
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the promising anticancer activity of palladium(II)–aryl complexes bearing diphosphine ligands: a structure–activity relationship analysis†
Giovanni
Tonon
 a,
Matteo
Mauceri
a,
Enrico
Cavarzerani
a,
Rachele
Piccolo
a,
Claudio
Santo
a,
Nicola
Demitri
b,
Laura
Orian
c,
Pablo A.
Nogara
d,
João Batista T.
Rocha
d,
Vincenzo
Canzonieri
ef,
Flavio
Rizzolio
*ae,
Fabiano
Visentin
*a and
Thomas
Scattolin
*c
a,
Matteo
Mauceri
a,
Enrico
Cavarzerani
a,
Rachele
Piccolo
a,
Claudio
Santo
a,
Nicola
Demitri
b,
Laura
Orian
c,
Pablo A.
Nogara
d,
João Batista T.
Rocha
d,
Vincenzo
Canzonieri
ef,
Flavio
Rizzolio
*ae,
Fabiano
Visentin
*a and
Thomas
Scattolin
*c
aDipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari, Campus Scientifico Via Torino 155, 30174 Venezia-Mestre, Italy. E-mail: fvise@unive.it; flavio.rizzolio@unive.it
bElettra – Sincrotrone Trieste, S.S. 14 Km 163.5 in Area Science Park, 34149 Basovizza, Trieste, Italy
cDipartimento di Scienze Chimiche, Università degli Studi di Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: thomas.scattolin@unipd.it
dDepartamento de Bioquímica e Biologia Molecular, Centro de Ciências Naturais e Exatas, Universidade Federal de Santa Maria, Santa Maria, RS, Brazil
ePathology Unit, Centro di Riferimento Oncologico di Aviano (C.R.O.) IRCCSvia Franco Gallini 2, 33081, Aviano, Italy. E-mail: flavio.rizzolio@unive.it
fDepartment of Medical, Surgical and Health Sciences, Università degli Studi di Trieste, Strada di Fiume 447, Trieste, Italy
First published on 17th April 2024
Abstract
In continuation of our previous works on the cytotoxic properties of organopalladium compounds, in this contribution we describe the first systematic study of the anticancer activity of Pd(II)–aryl complexes. To this end, we have prepared and thoroughly characterized a wide range of palladium derivatives bearing different diphosphine, aryl and halide ligands, developing, when necessary, specific synthetic protocols. Most of the synthesized compounds showed remarkable cytotoxicity towards ovarian and breast cancer cell lines, with IC50 values often comparable to or lower than that of cisplatin. The most promising complexes ([PdI(Ph)(dppe)] and [PdI(p-CH3-Ph)(dppe)]), characterized by a diphosphine ligand with a low bite angle, exhibited, in addition to excellent cytotoxicity towards cancer cells, low activity on normal cells (MRC5 human lung fibroblasts). Specific immunofluorescence tests (cytochrome c and H2AX assays), performed to clarify the possible mechanism of action of this class of organopalladium derivatives, seemed to indicate DNA as the primary cellular target, whereas caspase 3/7 assays proved that the complex [PdI(Ph)(dppe)] was able to promote intrinsic apoptotic cell death. A detailed molecular docking analysis confirmed the importance of a diphosphine ligand with a reduced bite angle to ensure a strong DNA–complex interaction. Finally, one of the most promising complexes was tested towards patient-derived organoids, showing promising ex vivo cytotoxicity.
Introduction
Palladium(II)–aryl complexes represent a fascinating and important class of organometallic compounds that have found extensive applications in the field of homogeneous catalysis.1–19 These palladium compounds are also valuable synthons in organometallic chemistry, enabling the formation of novel organopalladium derivatives with interesting properties.20–23 Beyond their synthetic applications, Pd(II)–aryl derivatives have found utility in the field of materials science. For instance, they are employed in the fabrication of organic light-emitting diodes (OLEDs) and other electronic devices.24,25 In addition, the ability to precisely control the arrangement of aryl groups through Pd-catalyzed processes enables the fine-tuning of the electronic and optical properties of these materials.As far as the application of Pd(II)–aryl complexes in medicinal chemistry is concerned, it is curious to note that, excluding studies on cyclopalladates26 and a couple of contributions dealing with Pd(II) complexes bearing perfluorinated aryl ligands,27,28 no systematic study on the antitumor activity of Pd(II) complexes with classical monodentate aryl fragments is found in the literature.
For this reason, with the aim of filling this gap and encouraged by some recent studies conducted on the promising anticancer activity of organopalladium compounds,29–36 in this work we propose the synthesis and study of the antiproliferative activity of Pd(II)–aryl complexes bearing diphosphine ancillary ligands (Fig. 1). In vitro tests on ovarian and breast cancer cell lines allowed us to select one of the most promising complexes and to carry out a preliminary study on its mechanism of action, as well as to determine its cytotoxicity on patient-derived organoids. Finally, considering the biotarget identified for this important class of organopalladium compounds, we propose a structure–activity relationship (SAR) analysis using a molecular docking approach. This method allows us to evaluate, in a qualitative and intuitive way, the influence of each ligand bonded to palladium on the antitumor activity observed in vitro.
 | ||
| Fig. 1 Relevant examples of organopalladium compounds with promising anticancer activity and target complexes of this work.29 | ||
Results and discussion
Synthesis of Pd(II)–aryl complexes
Most of the Pd(II)–aryl complexes used in this work were synthesized through the ligand exchange reaction between [PdI(tmeda)(p-R-Ph)] precursors 1a–d (tmeda = N,N,N′,N′-tetramethylethylenediamine; R = H, CH3, CF3, NO2) and five different diphosphine ligands (dppe = 1,2-bis(diphenylphosphino)ethane, dppp = 1,2-bis(diphenylphosphino)propane, dppf = 1,1′-bis(diphenylphosphino)ferrocene, dppbz = 1,2-bis(diphenylphosphino)benzene and DPEphos = bis[(2-diphenylphosphino)phenyl] ether). While the diphosphines are all commercially available, the [PdI(tmeda)(p-R-Ph)] precursors 1a–d were instead prepared according to the protocol reported in the literature.21,22Following a slightly modified procedure with respect to that described by Hartwig and colleagues,21 substitution of the tmeda ligand with the selected diphosphines occurred at room temperature in a few minutes, using anhydrous dichloromethane as a solvent. The only exception is complexes containing dppbz (5a–b), for which the exchange reaction is definitely slower and is completed within 1 hour. A more detailed kinetic study of this process is reported in the ESI.†
Scheme 1 shows the general reaction for the synthesis of the target complexes and the yields obtained. It should be noted that not all aryl fragment/diphosphine ligand combinations led to the isolation of the complexes of interest with sufficient purity. For this reason, only the compounds obtained in a pure form are reported in Scheme 1.
 | ||
| Scheme 1 Synthetic procedure to target Pd(II)–aryl complexes. | ||
While complexes 2b–d, 3a and 4b–d were previously reported by other research groups,21,37–40 six of the synthesized complexes ([PdI(p-NO2-Ph)(dppf)] 4a, [PdI(p-NO2-Ph)(dppe)] 2a, [PdI(p-NO2-Ph)(dppbz)] 5a, [PdI(p-NO2-Ph)(DPEphos)] 6a, [PdI(p-CF3-Ph)(dppbz)] 5b and [PdI(p-CF3-Ph)(DPEphos)] 6b) have never been published before. Therefore, these derivatives were exhaustively characterized by 1H, 31P, 19F, 13C NMR and FT-IR spectroscopy (Fig. S1–65 in ESI†). Moreover, in the case of complexes 4a and 6a, it was possible to unequivocally confirm their structure by XRD analysis of suitable crystals obtained by slow evaporation of diethyl ether in a dichloromethane solution of the Pd(II) complex (Fig. 2). Conversely, the correct outcome of the synthesis of the Pd(II)–aryl derivatives already reported in the literature was confirmed by 1H, 31P and 19F NMR analyses.21,37–40
 | ||
| Fig. 2 X-ray molecular structures of 4a (right) and 6a (left) are presented, showing thermal displacement ellipsoids at the 50% probability level with hydrogen atoms and solvent molecules omitted for clarity. | ||
Going into more detail about the characterization of the synthesized complexes, in all 1H NMR spectra the disappearance of the peaks of coordinated tmeda and the appearance of the peaks of the aromatic and aliphatic protons of the coordinated diphosphine are detectable. These latter signals are shifted and doubled compared to those of the free diphosphine, due to the presence of two different ligands in the Pd(II) coordination sphere (iodide and aryl fragment). Coordination of the chelating diphosphine is confirmed by the 31P NMR spectra, in which it is possible to observe the presence of two doublets, caused by the coupling of the two non-equivalent phosphorus nuclei, localised at higher chemical shifts compared to the singlet observable in the case of the free diphosphine. This shift is attributable to the strong σ-donor character of the diphosphines used and their coordination on the metal centre.
With the aim of evaluating not only the effect of the aryl and diphosphine fragments, but also the role of the halide ligand on the cytotoxicity of the target complexes, we wondered which could be the most promising synthetic route to Pd(II)–aryl complexes bearing chloride and bromide ligands.
After some preliminary tests we chose to prepare a Pd(II) precursor bearing 8-(tert-butylthio)quinoline (TtBQ) as an ancillary ligand. More precisely, we opted for [PdI(TtBQ)(p-NO2-Ph)] (7-I) as a model compound.41
Based on studies previously conducted by our group, the presence of an N^S ancillary ligand such as thioquinoline derivatives, enables the replacement of the iodide ligand with a bromide or a chloride by the simple addition of a stoichiometric amount of IBr or ICl, respectively.42 This reaction is particularly interesting as it avoids the use of silver-based dehalogenating agents, which are light-sensitive and often difficult to remove completely. In addition, the cationic intermediates that form before the entry of the new halide are generally not very stable, leading to very low yields or even preventing the isolation of the product of interest.
However, it is important to point out that the halide metathesis carried out with IBr or ICl is selective only in the case of Pd(II) complexes bearing N^S ligands.42 In fact, when P^P ligands are used, the same process leads to mixtures of products, the identities of which are difficult to establish.
Gratifyingly, the addition of IBr or ICl to complex 7-I allowed us to synthesize complexes [PdBr(TtBQ)(p-NO2-Ph)] (7-Br) and [PdCl(TtBQ)(p-NO2-Ph)] (7-Cl), respectively, in high yields and purity (see Scheme 2).
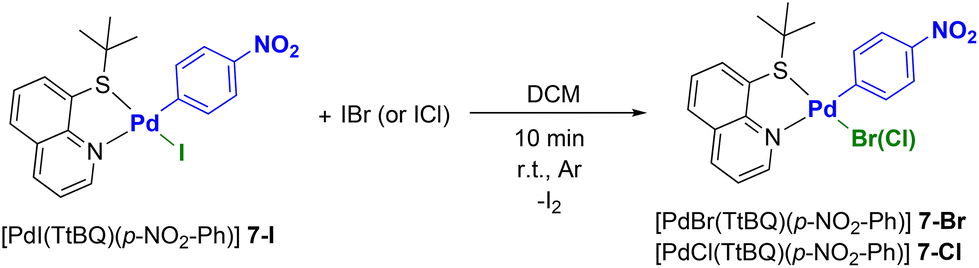 | ||
| Scheme 2 Halide-metathesis reactions. | ||
These complexes have been characterized by 1H, 13C NMR, and IR spectroscopy. For example, in the case of complex [PdCl(p-NO2-Ph)(TtBQ)] (7-Cl) it is possible to detect one singlet at 1.23 ppm ascribable to tert-butyl protons and one multiplet at 7.68–7.80 ppm attributable to H3, H6 quinoline protons and two of the phenyl protons. A further multiplet in the 7.86–7.91 region is attributable to the two remaining phenyl protons. H7 and H5 appear in the form of two very close doublets of doublets, located at 8.08 ppm (J = 7.5 Hz, 1.3 Hz) and 8.11 ppm (J = 8.4 Hz, 1.3 Hz), while H4 generates a doublet of doublets (J = 8.4 Hz, 1.6 Hz) at 8.46 ppm.
Finally, the most diagnostic signal corresponds to H2 (9.78 ppm, dd, J = 4.9 Hz, 1.6 Hz), which confirms the presence of the chloride ligand. In fact, this signal is present at 9.95 and 10.12 ppm in the complexes [PdBr(p-NO2-Ph)(TtBQ)] (7-Br) and [PdI(p-NO2-Ph)(TtBQ)] (7-l), respectively.
To further confirm halide metathesis, it was possible to obtain the structure of complexes 7-Cl and 7-Br by means of XRD analysis (Fig. 3). It is interesting to note that between the two possible isomers, the isolated complexes are always characterized by the sulfur atom in the trans position with respect to the halide ligand. This behavior can be justified by the different trans-influences of the ligands. In fact, the aryl fragment is positioned trans with respect to the less trans-labilizing ligand of the thioquinoline ligand (nitrogen atom).
 | ||
| Fig. 3 X-ray molecular structures of 7-Cl (right) and 7-Br (left) are presented, showing thermal displacement ellipsoids at the 50% probability level with hydrogen atoms and solvent molecules omitted for clarity. | ||
The thioquinoline ligand used (TtBQ) has the further advantage of being sufficiently labile to be replaced by diphosphine ligands. In particular, using dppf as a model ligand, complexes [PdBr(dppf)(p-NO2-Ph)] (4a-Br) and [PdCl(dppf)(p-NO2-Ph)] (4a-Cl) were synthesized (Scheme 3), and constitute the bromide and chloride congeners of complex [PdI(dppf)(p-NO2-Ph)] (4a), the synthesis of which was discussed at the beginning of this section.
 | ||
| Scheme 3 Synthetic procedure for Pd(II)–aryl complexes bearing dppf and bromide (or chloride) ligands. | ||
The identity of both complexes was ascertained by careful analysis of their 1H, 31P, 13C NMR and IR spectra. As far as the 1H NMR spectra are concerned, they obviously do not show substantial differences compared to that of complex [PdI(dppf)(p-NO2-Ph)] (4a) in terms of the number and multiplicity of signals. However, significant differences are observed concerning the chemical shifts of some peaks.
As regards the 31P NMR spectra, a general trend is observed for both phosphorus signals: as the electronegativity of the halogen increases, the signals relating to the phosphorus nuclei shift to higher chemical shifts. In fact, chloride exerts a lower trans-influence than iodide and this involves greater electron donation by the phosphorus of the phosphine, with the consequent shift to higher chemical shifts of the signal relating to the phosphorus nucleus trans with respect to the halide. Notably, in the case of complexes 4a-Cl and 4a-Br it was possible to confirm the proposed structures by XRD analysis (Fig. 4).
 | ||
| Fig. 4 X-ray molecular structures of 4a-Cl (right) and 4a-Br (left) are presented, showing thermal displacement ellipsoids at the 50% probability level with hydrogen atoms and solvent molecules omitted for clarity. | ||
Spectroscopic data for the synthesized Pd(II)–aryl complexes are summarized in Table S8 of the ESI.†
Structural characterization of Pd(II)–aryl complexes
Complexes 7-Br, 7-Cl, 4a, 4a-Br, 4a-Cl and 6a have been crystallized, characterized through XRD and show square planar Pd(II) coordination spheres (Tables S1–7 in ESI†). A change of the chelating ligand has an impact on the Pd(II)–C bond. Lengthening of the Pd–C bond is found when the phosphine is in the trans-position, reflecting the different electronic properties of P and N atoms. Minor differences can be observed as a consequence of halogen substitutions.All the crystalline forms bear one crystallographically independent neutral Pd(II) complex each. A comparison of phosphine-based complexes (4a, 4a-Br, 4a-Cl and 6a) shows almost perfectly superimposable atomic positions (Fig. S70 in ESI† – R.M.S.D. < 0.6 Å). Replacement of pentacene is efficiently compensated for by proper phosphine phenyl ring stacking in 6a (dπ–π = 3.627(2) Å between ring centroids with 0.79 Å slippage). Identical conformations are also found for the thioquinoline derivatives 7-Br and 7-Cl (Fig. S71 in ESI† – R.M.S.D. ∼0.1 Å). Crystal packing shows hydrophobic contacts among neighbouring molecules, involving weak intermolecular π⋯π and CH⋯π interactions, among neighbouring aromatic rings. Solvent molecules were found in the crystal packing of 6a (i.e. heavily disordered dichloromethane and diethyl ether).
Antiproliferative activity on human cancer and normal cell lines
With the aim of investigating the potential anticancer activity of our Pd(II)–aryl complexes, a panel of four different human tumor cell lines (ovarian cancer A2780, with its cisplatin resistant clone A2780cis, high-grade serous ovarian cancer OVCAR-5 and triple-negative breast cancer MDA-MB231) and MRC-5 normal cells (human lung fibroblasts) were treated for 96 hours with the synthesized compounds and cisplatin (positive control).Importantly, all the synthesized complexes are soluble when the DMSO stock solution (2.5 mM) is diluted in the culture medium to final concentrations of 100 μM, 10 μM, 1 μM, 0.1 μM, 0.01 μM, and 0.001 μM.
In the preliminary phase, we monitored the stability of the Pd(II)–aryl complexes in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 D2O/DMSO-d6 solution by NMR spectroscopy. After 24 hours no significant changes to the spectra are detectable, indicating that the complexes retain their structural integrity. Particularly diagnostic are the peaks in the 31P NMR spectra, which are almost superimposable on those recorded in chlorinated solvents (see Fig. S54 in ESI†). This aspect suggests that the hydrolysis of the Pd(II)–halide bond does not occur, as the formation of aquo species would lead to a significant change in the 31P NMR spectra. The only exception is complex [PdI(p-CH3-Ph)(dppf)] (4d), which decomposes rapidly under such conditions and was therefore not considered for biological tests.
:1 D2O/DMSO-d6 solution by NMR spectroscopy. After 24 hours no significant changes to the spectra are detectable, indicating that the complexes retain their structural integrity. Particularly diagnostic are the peaks in the 31P NMR spectra, which are almost superimposable on those recorded in chlorinated solvents (see Fig. S54 in ESI†). This aspect suggests that the hydrolysis of the Pd(II)–halide bond does not occur, as the formation of aquo species would lead to a significant change in the 31P NMR spectra. The only exception is complex [PdI(p-CH3-Ph)(dppf)] (4d), which decomposes rapidly under such conditions and was therefore not considered for biological tests.
The antiproliferative activity results for the tested compounds are reported in Table 1 in terms of half inhibitory concentrations (IC50) values.
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| A2780 | A2780cis | OVCAR-5 | MDA-MB-231 | MRC-5 | |
| Data after 96 h of incubation. Stock solutions in DMSO for all complexes; stock solutions in H2O for cisplatin. A2780 (cisplatin-sensitive ovarian cancer cells), A2780cis (cisplatin-resistant ovarian cancer cells), OVCAR-5 (high-grade serous ovarian cancer cells), MDA-MB-231 (triple-negative breast cancer), MRC-5 (normal lung fibroblasts). | |||||
| Cisplatin | 1.1 ± 0.1 | 11 ± 3 | 2 ± 1 | 28 ± 3 | 3.9 ± 0.6 |
| [PdI(p-NO2-Ph)(dppe)] (2a) | 4.1 ± 0.4 | 3.3 ± 0.2 | >100 | 7 ± 1 | >100 |
| [PdI(p-CF3-Ph)(dppe)] (2b) | 3.26 ± 0.04 | 3.0 ± 0.5 | 10 ± 2 | 3 ± 1 | 19 ± 2 |
| [PdI(Ph)(dppe)] (2c) | 3.1 ± 0.2 | 5 ± 1 | 5 ± 1 | 4.7 ± 0.4 | >100 |
| [PdI(p-CH3-Ph)(dppe)] (2d) | 1.8 ± 0.3 | 7 ± 1 | 6 ± 1 | 2 ± 1 | >100 |
| [PdI(p-NO2-Ph)(dppp)] (3a) | 3.1 ± 0.8 | 2.9 ± 0.7 | 2.4 ± 0.1 | 3 ± 1 | 5.4 ± 0.5 |
| [PdI(p-CF3-Ph)(dppp)] (3b) | 3.8 ± 0.2 | 3.1 ± 0.5 | 8.0 ± 0.4 | 4 ± 1 | 4.1 ± 0.2 |
| [PdI(p-NO2-Ph)(dppf)] (4a) | 8 ± 1 | 3.5 ± 0.4 | >100 | 22 ± 4 | >100 |
| [PdBr(p-NO2-Ph)(dppf)] (4a-Br) | 4.6 ± 0.5 | 3.1 ± 0.2 | >100 | 30 ± 10 | >100 |
| [PdCl(p-NO2-Ph)(dppf)] (4a-Cl) | 2.7 ± 0.3 | 1.5 ± 0.3 | >100 | 90 ± 50 | >100 |
| [PdI(p-CF3-Ph)(dppf)] (4b) | 14 ± 3 | 4.3 ± 0.6 | 40 ± 7 | 11 ± 2 | >100 |
| [PdI(Ph)(dppf)] (4c) | 2.1 ± 0.3 | 1.4 ± 0.3 | 6.6 ± 0.4 | 5 ± 1 | 3.2 ± 0.3 |
| [PdI(p-NO2-Ph)(dppbz)] (5a) | 17 ± 2 | 6 ± 1 | 40 ± 20 | >100 | >100 |
| [PdI(p-CF3-Ph)(dppbz)] (5b) | 3.9 ± 0.2 | 4.2 ± 0.4 | 12 ± 1 | 4 ± 1 | 4.26 ± 0.05 |
| [PdI(p-NO2-Ph)(DPEphos)] (6a) | 7.5 ± 0.7 | 1.0 ± 0.2 | >100 | 10 ± 3 | >100 |
| [PdI(p-CF3-Ph)(DPEphos)] (6b) | 3.1 ± 0.4 | 0.8 ± 0.1 | >100 | >100 | >100 |
On the basis of these IC50 values it is possible to draw some interesting conclusions.
As far as the A2780 and A2780cis cell lines (cisplatin-sensitive and cisplatin-resistant ovarian cancer, respectively) are concerned, the IC50 values fall in the low micromolar range and there are no marked differences between the tested compounds. It seems that among the complexes with the formula [PdX(p-NO2-Ph)(dppf)] (X = Cl, Br, I), the most active derivative is that with a chloride ligand. As regards the effect of the group in the para position of the aryl fragment, no significant trends are observed in the two lines. In the A2780 cell line, all compounds exhibit a cytotoxicity comparable with that of cisplatin, with the exception of complex [PdI(p-NO2-Ph)(dppbz)] (5a), for which it is more than an order of magnitude lower. Interestingly, in the cisplatin-resistant ovarian cancer cell line (A2780cis), all compounds are more active than cisplatin by up to an order of magnitude. Given that the IC50 values obtained on the A2780 and A2780cis cell lines are comparable in all tested compounds, while those of cisplatin differ by an order of magnitude (1.1 μM vs. 11 μM), we can reasonably assume that the mechanism of action of our complexes is different from that of platinum-based drugs.
In the OVCAR-5 (high-grade serous ovarian cancer) cell line, most compounds exhibit higher IC50 values with respect to those obtained on the A2780 cell line. In particular, some complexes were found to be substantially inactive (IC50 > 100, 2a, 4a, 4a-Cl, 4a-Br and 6a–b), others were active but much less than cisplatin (4b and 5a), while some were comparable with the reference drug (2b–d, 3a–b, 4c and 5b). The latter complexes are mostly characterized by the presence of diphosphine ligands with a reduced bite angle (dppe, dppp and dppbz).
Even in the triple-negative breast cancer cell line (MDA-MB-231), most of the compounds tested that exhibited a remarkable cytotoxicity (up to an order of magnitude higher than cisplatin) are those bearing diphosphine ancillary ligands with a low bite angle. Notably, the three complexes that are inactive against this type of cancer cell (4a-Cl, 5a and 6b) are characterized by the presence of DPEphos, dppbz and dppf diphosphines, and aryl ligands with electron-withdrawing substituents. Contrary to the trend observed for the A2780 and A2780cis cell lines, it seems that among the compounds with the formula [PdX(p-NO2-Ph)(dppf)], the most active are those bearing an iodide ligand.
In the case of the MRC-5 non-tumor cell line, cisplatin shows a very low IC50 (3.9 μM), which is substantially comparable to the results obtained on the four cancer cell lines, thus confirming the poor selectivity of this reference drug. Conversely, most of our complexes are inactive (IC50 > 100 μM) towards these normal cells, thus indicating a certain in vitro selectivity. This interesting result was not observed in the case of complexes 2b, 3a–b, 4c and 5b.
Overall, the most promising complexes are those that are active on all tumor cell lines investigated and, at the same time, inactive towards non-tumor cells (MRC-5). Among all the compounds tested, 2c and 2d are the only ones that fully satisfy these characteristics. Complex [PdI(p-CF3-Ph)(dppf)] (4b) is also quite promising, even if it exhibits a relatively low cytotoxicity on the OVCAR-5 line (IC50 = 40 ± 7 μM). The two most interesting complexes (2c and 2d) are characterized by the presence of a phosphine with a low bite angle (dppe) and by electron-rich aryl fragments (phenyl or para-tolyl).
Antiproliferative activity on patient-derived organoids
Encouraged by the results obtained on ovarian cancer cell lines, we envisioned whether further experiments on more complex biological models could furnish us with significant insights into the real efficacy of the compounds synthesized in this study. Within this framework, organoids are lab-built mini-organs that can act as models to summarise cancer development.43 The emergence of innovative organoid biobanks represents the forefront of ex vivo drug testing.44 Indeed, both innate and acquired chemoresistance, coupled with tumor heterogeneity, stand as formidable barriers to therapeutic success for ovarian cancer, necessitating innovative preclinical models to simulate this complexity.45A few pioneering groups in this field are actively developing animal and ex vivo organoid models of ovarian cancer to more accurately replicate the responses observed in clinical patients.46 It is crucial to remember that within the spectrum of ovarian cancers, the high-grade serous subtype (HGSOC) prevails as the most common and provides the lowest five-year survival rate.45 Furthermore, approximately 30% of HGSOC patients develop ascites, comprising free-floating cells accountable for intraperitoneal metastasis.47 Such patients pose challenges for conventional chemotherapy, often resorting to paracentesis for symptom relief.48
Given these challenges, existing lines of therapy are not effective, underscoring the pressing need for novel drugs to surmount innate or acquired resistance, which diminishes treatment effectiveness and, in turn, amplifies toxicity levels. Therefore, taking advantage of organoid biotechnology, we selected two patient-derived tumoroids (PDTO-1 and PDTO-2), originating from high-grade serous ovarian cancer (HGSOC) and recently isolated and characterized by our research group.49 Notably, from immunohistochemistry (IHC) analyses, the established organoids captured the histological characteristics of the primary tumors. In particular, hematoxylin as well as PAX8, WT-1, and CA-125 HGOC markers were evaluated by the pathologist.
The selected organoids were chosen to evaluate the effectiveness of one of the most promising Pd(II)–aryl complexes reported in this work (2c). The IC50 values reported in Table 2 demonstrate that complex 2c exhibits good/moderate activity even in these more complex biological models. In more detail, the cytotoxicity of complex 2c is comparable (same order of magnitude) to that of carboplatin, which is the reference compound for standard clinical therapy.
| Compound | IC50 (μM) | |
|---|---|---|
| PTDO-1 | PTDO-2 | |
| Carboplatin | 8 ± 2 | 9 ± 4 |
| 2c | 37 ± 9 | 20 ± 9 |
Mechanism of cell death
With the aim of determining the main biological target of the Pd(II)–aryl complexes reported in this work, we carried out a detailed study on OVCAR-5 cells using immunofluorescence techniques and [PdI(Ph)(dppe)] (2c) as a model compound.Based on our experience from studying the mechanism of action of organopalladium compounds, we preliminarily investigated cytochrome c release, DNA damage and the activation of caspases 3/7. To strengthen the data, the biological experiments were conducted by a kinetic analysis at different timepoints.
We initially wondered whether our Pd(II)–aryl complexes could cause early mitochondrial damage to tumor cells. To this end, we first investigated the release of cytochrome c. The cytochrome c immunofluorescence assay is widely utilized in cell biology to investigate the subcellular localization and dynamics of cytochrome c within cells.50 It should be remembered that cytochrome c is a crucial component of the mitochondrial electron transport chain and is involved in cellular respiration. However, during programmed cell death such as apoptosis, cytochrome c is released from the mitochondria into the cytoplasm. Alterations in cytochrome c distribution can function as an indicator of mitochondrial dysfunction and cellular death mechanisms, making this assay indispensable in research concerning apoptosis and mitochondrial activity. Following a well-established protocol, OVCAR-5 cells were treated with 2c (5 and 10 μM) and cisplatin (10 μM as positive control) for 6, 24 and 48 hours.
The collected images showed the release of cytochrome c in the case of cisplatin after 6 hours, although it was more evident at 24 and 48 hours, thus confirming an apoptosis pathway for this clinical drug (Fig. 5). In the case of complex [PdI(Ph)(dppe)] (2c), a significant release of cytochrome c was detected after 24 and 48 hours at both concentrations (5 μM and 10 μM).
 | ||
| Fig. 5 Immunofluorescence analysis of cytochrome c release after 6, 24 and 48 h of treatment with complex 2c. OVCAR-5 cancer cells were treated with complex 2c at concentrations of 5 and 10 μM. Arrows indicate cytochrome c release. Positive control: cisplatin 10 μM. Magnification 100×, scale bar 20 μm. | ||
Following this assay, our attention turned towards examining DNA damage through the γH2AX immunofluorescence assay. The gamma H2AX assay specifically focuses on the phosphorylation of histone H2AX at serine 139 in response to double-strand breaks (DSBs) in the DNA molecule.51 The term “gamma” refers to the gamma isoform of H2AX, which is a subtype of the histone H2A family. The gamma H2AX assay provides a sensitive and quantitative measure of DSBs, allowing researchers to evaluate the extent of DNA damage and the effectiveness of DNA repair mechanisms.52
One of the significant advantages of the gamma H2AX assay is its sensitivity. Even low levels of DNA damage can be detected, making it an invaluable tool for assessing the impact of various genotoxic agents.
As shown in Fig. 6, the cells treated with compound 2c showed a marked phosphorylation of Ser139 of histone H2AX after 6, 24 and 48 hours. This result suggests that DNA is likely to be a major molecular target of our Pd(II)–aryl complexes. Notably, even in the case of cisplatin the phosphorylation of histone H2AX is clearly evident, thus confirming the well-known interaction between cisplatin and DNA (DNA platination).
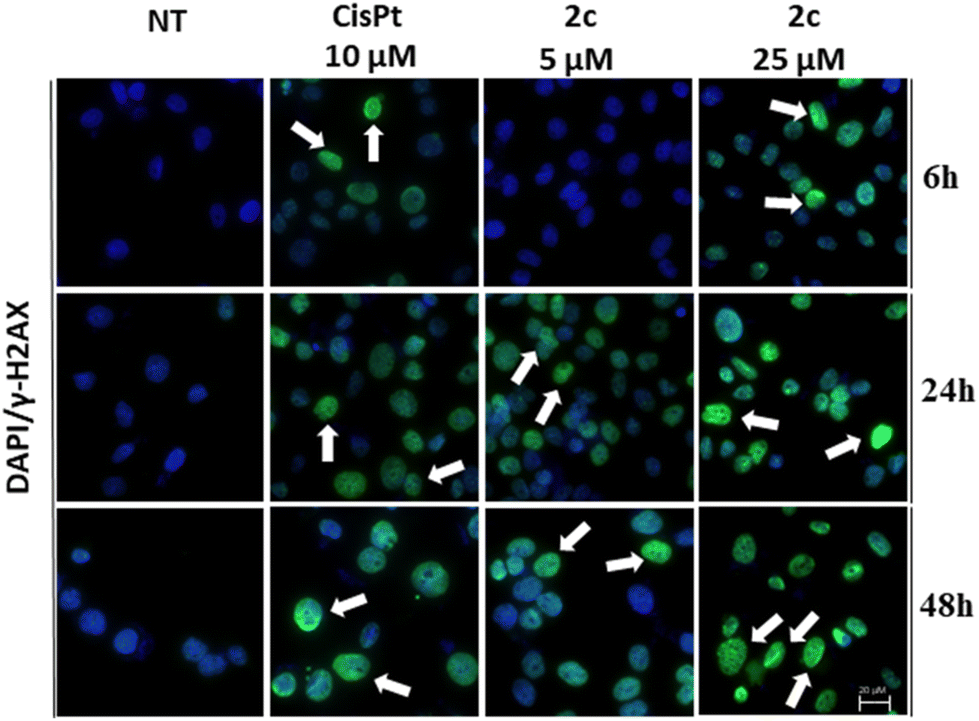 | ||
| Fig. 6 Assay on DNA damage after 6, 24 and 48 h of treatment with complex 2c. OVCAR-5 cancer cells were treated with complex 2c at concentrations of 5 and 25 μM. Arrows indicate DNA damage. Positive control: cisplatin 10 μM. Magnification 100×, scale bar 20 μm. | ||
Finally, we investigated the activation of caspases 3 and 7 to verify whether tumor cell death followed an apoptotic pathway. As a matter of fact, the caspases 3/7 assay is a well-established method in cell biology to assess apoptosis, a programmed cell death process crucial for maintaining tissue homeostasis and eliminating damaged or unwanted cells.53 Caspases 3 and 7 are key executioner caspases that play a central role in the execution phase of apoptosis by cleaving various cellular substrates, leading to cell dismantling.
The obtained data (Fig. 7) show a marked activation of caspases 3 and 7 for both cisplatin (24 and 48 hours) and complex 2c (at all timepoints with a 10 μM concentration).
 | ||
| Fig. 7 Activation of caspases 3/7 as markers of apoptosis after 3, 6, 24 and 48 h of treatment with complex 2c. OVCAR-5 cancer cells were treated with complex 2c at concentrations of 5 μM and 10 μM. Results showed that the complex induced apoptosis in the OVCAR-5 cancer cell line. Positive control: cisplatin 10 μM. p-value was calculated vs. NT (untreated samples). (p-values: * ≤ 0.05, ** ≤ 0.01, *** ≤ 0.001, **** ≤ 0.0001.) | ||
These results suggest that in the case of complex 2c, cell death follows an intrinsic apoptotic pathway.
Molecular docking analysis of structure–activity relationships
Aiming at better understanding the interaction between the Pd(II)–aryl complexes and DNA, molecular docking simulations were carried out. Three complexes have been considered: [PdI(Ph)(dppe)] (2c), [PdI(Ph)(DPEphos)] (6c*), and [PdI(p-CF3-Ph)(DPEphos)] (6b): the first and third show remarkable differences in biological activity and structurally exhibit a large difference in the diphosphine bite angle (86° vs. 102°, measured on the optimized molecular geometries). The second complex is a model that was considered because it has a close structural analogy to the third (both have a bite angle of 102°), but it lacks the CF3 groups and so in principle the comparison allows us to separate the role of the bite angle from other relevant effects due to the ring substituents.First, the in silico analysis demonstrates that the major groove region of DNA and the hydrophobic contacts with nucleotide residues are the main types of interactions for all three complexes (Fig. 8); this is in agreement with previous studies involving other Pd derivatives.54
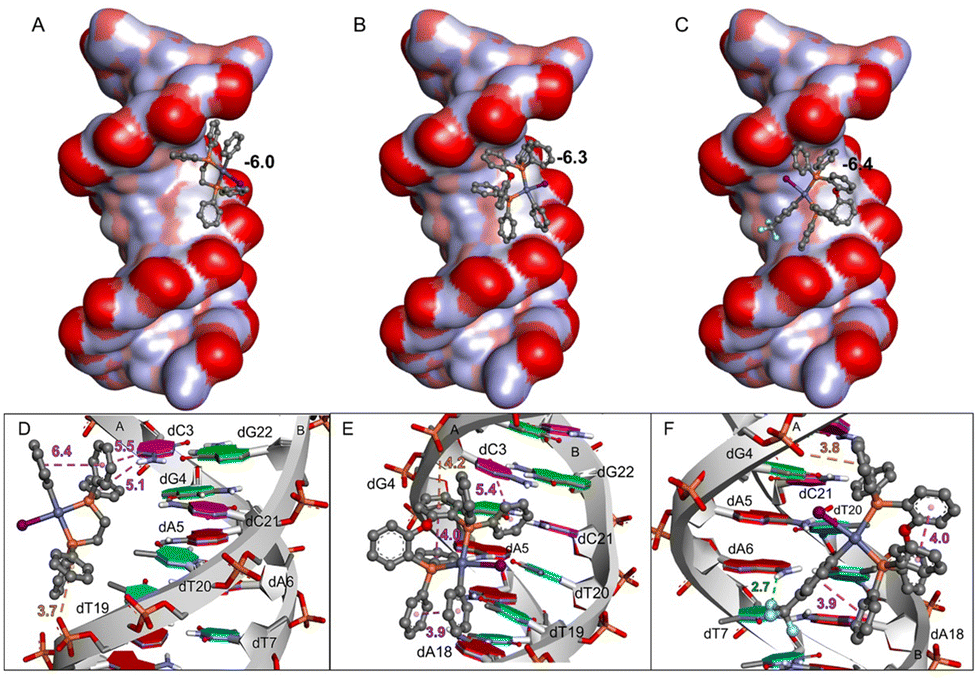 | ||
| Fig. 8 DNA interactions with Pd(II) complexes (A) [PdI(Ph)(dppe)], (B) [PdI(Ph)(DPEphos)], and (C) [PdI(p-CF3-Ph)(DPEphos)]. Close-up view of the binding pose and interactions between DNA (PDB ID 1BNA) and (D) [PdI(Ph)(dppe)], (E) [PdI(Ph)(DPEphos)], and (F) [PdI(p-CF3-Ph)(DPEphos)]. At the top, DNA is represented by a surface model colored by atom charges, and the Pd(II) complexes are shown as ball-and-stick models. The predicted binding energies (ΔG) for [PdI(Ph)(dppe)] (A), [PdI(Ph)(DPEphos)] (B), and [PdI(p-CF3-Ph)(DPEphos)] (C) were −6.0 kcal mol−1, −6.3 kcal mol−1 and −6.4 kcal mol−1, respectively. At the bottom, DNA is shown by the strands and rings in red, blue, pink, and green indicate deoxyadenosine (dA), deoxythymidine (dT), deoxycytidine (dC), and deoxyguanosine (dG) residues, respectively. Hydrophobic (π–π), H-bond, and electrostatic (anion–π) interactions are represented by purple, green, and orange dashed lines with their respective distances in Å. | ||
Binding is thermodynamically favourable (ΔG ≈ −6 kcal mol−1) in all cases and the energy values are very similar. Interestingly, the phenyl groups of [PdI(Ph)(dppe)] (2c) point directly into the groove, interacting mostly with deoxycytidine (dC) and deoxythymidine (dT) residues, while the Pd(II)–iodide moiety points outside this region. Conversely, the less reactive complexes ([PdI(Ph)(DPEphos)] (6c*) and [PdI(p-CF3-Ph)(DPEphos)] (6b)) present a different binding pose, due to steric hindrance caused by the Ph–O–Ph moiety, which imposes a larger bite angle. These molecules have intramolecular hydrophobic interactions, between the phenyl groups, and the Pd–I group is oriented parallel to the DNA. As shown in Fig. 9, [PdI(Ph)(dppe)] (2c) fits well into the major groove of DNA, with the hydrophobic region interacting with the DNA bases and the polar region (Pd–I moiety) being guided to the solvent-accessible area (water in the biological environment). On the other hand, [PdI(Ph)(DPEphos)] (6c*) and [PdI(p-CF3-Ph)(DPEphos)] (6b) show less favourable interactions with DNA. The Pd–I moiety of [PdI(Ph)(DPEphos)] (6c*) binds in the hydrophobic region of the DNA groove, and the iodide ligand of [PdI(p-CF3-Ph)(DPEphos)] (6b) interacts with the negative phosphate group of dG4, i.e., the two negative regions are close (3.9 Å). In addition, the Pd complexes show Pd⋯O interactions (from 6 to 8 Å) involving the metal center and the oxygen atom from the phosphodiester group of the nucleotide residues. In fact, it has already been reported that Pd is able to interact both with the phosphate and the nitrogen bases of DNA.55 In conclusion, complex [PdI(Ph)(dppe)] (2c) has the most favourable interactions with DNA, which may explain its remarkable biological activity.
 | ||
| Fig. 9 Surface models of the binding pose of (A) [PdI(Ph)(dppe)], (B) [PdI(Ph)(DPEphos)], and (C) [PdI(p-CF3-Ph)(DPEphos)] complexes with DNA. The surfaces are colored to indicate atom partial charges: red, blue, and grey indicate negative, positive, and neutral (or hydrophobic) regions. | ||
Conclusions
In conclusion, a synthetic protocol for a wide range of Pd(II)–aryl complexes, bearing different diphosphine, halide and aryl ligands was developed, profitably modifying some procedures reported in the literature. In particular, the way to introduce chloride or bromide ligands into the palladium coordination sphere was original and involved the passage through an intermediate species equipped with a chelating thioquinoline ligand, from which it was possible to obtain the fast and complete substitution of the coordinated iodide with bromide or chloride by adding IBr or ICl, respectively.A good portion of the prepared compounds are unpublished and for this reason their exhaustive characterization is provided and based on NMR, IR and HRMS analyses. Moreover, in some cases the solid-state structures were also defined by single crystal X-ray diffractometry.
In the case of complex [PdI(p-NO2-Ph)(dppbz)] (5a), a kinetic and computational study allowed us to determine the kinetic constant and the mechanism for the formation of this complex.
This synthetic effort has provided us with a sufficient number of Pd(II)–aryl complexes to propose the first systematic study on the anticancer properties of this class of organopalladium compounds. These were tested on a selection of ovarian and breast cancer cell lines, showing IC50 values often comparable to, or even better than, cisplatin, which was taken as a reference metallodrug. Of particular significance is the fact that complexes 2c and 2d exhibited excellent cytotoxicity towards cancer cells and, at the same time, low activity on normal ones (human lung fibroblasts). These compounds contain a diphosphine ligand with a low bite angle (dppe) and electron-rich aryl fragments (phenyl or para-tolyl). As a result of these findings, [PdI(Ph)(dppe)] (2c) was chosen as a model for studying the mechanism of action of the complexes reported in this work. Immunofluorescence tests (cytochrome c and H2AX assays) seem to indicate DNA as the primary cellular target. Moreover, the activation of caspases 3 and 7 suggests that cell death occurs through an apoptotic pathway.
Remarkably, a detailed molecular docking study confirmed that the interaction between DNA and our Pd(II)–aryl complexes was stronger when diphosphine ligands with low bite angles were employed. In contrast, aryl and halide ligands appear to make a less significant contribution to the stability of the complex–DNA adduct and therefore to the antitumor activity of the synthesized complexes.
Finally, complex [PdI(Ph)(dppe)] (2c) showed promising cytotoxicity even on more complex and realistic biological models such as 3D organoids obtained from tumoral tissues of different real patients. Its activity is comparable to that of carboplatin, which is the reference compound for standard clinical therapy.
We believe that the high selectivity observed in vitro and the efficacy for the ex vivo models are good premises in view the of future clinical application of some compounds described in this paper. However, further studies aimed primarily at evaluating their in vivo efficacy are ongoing in our laboratories.
Experimental section
Solvents and reagents
All syntheses were carried out under an inert atmosphere using standard Schlenk techniques. The solvent CH2Cl2 was distilled over P2O5 and stored under a N2 atmosphere. All other solvents and chemicals were commercial grade products and used as purchased. Complexes 1a–d21,22 and 7-I41 were synthesized according to published protocols.NMR, UV-Vis and IR measurements
1D NMR and 2D NMR spectra were recorded on Bruker 300 or 400 Avance spectrometers. Chemical shift values (ppm) are given relative to TMS (1H and 13C), H3PO4 (31P) and CCl3F (19F).IR spectra were recorded on a PerkinElmer Spectrum One spectrophotometer. HRMS spectra were recorded on a Bruker Compact Q-TOF instrument. Mass spectra were recorded in positive mode.
Computational details
All calculations were performed by using DFT, as implemented in the ORCA 4.2 suite of ab initio quantum chemistry programs.56 Geometry optimizations were performed with the B97M-D3BJ functional57 by using the double-ζ-quality def2-SVP58 basis set that included relativistic core potentials for Pd.Solvent effects (dichloromethane, ε = 8.93) were included using CPCM. More accurate single-point energies were computed from the optimized geometries by using ωB97M-V59 DFT and the triple-ζ-quality def2-TZVPP58 basis set. Vibrational frequencies were computed at the B97M-D3BJ/def2-SVP level of theory to derive the Gibbs free energy.
The DNA blind docking studies were carried out using the AutoDock Vina 1.1.1 program,60 using the B-DNA dodecamer crystallographic structure from the Protein Data Bank – PDB (ID 1BNA, sequence d(CGCGAATTCGCG)2), according to previous studies.61 The structures of the Pd complexes were obtained by full geometry optimizations using the BLYP potential62 combined with a Slater triple zeta quality basis set with two polarization functions. The small core approximation was used and scalar relativistic effects were included using the ZORA approximation.63 This level of theory, here denoted ZORA-BLYP/TZ2P, gave accurate results for compounds with heavy nuclei.64 The DFT calculations were carried out using ADF2019.65 Hirshfeld partial charges computed at the ZORA-BLYP/TZ2P level were used in the docking simulation and the DNA macromolecule was prepared using the Chimera 1.8 software.66 Since AutoDock Vina does not recognize the Pd atom, it was replaced by Zn for docking simulations, but it retained all the properties of this metal center obtained from the quantum mechanical results. It was used with an exhaustiveness of 50, and the grid box was positioned in the center of the DNA structure (coordinates xyz: 14.75, 20.98, and 9.23; size: 50 × 50 × 50 Å). As a model of the binding pose, the ligand's conformers with the lowest predicted binding free energy (ΔG) were selected from the most populated cluster.
Synthesis of Pd–aryl complexes
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 2.18–2.51 (m, 4H, PCH2), 7.27–7.56 (m, 20H, aryl–H), 7.84–7.91 (m, 4H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm) δ: 25.2 (CH2, dd, JC–P = 25.9 Hz, 13.2 Hz, PCH2), 29.7 (CH2, dd, JC–P = 30.0 Hz, 20.8 Hz, PCH2), 120.3, 120.4, 128.3, 128.9, 129.1, 129.1, 129.2, 129.3, 130.4, 130.9, 131.3, 131.4, 131.9, 131.9, 133.0, 133.2, 133.8, 133.9, 137.7, 137.8, 137.8, 137.8, 144.7, 172.0, 173.8.
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: 37.5 (d, JP–P = 26.1 Hz), 51.0 (d, JP–P = 26.1 Hz).
IR (KBr pellet, cm−1): νNO2 = 1335, 1497.
HRMS calcd for [C32H28INNaO2P2Pd]+: 775.9579; found: 775.9577.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 2.17–2.46 (m, 4H, PCH2), 6.93 (pseudo d, 2H, JH–H = 7.0 Hz, aryl–H), 7.20 (pseudo t, 2H, JH–H = 7.9 Hz, aryl–H), 7.32–7.37 (m, 8H, aryl–H), 7.45–7.52 (m, 8H, aryl–H), 7.89 (pseudo t, 4H, JH–H = 8.2 Hz, aryl–H).
31P{1H} NMR (162 MHz, CDCl3, T = 298 K, ppm) δ: 35.5 (d, JP–P = 27.2 Hz), 50.2 (d, JP–P = 27.2 Hz).
19F{1H} NMR (377 MHz, CDCl3, T = 298 K, ppm) δ: −61.9.
Data are in agreement with the reported values.38
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 2.13–2.46 (m, 4H, PCH2), 6.62–6.75 (m, 3H, aryl–H), 7.09 (pseudo t, 2H, JH–H = 7.8 Hz, aryl–H), 7.29–7.48 (m, 16H, aryl–H), 7.87–7.93 (m, 4H, aryl–H).
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: 34.0 (d, JP–P = 27.9 Hz), 48.9 (d, JP–P = 27.9 Hz).
Data are in agreement with the reported values.40
1H NMR (300 MHz, CDCl,3T = 298 K, ppm) δ: 2.10 (s, 3H, CH3), 2.13–2.45 (m, 4H, PCH2), 6.57–6.61 (m, 2H, aryl–H), 6.90–6.96 (m, 2H, aryl–H), 7.28–7.48 (m, 16H, aryl–H), 7.86–7.93 (m, 4H, aryl–H).
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: 33.9 (d, JP–P = 27.6 Hz), 48.7 (d, JP–P = 27.6 Hz).
Data are in agreement with the reported values.40
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 1.83–2.04 (m, 2H, CH2), 2.40–2.47 (m, 2H, PCH2), 2.53–2.60 (m, 2H, PCH2), 7.12–7.25 (7H, aryl–H), 7.27–7.46 (13H, aryl–H), 7.75–7.80 (4H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm) δ: 19.1 (CH2, d, JC–P = 2.3 Hz, CH2), 26.7 (CH2, dd, JC–P = 21.1 Hz, 3.8 Hz, PCH2), 28.0 (CH2, dd, JC–P = 24.9 Hz, 7.3 Hz, PCH2), 120.2, 120.4, 128.5, 128.7, 128.8, 129.8, 130.4, 130.7, 131.0, 132.0, 132.5, 133.0, 133.2, 133.7, 133.8, 137.1, 144.1, 171.8, 173.5.
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: −9.9 (d, JP–P = 52.6 Hz), 9.7 (d, JP–P = 52.6 Hz).
IR (KBr pellet, cm−1): νNO2 = 1334, 1495.
Data are in agreement with the reported values.37
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 1.80–2.02 (m, 2H, CH2), 2.39–2.46 (m, 2H, PCH2), 2.52–2.59 (m, 2H, PCH2), 6.73–6.76 (m, 2H, aryl–H), 7.02–7.08 (m, 2H, aryl–H), 7.11–7.17 (m, 4H, aryl–H), 7.22–7.35 (m, 7H, aryl–H), 7.42–7.48 (m, 5H, aryl–H), 7.76–7.83 (m, 4H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 19.2 (CH2, d, JC–P = 2.9 Hz, CH2), 26.9 (CH2, dd, JC–P = 19.9 Hz, 4.2 Hz, PCH2), 28.2 (CH2, dd, JC–P = 25.9 Hz, 7.1 Hz, PCH2), 124.1 (C, q, JC–F = 31.3 Hz, p-aryl–C), 125.1 (C, q, JC–F = 271.3 Hz, CF3), 122.8, 122.9, 122.9, 123.0, 128.4, 128.6, 128.6, 128.7, 130.2, 130.5, 130.5, 130.7, 130.7, 130.9, 132.3, 132.8, 133.0, 133.2, 133.7, 133.9, 136.9, 136.9, 136.9, 162.0, 163.8.
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: −10.3 (d, JP–P = 53.3 Hz), 10.3 (d, JP–P = 53.3 Hz).
19F{1H} NMR (377 MHz, CDCl3, T = 298 K, ppm) δ: −61.9.
Data are in agreement with the reported values.37
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 3.67 (q, 2H, JH–H = 1.8 Hz, Fc–H), 4.17 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.53 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.70 (q, 2H, JH–H = 2.1 Hz, Fc–H), 7.10–7.16 (m, 4H, aryl–H), 7.18–7.24 (m, 2H, aryl–H), 7.31–7.40 (m, 8H, aryl–H), 7.49–7.52 (m, 6H, aryl–H), 7.98–8.05 (m, 4H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 72.4 (CH, d, JC–P = 5.0 Hz, Fc-CH), 73.8 (CH, d, JC–P = 7.4 Hz, Fc-CH), 74.6 (CH, d, JC–P = 8.2 Hz, Fc-CH), 76.2 (CH, d, JC–P = 12.0 Hz, Fc-CH), 120.5, 120.7, 128.2, 128.3, 128.4, 128.4, 130.7, 130.7, 130.9, 130.9, 132.1, 132.8, 133.0, 133.5, 133.9, 134.1, 135.6, 135.7, 137.1, 144.2.
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: 9.3 (d, JP–P = 32.0 Hz), 26.2 (d, JP–P = 32.0 Hz).
IR (KBr pellet, cm−1): νNO2 = 1337, 1502.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 3.69 (q, 2H, JH–H = 1.8 Hz, Fc–H), 4.15 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.50 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.67 (q, 2H, JH–H = 2.0 Hz, Fc–H), 6.72–6.76 (m, 2H, aryl–H), 7.06–7.16 (m, 6H, aryl–H), 7.30–7.38 (m, 6H, aryl–H), 7.47–7.52 (m, 6H, aryl–H), 7.99–8.06 (m, 4H, aryl–H).
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: 8.7 (d, JP–P = 33.5 Hz), 26.5 (d, JP–P = 33.5 Hz).
19F{1H} NMR (377 MHz, CDCl3, T = 298 K, ppm) δ: −61.9.
Data are in agreement with the reported values.21
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 3.70 (q, 2H, JH–H = 1.8 Hz, Fc–H), 4.13 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.47 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.64 (q, 2H, JH–H = 2.1 Hz, Fc–H), 6.40–6.45 (m, 1H, aryl–H), 6.51–6.57 (m, 2H, aryl–H), 6.90–6.97 (m, 2H, aryl–H), 7.09–7.15 (m, 4H, aryl–H), 7.28–7.40 (m, 6H, aryl–H), 7.45–7.50 (m, 6H, aryl–H), 8.00–8.07 (m, 4H, aryl–H).
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: 8.0 (d, JP–P = 34.5 Hz), 26.4 (d, JP–P = 34.5 Hz).
Data are in agreement with the reported values.40
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 2.01 (s, 3H, CH3), 3.72 (q, 2H, JH–H = 1.8 Hz, Fc–H), 4.12 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.45 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.63 (q, 2H, JH–H = 2.1 Hz, Fc–H), 6.39 (pseudo d, 2H, JH–H = 7.6 Hz, aryl–H), 6.78 (pseudo td, 2H, JH–H = 7.9 Hz, 2.4 Hz, aryl–H), 7.12 (pseudo td, 4H, JH–H = 7.8 Hz, 2.3 Hz, aryl–H), 7.29–7.39 (m, 7H, aryl–H), 7.46–7.48 (m, 5H, aryl–H), 8.01–8.06 (m, 4H, aryl–H).
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 7.23–7.73 (28H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 120.1, 120.3, 128.6, 128.9, 129.0, 129.0, 129.1, 129.3, 130.9, 131.2, 131.2, 131.4, 131.6, 131.6, 132.0, 132.2, 132.3, 132.6, 133.2, 133.4, 133.6, 133.8, 134.1, 134.3, 134.6, 134.8, 137.9, 137.9, 138.0, 144.9.
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: 45.1 (d, JP–P = 26.4 Hz), 51.2 (d, JP–P = 26.4 Hz).
IR (KBr pellet, cm−1): νNO2 = 1339, 1500.
HRMS calcd for [C36H28INNaO2P2Pd]+: 823.9581; found: 823.9584.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 6.93–7.72 (28H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 122.7, 122.8, 122.8, 122.9, 123.5, 124.7, 125.2, 126.5, 127.1, 127.2, 128.8, 128.8, 129.0, 129.7, 131.0, 131.0, 131.2, 131.3, 131.3, 131.7, 132.1, 132.2, 132.4, 132.4, 133.3, 133.4, 133.6, 133.9, 134.1, 134.3, 134.6, 134.8, 137.5, 137.6, 137.6, 162.5, 164.3.
31P{1H} NMR (162 MHz, CDCl3, T = 298 K, ppm) δ: 43.8 (d, JP–P = 27.4 Hz), 51.1 (d, JP–P = 27.4 Hz).
19F{1H} NMR (377 MHz, CDCl3, T = 298 K, ppm) δ: −61.8.
HRMS calcd for [C37H28F3INaP2Pd]+: 846.9604; found: 846.9619.
1H NMR (300 MHz, CDCl3, T = 233 K, ppm) δ: 6.54–7.89 (m, 32H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 120.2, 123.4, 123.9, 128.0, 128.1, 130.2, 133.9, 135.6, 137.8, 144.1, 158.5, 158.6.
31P{1H} NMR (121 MHz, CDCl3, T = 233 K, ppm) δ: 5.2 (d, JP–P = 33.0 Hz), 9.7 (d, JP–P = 33.0 Hz).
IR (KBr pellet, cm−1): νNO2 = 1336, 1499.
1H NMR (300 MHz, CD2Cl2, T = 233 K, ppm) δ: 6.41–7.91 (m, 32H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 233 K, ppm) δ: 117.7, 117.8, 123.0, 123.1, 123.4, 123.5, 123.5, 123.8, 123.8, 123.9, 124.0, 124.0, 124.1, 125.2, 125.3, 126.7, 127.7, 127.8, 128.0, 128.1, 128.2, 129.8, 130.0, 131.4, 132.6, 133.5, 135.4, 158.1, 158.2, 158.3, 158.3.
31P{1H} NMR (121 MHz, CD2Cl2, T = 233 K, ppm) δ: 4.3 (d, JP–P = 34.0 Hz), 10.4 (d, JP–P = 34.0 Hz).
19F{1H} NMR (377 MHz, CD2Cl2, T = 298 K, ppm) δ: −61.9.
IR (KBr pellet, cm−1): νNO2 = 1336, 1499.
HRMS calcd for [C43H32F3INaOP2Pd]+: 938.9868; found: 938.9887.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 1.23 (s, 9H, t-Bu), 7.66–7.79 (m, 4H, H3, H6, Ph), 7.85–7.90 (m, 2H, Ph), 8.08 (dd, 1H, JH–H = 7.3 Hz, 1.3 Hz, H7), 8.10 (dd, 1H, JH–H = 8.0 Hz, 1.3 Hz, H5), 8.45 (dd, 1H, JH–H = 8.3 Hz, 1.6 Hz, H4), 9.95 (pseudo d, 1H, JH–H = 4.9 Hz, H2).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm) δ: 30.7 (CH3, t-Bu), 59.5 (C, t-Bu), 120.7 (CH, Ph), 123.6 (CH, C3), 127.4 (CH, C6), 129.6 (C, C4a), 130.3 (C, C8), 131.8 (CH, C5), 137.7 (CH, C7), 138.9 (CH, Ph), 139.1 (CH, C4), 145.7 (C, Ph), 148.8 (C, C8a), 152.2 (C, Ph), 155.1 (CH, C2).
IR (KBr pellet, cm−1): νNO2 = 1338, 1500.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 1.23 (s, 9H, t-Bu), 7.68–7.80 (m, 4H, H3, H6, Ph), 7.86–7.91 (m, 2H, Ph), 8.08 (dd, 1H, JH–H = 7.5 Hz, 1.3 Hz, H7), 8.11 (dd, 1H, JH–H = 8.4 Hz, 1.3 Hz, H5), 8.46 (dd, 1H, JH–H = 8.4 Hz, 1.6 Hz, H4), 9.78 (dd, 1H, JH–H = 4.9 Hz, 1.6 Hz, H2).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm) δ: 30.6 (CH3, t-Bu), 59.4 (C, t-Bu), 120.8 (CH, Ph), 123.4 (CH, C3), 127.4 (CH, C6), 129.5 (C, C4a), 130.2 (C, C8), 131.8 (CH, C5), 137.5 (CH, C7), 138.2 (CH, Ph), 139.2 (CH, C4), 145.7 (C, Ph), 148.7 (C, C8a), 153.5 (CH, C2), 153.8 (C, Ph).
IR (KBr pellet, cm−1): νNO2 = 1337, 1499.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 3.58 (q, 2H, JH–H = 1.9 Hz, Fc–H), 4.19 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.54 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.73 (q, 2H, JH–H = 2.1 Hz, Fc–H), 7.09–7.26 (m, 6H, aryl–H), 7.30–7.51 (m, 14H, aryl–H), 8.02–8.08 (m, 4H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 72.5 (CH, d, JC–P = 5.0 Hz, Fc–H), 74.0 (CH, d, JC–P = 7.8 Hz, Fc–H), 74.7 (CH, d, JC–P = 8.1 Hz, Fc–H), 76.4 (CH, d, JC–P = 12.3 Hz, Fc–H), 121.1, 121.2, 128.2, 128.4, 128.5, 128.6, 130.6, 130.7, 131.0, 131.0, 132.1, 132.2, 132.6, 132.9, 134.0, 134.1, 135.1, 135.2, 135.2, 135.4, 144.3.
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: 11.3 (d, JP–P = 31.0 Hz), 31.5 (d, JP–P = 31.0 Hz).
IR (KBr pellet, cm−1): νNO2 = 1339, 1504.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 3.62 (q, 2H, JH–H = 1.8 Hz, Fc–H), 4.18 (t, 2H, JH–H = 1.8 Hz, Fc–H), 4.54 (t, 2H, JH–H = 1.9 Hz, Fc–H), 4.72 (q, 2H, JH–H = 2.1 Hz, Fc–H), 7.10–7.24 (m, 6H, aryl–H), 7.30–7.42 (m, 8H, aryl–H), 7.47–7.52 (m, 6H, aryl–H), 8.00–8.07 (m, 4H, aryl–H).
13C{1H} NMR (75 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 72.5 (CH, d, JC–P = 4.9 Hz, Fc–H), 73.9 (CH, d, JC–P = 7.6 Hz, Fc–H), 74.6 (CH, d, JC–P = 8.2 Hz, Fc–H), 76.3 (CH, d, JC–P = 12.3 Hz, Fc–H), 121.0, 121.1, 128.2, 128.4, 128.5, 130.6, 130.7, 131.0, 131.0, 132.1, 132.4, 132.8, 132.9, 134.0, 134.1, 135.3, 135.5, 135.6, 135.7, 144.2.
31P{1H} NMR (121 MHz, CDCl3, T = 298 K, ppm) δ: 10.4 (d, JP–P = 31.2 Hz), 30.3 (d, JP–P = 31.2 Hz).
IR (KBr pellet, cm−1): νNO2 = 1338, 1503.
Cell viability assay
Four cancer cells lines (A2780, A2780cis, OVCAR-5, MDA-MB-231) and one non-tumoral cell type (MRC-5) were employed and grown in accordance with the supplier's instructions and maintained at 37 °C under a humidified atmosphere of 5% of CO2. A defined number of cells (A2780 1000 cells per well (cpw), A2780cis 2500 cpw, OVCAR-5 2000 cpw, MDA-MB-231 1000 cpw and MRC-5 8000 cpw) were seeded in 96 wells and after 24 h treated with six different concentrations of Pd(II) complexes (0.001, 0.01, 0.1, 1, 10, 100 μM). 96 h after treatment, the cell viability was measured with a CellTiter glow assay (Promega, Madison, WI, USA) with BioTek Synergy H1. IC50 values were calculated from logistic dose–response curves. Averages were obtained from experiments carried out in triplicate and error bars are standard deviations.Organoid cultures
Specimens underwent complete anonymization prior to the derivation of organoids. Nonetheless, participants provided informed consent for research utilization of the samples, facilitated through the biobank at the National Cancer Institute (CRO) in Aviano. Primary tumor samples were subjected to a pre-processing incubation in Dulbecco's modified Eagle's medium/nutrient mixture F-12 ham supplemented with a cocktail of antimicrobial agents (levofloxacin 100 μg mL−1, vancotex 25 μg mL−1, ciproxin 5 μg mL−1, gentamicin 200 μg mL−1, and fungizone 5 μg mL−1) for 30 minutes. Subsequently, tissues were meticulously minced to 0.5–1 mm3 fragments, treated with a 4 mg mL−1 solution of collagenase IV (Gibco, Massachusetts, USA), and incubated at 37 °C, avoiding exceeding 45-minute enzyme exposure. Mechanical disaggregation was achieved via pipetting. Following centrifugation at 1000 rpm for 10 minutes, the cellular aggregates were resuspended in Cultrex RGF BME, Type 2 (Bio-techne, Minnesota, USA), and seeded in 24-well culture plates for maintenance. Once the Cultrex matrix solidified, each well received 500 μL of organoid medium, refreshed tri-weekly as delineated by Kopper et al.46 The organoids were then maintained in a controlled environment at 37 °C under a 5% CO2 atmosphere.Organoids’ half-maximal inhibitory concentration (IC50)
Clusters of PDOs were mixed in an appropriate volume of Cultrex RGF BME, Type 2 (Bio-techne, Minnesota, USA) and 2 μL of this mixture were seeded in 96-well plates and treated with six different concentrations of carboplatin (0.032, 0.16, 0.8, 4, 20, 100 μM) and compound 2c (0.016, 0.08, 0.4, 2, 10, 50 μM) in four replicates. After 96 h, cell viability was measured using CellTiter-Glo 3D (Promega, Madison, WI, USA) with BioTek Synergy H1. Logistic dose–response curves were used to calculate IC50 using GraphPad Prism (La Jolla, CA, US).DNA damage assay
OVCAR-5 cells were seeded (20000 cells per well) on chamber slides. After overnight culture at 37 °C and 5% CO2, cells were treated with different concentrations of compound 2c (5 and 10/25 μM) or cisplatin (10 μM) for 6, 24, and 48 h. Cells were fixed in 4% paraformaldehyde/PBS (20 min, RT), permeabilized with 0.3% Triton X-100/PBS (15 min, RT) and blocked in 8% BSA/PBS (1 h, RT). Cells were incubated with rabbit monoclonal anti-Ser139γH2A.X antibody (Cell Signaling Cat. #9718 Burlington, MA, USA, 1:100 dilution in 1% BSA/PBS, at 4 °C, overnight). Samples incubated with Ser139γH2A.X were labeled with secondary antibody (Goat anti-Rabbit Alexa Fluor™ 488, 1:1000 dilution, RT, 1 h) obtained from Cell Signaling Technology (Cat. #A32731; Danvers, MA, US). To visualize nuclei, cells were stained with 1 mg mL−1 DAPI (in PBS, RT, 1 min). Cells were washed three times with PBS after all incubations. All the chamber slides were mounted with fluorSave™ reagent (Cat. #345789; Millipore: Burlington, MA, USA).
Cells were examined with a NIKON Eclipse TI2 (equipped with X-Light V2 L-FOV spinning disk and lumencore lamp) fluorescence microscope with an X-Cite 120 PC Q lamp and the images were analysed with NIS software.
Caspases 3/7 assay
The activity of caspases 3/7 on OVCAR-5 cancer cells was determined by Caspase-Glo™ 3/7 Assay (Cat. #G8091; Promega: Madison, WI, USA) following the standard protocol for cells cultured in a 96-well plate. 2000 cells per well were seeded on 96-well plates. After incubation overnight at 37 °C (5% CO2), cells were treated at IC50 concentrations. After 3, 6, 24 and 48 hours of treatment, the caspase activation was measured with the Caspase-Glo™ 3/7 Assay with BioTek Synergy H1. Data are normalized to heathy cells for each time point.Crystal structure determination
Data for crystals of 7-Br, 7-Cl, 4a, 4a-Br, 4a-Cl and 6a were collected at the XRD2 beamline of the Elettra Synchrotron, Trieste (Italy),67 using a monochromatic wavelength of 0.620 Å, at 100 K or 298 K. The data sets were integrated, scaled, and corrected for Lorentz absorption and polarization effects using the XDS package.68 The structures were solved by direct methods using the SHELXT program69 and refined using full-matrix least-squares implemented in SHELXL-2019/3.70 Thermal motions for all non-hydrogen atoms were treated anisotropically and hydrogen atoms were included at calculated positions, riding on their carrier atoms. Geometric restraints (DFIX, DANG, FLAT) were used to properly model disordered and poorly defined fragments. The Coot program was used for structure building.71 Pictures were prepared using Ortep372 and Pymol73 software. Crystal data are given in Table S1 in the ESI.† Crystallographic data were deposited with the Cambridge Crystallographic Data Centre and allocated the deposition numbers CCDC 2324443 (6a at 100 K), 2324442 (4a at 100 K), 2324438 (4a-Br at 298 K), 2324439 (7-Cl at 100 K), 2324440 (7-Br at 298 K) and 2324441 (4a-Cl at 298 K).†Author contributions
Fabiano Visentin, Giovanni Tonon, Laura Orian, Flavio Rizzolio, Thomas Scattolin: conception and design of study; Giovanni Tonon, Matteo Mauceri, Rachele Piccolo, Claudio Santo, Nicola Demitri, Laura Orian, Pablo A. Nogara, João Batista T. Rocha: acquisition of data; Giovanni Tonon, Matteo Mauceri, Enrico Cavarzerani, Rachele Piccolo, Claudio Santo, Nicola Demitri, Laura Orian, Pablo A. Nogara, João Batista T. Rocha, Flavio Rizzolio, Fabiano Visentin and Thomas Scattolin: analysis and/or interpretation of data; Thomas Scattolin, Laura Orian, Giovanni Tonon, Flavio Rizzolio: project management, fund acquisition, writing, review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding: this research was funded by the Fondazione AIRC per la Ricerca sul Cancro, IG23566.References
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS PubMed.
- J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed.
- C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed.
- C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef CAS PubMed.
- I. P. Beletskaya, F. Alonso and V. S. Tyurin, Coord. Chem. Rev., 2019, 385, 137 CrossRef CAS.
- S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Green Chem., 2019, 21, 381 RSC.
- D. Shen, Y. Xu and S. Shi, J. Am. Chem. Soc., 2019, 141, 14938 CrossRef CAS PubMed.
- N. Angello, V. Rathore, W. Beker, A. Wołos, E. R. Jira, R. Roszak, T. Wu, C. M. Schroeder, A. Aspuru-Guzik, B. A. Grzybowski and M. D. Burke, Science, 2022, 378, 399 CrossRef CAS PubMed.
- J. L. Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef PubMed.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed.
- D. Roy and Y. Uozumi, Adv. Synth. Catal., 2018, 360, 602 CrossRef CAS.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118 CrossRef CAS PubMed.
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, J. Organomet. Chem., 2018, 861, 17 CrossRef CAS.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Science, 2018, 360, 186 CrossRef CAS PubMed.
- C. Cordovilla, C. Bartolomé, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 3040 CrossRef CAS.
- Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981 CrossRef CAS PubMed.
- F. Villalba and A. C. Albéniz, Dalton Trans., 2022, 51, 14847 RSC.
- M. Yamashita, J. V. Cuevas-Vicario and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 16347 CrossRef CAS PubMed.
- D. Kruis, B. A. Markies, A. J. Canty, J. Boersma and G. Van Koten, J. Organomet. Chem., 1997, 532, 235 CrossRef CAS.
- D. Barafiano and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 2937 CrossRef.
- Z. Zhu, C. D. Park, K. Klimes and J. Li, Adv. Opt. Mater., 2019, 7, 1801518 CrossRef.
- J. Yang, X. Feng, N. Li, J. Li, X. Song, M. Li, G. Cui, J. Zhang, C. Jiang, C. Yang and K. Li, Sci. Adv., 2023, 9, eadh0198 CrossRef CAS PubMed.
- A. R. Kapdi and I. J. Fairlamb, Chem. Soc. Rev., 2014, 43, 4751 RSC.
- C. Cullinane, G. B. Deacon, P. R. Drago, A. P. Erven, P. C. Junk, J. Luu, G. Meyer, S. Schmitz, I. Ott, J. Schur, L. K. Webster and A. Klein, Dalton Trans., 2018, 47, 1918 RSC.
- J. Ruiz, N. Cutillas, C. Vicente, M. D. Villa, G. López, J. Lorenzo, F. X. Aviles, V. Moreno and D. Bautista, Inorg. Chem., 2005, 44, 7365 CrossRef CAS PubMed.
- T. Scattolin, V. A. Volonshki, F. Visentin and S. P. Nolan, Cell Rep. Phys. Sci., 2021, 2, 100446 CrossRef CAS.
- T. Scattolin, I. Pessotto, E. Cavarzerani, V. Canzonieri, L. Orian, N. Demitri, C. Shmidt, A. Casini, E. Bortolamiol, F. Rizzolio, F. Visentin and S. P. Nolan, Eur. J. Inorg. Chem., 2022, e202200 Search PubMed.
- T. Scattolin, E. Bortolamiol, F. Visentin, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio and A. Togni, Chem. – Eur. J., 2020, 26, 1 CrossRef PubMed.
- T. Scattolin, L. Canovese, N. Demitri, R. Gambari, I. Lampronti, C. Santo, F. Rizzolio, I. Caligiuri and F. Visentin, Dalton Trans., 2018, 47, 13616 RSC.
- T. Scattolin, E. Bortolamiol, I. Caligiuri, F. Rizzolio, N. Demitri and F. Visentin, Polyhedron, 2020, 186, 114607 CrossRef CAS.
- T. Scattolin, E. Bortolamiol, F. Rizzolio, N. Demitri and F. Visentin, Appl. Organomet. Chem., 2020, e5876 CrossRef CAS.
- T. Scattolin, A. Piccin, M. Mauceri, F. Rizzolio, N. Demitri, V. Canzonieri and F. Visentin, Polyhedron, 2021, 207, 115381 CrossRef CAS.
- A. Madabeni, T. Scattolin, E. Bortolamiol, F. Visentin and L. Orian, Organometallics, 2024 DOI:10.1021/acs.organomet.3c00514.
- L. Fiebig, N. Schlörer, H. G. Schmalz and M. Schäfer, Chem. – Eur. J., 2014, 20, 4906 CrossRef CAS PubMed.
- D. Barañano and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 2937 CrossRef.
- E. Shirakawa and T. Hiyama, J. Organomet. Chem., 1999, 576, 169 CrossRef CAS.
- G. Mann, D. Barañano, J. F. Hartwig, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120, 9205 CrossRef CAS.
- L. Canovese, F. Visentin, C. Biz, T. Scattolin, C. Santo and V. Bertolasi, Polyhedron, 2015, 102, 94 CrossRef CAS.
- T. Scattolin, F. Visentin, C. Santo, V. Bertolasi and L. Canovese, Dalton Trans., 2016, 45, 11560 RSC.
- J. Kim, B.-K. Koo and J. A. Knoblich, Nat. Rev. Mol. Cell Biol., 2020, 21, 571 CrossRef CAS PubMed.
- (a) S. Palazzolo, M. Hadla, C. R. Spena, I. Caligiuri, R. Rotondo, M. Adeel, V. Kumar, G. Corona, V. Canzonieri, G. Toffoli and F. Rizzolio, Cancers, 2019, 11, 1997 CrossRef CAS PubMed; (b) C. Granchi, G. Bononi, R. Ferrisi, E. Gori, G. Mantini, S. Glasmacher, G. Poli, S. Palazzolo, I. Caligiuri, F. Rizzolio, V. Canzonieri, T. Perin, J. Gertsch, A. Sodi, E. Giovannetti, M. Macchia, F. Minutolo, T. Tuccinardi and A. Chicca, Eur. J. Med. Chem., 2021, 209, 112857 CrossRef CAS PubMed; (c) F. Duzagac, G. Saorin, L. Memeo, V. Canzonieri and F. Rizzolio, Cancers, 2021, 13, 1 CrossRef PubMed.
- D. D. Bowtell, S. Böhm, A. A. Ahmed, P. J. Aspuria, R. C. Bast, V. Beral, J. S. Berek, M. J. Birrer, S. Blagden, M. A. Bookman, J. D. Brenton, K. B. Chiappinelli, F. C. Martins, G. Coukos, R. Drapkin, R. Edmondson, C. Fotopoulou, H. Gabra, J. Galon, C. Gourley, V. Heong, D. G. Huntsman, M. Iwanicki, B. Y. Karlan, A. Kaye, E. Lengyel, D. A. Levine, K. H. Lu, I. A. McNeish, U. Menon, S. A. Narod, B. H. Nelson, K. P. Nephew, P. Pharoah, D. J. Powell, P. Ramos, I. L. Romero, C. L. Scott, A. K. Sood, E. A. Stronach and F. R. Balkwill, Nat. Rev. Cancer, 2015, 15, 668 CrossRef CAS PubMed.
- (a) R. Perets, G. A. Wyant, K. W. Muto, J. G. Bijron, B. B. Poole, K. T. Chin, J. Y. Chen, A. W. Ohman, C. D. Stepule, S. Kwak, A. M. Karst, M. S. Hirsch, S. R. Setlur, C. P. Crum, D. M. Dinulescu and R. Drapkin, Cancer Cell, 2013, 24, 751 CrossRef CAS PubMed; (b) S. J. Hill, B. Decker, E. A. Roberts, N. S. Horowitz, M. G. Muto, M. J. Worley, C. M. Feltmate, M. R. Nucci, E. M. Swisher, H. Nguyen, C. Yang, R. Morizane, B. S. Kochupurakkal, K. T. Do, P. A. Konstantinopoulos, J. F. Liu, J. V. Bonventre, U. A. Matulonis, G. I. Shapiro, R. S. Berkowitz, C. P. Crum and A. D. D'Andrea, Cancer Discovery, 2018, 8, 1404 CrossRef CAS PubMed; (c) O. Kopper, C. J. de Witte, K. Lõhmussaar, J. E. Valle-Inclan, N. Hami, L. Kester, A. V. Balgobind, J. Korving, N. Proost, H. Begthel, L. M. van Wijk, S. A. Revilla, R. Theeuwsen, M. van de Ven, M. J. van Roosmalen, B. Ponsioen, V. W. H. Ho, B. G. Neel, T. Bosse, K. N. Gaarenstroom, H. Vrieling, M. P. G. Vreeswijk, P. J. van Diest, P. O. Witteveen, T. Jonges, J. L. Bos, A. van Oudenaarden, R. P. Zweemer, H. J. G. Snippert, W. P. Kloosterman and H. Clevers, Nat. Med., 2019, 25, 838 CrossRef CAS PubMed.
- E. Smolle, V. Taucher and J. Haybaeck, Anticancer Res., 2014, 34, 1553 CAS.
- E. Kipps, D. S. P. Tan and S. B. Kaye, Nat. Rev. Cancer, 2013, 13, 273 CrossRef CAS PubMed.
- E. Cavarzerani, I. Caligiuri, M. Bartoletti, V. Canzonieri and F. Rizzolio, Front. Bioeng. Biotechnol., 2023, 11, 1135374 CrossRef PubMed.
- M. Ott, J. D. Robertson, V. Gogvadze, B. Zhivotovsky and S. Orrenius, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1259 CrossRef CAS PubMed.
- I. H. Ismail, T. I. Wadhra and O. Hammarsten, Nucleic Acids Res., 2007, 35, e36 CrossRef PubMed.
- M. Fragkos, J. Jurvansuu and P. Beard, Mol. Cell. Biol., 2009, 29, 2828 CrossRef CAS PubMed.
- M. Brentnall, L. Rodriguez-Menocal, R. L. De Guevara, E. Cepero and L. H. Boise, BMC Cell Biol., 2013, 14, 32 CrossRef CAS PubMed.
- (a) A. Heydari and H. Mansouri-Torshizi, RSC Adv., 2016, 6, 96121 RSC; (b) K. Karami, M. Rafiee, Z. M. Lighvan, M. Zakariazadeh, A. Yeganeh-Faal, S. Esmaeili and A. A. Momtazi-Borojeni, J. Mol. Struct., 2018, 1154, 480 CrossRef CAS.
- (a) C. K. S. Pillai and U. S. Nandi, Biochim. Biophys. Acta, 1977, 474, 11 CrossRef CAS PubMed; (b) A. G. Tikhomirov, N. A. Ivanova, O. S. Erofeeva, L. B. Gorbacheva and I. A. Efimenko, Russ. J. Coord. Chem., 2003, 29, 489 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2015, 142, 074111 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2016, 144, 214110 CrossRef PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 31, 455 CrossRef PubMed.
- (a) R. A. Hussain, A. Badshah, M. Sohail, B. Lal and K. Akbar, J. Mol. Struct., 2013, 1048, 367 CrossRef CAS; (b) A. Zianna, G. D. Geromichalos, A.-M. Fiotaki, A. G. Hatzidimitriou, S. Kalogiannis and G. Psomas, Pharmaceuticals, 2022, 15, 886 CrossRef CAS PubMed; (c) N. Shahabadi, L. Ghaffari, Z. Mardani and F. Shiri, Biol. Trace Elem. Res., 2021, 200, 1988–2000 CrossRef PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS.
- (a) M. Bortoli, S. M. Ahmad, T. A. Hamlin, F. M. Bickelhaupt and L. Orian, Phys. Chem. Chem. Phys., 2018, 20, 27592 RSC; (b) M. Dalla Tiezza, F. M. Bickelhaupt and L. Orian, ChemistryOpen, 2019, 8, 143 CrossRef CAS PubMed; (c) M. Dalla Tiezza, F. M. Bickelhaupt and L. Orian, ChemPhysChem, 2018, 19, 1766 CrossRef CAS PubMed; (d) L. Orian, L. P. Wolters and F. M. Bickelhaupt, Chem. – Eur. J., 2013, 40, 12227 Search PubMed; (e) L. Orian, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2014, 15, 219 CrossRef CAS PubMed; (f) A. Madabeni, M. Bortoli, P. A. Nogara, J. B. T. Rocha and L. Orian, Inorg. Chem., 2021, 60, 4646 CrossRef CAS PubMed; (g) L. Orian, W.-J. van Zeist and F. M. Bickelhaupt, Organometallics, 2008, 27, 4028 CrossRef CAS.
- (a) E. J. Baerends, D. E. Ellis and P. Ros, Chem. Phys., 1973, 2, 41 CrossRef CAS; (b) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- (a) E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS PubMed; (b) P. A. Nogara, F. B. Omage, G. R. Bolzan, C. P. Delgado, L. Orian and J. B. T. Da Rocha, J. Mol. Model., 2022, 28, 354 CrossRef CAS PubMed.
- A. Lausi, M. Polentarutti, S. Onesti, J. R. Plaisier, E. Busetto, G. Bais, L. Barba, A. Cassetta, G. Campi, D. Lamba, A. Pifferi, S. C. Mande, D. D. Sarma, S. M. Sharma and G. Paolucci, Eur. Phys. J. Plus, 2015, 130, 1 CrossRef.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- P. Emsley, B. Lohkamp, W. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- L. Schrodinger, 2015, https://www.pymol.org.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2324438–2324443. For ESI and crystallographic data in CIF or other electronic formats see DOI: https://doi.org/10.1039/d4dt00919c |
| This journal is © The Royal Society of Chemistry 2024 |