
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and catalytic properties of palladium(II) complexes with P,π-chelating ferrocene phosphinoallyl ligands and their non-tethered analogues†
Karel
Škoch‡
ab,
Jakub
Antala‡
a,
Ivana
Císařová
a and
Petr
Štěpnička
 *a
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 40 Prague, Czech Republic. E-mail: stepnic@natur.cuni.cz
bInstitute of Inorganic Chemistry of the Czech Academy of Sciences, Husinec-Řež, Czech Republic
First published on 1st May 2024
Abstract
Hybrid phosphines usually combine a phosphine moiety with another heteroatom secondary donor group in their structures while compounds equipped with hydrocarbyl π–donor moieties remain uncommon. This contribution reports the synthesis and structural characterization of the first P/π-allyl-chelating complexes that were obtained using the structurally flexible and redox-active ferrocene unit as the scaffold, viz. [PdCl(R2PfcCHCHCH2-η3:κP)] (1R; R = Ph and cyclohexyl (Cy); fc = ferrocene-1,1′-diyl). These compounds were synthesized from the respective phosphinoferrocene carboxaldehydes R2PfcCHO via reaction with vinylmagnesium bromide to generate 1-(phosphinoferrocenyl)allyl alcohols, which were subsequently acetylated. The resulting allyl acetates reacted smoothly with [Pd2(dba)3]/[Et3NH]Cl (dba = dibenzylideneacetone) to produce the target compounds. Complexes 1R and their nontethered analogues [PdCl(η3-C3H5)(FcPR2-κP)] (5R; Fc = ferrocenyl) were evaluated as pre-catalysts for the Pd-catalysed allylic amination of cinnamyl acetate with aliphatic amines and Suzuki–Miyaura-type cross-coupling of 4-tolylboronic acid with benzoyl chloride. In these reactions, better results were achieved with compounds 5R (particularly with 5Ph), presumably because they form more stable LPd(0)-type catalysts.
Introduction
Phosphine and π-allyl donors are often found together in organometallic molecules, which corresponds to the frequent involvement of π-allyl intermediates in transition metal-mediated transformations of organic molecules.1 However, these two moieties are rarely the parts of one ditopic ligand.2 Indeed, several complexes that feature chelating phosphinoallyl ligands have been reported, but these compounds typically arose from transformations of organic substituents at the phosphorus atom (usually cyclic) in the coordination sphere of the ligated metal.3 None of these ligands contained the sterically and electronically distinct ferrocene moiety.4,5This inspired us to design a rational method to prepare compounds in which the phosphine and π–allyl moieties are present in one chelating ligand. As the central scaffold, we chose the ferrocene moiety the conformational freedom of which can help properly position the two donor moieties via rotation of the cyclopentadienyl rings.6 Compounds of this kind would complement the numerous Pd(allyl) complexes with chelating phosphinoferrocene auxiliary ligands4c–e,7 and, mainly, expand the family of ferrocene-based P,C-donor ligands that remain limited to phosphinoferrocene alkenes8 and alkynes,9 isocyanide Ph2PfcNC (fc = ferrocene-1,1′-diyl),10 carbenes with P-chelating11 phosphinoferrocene substituents,12 and compounds featuring 1′-(diphenylphosphino)ferrocene-1-yl as an anionic P,C-ligand.13
In this contribution, we describe the synthesis of palladium(II) complexes with chelating phosphinoferrocenyl-substituted η3-allyl ligands (compounds 1R in Scheme 1), and their evaluation as defined pre-catalysts for Pd-catalysed allylic amination and Suzuki–Miyaura cross-coupling, in comparison with the analogous conventional complexes 5R, in which the two functional moieties are not mutually connected.
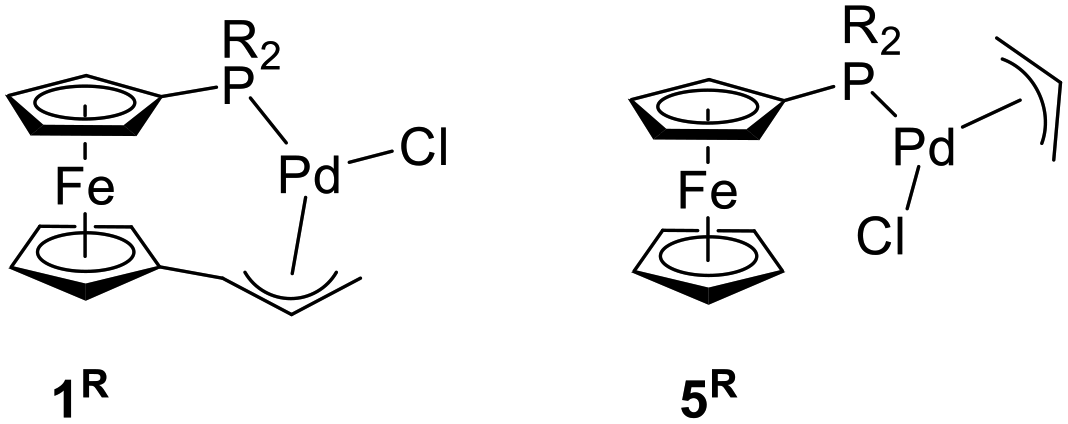 | ||
| Scheme 1 Chelating palladium(II) phosphinoallyl complexes 5R and their non-tethered counterparts 1R [R = phenyl and cyclohexyl (Cy)]. | ||
Results and discussion
Synthesis and characterisation
The synthesis of complexes 1R is outlined in Scheme 2. In the first step, the respective aldehydes 2R were treated with vinylmagnesium bromide in THF to produce racemic, phosphinoferrocenyl-substituted allyl alcohols 3R. These alcohols were isolated by column chromatography and obtained as viscous orange oils in good yields (>80%).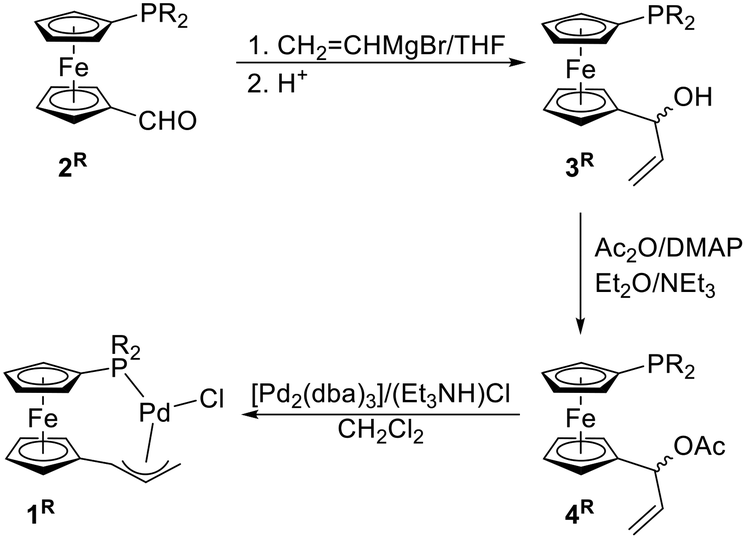 | ||
| Scheme 2 Synthesis of the chelating phosphinoallyl complexes 1R. Legend: R = Ph, Cy (cyclohexyl); DMAP = 4-(dimethylamino)pyridine, dba = dibenzylideneacetone. | ||
The alcohols were subsequently acetylated with acetic anhydride in the presence of 4-(dimethylamino)pyridine (DMAP)14 and triethylamine in diethyl ether. The acylation reactions proceeded selectively with full conversion of the starting material, but the resulting acetates 4R were unstable and could not be efficiently purified. When purification of 4Ph was performed by column chromatography over silica gel or alumina, extensive decomposition occurred, which markedly reduced the yield of the acetyl derivative. Compound 4Cy was even less stable and rapidly decomposed during chromatography as well as in chloroform solution. Fortunately, crude acetates 4R were sufficiently pure for use in the following step, in which minor impurities did not impede complex formation. The crude acetates were treated with [Pd2(dba)3] (1 equiv. of Pd) and triethylammonium chloride as the source of chloride ions in dichloromethane. Gratifyingly, oxidative additions15 of 4R across the Pd(0) precursor proceeded rapidly (full conversion was reached within approximately 5 min, as indicated by NMR analysis; see ESI†) and with complete anion exchange to afford complexes 1R, which were isolated as air-stable, orange crystalline solids with approximately 70% yields by column chromatography.
Alternatively, HCl (as a solution in Et2O) or solid LiCl (5 equiv.) were utilised as the chloride source for the last step. While the reaction in the presence of HCl proceeded cleanly and quickly, the reaction with LiCl was much slower, presumably due to the limited solubility of the ionic halide in the reaction mixture. The palladation could be achieved also with [Pd(PPh3)4], but the reaction took longer and was less selective. The [Pd2(dba)3]/[Et3NH]Cl combination thus appeared optimal in terms of the reaction yield and ease of product isolation.
Except for the easily decomposing 4Cy, complexes 1R and all intermediates were fully characterized by multinuclear NMR and IR spectroscopy, ESI mass spectrometry, and elemental analysis (conventional or from HR MS), and the structures of 1R were corroborated by X-ray diffraction analysis.
Notably, the ferrocene CH groups in all compounds were diastereotopic due to the presence of stereogenic carbon atoms in 3R and 4R, and due to the fixed conformation of 1R, which renders the ferrocene moiety axially chiral. As a result, eight signals of ferrocene CH groups were observed in the 1H and 13C{1H} NMR spectra, in addition to resonances corresponding to the phosphine substituents16 and the allyl group.17 A low-field shift of the 31P NMR signals (cf. δP −16.6/35.2 for 4Ph/1Ph and −7.5/56.4 for 4Cy/1Cy) and a change in the NMR signature of the allyl moiety suggested the coordination of both “functional” substituents. The π-coordinated allyl moieties18 generated four characteristic multiplets (ABCDX spin system; A–D = 1H, X = 31P) and three phosphorus-coupled doublets in the 1H and 13C{1H} NMR spectra of 1R, respectively.
The molecular structure of 1Ph is displayed in Fig. 1 along with selected geometric parameters; the structure of solvated 1Cy is presented in ESI.† The cyclopentadienyl rings in the molecule of 1Ph adopted a staggered conformation with τ = −38.6(2)° (τ is the torsion angle C1–Cg1–Cg2–C6, where Cg1 and Cg2 are the centroids of rings C(1–5) and C(6–10), respectively) and were tilted by 7.2(2)°. The allyl moiety C(23–25) and its bonding cyclopentadienyl ring C(1–5) were mutually twisted by 55.4(4)°, while the interplanar angle between the allyl moiety and the {Pd, P,Cl} plane was 67.1(4)°.19 The Pd–Cterminal distances in the η3-coordinated allyl moiety were practically identical and slightly longer than the Pd–Cmeso bond. Thus, the tethered structure eliminated the characteristic differentiation of the allylic termini by trans influence of the remaining ligands (P vs. Cl),20 which was observed in the structures of complexes [(η3-C3H5)PdCl(Ph2PfcX-κP)], where X = CO2H, CONH2, C(O)NH(CH2)2NHC(O)NHR (R = H and Et) and CH2NHC(O)NHPh.21
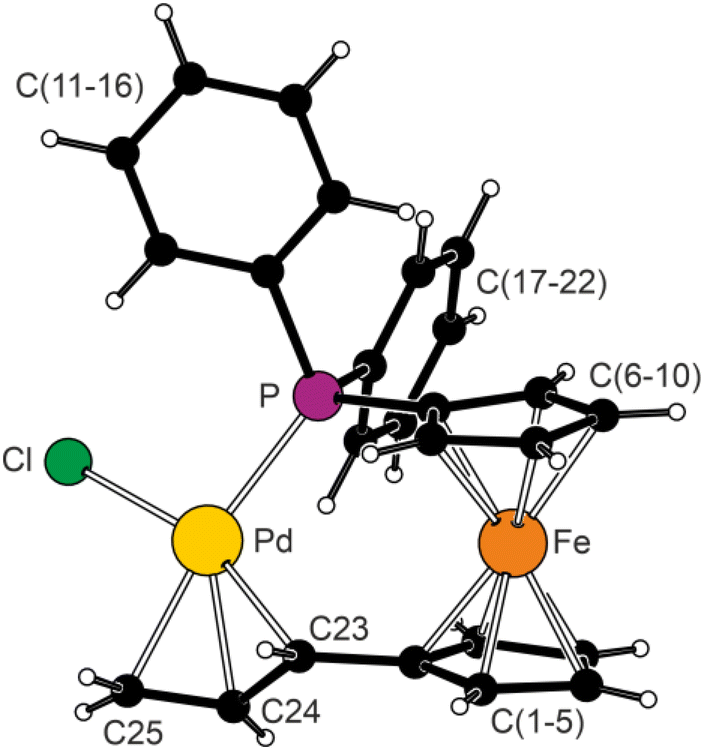 | ||
| Fig. 1 Molecular structure of 1Ph. Selected distances and angles (in Å and °): Fe–C(1–10) range 2.015(2)–2.075(3), Pd–Cl 2.3671(9), Pd–P 2.3324(6), Cl–Pd–P 97.85(3), Pd–C23 2.181(3), Pd–C24 2.159(3), Pd–C25 2.189(3), C23–C24–C25 122.2(3), C1–C23 1.494(5), C1–C23–C24 120.9(3), P–C6 1.804(2), P–C11 1.829(2), P–C17 1.830(2), C6–P–C11 100.5(1), C6–P–C17 103.8(1), and C11–P–C17 104.4(1). | ||
With an aim of investigating the catalytic properties of 1R, we also prepared complexes [(η3-C3H5)PdCl(FcPR2-κP)] (5R, R = Ph, Cy), which are close structural analogues of the tethered compounds. Compounds 5R were obtained by cleavage of the dimeric precursor [(η3-C3H5)Pd(μ-Cl)]2 with the stoichiometric amount of the respective phosphine (Scheme 3), isolated as orange-crystalline solids in approximately 90% yields by crystallization, and characterized similarly to 1R (see ESI†).22
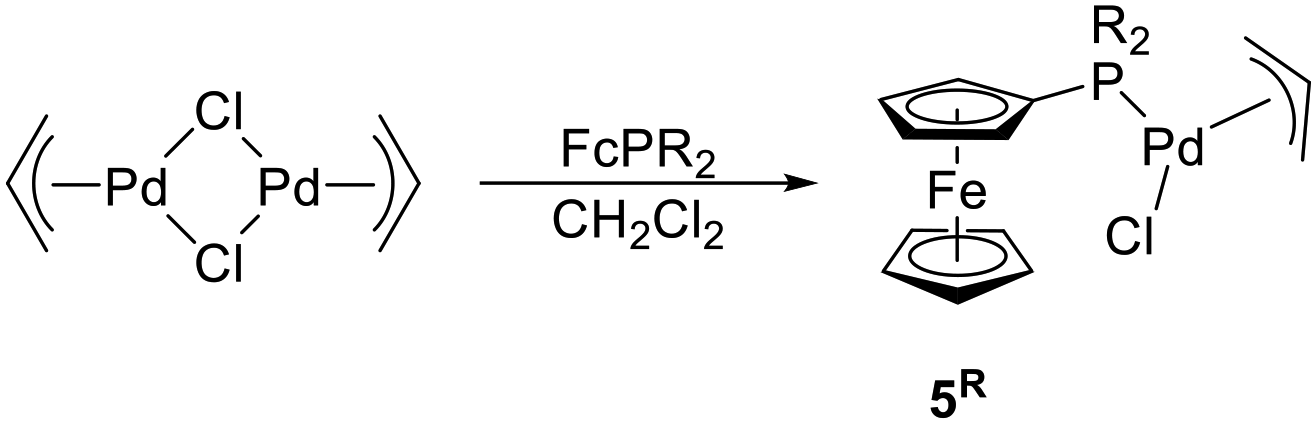 | ||
| Scheme 3 Synthesis of non-tethered complexes 5R [R = Ph, and Cy; Fc = ferrocenyl]. | ||
Lacking conjugation between the π-allyl moiety and the ferrocene unit, compounds 5R (rusty orange) were significantly lighter in colour than 1R (orange-red). This difference was manifested in the UV-vis spectra (Fig. 2), in which the representative compound 1Ph showed an absorption band at approximately 454 nm, while 5Ph displayed a less intense, tailing band at higher energies. Both bands were bathochromically shifted with respect to ferrocene itself (maximum at 440 nm with a shoulder at 528 nm in ethanol).23
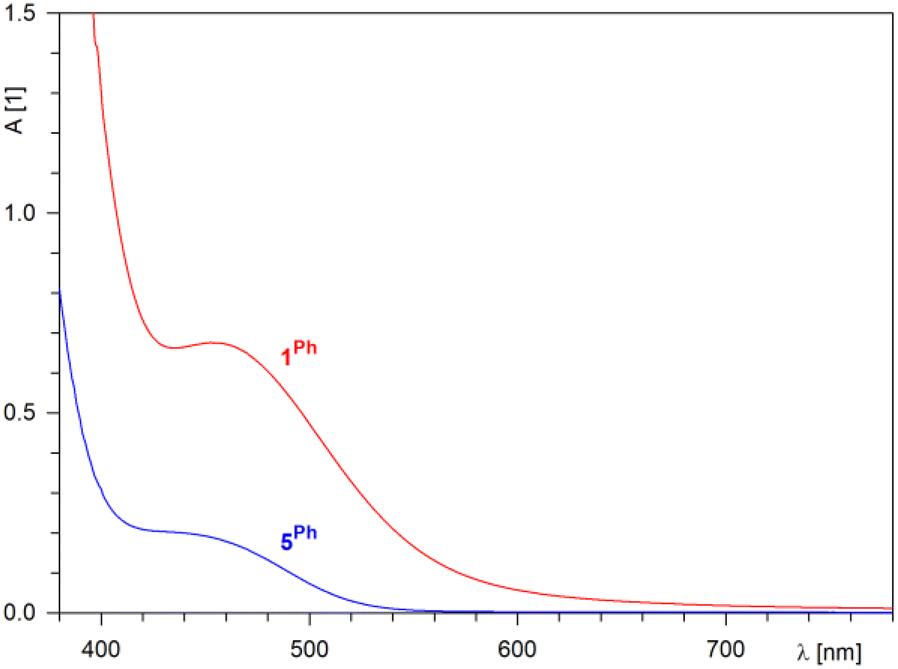 | ||
| Fig. 2 Electronic absorption spectra in the visible region of 1Ph (red) and 5Ph (c = 1 mM in dichloromethane, optical path 10 mm). | ||
In addition to structural characterization, compounds 1R and 5R were studied by cyclic voltammetry on a glassy carbon disc electrode using dichloromethane as the solvent and [Bu4N][PF6] as the supporting electrolyte. In the accessible potential range, complexes 1R showed reversible oxidation followed by a weaker irreversible oxidation step,24 and irreversible multielectron reduction (Fig. 3 and S3†). The primary oxidation, which probably results from a one-electron oxidation of the ferrocene unit, was observed at 0.30 V for 1Ph and 0.21 V for 1Cy relative to the ferrocene reference.25 The lower redox potential determined for the latter compound corresponded to the presence of a stronger donating dicyclohexylphosphine moiety (cf. the Hammett σp constants of −0.01 and −0.15 for the phenyl and cyclohexyl groups, respectively).26
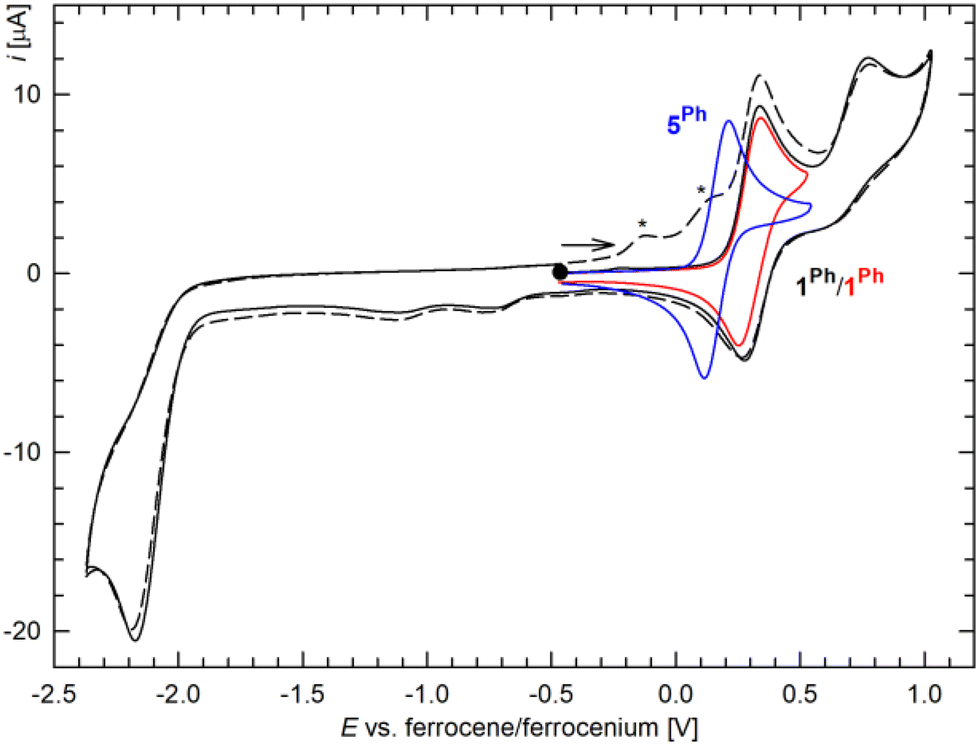 | ||
| Fig. 3 Cyclic voltammograms of 1Ph (red and black lines) and 5Ph (blue line) recorded at the glassy carbon disk electrode in 0.1 M [Bu4N][PF6]/CH2Cl2 (scan rate: 100 mV s−1). The scan direction is indicated by an arrow, and the second scan is shown as a dashed line (asterisks indicate peaks due to species generated electrochemically). | ||
Irreversible27 reduction, which occurred at approximately −2.2 V for 1Ph and −2.5 V for 1Cy, produced redox-active species, generating waves in the anodic region; these waves were not observed without prior reduction. This response can be rationalized as a two-electron reduction of the complexes into unstable Pd(0) species that are oxidized (or their decomposition products resulting via an EC process) during subsequent scanning.28 Also in this case, the reduction of 1Cy was shifted towards more negative potentials and thus occurred at the onset of base electrolyte decomposition.
Conversely, compounds 5R displayed only one reversible redox transition in the potential window investigated (Fig. 3). These redox waves, attributed to the ferrocene/ferrocenium redox couple, were observed at 0.17 and 0.12 V for 5Ph and 5Cy, respectively. The redox potentials determined for 5R were thus lower than that of 1R, suggesting that the ferrocene unit in the non-tethered compounds contains a relatively higher electron density. Simultaneous coordination of the phosphine and the conjugated allyl substituents, such as in 1R, apparently leads to a more pronounced decrease in the electron density at the ferrocene core than that of 5R, in which the π-allyl ligand remains separated.
Catalytic evaluation
The Pd-allyl complexes easily convert to active Pd(0) catalysts.29 For instance, complexes [Pd(η3-C3H5)(η5-C5H5)] and [Pd(η3-1-PhC3H4)(η5-C5H5)] were shown to react with phosphines (L) to produce species of the type [PdLn] (n = 2, 3), which proved to be active cross-coupling catalysts.30 More recent reports demonstrated that similar Pd(II)-to-Pd(0) transformations are also possible with the more stable and easier-to-access [PdL(Cl)(η3-allyl)] complexes (allyl = various π-coordinated allyl ligands, including cyclic ones, L = phosphine or NHC ligands).31The catalytic properties of complexes 1R and 5R were compared using Pd-catalysed allylic amination (Tsuji–Trost reaction) and Suzuki–Miyaura-type cross-coupling of aryl boronic acids with acyl chlorides that are simple but useful in practice (for experimental details, see ESI†). The former reaction, which proceeds via π-allyl intermediates, represents a general and helpful method for producing synthetically valuable allylamines (usually in enantioselective manner).32 In a similar vein, the Suzuki–Miyaura cross-coupling of boronic acids with acyl chlorides is a practical, selective, and functional group-tolerant alternative33 to the conventional methods of ketone synthesis as well as carbonylative Suzuki–Miyaura reaction that employs hazardous CO as the reagent.34
In particular, compounds 1R and 5R were applied as defined (pre)catalysts for the amination of cinnamyl acetate (6) with selected aliphatic secondary amines (Scheme 4). Initial experiments, performed to optimize the reaction conditions, employed morpholine as the nucleophile (1 equiv.) and 5Ph as the pre-catalyst in different solvents (THF, toluene, CH2Cl2, and MeCN). The results showed that the best yields and selectivity towards the linear amination product 7a were obtained in MeCN (Table 1, entries 1–4 and 7) and that at least 1 mol% of the Pd catalyst was necessary to achieve good conversions (at 50 °C for 20 h).
 | ||
| Scheme 4 The model allylic amination reaction [HNR2 = morpholine (a), diethylamine (b)]. | ||
| Entry | Catalyst [loadingb] | Solvent | Conversion [%] |
7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 :8 |
Yield of 7 [%] |
|---|---|---|---|---|---|
|
a Conditions: 6:HNR2 = 1:1, c(6) = 0.25 M, 50 °C, 20 h. The results in entries 5–12 are an average of two independent runs. Isolated yields are given.
b mol% Pd.
|
|||||
| HNR2 = morpholine (a) | |||||
| 1 | 5Ph [1] | THF | 99 | 94:6 |
80 |
| 2 | 5Ph [0.5] | THF | 45 | 91:9 |
45 |
| 3 | 5Ph [1] | Toluene | 85 | 92:8 |
76 |
| 4 | 5Ph [1] | CH2Cl2 | 73 | 98:2 |
68 |
| 5 | 1Ph [1] | MeCN | 53 | 91:9 |
47 |
| 6 | 1Cy [1] | MeCN | 7 | 70:30 |
5 |
| 7 | 5Ph [1] | MeCN | 77 | 99:1 |
69 |
| 8 | 5Cy [1] | MeCN | 8 | 82:18 |
5 |
| HNR2 = HNEt2 (b) | |||||
| 9 | 1Ph [1] | MeCN | 38 | >99:1 |
35 |
| 10 | 1Cy [1] | MeCN | 10 | >99:1 |
9 |
| 11 | 5Ph [1] | MeCN | 64 | >99:1 |
60 |
| 12 | 5Cy [1] | MeCN | 10 | >99:1 |
9 |
A comparison of the catalysts (entries 5–8) revealed that complex 5Ph generated the best results in terms of the reaction yield (69% of isolated 7a) and selectivity (7a:8a = 99:1). Its tethered analogue 1Ph afforded a lower yield and selectivity (91:9), and compounds bearing cyclohexyl substituents at the phosphorus provided an even poorer conversion and decreased selectivity. A similar reactivity trend was observed when diethylamine was used as the nucleophile (entries 9–12). In this case, however, all the catalysts produced only linear amine 7b; no signals due to the branched isomer 8b were detected in the 1H NMR spectra of the crude products.
Next, we focused on the Suzuki–Miyaura cross-coupling of 4-tolylboronic acid (9) with benzoyl chloride (10, 1.2 equiv.) in the presence of Na2CO3 (Scheme 5). The reaction was performed in a C6D6–H2O mixture (1:1) at 50 °C for 1 h, and the product mixture was analysed by NMR spectroscopy using anisole as a standard.21a
 | ||
| Scheme 5 Suzuki–Miyaura cross-coupling of 4-tolylboronic acid (9) with benzoyl chloride (10). | ||
The results compiled in Table 2 show that the most active catalysts were obtained from the non-tethered complexes 5Ph and 5Cy, which did not differ practically under the reaction conditions applied. Complexes 1R generated lower conversions, and 1Cy performed worst in the series of tested complexes. A similar reaction in toluene and with 0.5 mol% of 5Ph produced the coupling product 11 in 91% isolated yield.
|
a Conditions: 1.25 mmol of 9, 1.25 mmol of Na2CO3 and 1.5 mmol of 10 were reacted in 6 mL of a 1:1 C6D6:H2O mixture at 50 °C for 1 h. NMR yields are given as an average of two independent runs.
|
||||
|---|---|---|---|---|
| Entry | 1 | 2 | 3 | 4 |
| Catalyst | 1Ph | 1Cy | 5Ph | 5Cy |
| Yield of 11 [%] | 59 | 30 | 72 | 72 |
Conclusions
Two palladium phosphinoferrocene-allyl complexes were synthesized as the first compounds that feature ditopic, η3:κP-chelating phosphinoallyl ligands and were prepared rationally. In their synthesis, advantage was taken of the well-defined yet adaptable stereochemistry offered by the ferrocene skeleton. These compounds, which were fully characterized including structure determination, expand the family of the rare ferrocene P,C-donors (vide supra).Preliminary catalytic tests in Pd-catalysed allylic amination and Suzuki–Miyaura reactions using the chelating Pd(II)-phosphinoallyl complexes and their conventional, non-tethered counterparts corroborated the generally favourable catalytic properties of Pd–allyl complexes, which convert into active catalysts even at relatively low temperatures (50 °C in the present case). For both reactions, however, the best results (in terms of reaction yield and selectivity) were achieved with the conventional (pre)catalyst 5Ph. Very likely, the more electron-rich, non-tethered compounds are more easily “activated” via loss of the allyl ligand and/or give rise to more stable “LPd(0)” catalysts (L = phosphine) than their P,allyl-chelating analogues. The “activation” of 1R may result in species with a free reactive allyl moiety, which can undergo further transformations and thus limit the catalyst lifetime. Possible decomposition pathways include the formation of species with uncoordinated allyl groups via reactions with an external nucleophile or even with the phosphine group from the same molecule35 (intramolecular process; this reaction may account for the low stability of 4R containing the easily leaving acetate groups). On the whole, our results indicate that the all-in-one approach to the design of (pre)catalysts need not be always beneficial.
Experimental
Materials and methods
All manipulations were performed under a dinitrogen atmosphere using standard Schlenk techniques and oven-dried glassware. Compounds 2Ph,362Cy,37 (diphenylphosphino)- and (dicyclohexylphosphino)ferrocene38 were prepared following procedures described in the literature. Other chemicals were purchased from commercial suppliers (Sigma-Aldrich, TCI) and used as received. Diethyl ether and triethylamine were distilled from sodium metal. Dry and deoxygenated tetrahydrofuran and dichloromethane were obtained from a PureSolv MD5 solvent purification system (Innovative Technology, USA). Solvents used for column chromatography and crystallizations were utilised without any additional purification (analytical grade, Lach-Ner, Czech Republic).NMR spectra were measured with a Varian UNITY Inova 400, a Bruker Avance III 400, or a JEOL Delta 600 spectrometer at 25 °C. Chemical shifts (δ in ppm) are given relative to the internal SiMe4 (1H and 13C) and to the external 85% aqueous H3PO4 (31P). IR spectra were recorded on a Nicolet iS50 (Thermo Fisher Scientific) instrument over the 400–4000 cm−1 range. UV-vis spectra were recorded on a Unicam UV 300 spectrometer. Electrospray ionization mass spectra (ESI MS) were acquired with a Compact QTOF-MS spectrometer (Bruker Daltonics). The samples were dissolved in HPLC-grade methanol. Elemental analyses were performed with a PE 2400 Series II CHNS/O Elemental Analyser (PerkinElmer). The amount of residual solvent (if present) was corroborated by NMR analysis.
Electrochemical measurements were performed at room temperature (22 °C) with an μAUTOLAB III instrument (Eco Chemie, The Netherlands) using a standard three-electrode electrochemical cell equipped with a glassy carbon disc (2 mm diameter) working electrode, a platinum auxiliary electrode, and an Ag/AgCl (3 M KCl) reference electrode. The samples were dissolved in anhydrous dichloromethane to generate a solution with 1 mM of the analysed compound and 0.1 M [Bu4N][PF6] (Sigma–Aldrich). The solutions were deaerated with argon and maintained under an argon flow during the measurement. Decamethylferrocene (Alfa–Aesar) was used as a reference in the final scans, and the redox potentials were converted into the ferrocene/ferrocenium scale by subtracting 0.548 V.39
Synthesis of 3Ph
Aldehyde 2Ph (796 mg, 2.0 mmol) was dissolved in dry THF (25 mL), and the solution was cooled to approximately −75 °C with a dry ice/i-PrOH cooling bath. After temperature equilibration, vinylmagnesium bromide solution was introduced (2.5 mL of 1.0 M solution in THF, 1.25 equiv.) while stirring, whereupon the initial orange-red reaction mixture turned orange-yellow. After 10 min, the cooling bath was removed, and the reaction mixture was stirred at room temperature for an additional 30 min before quenching with saturated aqueous NH4Cl (10 mL) and diethyl ether (10 mL). The organic layer was separated, washed successively with water and brine, dried over MgSO4, and evaporated, leaving the crude product as a yellow viscous oil. This product was purified by chromatography over a short silica gel column using hexane/diethyl ether (1:1) as the eluent. A single yellow band was collected and evaporated to afford 3Ph as an orange-yellow, highly viscous oil. Yield: 812 mg (95% yield).
1H NMR (CDCl3): δ 2.15 (dd, J = 5.0 Hz, 2.1 Hz, 1H, CHOH), 4.06 (virtual t, J′ = 1.9 Hz, 2H, fc), 4.11–4.13 (m, 2H, fc), 4.14–4.16 (m, 2H, fc), 4.39–4.41 (m, 2H, fc), 4.74 (ddt, J = 5.0 Hz, 1.4 Hz, 1.2 Hz. 1H, CHOH), 5.11 (ddd, J = 10.3 Hz, 1.5, 1.4 Hz, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.22 (ddd, J = 17.1 Hz, 1.5 Hz, 1.4 Hz, 1H, CH2), 5.98 (ddd, J = 17.1 Hz, 10.3 Hz, 6.1 Hz, 1H, CHCH2), 7.30–7.40 (m, 10H, PPh2). 13C{1H} NMR (CDCl3): δ 67.54 (d, JPC = 1 Hz, CH of fc), 67.62 (d, JPC = 1 Hz, CH of fc), 69.24 (d, JPC ≈ 1 Hz, CH of fc), 69.28 (d, JPC = 1 Hz, CH of fc), 70.54 (CHOH), 71.14 (d, JPC = 1 Hz, CH of fc), 71.17 (d, JPC = 1 Hz, CH of fc), 73.12 (d, JPC = 14 Hz, CH of fc), 73.43 (d, JPC = 15 Hz, CH of fc), 76.33 (d, JPC = 5 Hz, Cipso–P of fc), 92.29 (Cipso–C of fc), 114.93 (CH2), 128.15 (CHmeta of Ph), 128.22 (CHmeta of Ph), 128.61 (CHpara of Ph), 128.64 (CHpara of Ph), 133.51 (d, 2JPC = 20 Hz, CHortho Ph), 133.54 (d, 2JPC = 19 Hz, CHortho Ph), 138.59 (d, 1JPC = 9 Hz, Cipso of Ph), 138.62 (d, 1JPC = 9 Hz, Cipso of Ph), 139.61 (CHCH2). 31P{1H} NMR (CDCl3): δ −16.4 (s). IR (neat): νmax 3409 br m, 3069 m, 3052 m, 3001 m, 2860 w, 1478 s, 1434 s, 1308 m, 1231, 1161 s, 1095 m, 1026 s, 989 s, 925 m, 895 w, 830 m, 743 s, 697 s, 633 w, 570 w, 502 s, 455 m cm−1. ESI+ MS: m/z 449.1 ([M + Na]+), 465.1 ([M + O + Na]+). HRMS calc. for C25H23FeO2P ([M + O + Na]+) 465.0677; found 465.0676.
CH2), 5.22 (ddd, J = 17.1 Hz, 1.5 Hz, 1.4 Hz, 1H, CH2), 5.98 (ddd, J = 17.1 Hz, 10.3 Hz, 6.1 Hz, 1H, CHCH2), 7.30–7.40 (m, 10H, PPh2). 13C{1H} NMR (CDCl3): δ 67.54 (d, JPC = 1 Hz, CH of fc), 67.62 (d, JPC = 1 Hz, CH of fc), 69.24 (d, JPC ≈ 1 Hz, CH of fc), 69.28 (d, JPC = 1 Hz, CH of fc), 70.54 (CHOH), 71.14 (d, JPC = 1 Hz, CH of fc), 71.17 (d, JPC = 1 Hz, CH of fc), 73.12 (d, JPC = 14 Hz, CH of fc), 73.43 (d, JPC = 15 Hz, CH of fc), 76.33 (d, JPC = 5 Hz, Cipso–P of fc), 92.29 (Cipso–C of fc), 114.93 (CH2), 128.15 (CHmeta of Ph), 128.22 (CHmeta of Ph), 128.61 (CHpara of Ph), 128.64 (CHpara of Ph), 133.51 (d, 2JPC = 20 Hz, CHortho Ph), 133.54 (d, 2JPC = 19 Hz, CHortho Ph), 138.59 (d, 1JPC = 9 Hz, Cipso of Ph), 138.62 (d, 1JPC = 9 Hz, Cipso of Ph), 139.61 (CHCH2). 31P{1H} NMR (CDCl3): δ −16.4 (s). IR (neat): νmax 3409 br m, 3069 m, 3052 m, 3001 m, 2860 w, 1478 s, 1434 s, 1308 m, 1231, 1161 s, 1095 m, 1026 s, 989 s, 925 m, 895 w, 830 m, 743 s, 697 s, 633 w, 570 w, 502 s, 455 m cm−1. ESI+ MS: m/z 449.1 ([M + Na]+), 465.1 ([M + O + Na]+). HRMS calc. for C25H23FeO2P ([M + O + Na]+) 465.0677; found 465.0676.
Synthesis of 3Cy
Aldehyde 2Cy (1.64 g, 4.0 mmol) was dissolved in dry THF (50 mL), and the mixture was cooled with a dry ice/i-PrOH cooling bath. Next, vinylmagnesium bromide solution was added (5 mL of 1.0 M in THF, 5.0 mmol), causing a change in colour from red to orange. After the mixture was stirred for 30 min, the cooling bath was removed, and the mixture was left to react at room temperature for an additional 60 min. The reaction was terminated by adding saturated aqueous NH4Cl (10 mL) and diethyl ether (10 mL). The organic layer was separated, washed with water and brine, dried over MgSO4, and evaporated. The crude product (red oil) was purified by column chromatography over silica gel and eluted with hexane/diethyl ether (2:1). A single orange band was collected and evaporated to give 3Cy as an orange-red, highly viscous oil. Yield: 1.45 g (83%).
1H NMR (CDCl3): δ 1.02–1.35 (m, 11H, Cy), 1.62–1.96 (m, 11H, Cy), 2.98 (br, 1H, OH), 4.12, 4.15, 4.15, 4.15, 4.15, 4.16, 4.23, 4.31 (8 × m, 1H, fc), 4.93 (dt, 3JHH = 5.8 Hz, 4JHH ≈ 4JHH ≈ 1.4 Hz, 1H, CHOH), 5.12 (dt, 3JcisHH = 10.3 Hz, 2JHH ≈ 4JHH ≈ 1.4 Hz, 1H, CH2), 5.27 (dt, 3JtransHH = 17.1 Hz, 2JHH ≈ 4JHH ≈ 1.4 Hz, 1H, CH2), 6.03 (ddd, 3JtransHH = 17.1 Hz, 3JcisHH = 10.3 Hz, 3JHH = 5.8 Hz, CHCH2). 13C{1H} NMR (CDCl3): δ 26.39 (CH2 Cy), 26.40 (CH2 Cy), 27.26 (d, JPC = 2 Hz, CH2 Cy), 27.33 (CH2 Cy), 27.34 (d, JPC = 2 Hz, CH2 Cy), 27.44 (CH2 Cy), 30.10 (CH2 Cy), 30.11 (CH2 Cy), 30.21 (d, JPC = 5 Hz, CH2 Cy), 30.25 (d, JPC = 4 Hz, CH2 Cy), 30.36 (d, 1JPC = 10 Hz, CH Cy), 33.50 (d, 1JPC = 11 Hz, CH Cy), 67.61 (CH of fc), 67.79 (CH of fc), 69.10 (CH of fc), 69.12 (CH of fc), 69.75 (d, JPC = 2.9 Hz, CH of fc), 69.85 (d, JPC = 3 Hz, CH of fc), 71.71 (d, JPC = 10 Hz, CH of fc), 72.17 (d, JPC = 11 Hz CH of fc), 76.44 (d, 1JPC = 4 Hz, Cipso–P of fc), 92.65 (Cipso–C of fc), 70.65 (CHOH), 114.58 (CH2), 140.17 (CHCH2). 31P{1H} NMR (CDCl3): δ −7.0 (s). IR (neat): νmax 3409 br m, 3089 m, 2919 s, 2849 s, 1641 w, 1461 m, 1448 s, 1420 m, 1383 m, 1342 m, 1292 m, 1266 m, 1231 w, 1194 m, 1177 w, 1156 m, 1113 m, 1042 s, 1028 s, 990 s, 920 s, 887 m, 850 m, 828 s, 814 s, 790 m, 744 w, 718 w, 629 w, 507 s, 497 s, 443 w cm−1. ESI+ MS: m/z 439.2 ([M + H]+) and 477.2 ([M + K]+). HRMS calc. for C25H36FeOP ([M + H]+) 439.1848, found 439.1847.
Synthesis of 4Ph
Alcohol 3Ph (812 mg, 1.91 mmol) and 4-(dimethylamino)pyridine (11.6 mg, 0.096 mmol) were dissolved in dry diethyl ether (20 mL). Triethylamine (0.32 mL, 2.3 mmol) was added, and the mixture was cooled on ice before adding acetic anhydride (0.23 mL, 2.3 mmol) with continuous stirring. The cooling bath was removed, and the mixture was left to react at room temperature for 4 h. Next, the reaction was terminated by adding saturated aqueous NaHCO3 and diethyl ether (10 mL each). The organic layer was separated, washed with water (3 × 10 mL) and brine (10 mL), dried over MgSO4, and evaporated under vacuum to produce 4Ph as an orange-yellow viscous oil, which was used directly in the following step. Yield: 840 mg (94% yield). NOTE: Attempted purification of the crude product by column chromatography over silica gel or neutral alumina resulted in decomposition and substantial loss of the product. Fortunately, minor impurities present in the crude sample did not hinder the subsequent reaction step.
1H NMR (CDCl3): δ 2.04 (s, 3H, CH3), 4.04–4.08 (m, 4H, fc), 4.09 (m, 1H, fc), 4.18 (m, 1H, fc), 4.33–4.36 (m, 2H, fc), 5.19 (m, 1H, CH2), 5.25 (m, 1H, CH2), 5.99 (m, 1H, CHCH2) 6.01 (m, 1H, CHOAc), 7.28–7.38 (m, 10H, Ph). 13C{1H} NMR (CDCl3): δ 21.28 (CH3), 67.78 (CH of fc), 69.41 (CH of fc), 69.73 (d, JPC = 1 Hz, CH of fc), 69.85 (d, JPC = 1 Hz, CH of fc), 71.73 (d, JPC = 4 Hz, CH of fc), 71.78 (d, JPC = 4 Hz, CH of fc), 72.52 (CHOAc), 73.67 (d, JPC = 15 Hz, CH of fc), 73.77 (d, JPC = 14 Hz, CH of fc), 76.76 (Cipso–P of fc, partly obscured by the solvent resonance), 86.26 (Cipso–C of fc), 117.05 (CH2), 128.15 (d, JPC = 7 Hz, CH of Ph), 128.18 (d, JPC = 7 Hz, CH of Ph), 128.53 (CHpara of Ph), 128.55 (CHpara of Ph), 133.48 (d, JPC = 20 Hz, CH of Ph), 133.50 (d, JPC = 20 Hz, CH of Ph), 135.10 (CHCH2), 138.87 (d, 1JPC = 10 Hz, Cipso of Ph), 138.98 (d, 1JPC = 10 Hz, Cipso of Ph), 169.95 (CO). 31P{1H} NMR (CDCl3): δ −16.6 (s). IR (neat): νmax 3069 w, 3052 w, 1736 s (CO), 1370 s, 1308 w, 1232 vs., 1161 w, 1092 w, 1027 m, 998 w, 943 w, 891 w, 829 m, 787 w, 742 s, 697 s 633 w, 568 w, 502 s, 451 m cm−1. ESI+ MS: m/z 409.1 ([M − OAc]+), 507.1 ([M + O + Na]+). HRMS calc. for C27H25FeNaO3P ([M + O + Na]+) 507.0783, found 507.0784.
Synthesis of 4Cy
Alcohol 3Cy (1.34 g, 3.06 mmol) and 4-(dimethylamino)pyridine (18.3 mg, 0.15 mmol) were dissolved in dry diethyl ether (60 mL). Triethylamine (0.65 mL, 4.6 mmol) was added, and the mixture was cooled on ice before acetic anhydride (0.35 mL, 3.67 mmol) was introduced dropwise with stirring. After the addition, the cooling bath was removed, and the mixture was stirred at room temperature for 4 h. Then, the mixture was quenched with saturated aqueous NaHCO3 and diethyl ether (10 mL each). The organic layer was separated, washed with water (3 × 10 mL) and brine (10 mL), dried over MgSO4, and evaporated under reduced pressure. The resulting orange-yellow viscous oil of crude 4Cy was immediately used in the following step. The yield of 4Cy was 1.40 g (corresponds to a 95% yield if pure).
NOTE: Purification of the crude product by column chromatography over silica gel or neutral alumina resulted in extensive decomposition. The product was unstable as a neat material and in solution. Due to the low stability, satisfactory
13
C{
1
H} or 2D NMR spectra could not be obtained. The decomposition was followed by the
31
P NMR spectra, in which the dominant signal due to
4
Cy
at δ
P
−7.5 was replaced by three signals at δ
P
36.8, 34.9, and 33.5 in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :4:1 ratio over 8 h (see ESI†). This might result from a facile oxidation of the phosphine group to phosphine oxide and by elimination of the labile acetate group followed by the generation of a phosphonium salt through a reaction between the phosphine and formed carbocation. Fortunately, impurities in the freshly prepared
4
Cy
did not significantly interfere with the subsequent reaction step.
:4:1 ratio over 8 h (see ESI†). This might result from a facile oxidation of the phosphine group to phosphine oxide and by elimination of the labile acetate group followed by the generation of a phosphonium salt through a reaction between the phosphine and formed carbocation. Fortunately, impurities in the freshly prepared
4
Cy
did not significantly interfere with the subsequent reaction step.
1H NMR (CDCl3): δ selected resonances 2.07 (s, 3H, OAc), 4.11–4.16 (m, 6H, fc), 4.22 (m, 1H, fc), 4.24 (m, 1H, fc), 5.26, 5.34, 6.13, and 6.15 (4 × m, 1H, CHCHCH2). 31P{1H} NMR (CDCl3): δ −7.5 (s). ESI+ MS: m/z 421 ([M − OAc]+).
Synthesis of 1Ph
Crude acetate 4Ph (1.17 g, 2.5 mmol) was dissolved in dry dichloromethane (20 mL). Solid [Pd2(dba)3] (1.14 g, 1.25 mmol) was added, and the resulting deep purple-red mixture was stirred for 5 min before solid [Et3NH]Cl (516 mg, 3.75 mmol) was added. The reaction mixture was stirred for another 30 min, whereupon the colour changed to deep brown-red. The volatilities were removed under reduced pressure, and the black-red oil was taken up with a small amount of dichloromethane and transferred onto a short neutral alumina column packed in hexane/ethyl acetate (3:1). After elution with the same solvent mixture, a yellow-brown band containing dibenzylidenacetone and unidentified impurities was removed. Then, the mobile phase was changed to pure ethyl acetate to elute complex 1Ph as a red band. Subsequent evaporation under reduced pressure afforded pure 1Ph as an orange-red solid with a 962 mg (70%) yield. Crystals used for X-ray diffraction analysis were grown from ethyl acetate/hexane.
1H NMR (CDCl3): δ 3.47 (ddt, 3JHH = 13.5 Hz, 10.6 Hz, 4JHH = 1.0 Hz, 1H, CH2 allyl), 4.02 (tdd, J′ = 2.5, 1.3, 0.8 Hz, 1H, fc), 4.22 (tdd, J′ = 2.5, 1.2, 0.5 Hz, 1H, fc), 4.30 (tdd, J′ = 2.6, 1.4, 0.6 Hz, 1H, fc), 4.37 (m, 2H, CH2 allyl and CH of fc), 4.44 (tdd, J′ = 2.5, 1.3, 0.7 Hz, 1H, fc), 4.60 (dt, J′ = 2.6, 1.3, 0.5 Hz, 1H, fc), 4.78 (m, 2 H, two CH of fc), 5.20 (d, 3JHH = 11 Hz, 1H, fc-CH allyl), 6.10 (dddd, 3JHH = 13.5, 11.4, 7.6 Hz, 3JPH = 1.2 Hz, 1H, CHmeso allyl), 7.36–7.44 (m, 6H, PPh2), 7.66–7.72 (m, 2H, PPh2), 7.88–7.94 (m, 2H, PPh2). 13C{1H} NMR (CDCl3): δ 68.53 (CH of fc), 68.88 (d, JPC = 2 Hz, CH of fc), 70.01 (d, JPC = 6 Hz, CH of fc), 70.72 (d, 2JPC = 29 Hz, CH2 allyl), 71.36 (d, JPC = 7 Hz, CH of fc), 71.79 (CH of fc), 73.70 (d, JPC = 2 Hz, CH of fc), 81.74 (d, 2JPC = 5 Hz, fc-CH allyl), 88.34 (Cipso–C of fc), 88.51 (d, 1JPC = 47 Hz, Cipso–P of fc), 117.51 (d, 2JPC = 5 Hz, CHmeso allyl), 128.20 (d, 3JPC = 10 Hz, CHmeta of Ph), 130.37 (d, 4JPC = 2 Hz, CHpara of Ph), 132.55 (d, 1JPC = 40 Hz, Cipso of Ph), 134.00 (d, 1JPC = 40 Hz, Cipso of Ph), 134.22 (d, 2JPC = 10 Hz, CHortho of Ph), 134.63 (d, 2JPC = 13 Hz, CHortho of Ph). Two signals due to ferrocene CH are obscured by the solvent resonance. 31P{1H} NMR (CDCl3): δ 35.2 (s). IR (Nujol): νmax 3123 w, 2051 w, 1309 w, 1287 w, 1181 w, 1168 m, 1097 m, 1067 w, 1058 w, 1045 w, 1028 s, 997 w, 974 w, 911 w, 891 w, 834 m, 817 m, 804 w, 764 s, 752 s, 698 s, 640 w, 537 m, 507 s, 494 s, 479 m, 464 m, 443 w, 432 w cm−1. ESI+ MS: m/z 515.0 ([M − Cl]+), 551.0 ([M + H]+). HRMS calc. for C25H23ClFePPd ([M + H]+) 550.9597 found 550.9604. Anal. calc. for C25H22ClFePPd (550.0): C 54.48, H 4.02; found C 54.30, H 4.02%.
Synthesis of 1Cy
Freshly prepared, crude 4Cy (960 mg, 2.0 mmol) was dissolved in dichloromethane (20 mL). Solid [Pd2(dba)3] (915 mg, 1.0 mmol) was added, and the deep purple-red mixture was stirred for 5 min before solid [Et3NH]Cl (413 mg, 3.0 mmol) was introduced. The stirring was continued for 30 min, during which time the reaction mixture turned deep brown-red. Next, all volatiles were removed under reduced pressure, and the black-red oil was dissolved in a small amount of dichloromethane and transferred onto the top of a neutral alumina column packed in hexane/ethyl acetate (3:1). Elution with the same solvent mixture removed a yellow-brown band containing dibenzylidenacetone and unidentified impurities. Then, the eluent was changed to neat ethyl acetate to remove the red band due to 1Cy, which was collected and evaporated under reduced pressure to produce 1Cy as a red microcrystalline solid. Yield: 753 mg (67%). The single crystal used for structure determination was grown from ethyl acetate/hexane.
1H NMR (CDCl3): δ 0.85–2.33 (m, 21H, Cy), 2.84 (m, 1H, Cy), 3.13 (ddt, 3JtransHH = 13.3 Hz, 3JPH = 10.5 Hz, 2JHH ≈ 4JHH = 1.1 Hz, 1H, CH2 allyl), 4.01 (tt, 3JcisHH ≈ 3JPH = 7.8 Hz, 4JHH ≈ 2JHH = 1.0 Hz, 1H, CH2 allyl), 4.06, 4.07, 4.23, 4.25, 4.81, 4.87, 4.99, 5.02 (8 × m, 1H, fc), 5.24 (br d, 3JHH = 11.7 Hz, fc-CH allyl), 6.06 (dddd, 3JHHtrans = 13.3 Hz, 3JHH = 11.7 Hz, 3JcisHH = 7.6 Hz, 3JPH = 1.4 Hz, CHmeso allyl). 13C{1H} NMR (CDCl3): δ 26.10 (d, JPC = 1 Hz, CH2 Cy), 26.14 (d, JPC = 1 Hz, CH2 Cy), 26.64 (d, JPC = 9 Hz, CH2 Cy), 27.06 (d, JPC = 4 Hz, CH2 Cy), 27.14 (d, JPC = 3 Hz, CH2 Cy), 27.23 (d, JPC = 4 Hz, CH2 Cy), 27.27 (d, JPC = 4 Hz, CH2 Cy), 28.75 (d, JPC = 5 Hz, CH2 Cy), 28.98 (d, JPC = 4 Hz, CH2 Cy), 31.79 (d, JPC = 2 Hz, CH2 Cy), 33.59 (d, JPC = 19 Hz, CH Cy), 34.66 (d, JPC = 21 Hz, CH Cy), 66.89 (d, JPC = 4.3 Hz, CH of fc), 67.27 (CH of fc), 67.38 (CH of fc), 69.79 (CH of fc), 69.85 (d, JPC = 6 Hz, CH of fc), 73.46 (d, JPC = 11 Hz, CH of fc), 73.96 (d, JPC = 1 Hz, CH of fc), 75.70 (d, JPC = 5 Hz, CH of fc), 90.64 (Cipso–C of fc), 95.11 (d, 1JPC = 33.6 Hz, Cipso–P of fc), 67.67 (d, 2JPC = 28.7 Hz, CH2 allyl), 81.84 (d, 2JPC = 6.0 Hz, fc-CH allyl), 116.75 (d, 2JPC = 5.5 Hz, CHmeso allyl). 31P{1H} NMR (CDCl3): δ 56.4 (s). IR (Nujol): νmax 3109 w, 2957 s, 2845 m, 1743 w, 1444 m, 1391 w, 1291 w, 1265 w, 1238 w, 1191 m, 1162 m, 1108 w, 1049 m, 1027 m, 1001 m, 978 w, 913 m, 888 w, 850 s, 810 m, 800 m, 745 m, 732 m, 632 m, 518 s, 501 vs. 446 s, 410 s cm−1. ESI+ MS: m/z 527.1 ([M − Cl]+). HRMS calc. for C25H34FePPd ([M − Cl]+) 527.0789, found 527.0787. Anal. calc. for C25H34ClFePPd (563.2): C 53.31, H 6.08; found C 53.69, H 6.17%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Czech Science Foundation (project no. 23-06718S).References
- (a) R. H. Crabtree, The Organometallic Chemistry of Transition Metals. 5th ed, J. Wiley and Sons, Hoboken, 2009 Search PubMed; (b) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books, Sausalito, 2009 Search PubMed.
- Incorporation of phosphorus atoms (up to three) directly into the allylic system provides structurally unique phosphallyl ligands. For leading references, see: (a) R. Appel, W. Schuhn and F. Knoch, Angew. Chem., Int. Ed. Engl., 1985, 24, 420 CrossRef; (b) F. Mercier, J. Fisher and F. Mathey, Angew. Chem., Int. Ed. Engl., 1986, 25, 357 CrossRef; (c) R. Appel, W. Schuhn and F. Knoch, J. Organomet. Chem., 1987, 319, 345 CrossRef CAS; (d) R. Appel, W. Schuhn and M. Nieger, Angew. Chem., Int. Ed. Engl., 1988, 27, 416 CrossRef; (e) C. Hugel-Le Goff, F. Mercier, L. Ricard and F. Mathey, J. Organomet. Chem., 1989, 363, 325 CrossRef CAS; (f) M. Scheer, C. Kuntz, M. Stubenhofer, M. Linseis, R. F. Winter and M. Sierka, Angew. Chem., Int. Ed., 2009, 48, 2600 CrossRef CAS PubMed.
- (a) T. Arliguie, B. Chaudret, F. Jalon and F. Lahoz, J. Chem. Soc., Chem. Commun., 1988, 998 RSC; (b) T. Arliguie, B. Chaudret, F. A. Jalon, A. Otero, J. A. Lopez and F. J. Lahoz, Organometallics, 1991, 10, 1888 CrossRef CAS; (c) C. Six, B. Gabor, H. Görls, R. Mynott, P. Philipps and W. Leitner, Organometallics, 1999, 18, 3316 CrossRef CAS; (d) O. V. Ozerov, L. A. Watson, M. Pink and K. G. Caulton, J. Am. Chem. Soc., 2007, 129, 6003 CrossRef CAS PubMed; (e) M. C. Puerta, P. Valerga and M. D. Palacios, Inorg. Chem., 2008, 47, 8598 CrossRef CAS PubMed; (f) D. K. Nielsen and A. G. Doyle, Angew. Chem., Int. Ed., 2011, 50, 6056 CrossRef CAS PubMed; (g) S. Suseno and T. Agapie, Organometallics, 2013, 32, 3161 CrossRef CAS PubMed; (h) M. C. MacInnis, A. J. Ruddy, R. McDonald, M. J. Ferguson and L. Turculet, Dalton Trans., 2016, 45, 15850 RSC; (i) T. M. Hood and A. B. Chaplin, Dalton Trans., 2021, 50, 2472 RSC.
- (a) Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, VCH, Weinheim, 1995 Search PubMed; (b) Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, 2008 Search PubMed; (c) R. C. J. Atkinson, V. C. Gibson and N. J. Long, Chem. Soc. Rev., 2004, 33, 313 RSC; (d) R. Gómez Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef PubMed; (e) L. Cunningham, A. Benson and P. J. Guiry, Org. Biomol. Chem., 2020, 18, 9329 RSC; (f) P. Štěpnička, Dalton Trans., 2022, 51, 8085 RSC.
- For an example of a transition metal complex with ferrocenyl-substituted η3-allyl ligand, see: R. Wiedemann, R. Fleischer, D. Stalke and H. Werner, Organometallics, 1997, 16, 866 CrossRef CAS.
- In ferrocene itself, the cyclopentadienyls tend to retain a parallel arrangement but can rotate nearly freely along the molecular axis: (a) J. C. Green, Chem. Soc. Rev., 1998, 27, 263 RSC; (b) A. Haaland and J. E. Nilsson, Chem. Commun., 1968, 88 RSC.
- For representative examples of structurally authenticated complexes of the [Pd(η3-allyl)(A^B)]X type (A^B = chelating ferrocene ligand, X = anion), see: (a) T. Hayashi, A. Yamamoto, Y. Ito, E. Nishioka, H. Miura and K. Yanagi, J. Am. Chem. Soc., 1989, 111, 6301 CrossRef CAS; (b) A. Togni, C. Breutel, M. C. Soares, N. Zanetti, T. Gerfin, V. Gramlich, F. Spindler and G. Rihs, Inorg. Chim. Acta, 1994, 222, 213 CrossRef CAS; (c) H. C. L. Abbenhuis, U. Burckhardt, V. Gramlich, C. Köllner, P. S. Pregosin, R. Salzmann and A. Togni, Organometallics, 1995, 14, 759 CrossRef CAS; (d) A. Togni, U. Burckhardt, V. Gramlich, P. S. Pregosin and R. Salzmann, J. Am. Chem. Soc., 1996, 118, 1031 CrossRef CAS; (e) H. C. L. Abbenhuis, U. Burckhardt, V. Gramlich, A. Martelletti, J. Spencer, I. Steiner and A. Togni, Organometallics, 1996, 15, 1614 CrossRef CAS; (f) U. Burckhardt, V. Gramlich, P. Hofmann, R. Nesper, P. S. Pregosin, R. Salzmann and A. Togni, Organometallics, 1996, 15, 3496 CrossRef CAS; (g) R. Fernández-Galán, F. A. Jalón, B. R. Manzano, J. Rodríguez-de la Fuente, M. Vrahami, B. Jedlicka, W. Weissensteiner and G. Jogl, Organometallics, 1997, 16, 3758 CrossRef; (h) M. Widhalm, K. Mereiter and M. Bourghida, Tetrahedron: Asymmetry, 1998, 9, 2983 CrossRef CAS; (i) W.-P. Deng, S.-L. You, X.-L. Hou, L.-X. Dai, Y.-H. Yu, W. Xia and J. Sun, J. Am. Chem. Soc., 2001, 123, 6508 CrossRef CAS PubMed; (j) U. Nettekoven, M. Widhalm, H. Kalchhauser, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, J. Org. Chem., 2001, 66, 759 CrossRef CAS PubMed; (k) S.-L. You, X.-L. Hou, L.-X. Dai, Y.-H. Yu and W. Xia, J. Org. Chem., 2002, 67, 4684 CrossRef CAS PubMed; (l) M. Lotz, G. Kramer and P. Knochel, Chem. Commun., 2002, 2546 RSC; (m) T. Tu, Y.-G. Zhou, X.-L. Hou, L.-X. Dai, X.-C. Dong, Y.-H. Yu and J. Sun, Organometallics, 2003, 22, 1255 CrossRef CAS; (n) A. M. Johns, M. Utsunomiya, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 1828 CrossRef CAS PubMed; (o) L. Hintermann, F. Lang, P. Maire and A. Togni, Eur. J. Inorg. Chem., 2006, 1397 CrossRef CAS; (p) M. Lamač, J. Tauchman, I. Císařová and P. Štěpnička, Organometallics, 2007, 26, 5042 CrossRef; (q) M. Lamač, I. Císařová and P. Štěpnička, New J. Chem., 2009, 33, 1549 RSC; (r) N. Debono, A. Labande, E. Manoury, J.-C. Daran and R. Poli, Organometallics, 2010, 29, 1879 CrossRef CAS; (s) K. E. Thiesen, K. Maitra, M. M. Olmstead and S. Attar, Organometallics, 2010, 29, 6334 CrossRef CAS; (t) C. Chen, T. M. J. Anselment, R. Fröhlich, B. Rieger, G. Kehr and G. Erker, Organometallics, 2011, 30, 5248 CrossRef CAS; (u) M. Zábranský, W. Oberhauser, G. Manca, I. Císařová and P. Štěpnička, Organometallics, 2019, 38, 1534 CrossRef; (v) P. Vosáhlo, L. Radal, M. Labonde, I. Císařová, J. Roger, N. Pirio, J.-C. Hierso and P. Štěpnička, Organometallics, 2021, 40, 1934 CrossRef and ref. 8e.
- (a) P. Štěpnička and I. Císařová, Collect. Czech. Chem. Commun., 2006, 71, 215 CrossRef; (b) P. Štěpnička and I. Císařová, Inorg. Chem., 2006, 45, 8785 CrossRef PubMed; (c) R. T. Stemmler and C. Bolm, Synlett, 2007, 1365 CAS; (d) P. Štěpnička, J. Organomet. Chem., 2008, 693, 297 CrossRef; (e) P. Štěpnička, M. Lamač and I. Císařová, J. Organomet. Chem., 2008, 693, 446 CrossRef; (f) D. Schaarschmidt, A. Hildebrandt, S. Bock and H. Lang, J. Organomet. Chem., 2014, 751, 753 CrossRef.
- P. Štěpnička and I. Císařová, J. Organomet. Chem., 2006, 691, 2863 CrossRef and ref. 12q.
- K. Škoch, I. Císařová, J. Schulz, U. Siemeling and P. Štěpnička, Dalton Trans., 2017, 46, 10339 RSC and ref. 12o and 12p.
- S. Gaillard and J.-L. Renaud, Dalton Trans., 2013, 42, 7255 RSC.
- For representative examples, see: (a) I. R. Butler, W. R. Cullen, F. W. B. Einstein and A. C. Willis, Organometallics, 1985, 4, 603 CrossRef CAS; (b) H. Seo, H. Park, B. Y. Kim, J. H. Lee, S. U. Son and Y. K. Chung, Organometallics, 2003, 22, 618 CrossRef CAS; (c) S. Gishig and A. Togni, Organometallics, 2004, 23, 2479 CrossRef; (d) L. Meca, D. Dvořák, J. Ludvík, I. Císařová and P. Štěpnička, Organometallics, 2004, 23, 2541 CrossRef CAS; (e) S. Gischig and A. Togni, Organometallics, 2005, 24, 203 CrossRef CAS; (f) J.-C. Shi, P.-Y. Yang, Q. Tong, Y. Wu and Y. Peng, J. Mol. Catal. A: Chem., 2006, 259, 7 CrossRef CAS; (g) F. Visentin and A. Togni, Organometallics, 2007, 26, 3746 CrossRef CAS; (h) A. Labande, J.-C. Daran, E. Manoury and R. Poli, Eur. J. Inorg. Chem., 2007, 1205 CrossRef CAS; (i) J. Shi, P. Yang, Q. Tong and L. Jia, Dalton Trans., 2008, 938 RSC; (j) S. Gülcemal, A. Labande, J.-C. Daran, B. Çetinkaya and R. Poli, Eur. J. Inorg. Chem., 2009, 1806 CrossRef; (k) N. Debono, A. Labande, E. Manoury, J.-C. Daran and R. Poli, Organometallics, 2010, 29, 1879 CrossRef CAS; (l) J. Csizmadiová, M. Mečiarová, A. Almássy, B. Horváth and R. Šebesta, J. Organomet. Chem., 2013, 737, 47 CrossRef; (m) A. Labande, N. Debono, A. Sournia-Saquet, J.-C. Daran and R. Poli, Dalton Trans., 2013, 42, 6531 RSC; (n) P. Loxq, N. Debono, S. Gülcemal, J.-C. Daran, E. Manoury, R. Poli, B. Çetinkaya and A. Labande, New J. Chem., 2014, 38, 338 RSC; (o) K. Škoch, I. Císařová, F. Uhlík and P. Štěpnička, Dalton Trans., 2018, 47, 16082 RSC; (p) K. Škoch, J. Schulz, I. Císařová and P. Štěpnička, Organometallics, 2019, 38, 3060 CrossRef; (q) K. Škoch, P. Vosáhlo, I. Císařová and P. Štěpnička, Dalton Trans., 2020, 49, 1011 RSC.
- (a) I. R. Butler and W. R. Cullen, Organometallics, 1984, 3, 1846 CrossRef CAS; (b) J. Schulz, I. Císařová, R. Gyepes and P. Štěpnička, Dalton Trans., 2022, 51, 6410 RSC.
- (a) G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed. Engl., 1978, 17, 569 CrossRef; (b) D. J. Berry, C. V. Digiovanna, S. S. Metrick and R. Murugan, Arkivoc, 2001, 201 Search PubMed; (c) R. Murugan and E. F. V. Scriven, Aldrichimica Acta, 2003, 36, 21 CAS.
- (a) P. Fitton, M. P. Johnson and J. E. McKeon, Chem. Commun., 1968, 6 RSC; (b) T. Yamamoto, O. Saito and A. Yamamoto, J. Am. Chem. Soc., 1981, 103, 5600 CrossRef CAS; (c) T. Hayashi, T. Hagihara, M. Konishi and M. Kumada, J. Am. Chem. Soc., 1983, 105, 7767 CrossRef CAS; (d) C. Amatore, A. Jutand, G. Meyer and L. Mottier, Chem. – Eur. J., 1999, 5, 466 CrossRef CAS; (e) C. Amatore, C. Gamez and A. Jutand, Chem. – Eur. J., 2001, 7, 1273 CrossRef CAS PubMed.
- J. Schraml, M. Čapka and V. Blechta, Magn. Reson. Chem., 1992, 30, 544 CrossRef CAS.
- The 13C{1H} NMR spectra further displayed the characteristic signals due to ferrocene Cipso at δC 88.3 (C–CH2) and 88.5 (d, 1JPC = 47 Hz, C–P) for 1Ph, and at δC 90.6 (C–CH2) and 85.1 (d, 1JPC = 347 Hz, C–P) for 1Cy.
- P. S. Pregosin and R. Salzmann, Coord. Chem. Rev., 1996, 155, 35 CrossRef CAS.
- S. F. A. Kettle and R. Mason, J. Organomet. Chem., 1966, 5, 573 CrossRef CAS.
- T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335 CrossRef CAS.
- (a) H. Solařová, I. Císařová and P. Štěpnička, Organometallics, 2014, 33, 4131 CrossRef; (b) K. Škoch, I. Císařová and P. Štěpnička, Organometallics, 2015, 34, 1942 CrossRef See also; (c) J. Tauchman, I. Císařová and P. Štěpnička, Dalton Trans., 2011, 40, 11748 RSC.
- Complex 5Ph was already reported albeit without any synthetic details and proper characterization, see: V. S. Tolkunova, A. Z. Rubezhov, V. I. Bakhmutov and V. D. VIl'chevskaya, Russ. Chem. Bull., 1981, 30, 1826 CrossRef.
- (a) D. R. Scott and R. S. Becker, J. Chem. Phys., 1961, 35, 516 CrossRef CAS; (b) U. Salzner, J. Chem. Theory Comput., 2013, 9, 4064 CrossRef CAS PubMed.
- The second, irreversible oxidation is relatively weaker and seems to produce electrochemically-active species reflected as weak reductive waves during repeated scans.
- G. Gritzner and J. Kůta, Pure Appl. Chem., 1984, 56, 461 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- The reduction remained fully irreversible even at relatively higher scan rates (1 V s−1).
- (a) C. Amatore, M. Azzabi and A. Jutand, J. Organomet. Chem., 1989, 363, C41 CrossRef CAS; (b) C. Amatore, M. Azzabi and A. Jutand, J. Am. Chem. Soc., 1991, 113, 8375 CrossRef CAS.
- K. H. Shaughnessy, Isr. J. Chem., 2020, 60, 180 CrossRef CAS.
- (a) P. Leoni, Organometallics, 1993, 12, 2432 CrossRef CAS; (b) S. R. Stauffer, N. A. Beare, J. P. Stambuli and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 4641 CrossRef CAS PubMed; (c) D. B. Grotjahn, Y. Gong, L. Zakharov, J. A. Golen and A. L. Rheingold, J. Am. Chem. Soc., 2006, 128, 438 CrossRef CAS PubMed; (d) E. A. Mitchell and M. C. Baird, Organometallics, 2007, 26, 5230 CrossRef CAS; (e) D. M. Norton, E. A. Mitchell, N. R. Botros, P. G. Jessop and M. C. Baird, J. Org. Chem., 2009, 74, 6674 CrossRef CAS PubMed.
- For representative examples, see: (a) M. S. Viciu, F. R. Germaneau, O. Navarro-Fernandez, E. D. Stevens and S. P. Nolan, Organometallics, 2002, 21, 5470 CrossRef CAS; (b) O. Navarro, H. Kaur, P. Mahjoor and S. P. Nolan, J. Org. Chem., 2004, 69, 3173 CrossRef CAS PubMed; (c) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101 CrossRef CAS PubMed; (d) L. L. Hill, J. L. Crowell, S. L. Tutwiler, N. L. Massie, C. C. Hines, S. T. Griffin, R. D. Rogers, K. H. Shaughnessy, G. A. Grasa, C. C. C. Johansson Seechurn, H. Li, T. J. Colacot, J. Chou and C. J. Woltermann, J. Org. Chem., 2010, 75, 6477 CrossRef CAS PubMed; (e) C. C. C. Johansson Seechurn, S. L. Parisel and T. J. Colacot, J. Org. Chem., 2011, 76, 7918 CrossRef CAS PubMed; (f) A. J. DeAngelis, P. G. Gildner, R. Chow and T. J. Colacot, J. Org. Chem., 2015, 80, 6794 CrossRef CAS PubMed; (g) P. R. Melvin, A. Nova, D. Balcells, W. Dai, N. Hazari, D. P. Hruszkewycz, H. P. Shah and M. T. Tudge, ACS Catal., 2015, 5, 3680 CrossRef CAS; (h) P. R. Melvin, D. Balcells, N. Hazari and A. Nova, ACS Catal., 2015, 5, 5596 CrossRef CAS; (i) M. R. Espinosa, A. Doppiu and N. Hazari, Adv. Synth. Catal., 2020, 362, 5062 CrossRef CAS PubMed; (j) P. M. MacQueen, R. Holley, S. Ghorai and T. J. Colacot, Organometallics, 2023, 42, 2644 CrossRef CAS.
- Selected reviews: (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed; (b) M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689 CrossRef CAS PubMed; (c) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (d) O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J.-E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373 CrossRef PubMed.
- Selected reviews: (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) R. K. Dieter, Tetrahedron, 1999, 55, 4177 CrossRef CAS; (c) M. Blangetti, H. Rosso, C. Prandi, A. Deagostino and P. Venturello, Molecules, 2013, 18, 1188 CrossRef CAS PubMed; (d) J. Buchspies and M. Szostak, Catalysts, 2019, 9, 53 CrossRef.
- For an overview, see: X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986 RSC.
- In principle, the coordinated allyl group can enter into the standard allylic substitution, see: K. Troshin, C. Schindele and H. Mayr, J. Org. Chem., 2011, 76, 9391 CrossRef CAS PubMed and ref. 32.
- P. Štěpnička and T. Baše, Inorg. Chem. Commun., 2001, 4, 682 CrossRef.
- P. Štěpnička and I. Císařová, Dalton Trans., 2013, 42, 3373 RSC.
- (a) A. W. Smalley, Org. Prep. Proced. Int., 1978, 10, 195 CrossRef CAS; (b) P. Kübler and J. Sundermeyer, Dalton Trans., 2014, 43, 3750 RSC; (c) K. A. Ahrendt, R. G. Bergman and J. A. Ellman, Org. Lett., 2003, 5, 1301 CrossRef CAS PubMed.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of 5R, description of the catalytic experiments, additional structure diagrams and crystallographic parameters, cyclic voltammograms of 1Cy and 5Cy, and copies of the NMR spectra. CCDC 2331020 and 2331021. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00961d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |