gederaas, M. Lindgren and V. Sarli, Eur. J. Med. Chem., 2017, 141, 221–231 CrossRef CAS PubMed.
- A. T. S. Lo, N. S. Bryce, A. V. Klein, M. H. Todd and T. W. Hambley, JBIC, 2021, 26, 217–233 CrossRef CAS PubMed.
- M. W. Ndinguri, R. Solipuram, R. P. Gambrell, S. Aggarwal and R. P. Hammer, Bioconjugate Chem., 2009, 20, 1869–1878 CrossRef CAS PubMed.
- M. A. Medrano, M. Morais, V. F. C. Ferreira, J. D. G. Correia, A. Paulo, I. Santos, C. Navarro-Ranninger, A. A. Valdes, A. Casini, F. Mendes and A. G. Quiroga, Eur. J. Inorg. Chem., 2017, 2017, 1835–1840 CrossRef CAS.
- L. Ma, L. Li and G. Zhu, Inorg. Chem. Front., 2022, 9, 2424–2453 RSC.
- E. E. Aranda, T. A. Matias, K. Araki, A. P. Vieira, E. de Mattos, P. Colepicolo, C. P. Luz, F. L. N. Marques and A. M. daCostaFerreira, J. Inorg. Biochem., 2016, 165, 108–118 CrossRef CAS PubMed.
- C. Zhang, R. Guan, X. Liao, C. Ouyang, J. Liu, L. Ji and H. Chao, Inorg. Chem. Front., 2020, 7, 1864–1871 RSC.
- A. Petitjean and J. K. Barton, J. Am. Chem. Soc., 2004, 126, 14728–14729 CrossRef CAS PubMed.
- A. G. Weidmann and J. K. Barton, Inorg. Chem., 2014, 53, 7812–7814 CrossRef CAS PubMed.
- K. van der Schilden, F. Garcìa, H. Kooijman, A. L. Spek, J. G. Haasnoot and J. Reedijk, Angew. Chem., Int. Ed., 2004, 43, 5668–5670 CrossRef CAS PubMed.
- V. Ramu, M. R. Gill, P. J. Jarman, D. Turton, J. A. Thomas, A. Das and C. Smythe, Chem. – Eur. J., 2015, 21, 9185–9197 CrossRef CAS PubMed.
- Ş. Özalp-Yaman, P. de Hoog, G. Amadei, M. Pitié, P. Gamez, J. Dewelle, T. Mijatovic, R. Kiss and J. Reedijk, Chem. – Eur. J., 2008, 14, 3418–3426 CrossRef PubMed.
- N. J. Farrer and D. M. Griffith, Curr. Opin. Chem. Biol., 2020, 55, 59–68 CrossRef CAS.
- X. Yao, C. M. Tracy and U. Bierbach, Inorg. Chem., 2019, 58, 43–46 CrossRef CAS PubMed.
- J. Hennessy, B. McGorman, Z. Molphy, N. P. Farrell, D. Singleton, T. Brown and A. Kellett, Angew. Chem., Int. Ed., 2022, 61, e202110455 CrossRef CAS PubMed.
- P. D. O’Dowd, A. S. Guerrero, K. R. Alley, H. C. Pigg, F. O’Neill, J. Meiller, C. Hobbs, D. A. Rodrigues, B. Twamley, F. O’Sullivan, V. J. DeRose and D. M. Griffith, ACS Chem. Biol., 2024, 19, 875–885 CrossRef.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |