
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,2,3-Triazol-5-ylidene- vs. 1,2,3-triazole-based tricarbonylrhenium(I) complexes: influence of a mesoionic carbene ligand on the electronic and biological properties†
Corinne
Vanucci-Bacqué
 a,
Mariusz
Wolff
bc,
Béatrice
Delavaux-Nicot
d,
Abanoub Mosaad
Abdallah
ae,
Sonia
Mallet-Ladeira
f,
Charles-Louis
Serpentini
g,
Florence
Bedos-Belval
a,
Kar Wai
Fong
h,
Xiao Ying
Ng
h,
May Lee
Low
i,
Eric
Benoist
*a and
Suzanne
Fery-Forgues
*a
a,
Mariusz
Wolff
bc,
Béatrice
Delavaux-Nicot
d,
Abanoub Mosaad
Abdallah
ae,
Sonia
Mallet-Ladeira
f,
Charles-Louis
Serpentini
g,
Florence
Bedos-Belval
a,
Kar Wai
Fong
h,
Xiao Ying
Ng
h,
May Lee
Low
i,
Eric
Benoist
*a and
Suzanne
Fery-Forgues
*a
aSPCMIB, CNRS UMR 5068, Université de Toulouse III Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 9, France. E-mail: eric.benoist@univ-tlse3.fr; suzanne.fery-forgues@univ-tlse3.fr
bInstitut für Funktionelle Materialien und Katalyse, Universität Wien, Währinger Straße 38-42, 1090 Wien, Österreich
cInstitute of Chemistry, University of Silesia in Katowice, Szkolna 9th Street, 40-006 Katowice, Poland
dLaboratoire de Chimie de Coordination du CNRS, UPR 8241, 205 route de Narbonne, 31077 Toulouse Cedex 4, France
eNarcotic Research Department, National Center for Social and Criminological Research (NCSCR), Giza 11561, Egypt
fService Diffraction des Rayons X, Institut de Chimie de Toulouse, ICT-UAR 2599, Université de Toulouse III Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 9, France
gLaboratoire SOFTMAT, CNRS UMR 5623, Université de Toulouse III Paul Sabatier, 118 route de Narbonne, 31062 Toulouse cedex 9, France
hSchool of Postgraduate Studies, IMU University, Kuala Lumpur, Malaysia
iFaculty of Pharmaceutical Sciences, UCSI University, Kuala Lumpur, Malaysia
First published on 13th May 2024
Abstract
The tricarbonylrhenium complexes that incorporate a mesoionic carbene ligand represent an emerging and promising class of molecules, the solid-state optical properties of which have rarely been investigated. The aim of this comprehensive study is to compare three of these complexes with their 1,2,3-triazole-based analogues. The Hirshfeld surface analysis of the crystallographic data revealed that the triazolylidene derivatives are more prone to π–π interactions than their 1,2,3-triazole-based counterparts. The FT-IR and electrochemical data indicated a stronger electron donor effect from the organic ligand to the rhenium atom for triazolylidene derivatives, which was confirmed by DFT calculations. All compounds were phosphorescent in solution, where the 1,2,3-triazole-based complexes showed unusually strong dependence on dissolved oxygen. All compounds also emitted in the solid state, some of them exhibited marked solid-state luminescence enhancement (SLE) effect. The 1,2,3-triazole based complex Re-Phe even displayed astounding photoluminescence efficiency with quantum yield up to 0.69, and proved to be an excellent candidate for applications linked to aggregation-induced emission (AIE). Interestingly, one triazolylidene-based complex (Re-T-BOP) showed attractive antibacterial activity. This study highlights the potential of these new molecules for applications in the fields of photoluminescent and therapeutic materials, and provides the first bases for the design of efficient molecules in these research areas.
Introduction
Over the past few years, 1,2,3-triazol-5-ylidenes have developed as a burgeoning subclass of mesoionic carbene ligands.1–3 Their modular synthesis and impressive sigma electron-donating properties have made them excellent ligands for transition metals. Many of the resulting Ir(III),4 Au(III),5 Ru(II) and Os(II)6 complexes exhibit very attractive physicochemical and biological properties. Curiously, rhenium(I) complexes are completely under-represented in these studies. An exception is the pioneering work of Sarkar and co-workers. By comparing seven tricarbonylrhenium(I) complexes, this group has established through detailed FTIR spectroscopy and electrochemical investigations that triazolylidenes are indeed stronger electron donors than pyridine and triazoles,7 and that these phosphorescent complexes are excellent electrocatalysts for the reduction of CO2 to CO.8 If considering a broader perspective, other types of tricarbonylrhenium(I) N-heterocyclic carbenes (NHC) complexes have proven to be particularly valuable.9,10 Many of them are very efficient electrocatalysts and photocatalysts.10–14 They emit blue-green to red light in solution at room temperature15–21 and have been proposed as efficient circularly polarized luminescence emitters.22–24 According to Massi and co-workers, they exhibit a particular photochemical reactivity15,16 and can be used as photoinitiators of polymerization upon visible light.25 Regarding biological applications, these complexes are precursors for radiopharmaceuticals and diagnostic agents.26 They have been tested as luminescent markers for aminoacids,27 imaging probes to detect amyloid plaques of Alzheimer's disease,28,29 anticancer drugs30,31 and antibacterial agents.32 In light of these inspiring examples, this research field deserves to be more widely explored, especially when it comes to tricarbonylrhenium(I) complexes incorporating a 1,2,3-triazol-5-ylidene ligand, and to their solid-state photoluminescence (PL) and biological properties.Recently, our group has investigated tricarbonylrhenium(I) complexes with the aim to develop photoluminescent materials, which are actively sought after for applications in the fields of bioimaging, sensors, optoelectronic systems and security devices.33–38 The chloride ancillary ligand endows our complexes with good photochemical stability. The bidentate pyridyl-triazole (pyta) moiety, appended with an aromatic ring, allows the luminescence behavior to be finely tuned. The pyta moiety has excellent chelating ability and exists in various isomeric forms. It appeared that the complexes built with a 3-(2-pyridyl)-1,2,4-triazole ligand, e.g. the phenyl-substituted complex Re-[1,2,4]-Phe, the benzoxazole-substituted complex Re-[1,2,4]-BOP (Fig. 1) and their derivatives, display remarkable solid-state emission properties with emission quantum yields up to 0.59, rare solid-state luminescence enhancement (SLE)39–43 and, for some of them, mechanoresponsive luminescence (MRL).43–45 These properties seem to be promoted by a large phenyl-pyta dihedral angle. In contrast, complex Re-BOP that incorporates an almost planar organic ligand based on a 4-(2-pyridyl)-1,2,3-triazole moiety is only weakly emissive in the solid state,39 and there is no information on how other members of this series may behave. Consequently, it seemed instructive to compare a series of original complexes based on pyridyl-triazolylidene (pytrz), and more exactly pyridyl-1,2,3-triazol-5-ylidene, with their poorly-known pyridyl-1,2,3-triazole counterparts, in order to move forwards two steps at once. Variations of the electron-donating capacity of the ligand should impact the electronic properties in solution and biological properties. Changes of the molecular shape and intermolecular arrangement should strongly affect the solid-state PL properties.38 To this end, triazolylidene-based complexes Re-T-BOP and Re-T-Phe were synthesized, as well as complex Re-T-Tol that allows studying the influence of the methyl group in the ortho position of the phenyl ring. In fact, tiny modifications of the organic ligand are enough to change drastically the PL properties of the complexes.44 The triazole-based complexes Re-Phe and Re-Tol were prepared for the sake of comparison, in addition to the data already available about Re-BOP.39 The electronic properties of the new complexes were studied both experimentally and theoretically. Their comparison brings out unexpected information about the factors that govern the PL properties and highlights the exceptional potential of one of the molecules. To the best of our knowledge, the present study is the first one that considers the influence of mesoionic carbene ligands on the solid-state photoluminescence properties of rhenium(I) complexes. Additionally, some rhenium(I) complexes have been recently reported to be efficient antibacterial agents,46,47 a capability currently in great demand to address the growing danger raised by antibiotic-resistant bacteria.48 This prompted us to evaluate the antibacterial properties of our complexes, and one triazolylidene-based complex appeared as a promising candidate.
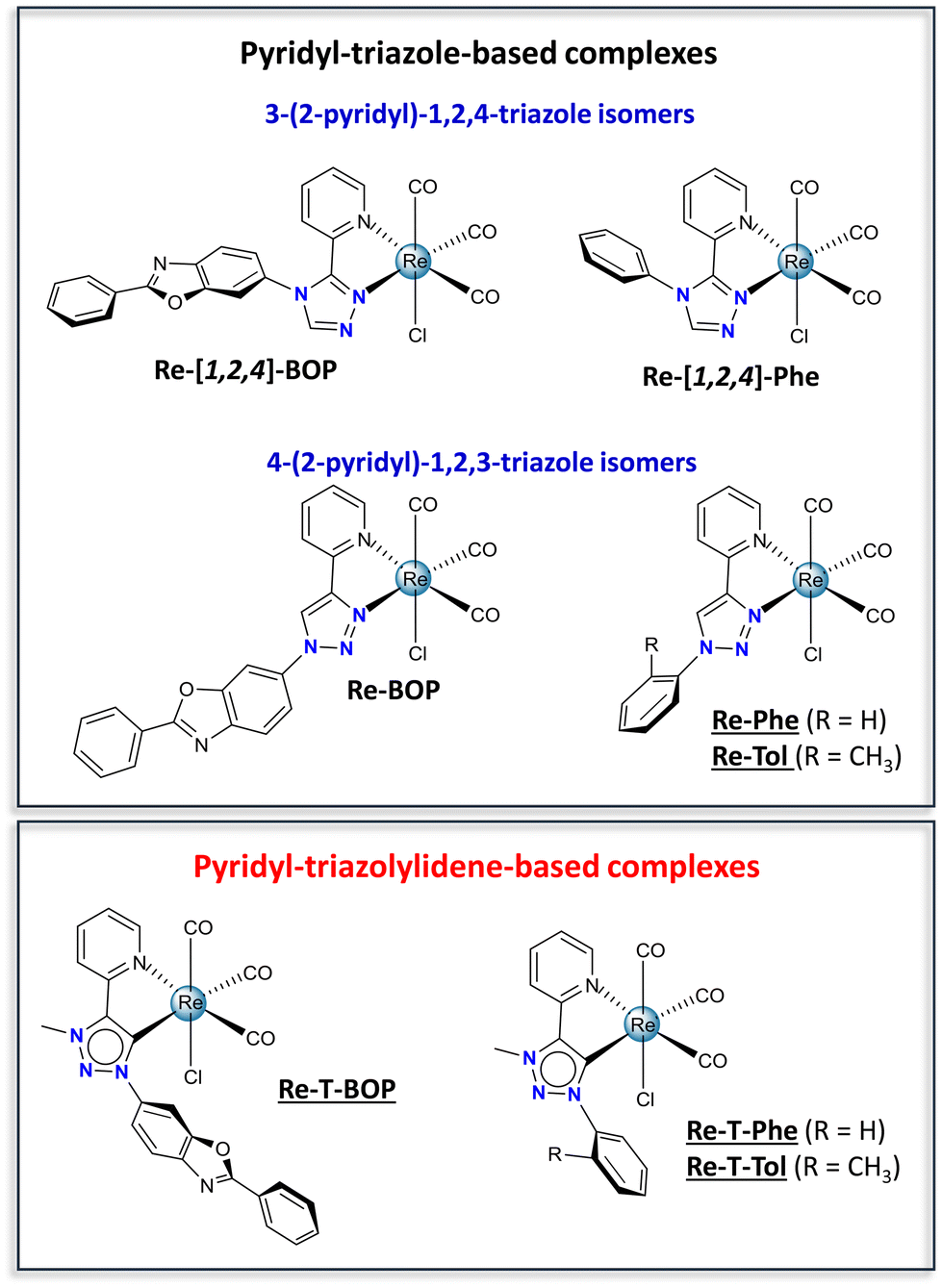 | ||
| Fig. 1 Chemical structures of the pyridyl-triazole-based complexes (Re-[1,2,4]-BOP and Re-BOP,39Re-[1,2,4]-Phe,42Re-Phe and Re-Tol), and pyridyl-triazolylidene-based complexes (Re-T-BOP, Re-T-Phe and Re-T-Tol). The new complexes synthesized for the present work are underlined. | ||
Results and discussion
Synthesis and characterization
The 2-pyridyl-1,2,3-triazole ligands were obtained via condensation of an azide derivative with 2-ethynylpyridine via a copper(I) catalyzed alkyne–azide cycloaddition using classical click reaction conditions. Specifically, 2-(1-phenyl-1H-1,2,3-triazol-4-yl)pyridine L-Phe was prepared following a described one-pot procedure, in which azidobenzene was generated in situ from iodobenzene.49 The o-tolyl derivative L-Tol was obtained in 77.5% yield starting from commercially available azido-2-methylbenzene. The benzoxazole derivative L-BOP was prepared as previously described.39 Subsequent reaction of the ligands L-Phe, L-PBO and L-Tol in refluxing methanol in the presence of [Re(CO)5Cl] afforded respectively the previously described tricarbonylrhenium complexes Re-Phe50 and Re-BOP,39 as well as the new complex Re-Tol in 86% yield (Scheme 1).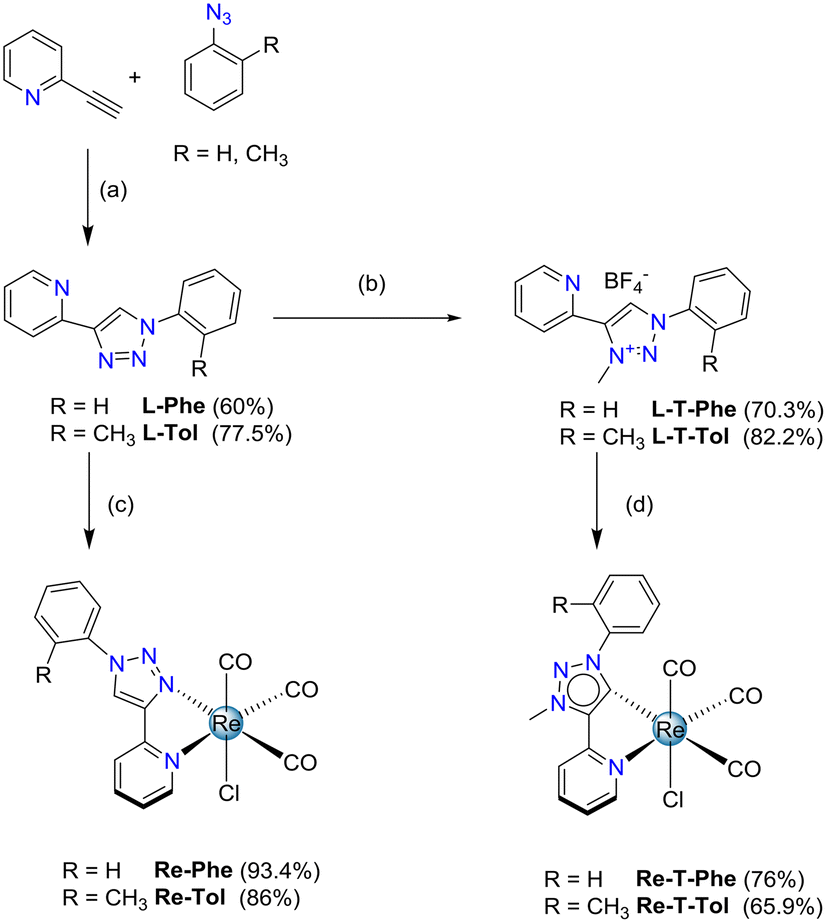 | ||
| Scheme 1 Synthesis of tricarbonylrhenium(I) complexes Re-Phe, Re-Tol, Re-T-Phe and Re-T-Tol. Conditions and reagents: (a) R = Me: CuSO4·5H2O, Na ascorbate, THF/H2O; (b) (i) m-chloro-perbenzoic acid (m-CPBA), CHCl3, reflux, 30 min; (ii) Me3OBF4, CH2Cl2, RT, 4 days; (iii) Mo(CO)6, EtOH, reflux, 1 h; (c) [Re(CO)5Cl], MeOH, 65 °C, 16 h; (d) [Re(CO)5Cl], excess of NEt3, toluene, 110 °C, 2.5 days. | ||
Concerning the methylated triazolylidene precursors, the triazolium salt L-T-Phe was selectively synthesized from L-Phe with good efficiency, using a 3-step described procedure that involves the protection of the pyridine nitrogen as an N-oxide, followed by a methylation using Meerwein's salt, and subsequent N–O bond cleavage in the presence of Mo(CO)6.51 Following a similar procedure, the triazolium salt L-T-Tol was obtained from L-Tol in 82.2% yield. However, this protocol failed when applied to the phenylbenzoxazole pyridyl-1,2,3-triazole ligand L-T-BOP, as the methylation step only afforded an unusable mixture of compounds. Thus, direct methylation of the ligand in the presence of methyl trifluoromethane sulfonate (MeOTf) was performed, leading to a mixture of the expected product L-T-BOP in 25% yield, along with the undesired product N-MP-BOP resulting from the methylation of the pyridine nitrogen, which occurred in 43% yield (Scheme 2). The syntheses of the tricarbonylrhenium complexes from the triazolium salts7 were finally accomplished in the presence of an excess of NEt3 in refluxing toluene for more than two days, leading to Re-T-Phe, Re-T-Tol and Re-T-BOP in good yields (76%, 65.9% and 69%, respectively).
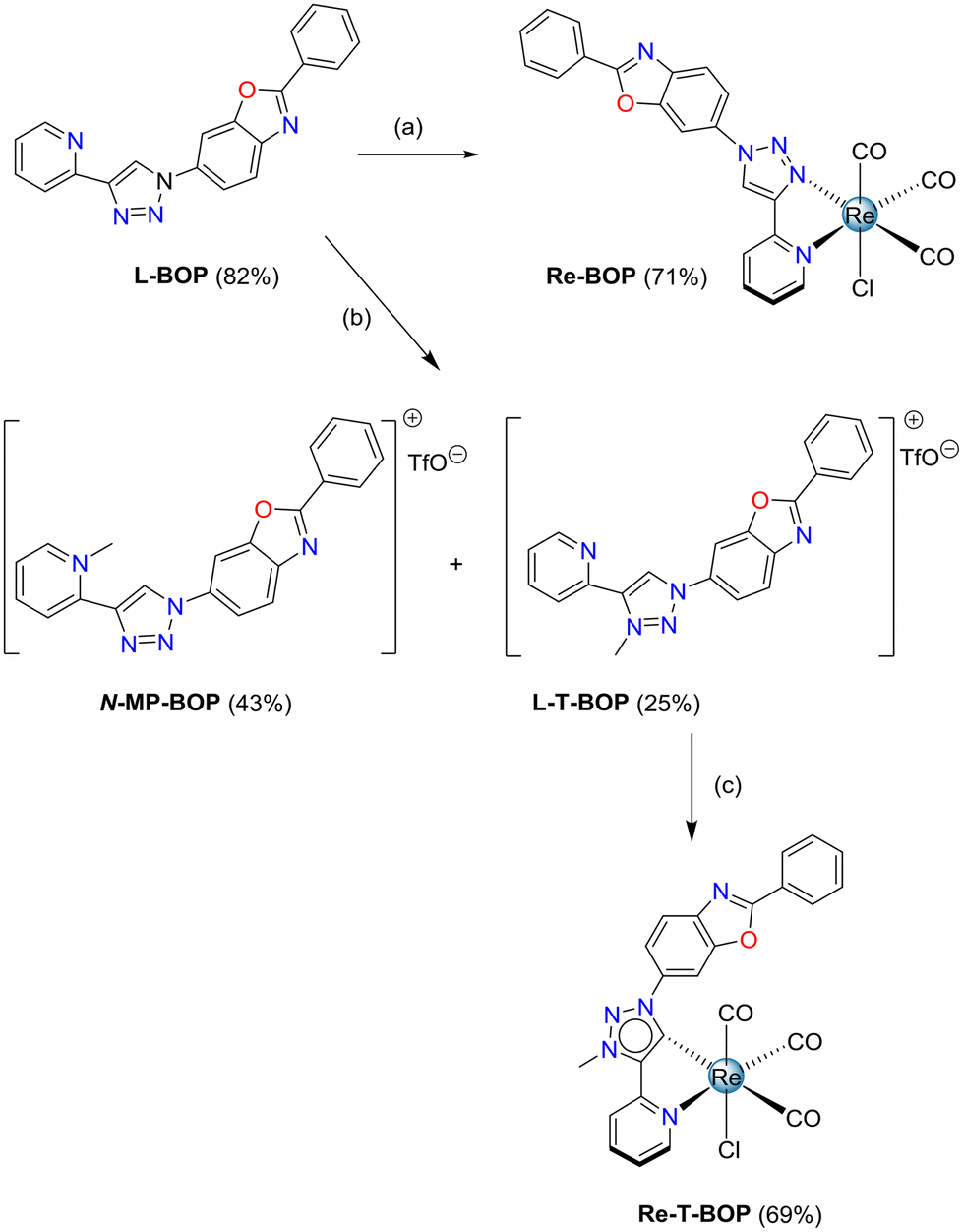 | ||
| Scheme 2 Synthesis of tricarbonylrhenium(I) complexes Re-BOP and Re-T-BOP. Conditions and reagents: (a) [Re(CO)5Cl], MeOH, 65 °C, 16 h; (b) reflux, MeOTf, CH2Cl2, 16 h; (c) [Re(CO)5Cl], NEt3 toluene, 110 °C, 2 days. | ||
The new compounds were characterized by 1H and 13C NMR spectroscopy (Fig. S1–8†), mass spectrometry (Fig. S9–12†), single crystal X-ray diffraction analysis (vide infra) and FT-IR spectroscopy (Table S1 and Fig. S13, 14†). It is noteworthy that the IR stretching bands of all complexes were split when using the attenuated total reflectance (ATR) technique on microcrystalline powders, which is not the case in solution. This effect, already noticed by us for closely related complexes, was attributed to crystallinity.41,43 According to Sarkar's group, the average of the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O values measured in solution accurately reflects the electron density on the metal center.7 In the present case, these values were found to be very close for the compounds of the same series, showing the weak effect of substitution. In contrast, they were significantly lower for the triazolylidene-based complexes (νCO ∼ 1939 cm−1) than for their triazole-based counterparts (νCO ∼ 1951 cm−1), indicating that the introduction of the mesoionic carbene leads to an increase in the overall donor capacity of the organic ligand, in good agreement with the literature.7 The purity of the complexes was checked by elemental analysis.
O values measured in solution accurately reflects the electron density on the metal center.7 In the present case, these values were found to be very close for the compounds of the same series, showing the weak effect of substitution. In contrast, they were significantly lower for the triazolylidene-based complexes (νCO ∼ 1939 cm−1) than for their triazole-based counterparts (νCO ∼ 1951 cm−1), indicating that the introduction of the mesoionic carbene leads to an increase in the overall donor capacity of the organic ligand, in good agreement with the literature.7 The purity of the complexes was checked by elemental analysis.
Crystal structures
X-Ray-quality crystals of Re-Phe, Re-T-Phe, Re-Tol and Re-T-Tol were grown from CH3CN, CH3Cl/pentane, CH2Cl2 and CH2Cl2/CH3CN, respectively. Selected crystallographic data are collected in Tables S2–4.†The coordination sphere of all complexes exhibits a quasi-octahedral geometry, as confirmed by the calculation of the distortion parameters (Table S5†). In every case, the rhenium atom is coordinated to three carbonyl groups in a fac configuration, one chlorine atom and the nitrogen atom of the pyridyl ring. It is also coordinated to the triazolylidene C(4) atom for the Re-T-Phe and Re-T-Tol complexes, and to one of the triazole nitrogen atoms for Re-Phe and Re-Tol. Distances and angles of the coordination sphere are quite close for the various compounds, and the pyridyl-triazolylidene and pyridyl-triazole moieties are almost planar. In contrast, significant differences appear when considering the molecular geometry. The most striking indicator is probably the dihedral angle α between the phenyl ring and the triazolylidene or triazole moiety. For instance, Re-T-Phe is a moderately twisted structure, whose α value is 42.8°. The α angle widens till 78.6° for Re-T-Tol due to steric hindrance of the methyl group borne by the phenyl with the triazolylidene ring, on the one hand, and the carbonyl ligand, on the other hand (Fig. 2). Triazole-based complexes, where the phenyl ring is far from the coordination sphere, are comparatively less twisted. Complex Re-Phe exhibits an angle of 34.6°, which increases to 54.2° for Re-Tol. It is instructive to compare the effect of the benzoxazole substituent. The X-ray structure of Re-T-BOP could not be obtained, but DFT calculations (vide infra) indicated that the angle in dichloromethane (DCM) solution should be around 55° (Fig. S31†). In contrast, in the triazole-based series, the presence of benzoxazole leads to planarization of the whole electron conjugated system for Re-BOP (α = 9°).39 Some substituents therefore play a very different role on the molecular geometry in both series.
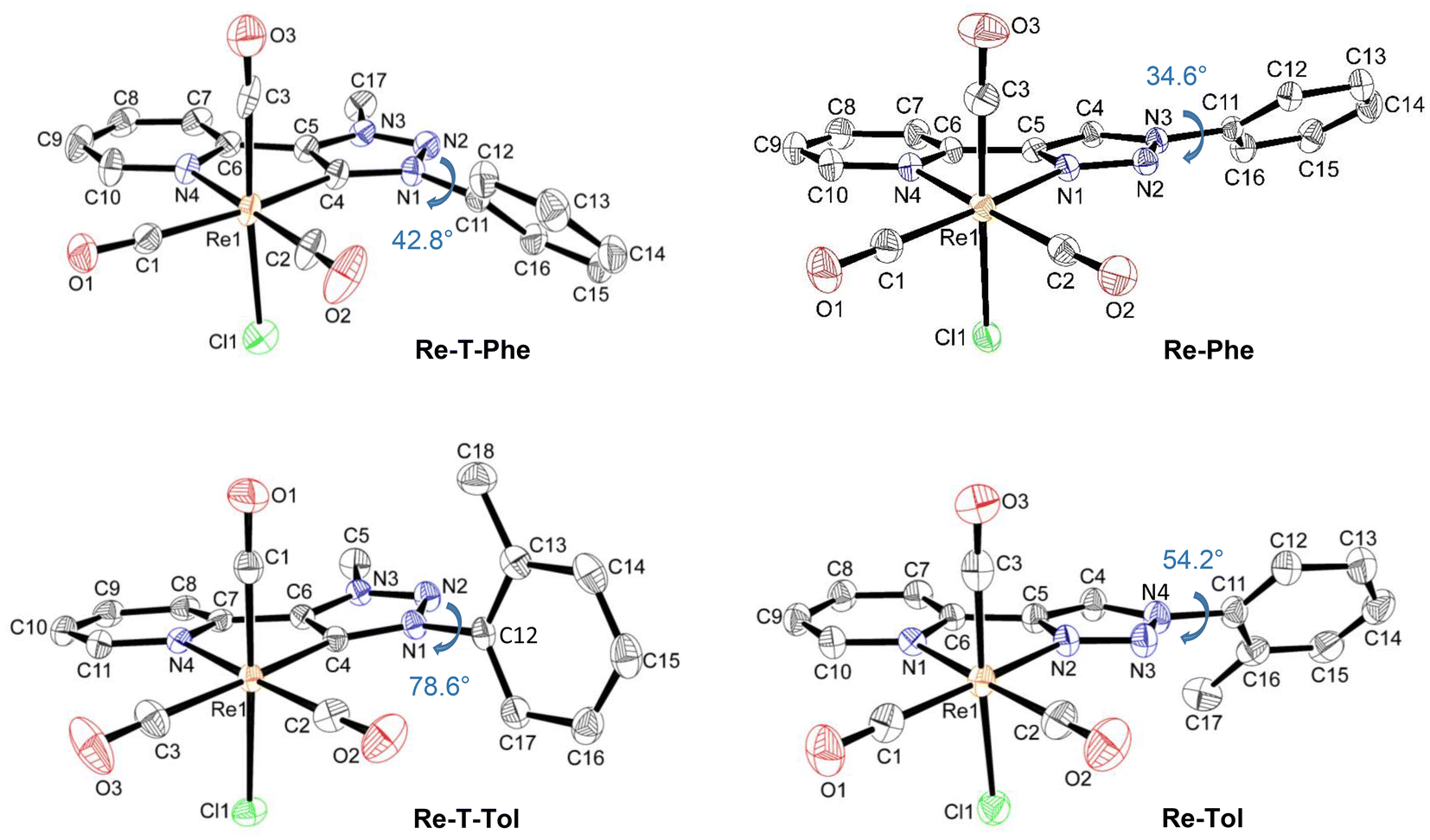 | ||
| Fig. 2 Asymmetric units of complexes Re-T-Phe, Re-T-Tol, Re-Phe and Re-Tol, with the dihedral angle α indicated in blue ink. Hydrogen atoms are not represented for the sake of clarity. Displacement ellipsoids are drawn at 30% probability. | ||
For all complexes, two enantiomers, which differ by the position of the organic ligand with respect to the chlorine atom, coexist in the crystal cell in identical proportions. The intermolecular arrangement varies from one complex to another (Fig. 3), as described in detail in the ESI (Tables S6–8 and Fig. S15–25†). Briefly, the molecules of the triazolylidene derivatives are displayed in two separate planes, at an angle of ∼40° for Re-T-Phe, and close to perpendicular for Re-T-Tol. The crystal structures are stabilized by C–H⋯O and C–H⋯Cl interactions (Table S6†). Specifically, intermolecular hydrogen bonds between the CH group of the phenyl ring and the oxygen atom of the equatorial carbonyl group induce the neighboring molecules of Re-T-Phe to assemble into an infinite one-dimensional chain, which expands along the c axis (Fig. S15†). For Re-T-Tol, the same type of chain results from interactions between the CH group of the pyridine ring and the chloride ligand of an adjacent molecule (Fig. S17†). Additionally, for both compounds, the packing arrangement promotes intermolecular C–H⋯π and π–π stacking interactions (Tables S7 and 8†), because the phenyl group of one molecule partly overlaps the pyridyl ring of a neighboring one (Cp–Cp < 3.68 Å) (Fig. 3, S16 and S18†).
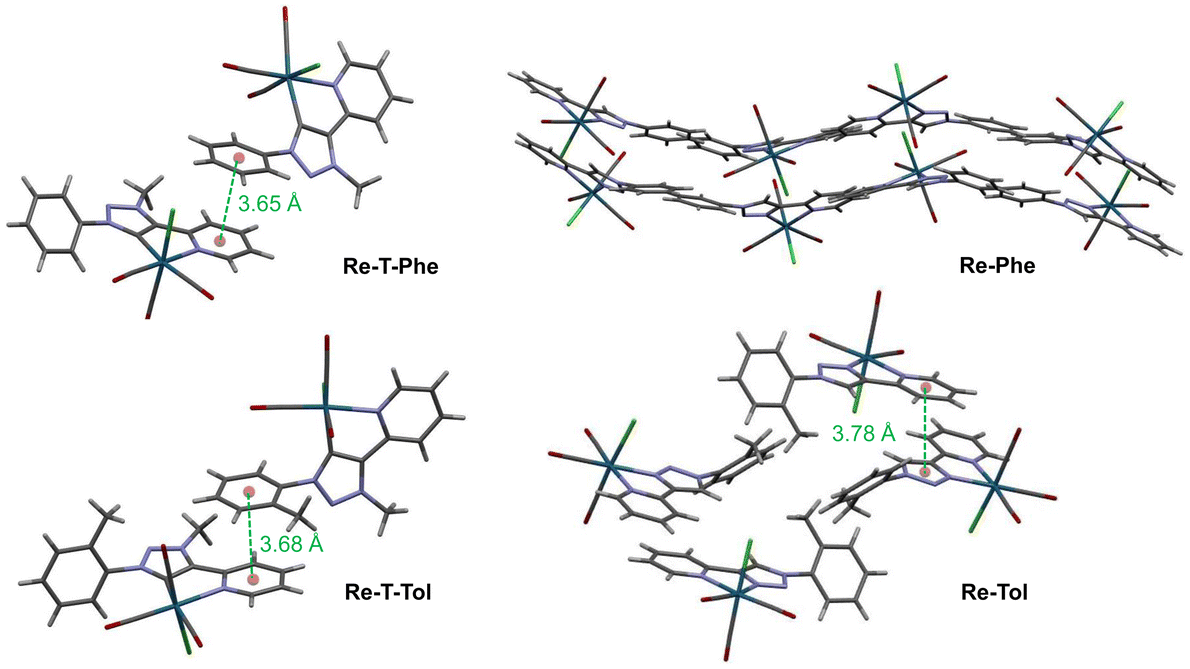 | ||
| Fig. 3 Crystal cells of Re-T-Phe, Re-T-Tol, Re-Phe and Re-Tol. The distances between centroids are indicated in green ink. | ||
Considering the triazole-based complexes Re-Phe and Re-Tol, the pyta moieties are situated on rather close planes, with their long axis oriented along four distinct directions. The neighboring molecules of Re-Tol interact with each other via C–H⋯Cl hydrogen bonds between the triazole and pyridine rings, and form a one-dimensional infinite chain along the c axis (Fig. S22†). Numerous C–H⋯O contacts induce the formation of a two-dimensional network in the ab plane (Fig. S23†). Re-Tol is further stabilized by π⋯π interactions between the pyridyl ring of one molecule and the triazole ring of a neighboring one (Cp–Cp = 3.78 Å) (Fig. S24†). Regarding Re-Phe, intermolecular C–H⋯O hydrogen bonds between one hydrogen atom of the pyridyl ring and one carbonyl oxygen atom connect the neighboring molecules, which form a one-dimensional infinite zig-zag chain along the b axis (Fig. S19†). The presence of three basic nitrogen atoms within a five-membered 1,2,3-triazole ring makes the C–H bond highly acidic, strongly enhancing its involvement in hydrogen bonding with chloride acceptors. The bifurcated hydrogen bond between the chloride ligand, on the one hand, and the C–H groups of the 1,2,3-triazole ring and pyridine ring of neighboring molecules, on the other hand, contributes to the formation of a two-dimensional network in the ac plane (Fig. S20†). Crystals of Re-Phe are further stabilized by C–H⋯π interactions (Fig. S21 and Table S7†), but they differ from those of the other analogues by the total absence of π⋯π interactions.
The Hirshfeld surface (HS) analysis52 was performed to visualize and quantify the intermolecular interactions in the crystal lattice of the four complexes. In the example given in Fig. 4a, HS were mapped over dnorm for Re-Phe and Re-T-Phe. The red spots mainly correspond to C–H⋯O and C–H⋯Cl short interatomic contacts. The white zones refer to intermolecular distances close to the sum of the van der Waals radii of the atoms considered; they indicate H⋯H interactions. The blue areas illustrate the domains where neighboring atoms are too far away to interact with each other. The comparison of the four molecules shows that the distribution of short contacts is indeed different, but the number and type of H-mediated interactions remain relatively close (Fig. S26†). In contrast, the overall molecular shape is very different. Re-Phe is the most angular molecule and the least able to offer flat surfaces where π–π stacking can take place. The shape index of the Hirshfeld surface (Fig. 4b) is a tool to visualize π–π stacking interactions by the presence of adjacent red and blue triangles. Blue triangles represent the convex region, which is formed due to the carbon atoms present in the molecule inside the surface, while red triangles represent concave regions due to the carbon atoms of the π-stacked molecule above it. Such adjacent triangles were not observed for Re-Phe, which confirms the absence of π–π stacking interactions, while this pattern can be found at least twice for the other complexes. The relative contributions of the various interactions to the total HS are shown in the 2D fingerprint plots given in Fig. S27–30.† The reciprocal C–O⋯H/H⋯O–C interactions give two sharp symmetrical spikes, with a contribution to the HS reaching 27.8% (for Re-T-Phe). The C⋯H/H⋯C interactions, which correspond to C–H⋯π interactions, appear as two symmetrical broad wings and contribute up to 23.4% (for Re-Phe). The H⋯H interactions appear in the middle of the scattered points, with an overall contribution varying between 15.1% and 23.5% (for Re-Phe and Re-T-Tol, respectively). The C–H⋯Cl interactions also generate two sharp spikes, and they contribute for ∼10%. Other interactions (N⋯H, C⋯N, C⋯O, O⋯O, C⋯C) have minor contributions. For the sake of comparison, the relative contributions of the various interactions to the HS are collected in Fig. 4c. Globally, triazole-based complexes show slightly more C⋯H interactions, and less C–H⋯Cl and O⋯H interactions, than their triazolylidene-based counterparts. Noticeably, π⋯π stacking interactions (C⋯C/C⋯C) are only 2.5% for Re-Phe, and between 4.2% and 5.8% for the other three complexes.
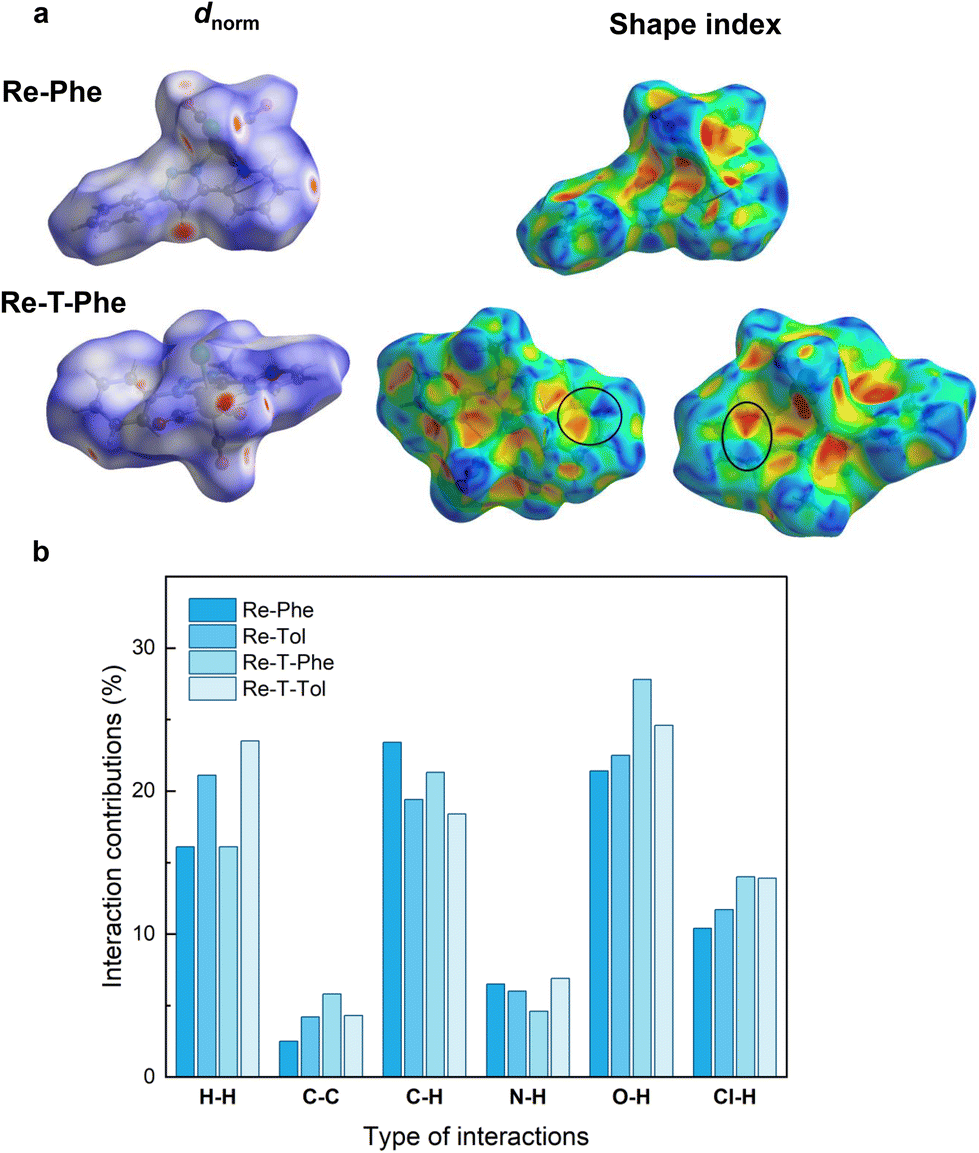 | ||
| Fig. 4 View of the Hirshfeld surface of Re-Phe and Re-T-Phe plotted over dnorm (a) and shape index (b). The adjacent red and blue triangles that reveal π⋯π stacking interactions are highlighted by black circles. (c) Percentage contributions of various interactions to the Hirshfeld surface for the four complexes. | ||
Electronic properties
Computational studies were made using the time-dependent density functional theory (TD-DFT) method considering the complexes in vacuum and in dichloromethane continuum (Tables S9–30 and Fig. S31–41†). Only the latter ones will be commented. The tolyl derivatives Re-T-Tol and Re-Tol were considered assuming that their properties are very close to those of the unsubstituted analogues. Complex Re-T-BOP was also studied to understand the influence of the BOP fragment; the results were compared to those previously acquired for Re-BOP.39 For the three new complexes, the calculated bond lengths and angles were in excellent agreement with the experimental data (Tables S9–11†). It is also the case for the FT-IR spectra (Fig. S41†). Regarding the composition of the frontier molecular orbitals (Tables S13–18, S25 and Fig. 5, S32–38†), the three highest occupied molecular orbitals (HOMO, HOMO−1 and HOMO−2) are localized on the rhenium atom and carbonyl ligands, and on the chlorine atom for the first two, as is commonly the case for tricarbonylrhenium(I) complexes. For Re-T-Tol and Re-Tol, the LUMO, L+1 and L+2 are centered on the pyta and pytrz moieties, respectively. Some differences appear for lower and higher orbitals. For example, if considering the orbitals centered on the organic ligand, the H−4 and H−5 orbitals of Re-T-Tol are concentrated on the phenyl ring, while electrons are more evenly distributed between the pyta moiety and the phenyl ring for the H−3, H−4 and H−5 orbitals of Re-Tol. Regarding Re-T-BOP, an important contribution of the PBO moiety is noticed for orbitals H−2, H−5 and H−6, as well as for L+1 and L+5 (H−2, H−4 and H−5, as well as L+1, L+2, L+3 and L+5 for Re-BOP39).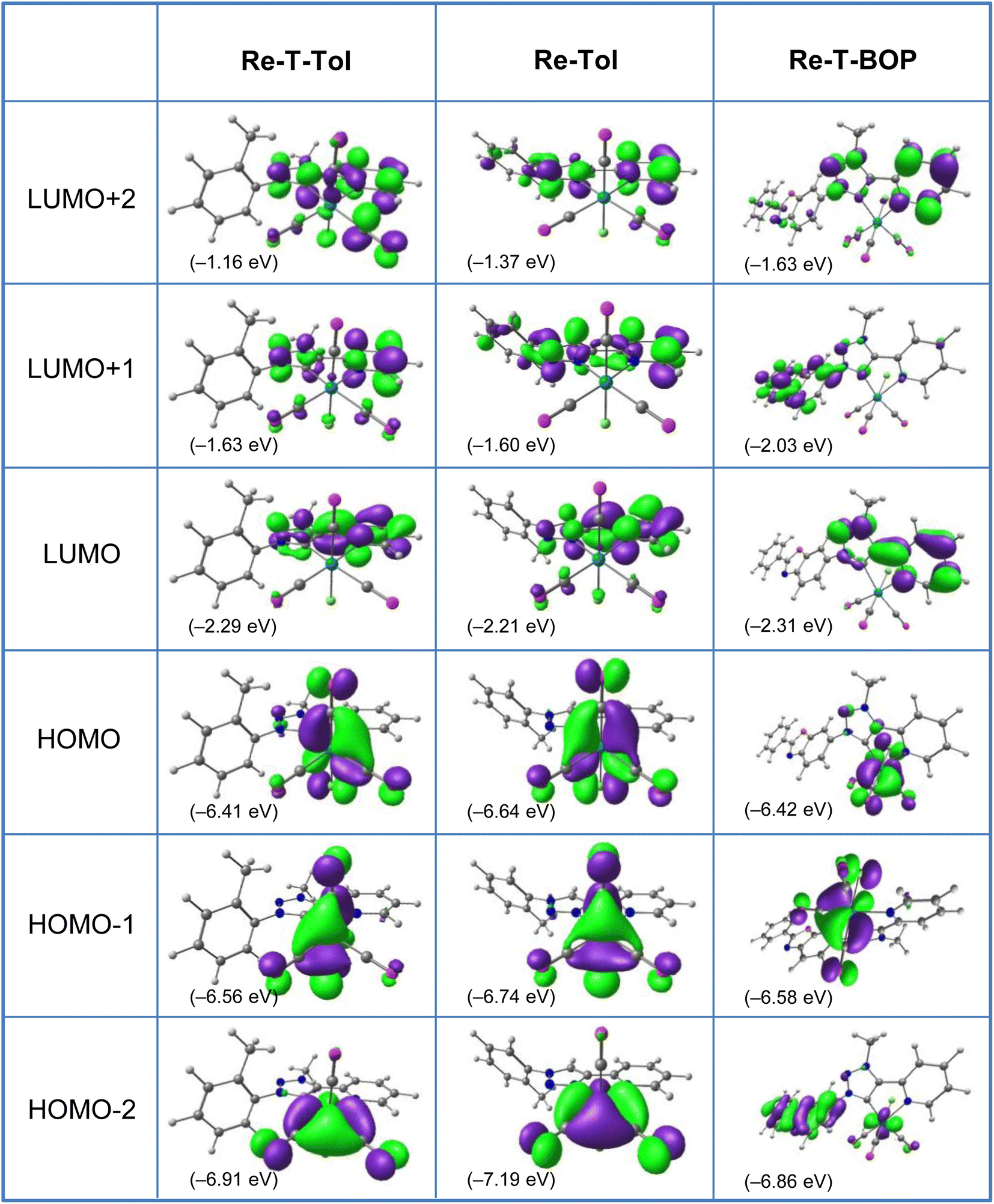 | ||
| Fig. 5 Isodensity plots (isovalue = 0.03 e bohr−3) and energy levels of the first frontier molecular orbitals, for complexes Re-T-Tol, Re-Tol and Re-T-BOP in DCM, according to DFT calculations at the PBE1PBE/LANL2DZ level of theory. | ||
For all compounds, the lowest energy transitions correspond to a shift of the electronic density from the coordination sphere to the organic ligand (Tables S19–24 and S26†). They are therefore of metal-to-ligand charge transfer (MLCT), halogen-to-ligand charge transfer (XLCT) and ligand-to-ligand charge transfer (LLCT) type. At low energy, the significantly-active transitions are H−1 → LUMO, expected at 366.2, 344.5, 364.8 and 347.4 nm for Re-T-Tol, Re-Tol, Re-T-BOP and Re-BOP, respectively. Higher-energy transitions are of various natures. Re-T-Tol is characterized by a strong MLCT transition around 244.9 nm, while Re-Tol exhibits a strong ILCT band around 236.7 nm. The transitions with the highest oscillator factor have intraligand charge transfer (ILCT) character for Re-T-BOP and Re-BOP.
Calculations allow to highlight differences in the electronic system of the two series of complexes in the ground state. For instance, according to the natural population analysis (NPA) (Table S29†) that indicates the charge transferred between the donor and acceptor moieties, the charge on the rhenium atoms is ∼−1.0 (e) in complexes Re-Tol and Re-BOP, vs. ∼−1.2 (e) in complexes Re-T-Tol and Re-T-BOP. The pyridyl nitrogen atom donates as much as 0.40 (e) to the Re atom in the first series of complexes, and 0.38 (e) in the second series. In triazole-based complexes, the triazole nitrogen atom also donates ∼0.19 (e), while the C(4) atom of the triazolylidene-based complexes is enriched with a charge of ∼0.12 (e). Consequently, there is better electron density delocalization from the organic ligand to the metal center for triazolylidene-based complexes. In every complex, the positively-charged carbon atom of the carbonyl ligands is found to accept as much as ∼0.76 (e) from the Re atom, while the chlorine atom Cl(1) gives it ∼0.47 (e).
The HOMO–LUMO gap, which reflects the chemical reactivity or stability of the molecules, was smaller for the triazolylidene-based complexes than for their triazole-based counterparts (∼4.12 eV for Re-T-Tol and Re-T-BOPvs. 4.43 eV for Re-Tol and 4.38 eV for Re-BOP 39) due to the increase of the HOMO energy level in the first case. The examination of the frontier molecular orbital descriptors (Table S30†), i.e. ionization potential, electron affinity, hardness, electronegativity, chemical potential, softness and electrophilicity index, allows to conclude that the triazolylidene-based complexes Re-T-Tol and Re-T-BOP are less kinetically stable and more chemically reactive than their triazole-based counterparts Re-Tol and Re-BOP.
Electrochemical properties
The electrochemical behavior of the new complexes Re-Tol, Re-T-Tol and Re-T-BOP was studied by cyclic voltammetry (CV) and Osteryoung square wave voltammetry (OSWV) measurements in DCM at room temperature. The results are summarized in Tables 1 and S31,† and Fig. 6, and selected curves are given in Fig. S42–50.† The new compound Re-Tol exhibited electrochemical characteristics close to those of Re-BOP.39 Thus, the first one-electronic oxidation process at 1.48 V was mainly assigned to the Re(I) oxidation, while the first reduction process around −1.60 V was ligand centered on the pyta moiety. These data also indicate that the pyridyl-1,2,3-triazole ligand is not very sensitive to the nature of its phenyl substituent. However, it is noteworthy that the first reduction process presented more reversibility at high scan rate for the Re-Tol compound than for Re-BOP (see below).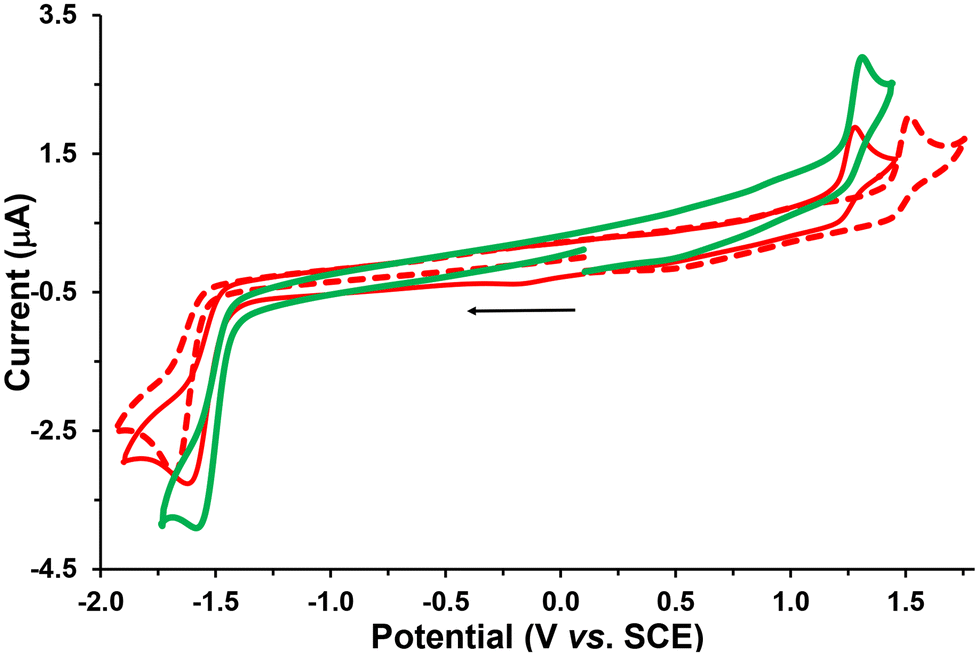 | ||
| Fig. 6 Cyclic voltammograms of the first oxidation and reduction processes of complexes Re-Tol (red dotted line), Re-T-Tol (red line), and Re-T-BOP (green line) on a Pt working electrode in CH2Cl2 + 0.1 M [nBu4N][BF4] at room temperature and at a scan rate of 0.2 V s−1 toward cathodic potentials. | ||
| Oxidation | Reduction | |||
|---|---|---|---|---|
| E 2 | E 1 | E 1 | E 2 | |
| a OSWVs were obtained using a sweep width of 20 mV, a frequency of 20 Hz, and a step potential of 5 mV. b Potential values in volts vs. SCE (Fc+/Fc is observed at 0.55 V ± 0.01 V vs. SCE). c Data from ref. 39. d Other oxidation processes at 1.69, 1.78, and 1.90 V. e More reversible process at 50 V s−1 (Fig. S44†). f More reversible process at 5 V s−1 (Fig. S46 and S47†). g More reversible process at 10 V s−1 (Fig. S50†). | ||||
| Re-Tol | 1.88 | 1.48 | −1.63e | — |
| Re-BOP | 1.79 | 1.48 | −1.60 | −1.89 |
| Re-T-Tol | 1.59 | 1.26 | −1.54f | — |
| Re-T-BOP | 1.62 | 1.27 | −1.51g | −1.85 |
The two new triazolylidene-based complexes Re-T-Tol and Re-T-BOP displayed distinct electrochemical properties. Indeed, in CV at 0.2 V s−1, two irreversible processes were detected at around 1.26 V in oxidation, and −1.52 V in reduction. These potential values clearly differed from those observed for our previously studied pyridyl-1,2,3-triazole- and pyridyl-1,2,4-triazole-based Re complexes.39–41,44 The first one-electron oxidation process occurred now at lower anodic potential (around −200 mV) when compared with those of their pyridyl-1,2,3-triazole-based counterparts. This is due to the increased electron density on the rhenium center, which is supported by theoretical calculations. These results also suggest that the triazolylidene ligands are likely better electron donors than the corresponding pyridyl-1,2,3-triazole ligands. The Re-T-BOP complex also presented a second reduction process around −1.9 V that was attributed to the reduction of the PBO moiety, thus confirming that the latter is not involved in the complexation process.39
Regarding now the literature data, the value of the first oxidation potential of the Re-T-Tol and Re-T-BOP complexes was close to that of the mesoionic carbene complexes synthesized by Suntrup et al.: 0.74–0.78 V range vs. FcH+/FcH.8 This value is not very sensitive to the nature of the R group grafted on the triazole ring, and the comparison with 1,2,3-triazole-based complexes shows that it correlates well with the overall donor strength of the corresponding organic ligands. The first reduction potential can be used as an approximation to gauge the π-acceptor capacity of our ligands, the triazolylidene ligand being a better π acceptor than the 1,2,3-triazole ligand.7 The first reduction potentials of the rhenium complexes reported in Table 1 maintain the same π acceptor character than their respective ligands.
The experimental data of Table 1 were also in good agreement with the TD-DFT calculations. In particular, the latter predict that (i) for Re-Tol and Re-BOP, the HOMO are Re-centered and very close in energy; (ii) for Re-T-Tol and Re-T-BOP, the (Re centered) HOMO and (triazolylidene-centered) LUMO present respectively the same energy level; (iii) the calculated HOMO–LUMO energy gap value Ecalc for the first series of compounds is superior to that of the second one (around 0.3 eV) as observed experimentally.
Considering further the Re-BOP and Re-T-BOP complexes, a different electrochemical behavior regarding the first reduction process was noted in CV when increasing the scan rate. This one became more reversible only for Re-T-BOP. This property is probably related to the different nature of the two compounds. Indeed, for Re-BOP, the LUMO and LUMO+1 energy levels only differ by 0.07 eV. Consequently, the first reduction potential detected at −1.60 V may originate from the contribution of both the LUMO and LUMO+1 energy levels involving respectively the π*(pyta) and π*(pyta) + π*(PBO) orbitals. In contrast, for Re-T-BOP, the energy difference between these levels is greater (0.28 eV) and allows an easier electrochemical assignment of this reduction process exclusively resulting from its π*(pytrz) orbital. This is reminiscent of the electrochemical first reduction behavior of Re-[1,2,4]-BOP, which presents a LUMO–LUMO+1 energy difference of 0.4 eV and a LUMO of exclusively π*(pyta) nature.39 In the latter case, the first reduction process becomes quasi-reversible at 1 V s−1. Concerning the Re-Tol and Re-T-Tol complexes, the LUMO–LUMO+1 energy difference is at least of 0.6 eV, and their LUMO is respectively of pure π*(pyta) or π*(pytrz) nature, suggesting that some reversibility can occur at high scan rate in CV for each compound, which is actually observed.
Finally, the values of the electrochemical HOMO–LUMO gap (Eelg) found for Re-Tol and the triazolylidene-based complexes, i.e. 3.0 and 2.7 eV, respectively, also fit very well with the calculated Ecalc gap values 3.0 and 2.8 eV, highlighting good correlations with the theoretical study (Table S31†).
UV-visible absorption and emission properties
Whatever the complex, no photochemical instability that could interfere with the measurements was detected during the spectroscopic study. The dilute DCM solutions of all complexes were almost colorless in daylight. The absorption spectra were in very good agreement with the calculated ones (Fig. S41†). All of them (Fig. 7a and Table 2) showed an intense high-energy band, attributed to a combination of ILCT and MLCT transitions, as well as a low-energy band of moderate intensity with MLCT character, almost not visible for Re-BOP. The absorption maxima of the triazolylidene-based complexes were slightly red-shifted with respect to triazole-based complexes. The absorptivity values of the high-energy band were markedly increased for both BOP derivatives, due to the extention of the conjugated electron system.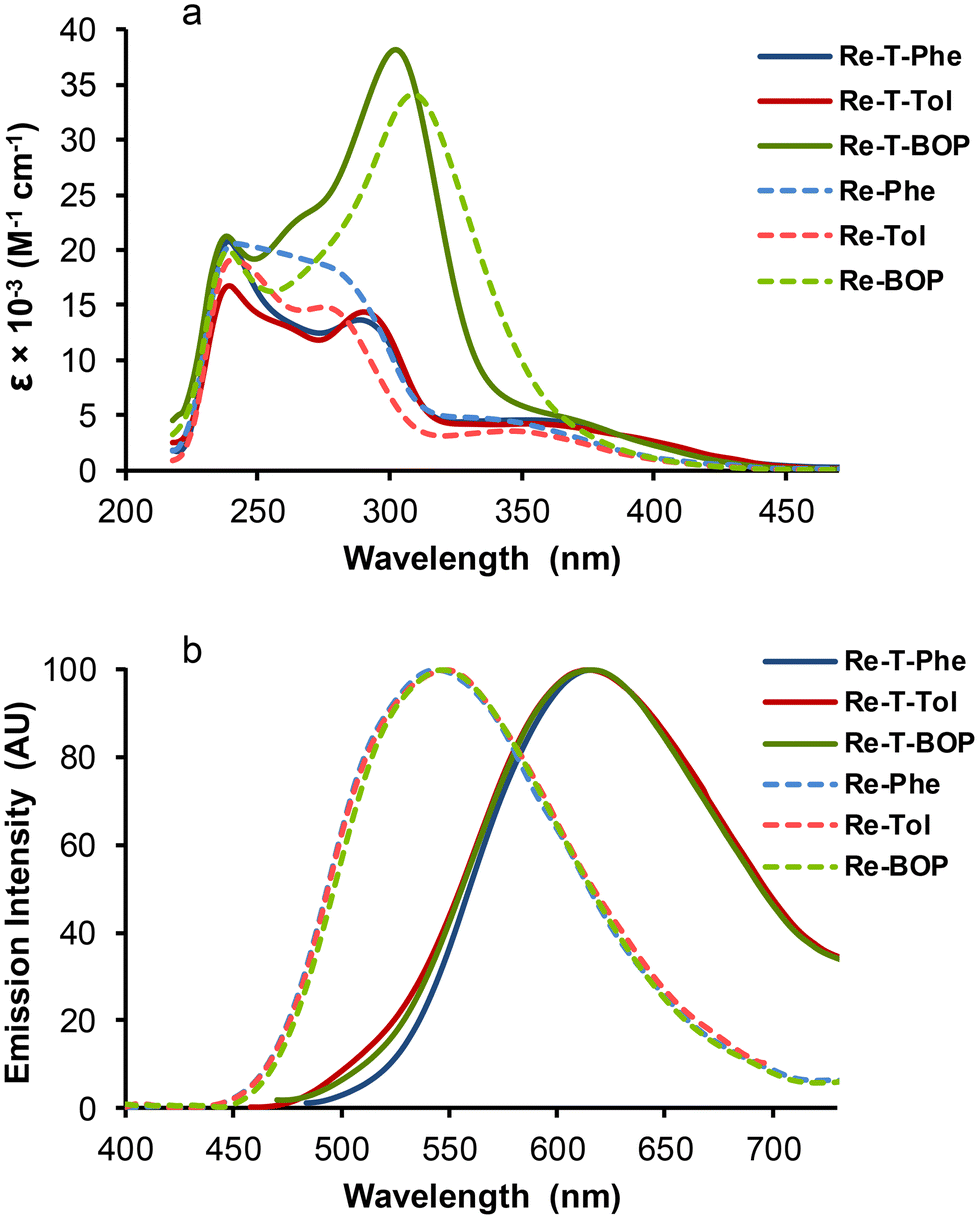 | ||
| Fig. 7 UV-vis absorption spectra (a) and normalized emission spectra (b) of complexes Re-T-Phe (blue solid line), Re-T-Tol (red solid line), Re-T-BOP (green solid line), Re-Phe (blue dotted line), Re-Tol (red dotted line) and Re-BOP (green dotted line) in undegassed DCM. Concentrations ∼3.3 × 10−5 M for absorption, 1.2 × 10−5 M for emission. Excitation in the low-energy absorption band. | ||
| Compound | DCM solutions | Powdersa | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | Φ P | τ (ns), (f), [χ2] | 〈τ〉 (ns) | k r (s−1) | k nr (s−1) | λ PL (nm) | Φ PL | τ PL (ns), (f), [χ2]b | 〈τPL〉 (ns) | |
| a Pristine powders, except for Re-Tol (ground powder). b Decays are triexponential, the full data are given in the ESI (Fig. S52†). c When not specified, data are from ref. 39. d This work. | ||||||||||||
| Re-T-Phe | 240 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
616 | 9.7 × 10−3 | 47 | 47 | 2.06 × 105 | 2.10 × 107 | 537 | 0.16 | 64 (0.03) | 692 |
| 288 | 13700 |
[1.70] | 711 (0.96) | |||||||||
| 354 | 4400 | [1.07] | ||||||||||
| Re-T-Tol | 240 | 16700 |
618 | 1.1 × 10−2 | 48 | 48 | 2.29 × 105 | 2.06 × 107 | 567 | 0.12 | 57 (0.04) | 543 |
| 290 | 14300 |
[1.19] | 570 (0.95) | |||||||||
| 354 | 4100 | [1.39] | ||||||||||
| Re-T-BOP | 238 | 21200 |
616 | 1.2 × 10−2 | 37 (0.71) | 89 | 1.35 × 105 | 1.11 × 107 | 560 | 0.04 | 73 (0.11) | 426 |
| 302 | 39200 |
217 (0.29) | 475 (0.88) | |||||||||
| 360 | 5300 | [1.32] | [1.18] | |||||||||
| Re-Phe | 242 | 20600 |
546 | 1.7 × 10−2 | 154 | 154 | 1.10 × 105 | 6.38 × 106 | 510 | 0.69 | 32 (<0.01) | 1154 |
| 278 | 18300 |
[1.08] | 1164 (0.99) | |||||||||
| 340 | 4400 | [1.28] | ||||||||||
| Re-Tol | 240 | 19100 |
546 | 1.6 × 10−2 | 145 | 145 | 1.10 × 105 | 6.79 × 106 | 528 | 0.07 | 107 (0.14) | 789 |
| 276 | 14800 |
[1.34] | 921 (0.84) | |||||||||
| 344 | 3400 | [1.53] | ||||||||||
| Re-BOP | 309 | 34100 |
546 | 1.5 × 10−2 | 4.79 (0.04) | 192 | 7.81 × 104 | 5.13 × 106 | 520d | 0.016 | 41.4 (0.01) | 410 |
| 192 (0.96) | 414 (0.99) | |||||||||||
| [1.16] | ||||||||||||
Regarding the emission properties in DCM solutions, the complexes can clearly be divided into two groups. When illuminated by UV light (365 nm), the triazolylidene-based complexes emitted very weak orange-red light, while triazole-based complexes emitted green light. All emission spectra showed a single unresolved band, peaking between around 616 nm for triazolylidene complexes and 546 nm for triazole-based complexes. For Re-T-Tol, Re-T-BOP and Re-Tol, the experimental emission maxima were close to those calculated by DFT and TD-DFT (Table S27†). Since calculations consider the involvement of the lowest 3MLCT state, the comparison of the experimental and theoretical values confirms that emission is indeed phosphorescence in every case. For triazolylidene-based complexes in aerated solutions, the quantum yields were quite low, around 1 × 10−2. The emission decays (Fig. S51†) were monoexponential for Re-T-Phe and Re-T-Tol, with lifetimes below 50 nanoseconds. For Re-T-BOP, a second lifetime at 217 ns with contribution around 29% could arise from the presence of conformers in solution. For the triazole-based complexes, the quantum yields were 1.5 fold higher, and the lifetimes were longer, between 145 and 192 ns, than for their triazolylidene-based analogues. Considering the whole set of compounds, the radiative rate constants were slightly higher, and the non-radiative rate constants were markedly higher for triazolylidene-based complexes. This suggests that these complexes are intrinsically a little more emissive than their triazole-based counterparts, but this trend is compensated by the promotion of thermal de-excitation pathways, as can be expected from the energy gap law.53
It is striking to see that the three triazolylidene derivatives, on the one hand, and the three 1,2,3-triazole derivatives, on the other hand, show great similarity in their emission properties in solution. This is explained by the fact that, within the same series of compounds, the first excited triplet state is almost the same for each derivative because the electron density is centered on the rhenium coordination sphere and does not involve the organic substituents borne by the pytrz or pyta moiety (Fig. S39†).
The effect of dissolved oxygen on the emission efficiency was investigated. Bubbling the complex solutions with argon for 4 min multiplies the emission intensity by 1.6, 2.9 and 1.3 for Re-T-Phe, Re-T-Tol and Re-T-BOP, respectively, like for the previously studied 1,2,4-triazole-based complexes. The short emission lifetimes of triazolylidene derivatives may explain the low sensitivity to oxygen quenching. In contrast, the emission was dramatically enhanced by 8.3, 12.1 and 13.4 for Re-Phe, Re-Tol and Re-BOP in the same conditions, so that the quantum yield changes by one order of magnitude. The longer lifetimes are one of the factors contributing to this effect, making the 1,2,3-triazole derivatives the most oxygen-sensitive compounds among all rhenium(I) complexes examined to date in the group.
All compounds were emissive in the solid state. Unlike solutions, where the spectra were identical by class of compounds, dramatic differences appeared between all compounds. The PL spectra covered a large range of wavelengths (Fig. 8a), with very different intensities (Fig. 8b). The microcrystalline pristine powders of the triazolylidene-based complexes emitted yellow to orange-yellow light, with moderate to low quantum yields (between 0.16 for Re-T-Phe to 0.04 for Re-T-BOP) (Table 2). Triazole-based complexes emitted green light. The emission of Re-Phe was exceptionally strong, with quantum yield around 0.69. These superior solid-state PL properties can be explained jointly by the absence of π–π staking in the crystal network and by the prevention of quenching by dissolved oxygen, to which the compound is very sensitive when in solution. Again, the BOP derivative was the weakest luminophore of the series. Curiously, pristine Re-T-Tol was virtually not emissive, but emission was induced by grinding the powder with a pestle in a mortar, which generally leads to partial amorphization of the sample, suggesting that the crystal packing mode is detrimental to light emission. The influence of mechanical stimulus was investigated by grinding the pristine powders of the other five complexes, but no significant differences were observed, ruling out any mechanoresponsive luminescence behavior for these compounds.
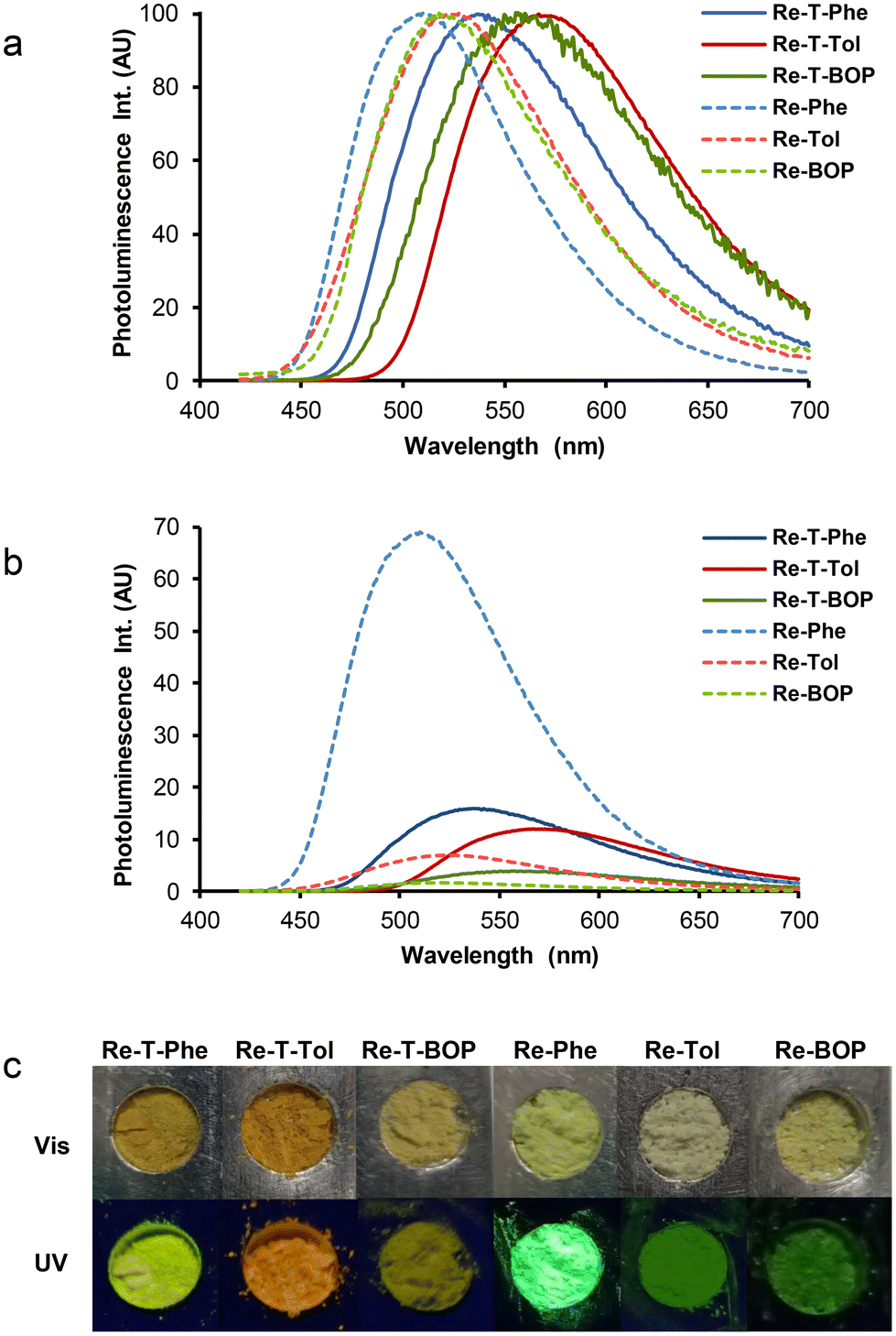 | ||
| Fig. 8 (a) Normalized emission spectra of complexes Re-T-Phe (blue solid line), Re-T-Tol (red solid line), Re-T-BOP (green solid line), Re-Phe (blue dotted line), Re-Tol (red dotted line) and Re-BOP (green dotted line) in the solid state (pristine microcrystalline powders, except for Re-Tol (ground powder)). (b) Same spectra with intensity at the maximum emission wavelength proportional to the quantum yield. λex = 370 nm. (c) Images of the powders examined in daylight (Vis) and illuminated at 365 nm (UV). | ||
All decays were found to be multiexponential (Fig. S52†). The preponderant contribution was associated to lifetimes between 414 and 1164 ns. Minor contributions with lifetimes of many tens of nanoseconds can be attributed to molecules experiencing a different environment. Very weak contributions associated to lifetimes in the nanosecond range are probably due to fluorescent impurities. The mean lifetimes were much longer than for the corresponding complexes in solution, as currently observed in the team for Re(I) complexes in the solid-state.
It was interesting to see if our molecules can be used as AIE-gens in aqueous medium, knowing that the formation of aggregates in this medium is not always compatible with good light emission. The AIE behavior was investigated using Re-T-Phe and Re-Phe that exhibit the best solid-sate emission properties in their respective series of complexes. The complexes were dissolved in acetonitrile, where they are very stable chemically and photochemically (no change in the UV-vis spectra was detected for 3 h upon irradiation with a 385 nm emitting diode). By increasing the water proportion in acetonitrile solutions of Re-T-Phe, the weak orange-red emission centered at 612 nm progressively became a strong green-yellow emission with maximum at 536 nm, and then the emission intensity decreased again (Fig. 9a and b). Under the same conditions, the green-yellow emission of Re-Phe at 544 nm was shifted to 508 nm with dramatic increase of intensity for water content (fw) above 70% (Fig. 9c and d). The intensity of the PL signal at the maximum wavelength of the most intense band was multiplied by 33 and 72, respectively. It is noteworthy that these emission maxima are similar to those of the solid compounds. In fact, these spectroscopic changes were accompanied by the appearance of small particles. The examination under the fluorescence microscope showed for Re-T-Phe the presence of rare, strongly-emissive rod-like microcrystals, which were about 200–150 μm × 5 μm (Fig. 9e). Suspensions of Re-Phe contained a large number of particles visible to the naked eye, which turned out to be agglomerates of very thin microfibers measuring 10–20 μm long (Fig. 9f). The AIE effect was therefore clear in both cases, but the superior emission properties of Re-Phe (Fig. 9g) make this compound the best candidate for AIE-related applications.
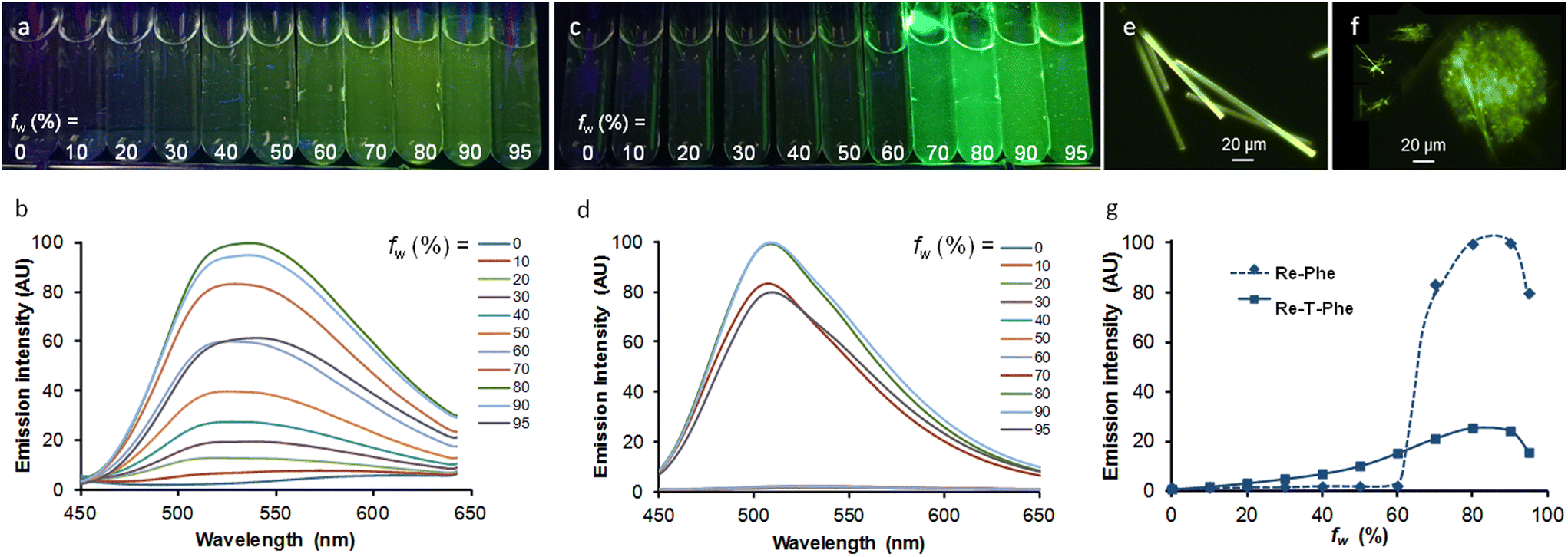 | ||
| Fig. 9 Samples of Re-T-Phe (a) and Re-Phe (c) at 3.3 × 10−5 M in acetonitrile solutions containing from 0 to 95% water (from left to right), illuminated at 365 nm. (b) and (d) Corresponding emission spectra, λex = 350 nm and 340 nm, respectively. Suspensions of Re-T-Phe (e) and Re-Phe (f) with fw = 80% observed under the fluorescence microscope. (g) Comparison of the PL intensity increase at 536 nm (Re-T-Phe) and 508 nm (Re-Phe) as a function of the water/acetonitrile ratio fw, after normalization with respect to emission quantum yield in acetonitrile solutions. All samples were observed 3 h after preparation. | ||
Antibacterial properties
Finally, the antibacterial activities of the complexes were tested. One Gram-positive strain, i.e. Staphylococcus aureus (S. aureus), and three Gram-negative strains, Pseudomonas aeruginosa (P. aeruginosa), Acinetobacter baumannii (A. baumannii) and Escherichia coli (E. coli), were selected as common causative agents of the most dangerous nosocomial infections. Both antibiotic susceptible (S) and multidrug-resistant (R) strains were used. The three triazolylidene complexes and their 1,2,3-triazole-based counterparts were studied, together with Re-[1,2,4]-Phe and Re-[1,2,4]-BOP for the sake of comparison. Given their low solubility in water, the complexes were previously dissolved in DMSO, before introduction and dilution into aqueous medium. The final concentration of complexes in each well ranged from 0.125 to 128 μM, and the highest concentration of DMSO was below 1.25%. The bacteria suspended in their culture medium were then put in contact with the complexes and incubated for 18 h in the dark. The minimum inhibitory concentration (MIC), i.e. the lowest concentration of a compound at which bacterial growth is completely inhibited, was measured. Results are collected in Table 3. Remarkably, antibacterial activity was observed only against S. aureus, whatever the strain is susceptible or resistant to conventional antibiotics. Among the active complexes, the triazolylidene-based derivative Re-T-BOP exhibited the stronger antibacterial activity, with a MIC value around 4 μM. This complex is the most lipophilic of all, but probably not enough to explain the difference in antibacterial activity. A connection between the antibacterial activity of all complexes and common drug likeliness parameters (i.e. topological polar surface area (TPSA), lipophilicity (miLogP) and molecular weight (MW))54,55 was sought. These parameters are typically determined for eukaryotic cells, but can be used to some extent for bacteria.56–58 In particular, the lipophilicity may facilitate drug penetration and cellular uptakes through interaction with bacterial membranes.59 Likewise, TPSA has been associated with bioavailability, and it is suggested that compounds with a TPSA greater than 140 tend to be poor at permeating cell membranes.56 In the present case, if considering the whole set of data, no obvious correlation was found with the drug likeliness parameters considered. The presence of the benzoxazole substituent in Re-T-BOP certainly brings a benefit, because this fragment is considered to be one of the most relevant biologically-active heterocycles, often incorporated in ligands to target a variety of receptors and enzymes in medicinal chemistry studies.60,61 However, the presence of benzoxazole in Re-BOP and Re-[1,2,4]-BOP is not sufficient to endow these compounds with an antibacterial activity. Besides, it is noteworthy that the presence of the methyl group in Re-Tol and Re-T-Tol modifies the antibacterial activity in comparison with their analogues Re-Phe and Re-T-Phe. Therefore, the activity could be related to the nature of the coordination sphere, and more precisely to the molecular geometry in space or flexibility. The examination of new complexes substituted by a methyl group in the meta and para positions could be instructive. It would allow validating our hypothesis by the analysis of a greater number of data. Of course, a deeper insight into the involved mechanism will also be necessary.| Compound | TPSA | miLogPa | MW (g mol−1) | MIC (μM) Non-irradiated samples | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S. aureus | P. aeroginosa | E. coli | A. baumannii | ||||||||
| (S) | (R) | (S) | (R) | (S) | (R) | (S) | (R) | ||||
| ATCC | ATCC | ATCC | ATCC | ATCC | ATCC | ATCC | ATCC | ||||
| 35923 | 43300 | 27853 | BAA-2108 | 25922 | BAA-196 | 19606 | BAA-1797 | ||||
| a Octanol–water partition coefficient logP calculated using Molinspiration desktop property calculator <https://www.molinspiration.com/services/logp.html>. | |||||||||||
| Re-T-Phe | 76.81 | 9.58 | 541.965 | 32 | 32 | >128 | >128 | >128 | >128 | >128 | >128 |
| Re-T-Tol | 76.81 | 9.66 | 555.992 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 |
| Re-T-BOP | 102.84 | 9.88 | 659.072 | 4 | 4 | >128 | >128 | >128 | >128 | >128 | >128 |
| Re-Phe | 76.81 | 9.49 | 527.938 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 |
| Re-Tol | 76.81 | 9.62 | 541.965 | 32 | 32 | >128 | >128 | >128 | >128 | >128 | >128 |
| Re-BOP | 102.84 | 9.85 | 645.045 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 |
| Re-[1,2,4]-Phe | 76.81 | 9.45 | 527.938 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 |
| Re-[1,2,4]-BOP | 102.84 | 9.82 | 645.045 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 |
| Gentamicin sulfate | 1 | 128 | 0.5 | 8 | 0.125 | >128 | 0.5 | >128 | |||
| Ampicillin | 0.25 | 32 | >128 | >128 | 16 | >128 | >128 | >128 | |||
In comparison with control antibiotics gentamicin and ampicillin, Re-T-Phe, Re-T-BOP and Re-Tol showed lower or comparable MIC values against multidrug resistant S. aureus ATCC 43300 strain demonstrating their antibacterial potentials. No compound was active against Gram-negative bacteria. Indeed, these bacteria have additional outer membrane, as well as an abundance of efflux pumps and highly selective porins, which make internalization of active compounds particularly difficult.62
Finally, a photochemical effect was investigated. Our complexes are not prone to photodecarbonylation. However, the presence of rhenium as a heavy atom induces fast population of the first excited triplet state, and the photogeneration of singlet oxygen could take place via energy transfer. In the present case, irradiation of the samples in the near UV does not lead to different results in most of the cases. Exceptions are Re-BOP and Re-T-Tol that show enhanced activity towards the antibiotic-susceptible strain of S. aureus (Table S32†).
Conclusions
In this comprehensive study, the properties of three new triazolylidene-based complexes were compared with those of their 1,2,3-triazole-based analogues, for which very little information was available. From an electronic point of view, the superior electron-donating properties of the mesoionic carbene ligand resulted in an increase in the charge density on the rhenium atom. The crystallographic data showed that small structural variations strongly influence the molecular conformation and intermolecular interactions in the solid state, and the Hirshfeld surface analysis allowed the latter to be finely quantified. The compounds of the two series were emissive in solution and in the solid state. Complex Re-Phe showed exceptional emission efficiency in the form of powder, and was a remarkable AIE-gen in aqueous medium. Although less efficient, our triazolylidene-based complexes proved to be photochemically stable, unlike the Re(I)-NHC complexes reported in the literature,15,16 and are therefore well suited for light-related applications. Until now, only 1,2,4-triazole-based complexes had shown attractive spectroscopic properties in the solid state. This work shows that some triazolylidene- and 1,2,3-triazole-based complexes must also be considered for potential applications in the fields of photoluminescent materials and AIE-gens. However, the mechanoresponsive emission behavior remains the prerogative of the 1,2,4-triazole-based series. It would be instructive to extend the solid-state emission study to other rhenium(I) complexes incorporating various mesoionic and N-heterocyclic carbenes.15–24,63–68Finally, one triazolylidene-based complex (Re-T-BOP) showed clear superiority over all other compounds as regards the antibacterial properties. It would be instructive to test the cytotoxicity of this molecule on eukaryotic cells to see if it could be used in therapy. A thorough examination of the mechanism of action would facilitate the further development of more effective molecules. This result confirms that triazolylidene-based Re(I) complexes must be considered as potential candidates in the highly demanding field of therapeutic chemistry.
Experimental section
General methods
All purchased chemicals were of the highest purity commercially available and used without further purification. Unless otherwise noted, all experiments were carried out under a nitrogen atmosphere. All reactions were monitored by TLC on silica gel Alugram® Xtra SIL G/UV254. Column chromatography was performed on Machery–Nagel silica gel. NMR spectra were recorded with a Bruker Avance 300 instrument. Chemical shifts are given in ppm and are referenced by using the residual signals of the solvent as internal standard. Signals are described as follow: s, singlet; d, doublet; t, triplet; m, multiplet. HRMS data were recorded on a Xero G2 QTOF (Waters) instrument. Infrared spectra were obtained on a Nexus Thermonicolet apparatus with DTGS as the detector. The microanalyses were made in the ‘Service d'analyse’ of LCC using a PerkinElmer 2400 series II analyzer.Synthesis
The syntheses of ligand L-T-Phe,51 and complexes Re-Phe50 and Re-BOP39 have been previously described.Tricarbonylrhenium(I) complex [Re(CO)3(L-T-Phe)Cl], (Re-T-Phe)
To a suspension of the ligand L-T-Phe (101 mg, 0.31 mmol) and [Re(CO)5Cl] (113 mg, 0.31 mmol) in toluene (15 mL) was added excess of Et3N (0.8 mL). The reaction mixture was refluxed for 60 h. After cooling to room temperature, the solvent was removed under reduced pressure. Silica gel column chromatography (CH2Cl2/acetone 9:1 v/v) afforded pure complex Re-T-Phe (127 mg, 76%) as a yellow solid.
1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.08 (dt, J = 5.5, 1.2 Hz, 1H), 8.34–8.22 (m, 2H), 7.97–7.87 (m, 2H), 7.81–7.67 (m, 3H), 7.64 (td, J = 5.7, 3.1 Hz, 1H), 4.68 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ(ppm): 198.5, 198.4, 190.1, 180.9, 155.2, 149.6, 149.1, 139.9, 138.2, 130.6, 129.8, 126.3, 124.9, 122.4, 39.8. HRMS (ESI) m/z 558.0479 ([M + NH4]+ calcd for C17H16N5O3Cl185Re: 558.0471), m/z 505.0443 ([M − Cl]+ calcd for C17H12N4O3185Re: 505.0439). IR (ATR) νCO: 2003, 1912, 1891, 1874, 1853 cm−1. Anal. calcd (%) for C17H12N4O3ClRe: C 37.68, H 2.23, N 10.34; found: C 37.40, H 1.59, N 10.06.
3-Methyl-4-(pyridin-2-yl)-1-(o-tolyl)-1H-1,2,3-triazol-3-ium tetrafluoroborate (L-T-Tol)
A mixture of triazole L-Tol (394 mg, 1.77 mmol) and m-CPBA (70%, 823 mg, 3.34 mmol) in CHCl3 (25 mL) was refluxed for 30 min. After cooling to room temperature, aqueous Na2S2O3 solution (0.5 M) was added and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with aqueous NaOH (1 M) and concentrated in vacuo. The obtained pyridine N-oxide was dissolved in anhydrous CH2Cl2 (10 mL) and Me3OBF4 (1.12 mg, 7.6 mmol) was added. The reaction mixture was stirred at room temperature for 4 days. The solvent was evaporated to dryness and the residue dissolved in EtOH (50 mL). Mo(CO)6 (501 mg, 1.9 mmol) was added and the reaction mixture was refluxed for 1 h. The solvent was removed under reduced pressure. Silica gel column chromatography (CH2Cl2/MeOH 9:1 v/v) afforded pure compound L-T-Tol (464 mg, 82.2%) as a solid.
1H NMR (300 MHz, DMSO-d6) δ 9.89 (s, 1H), 8.91 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H), 8.20 (td, J = 7.7, 1.7 Hz, 1H), 8.13 (dt, J = 7.9, 1.2 Hz, 1H), 7.78–7.55 (m, 5H), 4.69 (s, 3H), 2.38 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 150.2, 143.2, 140.5, 138.4, 134.2, 133.4, 132.2, 132.1, 131.2, 127.6, 126.3, 126.0, 124.7, 41.2, 17.1. HRMS (DCI-CH4) m/z 251.1285 ([M − BF4]+ calcd for C15H15N4: 251.1297).
Tricarbonylrhenium(I) complex [Re(CO)3(L-T-Tol)Cl], (Re-T-Tol)
To a suspension of the ligand L-T-Tol (89 mg, 0.263 mmol) and [Re(CO)5Cl] (96 mg, 0.265 mmol) in toluene (15 mL) was added excess of Et3N (0.8 mL). The reaction mixture was refluxed for 60 h. After cooling to room temperature, the solvent was removed under reduced pressure. Silica gel column chromatography (CH2Cl2/acetone 9:1 v/v) afforded pure complex Re-T-Tol (96 mg, 65.9%) as a yellow solid.
1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.04 (d, J = 5.5 Hz, 1H), 8.68–8.15 (m, 2H), 7.88–7.17 (m, 4H), 4.67 (s, 3H), 2.22 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ(ppm): 198.9, 197.5, 190.0, 155.3, 149.3, 149.2, 139.8, 137.7, 134.4, 131.2, 131.0, 127.1, 127.1, 126.2, 122.4, 39.1, 17.0. HRMS (ESI) m/z 577.0197 ([M + Na]+ calcd for C18H14N4O3NaCl185Re: 577.0182), m/z 519.0609 ([M − Cl]+ calcd for C18H14N4O3185Re: 519.0595). IR (ATR) νCO: 2019, 1917, 1888, 1868 cm−1. Anal. calcd (%) for C18H14N4O3ClRe: C 38.89, H 2.54, N 10.08; found: C 38.74, H 2.08, N 9.79.
3-Methyl-1-(2-phenylbenzo[d]oxazol-6-yl)-4-(pyridin-2-yl)-1H-1,2,3-triazol-3-ium trifluoromethanesulfonate (L-T-BOP)
To a solution of triazole L-BOP39 (79 mg, 0.233 mmol) in anhydrous CH2Cl2 (5 mL) was added MeOTf (31 μL, 0.283 mmol). The reaction mixture was refluxed overnight. The solvent was removed in vacuo and the crude product was purified by silica gel column chromatography (CH2Cl2/MeOH 9:1 v/v) to give the expected triazolium triflate L-T-BOP as a beige solid (30 mg, 25%) along with the N-methylpyridinium triflate derivative N-MP-BOP as a beige powder (51 mg, 43%).
For L-T-BOP: 1H NMR (300 MHz, DMSO-d6) δ(ppm): 10.24 (s, 1H), 8.92 (d, J = 4.7 Hz, 1H), 8.64 (d, J = 2.1 Hz, 1H), 8.33–8.20 (m, 4H), 8.18–8.09 (m, 2H), 7.78–7.64 (m, 4H), 4.73 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ(ppm): 165.3, 150.3, 150.2, 144.0, 143.0, 140.9, 138.5, 132.9, 131.7, 129.6, 128.6, 127.8, 126.1, 125.6, 124.6, 121.3, 118.8, 105.5, 41.3. HRMS (DCI-CH4) m/z 354.1364 ([M]+ calcd for C21H16N5O: 354.1355).
For 1-methyl-2-(1-(2-phenylbenzo[d]oxazol-6-yl)-1H-1,2,3-triazol-4-yl)pyridin-1-ium trifluoromethanesulfonate N-methylpyridinium, (N-MP-BOP)
1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.81 (s, 1H), 9.19–9.08 (m, 1H), 8.76–8.63 (m, 2H), 8.58 (t, J = 1.3 Hz, 1H), 8.27 (dd, J = 7.6, 2.0 Hz, 2H), 8.20–8.10 (m, 3H), 7.78–7.59 (m, 3H), 4.60 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ(ppm) = 164.4, 150.4, 147.5, 145.4, 145.1, 142.3, 139.2, 133.1, 132.6, 129.5, 128.0, 127.6, 127.4, 126.6, 125.9, 120.9, 118.2, 104.4, 48.2. HRMS (ESI) m/z 354.1355 ([M − OTf]+ calcd for C21H16N5O: 354.1356).Tricarbonylrhenium(I) complex [Re(CO)3(L-T-BOP)Cl], (Re-T-BOP)
To a suspension of the ligand L-T-BOP (32 mg, 0.0636 mmol) and [Re(CO)5Cl] (24 mg, 0.0663 mmol) in toluene (6 mL) was added an excess of Et3N (0.22 mL). The reaction mixture was refluxed for 48 h. After cooling to room temperature, the solvent was removed under reduced pressure. Silica gel column chromatography (CH2Cl2/MeOH 9.5:0.5 v/v) afforded pure compound (29 mg, 69%) as a yellow solid.
1H NMR (300 MHz, DMSO-d6) δ(ppm) = 9.10 (d, J = 5.4 Hz, 1H), 8.41 (d, J = 2.0 Hz, 1H), 8.34–8.25 (m, 4H), 8.15 (d, J = 8.5 Hz, 1H), 7.98 (dd, J = 8.5, 2.1 Hz, 1H), 7.68 (q, J = 6.0, 4.8 Hz, 4H), 4.72 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ(ppm) = 198.5, 198.4, 190.0, 181.6, 164.7, 155.2, 149.8, 149.6, 149.0, 143.0, 140.0, 135.3, 132.7, 129.5, 127.7, 126.4, 125.8, 122.5, 122.2, 120.6, 108.8, 39.3. HRMS (ESI) m/z 680.0265 ([M + Na]+ calcd for C24H15N5O4NaCl185Re: 680.0240), m/z 622.0667 ([M − Cl]+ calcd for C24H15N5O4185Re: 622.0654). IR (ATR) νCO: 2011, 1913, 1894, 1877 cm−1. Anal. calcd (%) for C24H15N5O4ClRe + CH2Cl2 (solvate): C 40.36, H 2.30, N 9.41; found: C 40.40, H 2.16, N 8.70.
2-(1-(o-Tolyl)-1H-1,2,3-triazol-4-yl)pyridine (L-Tol)
To a mixture of 1-azido-2-methylbenzene (1.05 g, 7.9 mmol), 2-ethynylpyridine (0.8 mL, 7.9 mmol) in a mixed solvent system (THF/H2O 1:2 v/v, 68 mL) was added sodium ascorbate (812 mg, 4.1 mmol) and CuSO4·5H2O (336 mg, 1.34 mmol). The reaction mixture was stirred at room temperature for 24 h, and then extracted with EtOAc. The combined organic layer was dried over Na2SO4 and concentrated in vacuo. Silica gel column chromatography (AcOEt/petroleum ether 3:7 v/v) afforded pure compound L-Tol (1.44 g, 77.5%) as an oil.
1H NMR (300 MHz, CDCl3) δ(ppm): 8.62 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 8.35 (s, 1H), 8.28 (dt, J = 8.0, 1.1 Hz, 1H), 7.83 (td, J = 7.8, 1.8 Hz, 1H), 7.48–7.33 (m, 4H), 7.30–7.22 (m, 1H), 2.30 (s, 3H). 13C NMR (75 MHz, CDCl3) δ(ppm): 150.1, 149.4, 148.1, 137.1, 136.4, 133.6, 131.6, 129.9, 126.9, 125.9, 123.6, 123.1, 120.5, 17.9. HRMS (DCI-CH4) m/z 237.1138 ([M + H]+ calcd for C14H13N4: 237.1140).
Tricarbonylrhenium(I) complex [Re(CO)3(L-Tol)Cl], (Re-Tol)
A mixture of ligand L-Tol (138 mg, 0.58 mmol) and [Re(CO)5Cl] (215 mg, 0.59 mmol) in methanol (5 mL) was stirred for 16 h at 65 °C. The mixture was cooled to room temperature and the precipitated product was filtered and washed with MeOH to yield a light yellow solid (272 mg, 86%).
1H NMR (300 MHz, DMSO-d6) δ(ppm): 9.61 (s, 1H), 9.02 (d, J = 5.4 Hz, 1H), 8.32–8.35 (m, 2H), 7.91–7.36 (m, 5H), 2.23 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ(ppm): 197.7, 196.7, 189.6, 153.3, 148.7, 148.6, 140.9, 135.0, 133.5, 131.9, 131.4, 127.5, 126.8, 126.3, 122.9, 17.2 HRMS (ESI) m/z 507.0476 ([M − Cl]+ calcd for C17H12N4O3187Re: 507.0467), m/z 560.0498 ([M + NH4]+ calcd for C17H16N5O3Cl187Re: 560.0499). IR (ATR) νCO: 2024, 1920, 1893, 1852, 1844 cm−1. Anal. calcd (%) for C17H12N4O3ClRe: C 37.68, H 2.23, N 10.34; found: C 37.20, H 1.36, N 10.21.
Crystallography
Crystallographic data were collected at low temperature (193 K) with an Oxford Instruments Cryostream 700+ Series device using MoKα radiation (wavelength = 0.71073 Å) on a Bruker APEX II Quazar diffractometer equipped with a 30 W air-cooled microfocus (Re-T-Phe and Re-Phe) and on a Bruker AXS D8-Venture diffractometer equipped with a multilayer TRIUMPH X-ray mirror and a Photon III-C14 detector (Re-T-Tol and Re-Tol). Phi- and omega-scans were used. The space group was determined on the basis of systematic absences and intensity statistics. An empirical absorption correction was employed.69 The structures were solved using an intrinsic phasing method (ShelXT).70 All non-hydrogen atoms were refined anisotropically using the least-square method on F2.71 Hydrogen atoms were refined isotropically at calculated positions using a riding model. Supplementary crystallographic data can be obtained free of charge from the Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/structures/.3D Hirshfeld surfaces (HS) and 2D fingerprint plots (FP)72 were generated with high resolution based on the crystallographic information file (CIF) using Crystal Explorer 17.5 software.73 HS were mapped over dnorm, shape-index and curvedness. The normalized contact distance (dnorm) is defined by di and de, which are the distances from the surface to the nearest atom interior and exterior to the surface, respectively. The vdW radii of atoms is defined according to eqn (1).
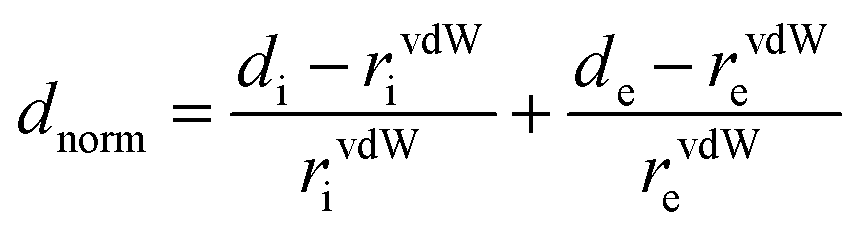 | (1) |
Spectroscopy
UV-visible absorption spectra and emission spectra in solutions were measured at 20 °C with a Xenius SAFAS spectrofluorometer, using cells of 1 cm optical pathway. All emission spectra were corrected. The emission quantum yields in solution (Φ) were determined using the classical formula:| Φx = (As × Ix × nx2 × Φs)/(Ax × Is × ns2) | (2) |
The rate constants for radiative (kr) and nonradiative (knr) decay were calculated using the following equation:
| kr = Φ/τ and knr = (1 − Φ)/τ | (3) |
Solid state spectra were recorded on a HORIBA Fluorolog 3-2iHR320 spectrofluorometer and were corrected. The absolute photoluminescence quantum yield values (ΦPL) were determined using the Xenius SAFAS spectrofluorometer provided with an integrating sphere, by a method based on the one developed by De Mello et al.,75 as described elsewhere.41 The error was estimated to be about 20%.
Emission decay curves were recorded using the time-correlated single photon counting method (TCSPC) on the Horiba Fluorolog 3-2 iHR320 spectrofluorometer equipped with a Nanoled-370 (λex = 371 nm). Emitted photons were detected at 90° by means of a Hamamatsu R928 photomultiplier. Emission was recorded near the maximum with a bandpass of 4 nm. The instrumental response was recorded directly before each decay curve, using the scattering of the sample at 371 nm. All analyses were recorded using the Datastation v2.7 software from Horiba. The decay curves were analyzed with reconvolution and global non-linear least-squares minimization method using DAS6 v6.8 software. The absorbance of solutions at λex was lower than 0.1. Solid state samples were deposited on a quartz holder.
Fluorescence microscopy was performed with a Leitz Laborlux D fluorescence microscope equipped with an Andor Luca camera (λex ∼ 450–490 nm, λem > 500 nm).
Computational details
The GAUSSIAN16 program package76 was employed for all calculations (the geometry optimization, the ground-state and excited-state electronic structures, and optical spectra) with the aid of the ChemCraft visualization program.77 The ground state (S0), the first excited state (S1) and the lowest triplet state (T1) geometries of Re-Tol, Re-T-Tol and Re-T-BOP, were fully optimized with the restricted and unrestricted density functional theory (R-DFT and U-DFT) methods using the Perdew–Burke–Ernzerhof PBE1PBE functional with no symmetry constraints.78 In all calculations, the “double-ζ” quality basis set LANL2DZ with Hay and Wadt's relative effective core potential ECP (outer-core [(5s25p6)] electrons and (5d6) valence electrons)79,80 was employed for the Re atom. The 6-311+G** basis set for H, C, N and O atoms was used.81 The solvent effect (dichloromethane, ε = 8.93) was simulated using the Self-Consistent Reaction Field (SCRF) under the Conductor Polarizable Continuum Model (CPCM).82–84 The vibrational frequencies calculations were performed using the optimized structural parameters of the complexes, to confirm that each optimized structure represents a local minimum on the potential energy surface and all eigenvalues are non-negative. To obtain a good agreement with the experimental vibrational frequencies in solid state, the theoretically calculated vibrational frequencies were scaled with a scaling factor of 0.9485. The optimized Cartesian coordinates of Re-Tol, Re-T-Tol and Re-T-BOP are included in the ESI part (see Tables S33–47†). On the basis of the optimized ground and excited state geometries, the absorption and emission properties were calculated by the time dependent density functional theory (TD-DFT) method at the PBE1PBE/LANL2DZ/6-31+G** level. These methods have already shown good agreement with experimental studies for different rhenium(I) complexes.85Electrochemistry
The electrochemical properties of the complexes were determined by cyclic voltammetry (CV) and Osteryoung square wave voltammetry (OSWV) in DCM. The solutions used during the electrochemical studies were typically 1 × 10−3 M in complex, and 0.1 M in supporting electrolyte. The supporting electrolyte [nBu4N][BF4] (Fluka, 99% electrochemical grade) was used as received and simply degassed under Ar. DCM was dried using an MB SPS-800 solvent purification system just prior to use. The measurements were carried out with an Autolab PGSTAT100 potentiostat controlled by GPES 4.09 software. Experiments were performed at room temperature in a homemade airtight three-electrode cell connected to a vacuum/Ar line. The reference electrode consisted of a saturated calomel electrode (SCE) separated from the solution by a bridge compartment. The counter electrode was a Pt wire of ca. 1 cm2 apparent surface. The working electrode was a Pt microdisk (0.5 mm diameter). Before each measurement, the solutions were degassed by bubbling Ar and the working electrode was polished with a polishing machine (Presi P230). Under these experimental conditions, Fc+/Fc is observed at +0.55 ± 0.01 V vs. SCE. OSWVs were obtained using an amplitude of 20 mV, a frequency of 20 Hz, and a step potential of 5 mV.Microbiology
The bacteria strains were obtained from the American Type Culture Collection (ATCC). Susceptible strains include Acinetobacter baumannii ATCC 19606, Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853 and Staphylococcus aureus ATCC 35923 whereas resistant strains include A. baumannii ATCC BAA-1797, E. coli ATCC BAA-196, P. aeruginosa ATCC BAA-2108 and S. aureus ATCC 43300.The minimum inhibitory concentration (MIC) of synthesized compounds against all eight bacteria were determined using the broth microdilution method adapted from Standard Operating Procedures (SOP) drafted by Monash University Facility for Anti-Infective Drug Development and Innovation (FADDI) and Clinical and Laboratory Standards Institute (CLSI) guidelines.86 Gentamicin and ampicillin were used as controls. Stock solutions of all compounds were prepared at a concentration of 1.024 × 10−2 M by dissolving the solid powders in dimethyl sulfoxide (DMSO). Two-fold serial dilution was carried out using cation-adjusted Mueller Hinton Broth (CAMHB) to obtain a final concentration ranging from 0.125 to 128 μM in the wells of 96 wells U-bottomed plate. The highest concentration of DMSO in the well was maintained at no more than 1.25%. Bacteria suspension was prepared following 0.5 McFarland standard. For quantification of the bacteria, absorbance was taken using Tecan microplate reader at a wavelength of 625 nm, and the optical density (OD625) values were adjusted to 0.07–0.08. Briefly, bacteria colonies were aseptically taken and resuspended into 1 mL of sterile normal saline (0.9% NaCl) to make bacteria suspension before checking for absorbance. Then, 200 μL of the optical density-adjusted bacteria suspension was added to 19.8 mL of 0.9% NaCl. Lastly, 100 μL of this bacteria suspension was added to each well containing 100 μL of the compounds and plates were incubated for 18 hours at 37 °C. To study the photodynamic therapy activities of the complexes, plates were incubated for 15 minutes after addition of compounds and irradiated for 10 minutes using LED light (365 nm) at a light density of 1.57 mW before incubation overnight. The light intensity was measured with a calibrated power meter through adjustment of distance between the LED light source and the 96-well plates. All assays were performed as at least duplicated experiments. Results were observed visually and tabulated after addition of resazurin.
Author contributions
Corinne Vanucci-Bacqué: investigation, writing (original draft); Mariusz Wolff: investigation, writing (original draft); Béatrice Delavaux-Nicot: investigation, writing (original draft); Abanoub Mosaad Abdallah: investigation; Sonia Mallet-Ladeira: investigation, writing (editing); Charles-Louis Serpentini: investigation; Florence Bedos-Belval: investigation, writing (editing), funding acquisition; Kar Wai Fong: investigation; Xiao Ying Ng: investigation; May Lee Low: investigation, writing (original draft), funding acquisition; Eric Benoist: conceptualization, writing (editing), project administration, funding acquisition; Suzanne Fery-Forgues: investigation, methodology, writing (original draft).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The France-Malaysia Collaboration Programme for Joint Research 2023 – 2nd call (MyTIGER) Matching Grant (Project code: MyTIGER-FPS-2023/001), initiated by the Embassy of France to Malaysia in collaboration with the Ministry of Higher Education (MoHE), is acknowledged for financial support. The postdoctoral fellowship of A. M. A. was supported by the French Embassy in Egypt (Campus France) and the Egyptian Science, Technology, and Innovation Funding Authority (STDF) (grant no. 46792). DFT calculations were carried out using resources provided by Wrocław Centre for Networking and Supercomputing (https://www.wcss.wroc.pl), Poland, Grant No. 18. We thank Dr Alix Sournia-Saquet and Mr Alain Moreau (LCC) for their help in electrochemical measurements.References
- R. Maity and B. Sarkar, Chemistry of Compounds Based on 1,2,3-Triazolylidene-Type Mesoionic Carbenes, J. Am. Chem. Soc. Au, 2022, 2, 22–57 CAS.
- Á. Vivancos, C. Segarra and M. Albrecht, Mesoionic and Related Less Heteroatom-Stabilized N-Heterocyclic Carbene Complexes: Synthesis, Catalysis, and Other Applications, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, 1H-1,2,3-Triazol-5-ylidenes: Readily Available Mesoionic Carbenes, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed.
- J. P. Byrne, L. Delgado, F. Paradisi and M. Albrecht, Carbohydrate-Functionalized Triazolylidene Iridium Complexes: Hydrogenation Catalysis in Water with Asymmetric Induction, ChemCatChem, 2022, 14, e202200086 CrossRef CAS PubMed.
- D. van der Westhuizen, C. A. Slabber, M. A. Fernandes, D. F. Joubert, G. Kleinhans, C. J. van der Westhuizen, A. Stander, O. Q. Munro and D. I. Bezuidenhout, A Cytotoxic Bis(1,2,3-triazol-5-ylidene)carbazolide Gold(III) Complex Targets DNA by Partial Intercalation, Chem. – Eur. J., 2021, 27, 8295–8307 CrossRef CAS PubMed.
- K. J. Kilpin, S. Crot, T. Riedel, J. A. Kitchen and P. J. Dyson, Ruthenium(II) and osmium(II) 1,2,3-triazolylidene organometallics: a preliminary investigation into the biological activity of ‘click’ carbene complexes, Dalton Trans., 2014, 43, 1443–1448 RSC.
- L. Suntrup, S. Klenk, J. Klein, S. Sobottka and B. Sarkar, Gauging Donor/Acceptor Properties and Redox Stability of Chelating Click-Derived Triazoles and Triazolylidenes: A Case Study with Rhenium(I) Complexes, Inorg. Chem., 2017, 56, 5771–5783 CrossRef CAS PubMed.
- L. Suntrup, F. Stein, J. Klein, A. Wilting, F. G. L. Parlane, C. M. Brown, J. Fiedler, C. P. Berlinguette, I. Siewert and B. Sarkar, Rhenium Complexes of Pyridyl-Mesoionic Carbenes: Photochemical Properties and Electrocatalytic CO2 Reduction, Inorg. Chem., 2020, 59, 4215–4227 CrossRef CAS PubMed . And ref. cited.
- C. Hille and F. E. Kühn, Cationic rhenium complexes ligated with N-heterocyclic carbenes – an overview, Dalton Trans., 2016, 45, 15–31 RSC.
- P. V. Simpson, M. Falasca and M. Massi, Properties and prospects for rhenium(I) tricarbonyl N-heterocyclic carbene complexes, Chem. Commun., 2018, 54, 12429–12438 RSC.
- S. Friães, S. Realista, H. Mourão and B. Royo, N-Heterocyclic and Mesoionic Carbenes of Manganese and Rhenium in Catalysis, Eur. J. Inorg. Chem., 2022, e202100884 CrossRef.
- L. Kearney, M. P. Brandon, A. Coleman, A. M. Chippindale, F. Hartl, R. Lalrempuia, M. Pižl and M. T. Pryce, Ligand-Structure Effects on N-Heterocyclic Carbene Rhenium Photo- and Electrocatalysts of CO2 Reduction, Molecules, 2023, 28, 4149 CrossRef CAS PubMed.
- T. P. Nicholls, L. K. Burt, P. V. Simpson, M. Massi and A. C. Bissember, Tricarbonyl rhenium(I) tetrazolato and N-heterocyclic carbene complexes: versatile visible-light-mediated photoredox catalysts, Dalton Trans., 2019, 48, 12749–12754 RSC.
- C. J. Stanton, C. W. Machan, J. E. Vandezande, T. Jin, G. F. Majetich, H. F. Schaefer, C. P. Kubiak, G. Li and J. Agarwal, Re(I) NHC Complexes for Electrocatalytic Conversion of CO2, Inorg. Chem., 2016, 55, 3136–3144 CrossRef CAS PubMed.
- J. G. Vaughan, B. L. Reid, S. Ramchandani, P. J. Wright, S. Muzzioli, B. W. Skelton, P. Raiteri, D. H. Brown, S. Stagni and M. Massi, The photochemistry of rhenium(I) tricarbonyl N-heterocyclic carbene complexes, Dalton Trans., 2013, 42, 14100–14114 RSC.
- P. V. Simpson, B. W. Skelton, P. Raiteria and M. Massi, Photophysical and photochemical studies of tricarbonyl rhenium(I) N-heterocyclic carbene complexes containing azide and triazolate ligands, New J. Chem., 2016, 40, 5797–5807 RSC.
- A. Bonfiglio, K. Magra, C. Cebrián, F. Polo, P. C. Gros, P. Mercandelli and M. Mauro, Red-emitting neutral rhenium(I) complexes bearing a pyridyl pyridoannelated N-heterocyclic carbene, Dalton Trans., 2020, 49, 3102–3111 RSC.
- X.-W. Li, H.-Y. Li, G.-F. Wang, F. Chen, Y.-Z. Li, X.-T. Chen, Y.-X. Zheng and Z.-L. Xue, Blue-Green Luminescent Rhenium(I) Tricarbonyl Complexes with Pyridine-Functionalized N-Heterocyclic Carbene Ligands Organometallics, Organometallics, 2012, 31, 3829–3835 CrossRef CAS.
- Z. Asbai, A. Bonfiglio, P. Mercandelli, F. Polo and M. Mauro, Cationic rhenium(I) complexes bearing a π-accepting pyridoannulated N-heterocyclic carbene ligand: Synthesis, photophysical, electrochemical and theoretical investigation, Polyhedron, 2021, 197, 115025 CrossRef CAS.
- G.-F. Wang, Y.-Z. Liu, X.-T. Chen, Y.-X. Zheng and Z.-L. Xue, Synthesis, structure and luminescent properties of rhenium(I) carbonyl complexes containing pyrimidine-functionalized N-heterocyclic carbenes, Inorg. Chim. Acta, 2013, 394, 488–493 CrossRef CAS.
- W.-M. Xue, M. C.-W. Chan, Z.-M. Su, K.-K. Cheung, S.-T. Liu and C.-M. Che, Spectroscopic and Excited-State Properties of Luminescent Rhenium(I) N-Heterocyclic Carbene Complexes Containing Aromatic Diimine Ligands, Organometallics, 1998, 17, 1622–1630 CrossRef CAS.
- E. S. Gauthier, L. Abella, E. Caytan, T. Roisnel, N. Vanthuyne, L. Favereau, M. Srebro-Hooper, J. A. G. Williams, J. Autschbach and J. Crassous, Modulation of Chiroptical and Photophysical Properties in Helicenic Rhenium(I) Systems: The Use of an N-(Aza[6]helicenyl)-NHC Ligand, Chem. – Eur. J., 2023, e202203477 CrossRef CAS PubMed.
- N. Saleh, M. Srebro, T. Reynaldo, N. Vanthuyne, L. Toupet, V. Y. Chang, G. Muller, J. A. G. Williams, C. Roussel, J. Autschbach and J. Crassous, enantio-Enriched CPL-active helicene–bipyridine–rhenium complexes, Chem. Commun., 2015, 51, 3754–3757 RSC.
- V. Giuso, C. Gourlaouen, M. Delporte-Pébay, T. Groizard, N. Vanthuyne, J. Crassous, C. Daniel and M. Mauro, Chiroptical activity of benzannulated N-heterocyclic carbene rhenium(I) tricarbonyl halide complexes: towards efficient circularly polarized luminescence emitters, Phys. Chem. Chem. Phys., 2024, 26, 4855–4869 RSC.
- H. Chen, Y. Zhang, A. Bonfiglio, F. Morlet-Savary, M. Mauro and J. Lalevée, Rhenium(I) N-Heterocyclic Carbene Complexes in Photoinitiating Systems for Polymerization upon Visible Light: Development of Photosensitive Resins for 3D and 4D Applications, ACS Appl. Polym. Mater., 2021, 3, 464–473 CrossRef CAS.
- C. Y. Chan, P. A. Pellegrini, I. Greguric and P. J. Barnard, Rhenium and Technetium Tricarbonyl Complexes of N-Heterocyclic Carbene Ligands, Inorg. Chem., 2014, 53, 10862–10873 CrossRef CAS PubMed.
- C. Y. Chan and P. J. Barnard, Rhenium complexes of bidentate, bis-bidentate and tridentate N-heterocyclic carbene ligands, Dalton Trans., 2015, 44, 19126–19140 RSC.
- C. Y. Chan, A. Noor, C. A. McLean, P. S. Donnelly and P. J. Barnard, Rhenium(I) complexes of N-heterocyclic carbene ligands that bind to amyloid plaques of Alzheimer’s disease, Chem. Commun., 2017, 53, 2311–2314 RSC.
- N. Wiratpruk, A. Noor, C. A. McLean, P. S. Donnelly and P. J. Barnard, Charge neutral rhenium tricarbonyl complexes of tridentate N-heterocyclic carbene ligands that bind to amyloid plaques of Alzheimer's disease, Dalton Trans., 2020, 49, 4559–4569 RSC.
- P. V. Simpson, I. Casari, S. Paternoster, B. W. Skelton, M. Falasca and M. Massi, Defining the Anti-Cancer Activity of Tricarbonyl Rhenium Complexes: Induction of G2/M Cell Cycle Arrest and Blockade of Aurora-A Kinase Phosphorylation, Chem. – Eur. J., 2017, 23, 6518–6521 CrossRef CAS PubMed.
- A. Domenichini, I. Casari, P. V. Simpson, N. M. Desai, L. Chen, C. Dustin, J. S. Edmands, A. van der Vliet, M. Mohammadi, M. Massi and M. Falasca, Rhenium N-heterocyclic carbene complexes block growth of aggressive cancers by inhibiting FGFR- and SRC-mediated signaling, J. Exp. Clin. Cancer Res., 2020, 39, 276 CrossRef CAS PubMed.
- D. Siegmund, N. Lorenz, Y. Gothe, C. Spies, B. Geissler, P. Prochnow, P. Nuernberger, J. E. Bandow and N. Metzler-Nolte, Benzannulated Re(I)–NHC complexes: synthesis, photophysical properties and antimicrobial activity, Dalton Trans., 2017, 46, 15269–15279 RSC.
- P. Alam, C. Climent, P. Alemany and I. R. Laskar, “Aggregation-induced emission” of transition metal compounds: Design, mechanistic insights, and applications, J. Photochem. Photobiol., C, 2019, 41, 100317 CrossRef CAS.
- L. Ravotto and P. Ceroni, Aggregation induced phosphorescence of metal complexes: From principles to applications, Coord. Chem. Rev., 2017, 346, 62–76 CrossRef CAS.
- V. Sathish, A. Ramdass, P. Thanasekaran, K.-L. Lu and S. Rajagopal, Aggregation-induced phosphorescence enhancement (AIPE) based on transition metal complexes—An overview, J. Photochem. Photobiol., C, 2015, 23, 25–44 CrossRef CAS.
- L. Ma, Y. Wang, X. Wang, Q. Zhu, Y. Wang, L. Li, H.-B. Cheng, J. Zhang and X.-J. Liang, Transition metal complex-based smart AIEgens explored for cancer diagnosis and theranostics, Coord. Chem. Rev., 2022, 473, 214822 CrossRef CAS.
- H. Shen, C. Xu, F. Sun, M. Zhao, Q. Wu, J. Zhang, S. Li, J. Zhang, J. W. Y. Lam and B. Z. Tang, Metal-Based Aggregation-Induced Emission Theranostic Systems, ChemMedChem, 2022, 17, e202100578 CrossRef CAS PubMed.
- J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Y. Park, Luminescence in Crystalline Organic Materials: From Molecules to Molecular Solids, Adv. Opt. Mater., 2021, 2002251 CrossRef CAS.
- J. Wang, B. Delavaux-Nicot, M. Wolff, S. Mallet-Ladeira, R. Métivier, E. Benoist and S. Fery-Forgues, The unsuspected influence of the pyridyl-triazole ligand isomerism upon the electronic properties of tricarbonyl rhenium complexes: an experimental and theoretical insight, Dalton Trans., 2018, 47, 8087–8099 RSC.
- J. Wang, A. Poirot, B. Delavaux-Nicot, M. Wolff, S. Mallet-Ladeira, J. P. Calupitan, C. Allain, E. Benoist and S. Fery-Forgues, Optimization of aggregation-induced phosphorescence enhancement in mononuclear tricarbonyl rhenium(I) complexes: the influence of steric hindrance and isomerism, Dalton Trans., 2019, 48, 15906–15916 RSC.
- A. Poirot, C. Vanucci-Bacqué, B. Delavaux-Nicot, N. Leygue, N. Saffon-Merceron, F. Alary, F. Bedos-Belval, E. Benoist and S. Fery-Forgues, Phenyl-pyta-tricarbonylrhenium(I) complexes: combining a simplified structure and steric hindrance to modulate the photoluminescence properties, Dalton Trans., 2021, 50, 13686–13698 RSC.
- A. Poirot, C. Vanucci-Bacqué, B. Delavaux-Nicot, C. Meslien, N. Saffon-Merceron, C.-L. Serpentini, F. Bedos-Belval, E. Benoist and S. Fery-Forgues, Using a diphenyl-bi-(1,2,4-triazole) tricarbonylrhenium(I) complex with intramolecular π–π stacking interaction for efficient solid-state luminescence enhancement, Dalton Trans., 2023, 52, 5453–5465 RSC.
- A. Poirot, C. Vanucci-Bacqué, B. Delavaux-Nicot, N. Saffon-Merceron, C.-L. Serpentini, N. Leygue, F. Bedos-Belval, E. Benoist and S. Fery-Forgues, Luminescent fac-[ReX(CO)3(phenyl-pyta)] (X = Cl, Br, I) complexes: influence of the halide ligand on the electronic properties in solution and in the solid state, Photochem. Photobiol. Sci., 2023, 22, 169–184 CrossRef CAS PubMed.
- A. Poirot, N. Leygue, B. Delavaux-Nicot, N. Saffon-Merceron, C. Allain, E. Benoist and S. Fery-Forgues, Tuning the photoluminescence properties of SLE- and MRL-active tricarbonylrhenium(I) complexes through minor structural changes of the organic ligand, J. Photochem. Photobiol., A, 2023, 445, 114982 CrossRef CAS.
- J. P. Calupitan, A. Poirot, J. Wang, B. Delavaux-Nicot, M. Wolff, M. Jaworska, R. Métivier, E. Benoist, C. Allain and S. Fery-Forgues, Mechanical Modulation of the Solid-State Luminescence of Tricarbonyl Rhenium(I) Complexes through the Interplay between Two Triplet Excited States, Chem. – Eur. J., 2021, 27, 4191–4196 CrossRef CAS PubMed.
- K. Schindler and F. Zobi, Anticancer and Antibiotic Rhenium Tri- and Dicarbonyl Complexes: Current Research and Future Perspectives, Molecules, 2022, 27, 539 CrossRef CAS PubMed.
- L. K. Wareham, R. K. Poole and M. Tinajero-Trejo, CO-releasing Metal Carbonyl Compounds as Antimicrobial Agents in the Post-antibiotic Era, J. Biol. Chem., 2015, 290, 18999–19007 CrossRef CAS PubMed.
- S. B. Zaman, M. A. Hussain, R. Nye, V. Mehta, K. T. Mamun and N. Hossain, A Review on Antibiotic Resistance: Alarm Bells are Ringing, Cureus, 2017, 9, e1403 Search PubMed.
- J. D. Crowley, P. H. Bandeen and L. R. Hanton, A one pot multi-component CuAAC “click” approach to bidentate and tridentate pyridyl-1,2,3-triazole ligands: Synthesis, X-ray structures and copper(II) and silver(I) complexes, Polyhedron, 2010, 29, 70–83 CrossRef CAS.
- T. Y. Kim, A. B. S. Elliott, K. J. Shaffer, C. J. McAdam, K. C. Gordon and J. D. Crowley, Rhenium(I) complexes of readily functionalized bidentate pyridyl-1,2,3-triazole “click” ligands: A systematic synthetic, spectroscopic and computational study, Polyhedron, 2013, 52, 1391–1398 CrossRef CAS.
- A. Bolje and J. Košmrlj, A Selective Approach to Pyridine Appended 1,2,3-Triazolium Salts, Org. Lett., 2013, 15, 5084–5087 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, Hirshfeld surface analysis, CrystEngComm, 2009, 11, 19–32 RSC.
- J. S. Wilson, N. Chawdhury, M. R. A. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Köhler and R. H. Friend, The Energy Gap Law for Triplet States in Pt-Containing Conjugated Polymers and Monomers, J. Am. Chem. Soc., 2001, 123, 9412–9417 CrossRef CAS PubMed.
- W. Zhu, Y. Wang, Y. Niu, L. Zhang and Z. Liu, Current Trends and Challenges in Drug-Likeness Prediction: Are They Generalizable and Interpretable?, Health Data Sci., 2023, 3, 0098 CrossRef PubMed.
- G. Bickerton, G. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Quantifying the chemical beauty of drugs, Nat. Chem., 2012, 4, 90–98 CrossRef CAS PubMed.
- N. Li, D. Liu, J. K. Dai, J. Y. Wang and J. R. Wang, Synthesis and In Vitro Antibacterial Activity of Quaternized 10-Methoxycanthin-6-one Derivatives, Molecules, 2019, 24, 1553 CrossRef CAS PubMed.
- G. Yuan, Y. Guan, H. Yi, S. Lai, Y. Sun and S. Cao, Antibacterial activity and mechanism of plant flavonoids to Gram-positive bacteria predicted from their lipophilicities, Sci. Rep., 2021, 11, 10471 CrossRef CAS PubMed.
- J. P. Ebejer, M. H. Charlton and P. W. Finn, Are the physicochemical properties of antibacterial compounds really different from other drugs?, J. Cheminf., 2016, 8, 30 Search PubMed.
- P. Y. Chung, R. E. Y. Khoo, H. S. Liew and M. L. Low, Antimicrobial and antibiofilm activities of Cu(II) Schiff base complexes against methicillin-susceptible and resistant Staphylococcus aureus, Ann. Clin. Microbiol. Antimicrob., 2021, 20, 67 CrossRef CAS PubMed.
- U. Kamal, N. M. Javed and K. Arun, Biological potential of benzoxazole derivatives: an updated review, Asian J. Pharm. Clin. Res., 2020, 13, 28–41 CrossRef CAS.
- C. S. Demmer and L. Bunch, Benzoxazoles and oxazolopyridines in medicinal chemistry studies, Eur. J. Med. Chem., 2015, 97, 778–785 CrossRef CAS PubMed.
- J. Sun, S. T. Rutherford, T. J. Silhavy and K. C. Huang, Physical properties of the bacterial outer membrane, Nat. Rev. Microbiol., 2022, 20, 236–248 CrossRef CAS PubMed.
- A. Baschieri, F. Monti, E. Matteucci, A. Mazzanti, A. Barbieri, N. Armaroli and L. Sambri, A Mesoionic Carbene as Neutral Ligand for Phosphorescent Cationic Ir(III) Complexes, Inorg. Chem., 2016, 55, 7912–7919 CrossRef CAS PubMed.
- S. Urinda, G. Das, A. Pramanik and P. Sarkar, Essential Role of Ancillary Ligand in Color Tuning and Quantum Efficiency of Ir(III) Complexes with N-Heterocyclic or Mesoionic Carbene Ligand: A Comparative Quantum Chemical Study, J. Phys. Chem. A, 2018, 122, 7532–7539 CrossRef CAS PubMed.
- E. Matteucci, F. Monti, R. Mazzoni, A. Baschieri, C. Bizzarri and L. Sambri, Click-Derived Triazolylidenes as Chelating Ligands: Achievement of a Neutral and Luminescent Iridium(III)–Triazolide Complex, Inorg. Chem., 2018, 57, 11673–11686 CrossRef CAS PubMed.
- R. E. Karmis, S. Carrara, A. A. Baxter, C. F. Hogan, M. D. Hulett and P. J. Barnard, Luminescent iridium(III) complexes of Nheterocyclic carbene ligands prepared using the ‘click reaction, Dalton Trans., 2019, 48, 9998–10010 RSC.
- M. A. Topchiy, S. A. Rzhevskiy, A. A. Ageshina, N. Y. Kirilenko, G. K. Sterligov, D. Y. Mladentsev, D. Y. Paraschuk, S. N. Osipov, M. S. Nechaev and A. F. Asachenko, Deep blue luminescent cyclometallated 1,2,3-triazol-5-ylidene iridium(III) complexes, Mendeleev Commun., 2020, 30, 717–718 CrossRef CAS.
- M. A. Topchiy, P. B. Dzhevakov, N. Y. Kirilenko, S. A. Rzhevskiy, A. A. Ageshina, V. N. Khrustalev, D. Y. Paraschuk, M. V. Bermeshev, M. S. Nechaev and A. F. Asachenko, Cyclometallated 1,2,3-triazol-5-ylidene iridium(III) complexes: synthesis, structure, and photoluminescence properties, Mendeleev Commun., 2019, 29, 128–131 CrossRef CAS.
- SADABS, Program for data correction, Bruker-AXS Search PubMed.
- G. M. Sheldrick, SHELXT-Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. A. Spackman and J. J. McKinnon, Fingerprinting intermolecular interactions in molecular crystals, CrystEngComm, 2002, 4, 378–392 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Reevaluation of absolute luminescence quantum yields of standard solutions using a spectrometer with an integrating sphere and a back-thinned CCD detector, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- J. C. De Mello, H. F. Wittmann and R. H. Friend, An improved experimental determination of external photoluminescence quantum efficiency, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- G. Zhurko and D. Zhurko, ChemCraft 1.6, 2011, https://www.chemcraftprog.com/index.html Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. J. Perdew and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- G. Velmurugan, B. K. Ramamoorthi and P. Venuvanalingam, Are Re(I) phenanthroline complexes suitable candidates for OLEDs? Answers from DFT and TD-DFT investigations, Phys. Chem. Chem. Phys., 2014, 16, 21157–21171 RSC.
- J. L. Grace, J. X. Huang, S.-E. Cheah, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, Antibacterial low molecular weight cationic polymers: dissecting the contribution of hydrophobicity, chain length and charge to activity, RSC Adv., 2016, 6, 15469–15477 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including NMR, HRMS and FTIR spectra, molecular views and crystallographic data, theoretical calculations, electrochemical experiments, photoluminescence spectra and decays. CCDC 2327668–2327671. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00922c |
| This journal is © The Royal Society of Chemistry 2024 |