
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
EPR spectroscopic characterisation of native CuII-binding sites in human serum albumin†
Katrin
Ackermann
 a,
Dongmei
Wu
b,
Alan J.
Stewart
*b and
Bela E.
Bode
*a
a,
Dongmei
Wu
b,
Alan J.
Stewart
*b and
Bela E.
Bode
*a
aEaStCHEM School of Chemistry, Biomedical Sciences Research Complex, and Centre of Magnetic Resonance, University of St Andrews, St Andrews, KY16 9ST, UK
bSchool of Medicine, Biomedical Sciences Research Complex, and Centre of Magnetic Resonance, University of St Andrews, St Andrews, KY16 9TF, UK. E-mail: beb2@st-andrews.ac.uk; ajs21@st-andrews.ac.uk
First published on 23rd July 2024
Abstract
Human serum albumin (HSA) is the most abundant plasma protein, which functions to transport a large range of ligands within the circulation. These interactions have important implications for human health and disease. The primary binding site for CuII ions on HSA is known to be the so-called amino-terminal CuII and NiII binding (ATCUN) motif. However, the number and identity of secondary binding sites is currently not understood. In this study, we harnessed a suite of contemporary electron paramagnetic resonance (EPR) spectroscopy methods to investigate recombinantly produced constructs of HSA bearing single-histidine knockouts, with the aim to characterise its endogenous CuII ion binding sites.
Introduction
Human serum albumin (HSA) is the most abundant protein in blood plasma with an average concentration of 600–700 μM.1 The mature protein has 585 amino acids and a molecular weight of 66.5 kDa, and has been characterised as a monomer with three distinct domains.1 HSA has a plethora of ligands, and functional relevance has been attributed to its binding and transport of hormones, drugs, fatty acids and particularly divalent metal ions.2–4 For some of these metal ions (e.g., CuII, ZnII, MgII, MnII, and CaII), HSA is involved in their transport to cells and tissues and aids in the regulation of cellular metal ion homeostasis,1,3,5 while for other metal ions (e.g., NiII, CdII, CoII) the interest in their binding to HSA is mainly of toxicological relevance.2,3 Increased fatty acid binding to HSA can have important implications in several human diseases,1,6 and can be intertwined with concomitant alterations of metal ion binding.1,5,7Free CuII ions have the potential to generate unwanted reactive oxygen species (ROS) and are therefore meticulously controlled, with HSA binding ∼10–15% of free CuII in the extracellular environment, while the rest is mainly bound by ceruloplasmin.8–10 The interaction of CuII with serum albumin has been studied for several decades,11 and led to the first characterisation of the ATCUN motif, an amino-terminal CuII- and NiII-binding structural feature (also called NTS for N-terminal binding site) involving a critical histidine residue in position three (H3), amongst other characteristics.9 The NTS has been identified as the site of highest (picomolar) affinity for CuII in HSA,12 and a study of the isolated N-terminal four residues of HSA (DAHK) in complex with CuII confirmed its equatorial coordination sphere involving surrounding nitrogen atoms in square-planar orientation and an apically bound water molecule, as well as the silencing effect of the coordination on the redox activity of CuII.13 However, due to the high structural flexibility of the native NTS in HSA, the ATCUN residues are mostly missing from high-resolution experimental structures, and a structure directly demonstrating CuII binding to the HSA ATCUN motif is not available.
Furthermore, the identity of secondary CuII-binding site(s) of HSA remains a matter of active research, with currently no CuII-HSA crystal structures available. Several high-resolution crystal structures of serum albumin with metal ions are available, including CaII,14 ZnII,7 and CoII.1 One candidate for a secondary CuII-binding site is the multi-metal binding site in HSA, previously introduced as CdII-binding site A,15 which was initially suggested to involve histidine residue H247 of HSA domain II and histidine residues H105 and H146 of domain I.15,16 Later mutagenesis studies demonstrated the involvement of H67 in addition to H247.3 However, relative affinities of this site with respect to CdII and CuII are being debated.15,16 For CoII, three binding sites have been proposed in HSA, including the ATCUN motif, site A, and the as yet unlocalised site B introduced in the CdII study,15 later proposed to involve H9 in domain I,1 yet the relevance of site B for CuII-binding is currently not clear. Histidine residue H288 has previously been implicated in binding of CoII and PtII in HSA,1,17 and in binding of ZnII in horse serum albumin (ESA),18 and thus serves as another candidate for a secondary CuII binding site.
Electron paramagnetic resonance (EPR) spectroscopy is exclusively and exquisitely sensitive to paramagnetic centres, such as stable nitroxide radicals introduced at particular sites in biomolecules using site-directed spin labelling,19 or paramagnetic metal ions.20–22 These can be bound to artificial metal ion binding sites such as the double-histidine motif in proteins,23 or to endogenous sites, potentially substituting the native diamagnetic ligand (e.g., ZnII), with a paramagnetic one (e.g., CuII).24 Due to this sensitivity, EPR is not limited by the size, shape, or complexity of the system under study, nor does it require crystallisation or isotope labelling, which makes it an attractive method for structural biology.
A suite of EPR techniques is available to probe local structural information around paramagnetic centres in metalloproteins, and to measure distances on the nanometre scale between those centres.24 The identity of the paramagnetic centre and the binding geometry around it can be characterised by continuous wave (CW) EPR.25 Hyperfine spectroscopy, such as electron spin echo envelope modulation (ESEEM)26–28 or the corresponding two-dimensional hyperfine sublevel correlation (HYSCORE)29 experiment provide information on weakly coupled magnetic nuclei, such as remote nitrogen atoms often attributed to histidine residues involved in metal ion coordination.28,30–33 CW EPR has been employed previously to characterise the ATCUN motif in HSA,34 and HYSCORE was used to study the NTS peptide of HSA (DAHK).13 To complement these hyperfine techniques, pulse dipolar EPR spectroscopy (PDS) experiments, such as the RIDME35 method (relaxation-induced dipolar modulation enhancement), are available to determine distances between paramagnetic centres on the nanometre scale.36,37
In this study, we aim to identify and characterise the primary and secondary CuII binding sites of HSA using a holistic approach based on a suite of EPR techniques in combination with recombinantly produced HSA constructs bearing single histidine-knockouts. No high-resolution experimental structure of the full-length protein complexed with CuII has been reported to date. This strategy can be employed to elucidate the role of individual histidine residues in CuII binding and obtain insights into the CuII coordination at individual sites. A better understanding of the occupation of these binding sites will provide a deeper insight into CuII homeostasis and allow identifying variations observed with fatty acid loading, with potential implications in disease.
Experimental
Cloning and site-directed mutagenesis
A synthetic gene containing the coding sequence of HSA (corresponding to residues 25–609 of the albumin preprotein sequence; NP_000468.1, Eurofins Genomics) was amplified and cloned into pPICZαB vector (EasySelect™ Pichia Expression Kit, Invitrogen) for expression in Pichia pastoris (Fig. S1†). The gene was inserted in-frame with the N-terminal α-factor secretion signal sequence. Mutant albumin constructs (H3A, H9A, H67A, H247A, H288A) were generated using the GeneArt™ site-directed mutagenesis system (Thermo Fisher Scientific) and Q5 High-Fidelity DNA polymerase (NEB) according to manufacturer protocols. Primers are given in Table S1.† Obtained mutated plasmids were transformed into DH5α™-T1R competent E. coli cells and incubated at 37 °C overnight on low salt Luria–Bertani plates containing 25 μg mL−1 Zeocin (InvivoGen). Successful mutagenesis was confirmed by DNA sequencing using the α-factor, the 5′ AOX1 and the 3′ AOX1 primers (Table S1†).Transformation of DNA into Pichia pastoris
Genomic DNA from resultant clones was extracted, linearised using Pme1 (NEB), and transformed into X-33 Pichia pastoris competent cells (Thermo Fisher Scientific) by electroporation. Briefly, 6.25 μg of linearised plasmids containing the respective inserts were mixed with 100 μL of X-33 cells and incubated on ice for 5 minutes. The mixtures were then transferred into pre-cooled 0.2 cm electroporation cuvettes. Cells were pulsed (voltage/time constant: 1500 V/5 ms), followed by addition of 1 mL of ice-cold 1 M sorbitol and incubated at 30 °C for 2 hours without shaking. 1 mL of YPD medium was added to the cell cultures and incubated at 30 °C for a further 3 hours without shaking. Cells were then spread on YPDS plates containing 100 μg mL−1 Zeocin and incubated at 30 °C for 2 days. To confirm successful integration into the Pichia pastoris genome, DNA was extracted (Yeast DNA extraction kit, Thermo Fisher Scientific) and assessed by PCR using the 5′ AOX1 and the 3′ AOX1 primers flanking the insertion site, followed by agarose gel electrophoresis.Protein expression and purification
Recombinant wild-type (WT) HSA and mutant forms generated by site-directed mutagenesis were expressed in Pichia pastoris and purified as described previously.1 Briefly, X-33 cells were grown overnight in 15–50 mL of sterile BMGY medium (buffered minimal glycerol medium containing histidine) containing 100 mg mL−1 zeocin at 28 °C with shaking at 200 rpm. This starter culture was used to inoculate 350–500 mL of sterile BMGY medium and grown overnight until the culture reached log phase growth (OD600 = 2–6). Cells were harvested and resuspended into 2–4 L of medium. This culture was grown for 5 days, and protein expression was induced by adding 0.5% methanol every 24 h.The supernatant containing the secreted overexpressed protein was obtained by centrifugation and concentrated before purification using a HiTrap Blue HP column (Cytiva) followed by size exclusion chromatography on a HiLoad Superdex-75 column (Cytiva). Final purity of the protein was assessed to be >95% by SDS-PAGE. Protein intact mass was confirmed by our in-house mass spectrometry service.
EPR sample preparation
WT (plasma-purified) HSA (Sigma-Aldrich; cat. no. A1887), recombinantly-produced WT HSA (rHSA) as well as mutant HSA variants were dialysed against Milli-Q water and lyophilised. Stock solutions of 1 mM HSA were prepared by resuspension in protonated or deuterated buffer A (50 mM Tris (Merck), 50 mM NaCl (Fisher Scientific), pH 7.4). Albumin concentration was determined by measuring the absorbance at 280 nm based on an extinction coefficient of 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 445 M−1 cm−1. Stock solutions of CuCl2 (Thermo Scientific Chemicals) at 1 mM or 5 mM concentration were prepared freshly in buffer A for protonated samples, or in D2O (Merck) for deuterated samples. Protonated and deuterated samples were prepared with a final volume of 20 and 70 μL, respectively, at 250 μM final protein concentration, with varying concentrations of CuCl2 (0 to 5 molar equivalents), and 50% (v/v) cryoprotectant (protonated (Alfa Aesar) or deuterated (Cortecnet) glycerol). Samples were immediately snap-frozen in liquid nitrogen. A detailed sample list can be found in Table S2.†
445 M−1 cm−1. Stock solutions of CuCl2 (Thermo Scientific Chemicals) at 1 mM or 5 mM concentration were prepared freshly in buffer A for protonated samples, or in D2O (Merck) for deuterated samples. Protonated and deuterated samples were prepared with a final volume of 20 and 70 μL, respectively, at 250 μM final protein concentration, with varying concentrations of CuCl2 (0 to 5 molar equivalents), and 50% (v/v) cryoprotectant (protonated (Alfa Aesar) or deuterated (Cortecnet) glycerol). Samples were immediately snap-frozen in liquid nitrogen. A detailed sample list can be found in Table S2.†
CW EPR
CW EPR spectra of deuterated samples were obtained at 120 K with a Bruker EMX 10/12 spectrometer running Xenon software and equipped with an ELEXSYS Super Hi-Q resonator at an operating frequency of ∼9.50 GHz with 100 kHz modulation. Temperature was controlled with an ER4141 VTM Nitrogen VT unit (Bruker) operated with liquid nitrogen. CW spectra of the pseudo-titration samples (0.5–5.0 molar equivalents of CuII added) were recorded using a 1600 G field sweep centred at 3100 G, a time constant of 20.48 ms, a conversion time of 20.62 ms, and 2667 points resolution. CW spectra of the recombinantly produced HSA samples with 2 molar equivalents of CuII were recorded using a 1800 G field sweep centred at 3100 G, a time constant of 20.48 ms, a conversion time of 20.62 ms, and 3000 points resolution. An attenuation of 10.0 dB (20 mW power) and a modulation amplitude of 3 G were used for all recordings. All CW spectra were phase- and background-corrected and the double integral was obtained using the Xenon software. Simulations and fitting of field-corrected CW EPR spectra (DPPH standard) were performed using the pepper and esfit functions in EasySpin.38Pulse EPR experiments
Pulse experiments were performed at X- (9–10 GHz) and at Q-band (34 GHz) frequencies, both operating on a Bruker ELEXSYS E580 spectrometer, with probe-heads supporting a split ring resonator (4118X-MS3) for X-band and a 3 mm cylindrical resonator (ER 5106QT-2w) for Q-band, respectively. Pulses were amplified by pulse travelling wave tube (TWT) amplifiers (Applied Systems Engineering) with nominal output of 1 kW and 150 W at X- and Q-band, respectively. Temperature was controlled via cryogen free variable temperature cryostats (Cryogenic Ltd) operating in the 3.5–300 K temperature range.Hyperfine spectroscopy (protonated samples)
3-Pulse ESEEM26–28 spectroscopy was performed at 30 K at X-band frequencies (∼9–10 GHz) and on the maximum of the field-swept spectrum with a pulse length of 16 ns for π/2 pulses and the inter-pulse delay τ set at the blind spot of the proton (∼220 ns corresponding to ∼3 times the inverse of the proton Larmor frequency, with τ1(H) ∼73 ns). The delay T, set at 280 ns, was incremented with a dwell time ΔT of 8 ns and a 4-step phase cycle was used. Four τ values were recorded, whereby the inter-pulse delay was incremented by 36 ns (∼0.5 × τ1(H)); the third τ was selected for further processing of the ESEEM spectra and for setting up the HYSCORE experiment (see below). Data were analysed by fitting an exponential decay background function to the (phase-corrected) raw data, subtracting the raw data by this background function, and then dividing the difference by the background function, thus retaining amplitude information after fast Fourier transformation (FFT) similar as described previously.39 The resulting trace was further subjected to a Hamming window, zero-filling and FFT, before obtaining the absolute (or magnitude) spectrum.HYSCORE33 spectroscopy was performed at 15 K at X-band frequencies (∼9–10 GHz) and on the maximum of the field-swept spectrum with a pulse length of 16 and 32 ns for π/2 and π, respectively, τ set at the blind spot of the proton (∼290 ns) as chosen from the 3-pulse ESEEM, t1 = t2 = 56 ns, and a 4-step phase cycle. Data were processed and analysed using HYSCOREAN,30 employing Hamming apodization, zero-filling, 3rd order polynomial background correction, and diagonal and anti-diagonal spectral symmetrisation, keeping a similar amount of noise for each spectrum by adjusting the minimum contour level percentage accordingly.
Relaxation time measurements (deuterated samples)
Relaxation time measurements were recorded at 30 K at Q-band frequencies (34 GHz) and at the field position corresponding to the maximum of the CuII field-swept spectrum. Transverse dephasing times T2 (or Tm) were determined from 2-pulse electron–spin echo decay experiments recorded with a τ of 200 ns, using 16 and 32 ns π/2 and π pulses, under the stretched exponential approximation. Longitudinal relaxation times T1 were estimated from 3-pulse inversion recovery measurements recorded with a τ of 800 ns, an 8 ns inversion pulse and 16 and 32 ns observer pulses (π/2 and π, respectively), under the biexponential approximation.RIDME measurements (deuterated samples)
The 5-Pulse RIDME experiments36 were recorded at 30 K at Q-band frequencies (34 GHz), using the pulse sequence (π/2–τ1–π–(τ1 + t)–π/2–Tmix–π/2–(τ2–t)–π–τ2–echo) at the field position corresponding to the maximum of the CuII field-swept spectrum with 8-step phase cycling, a τ1 of 400 ns, a τ2 of 2500 ns, a shot repetition time (SRT) of 500 μs, and a critically coupled resonator (high Q). Measurements were recorded with mixing times corresponding to 0.7 × T1 (longitudinal relaxation time as estimated under the biexponential approximation). RIDME data were analysed using the ComparativeDEERAnalyzer40,41 (CDA) 2.0 within DeerAnalysis2022.42 Full CDA reports are provided in the ESI and the underpinning data.†Results & discussion
In this study, we first investigated CuII binding to HSA using CW EPR. Together with hyperfine techniques, these EPR methods can also provide evidence on the chemical environment of the bound CuII, allowing some characterisation of the binding sites.To this end, a pseudo-titration series of plasma-purified HSA with varying amounts of CuII was generated, where each titration point is a distinct sample.24
Double integration of CW EPR spectra showed a linear increase of the CuII signal from 0.5 to the maximum of 5.0 molar equivalents of CuII added, suggesting that all added CuII was bound (Fig. 1A). There was also a small but measurable CuII-signal without addition of CuII (red point in Fig. 1A), corresponding to ∼1/3 to 1/4 binding site (as estimated from the double integral), which suggested that some CuII had been co-purified with HSA from plasma.
 | ||
| Fig. 1 (A) Double integration of CuII spectra for the HSA pseudo-titration series, normalised. (B) Selected individual CW EPR spectra, normalised and overlaid. Note the distinct, almost constant spectral component at ∼3420 G (see Inset) corresponding to a lower g value for the first (high affinity) binding site. (C) Overlay of normalised CW EPR spectra for recombinant WT HSA (rHSA) and the recombinantly produced HSA variant H3A, each with 2 molar equivalents of CuII. The H3A variant does not show the spectral component for the high affinity binding site observed in (B), strongly indicating that H3 is involved in CuII coordination of this site. (D) Structure of HSA showing locations of studied histidine residues. The cartoon was drawn from structural coordinates obtained from the RSCB Protein Data Bank (PDB ID: 5IJF18). Note that the sidechains of His3 and His9 were partially missing from the X-ray structure, and were modelled using Discovery Studio Visualizer v24.1.0.23298 (Dassault Systèmes Biova Corp.). For more details see Fig. S10.† | ||
Individual CW EPR spectra (Fig. 1B) revealed the presence of a distinct spectral component at approximately 3420 G already from the addition of 0.5 molar equivalents of CuII. Thus, this initial signal must be directly associated with the binding site of highest affinity, presumably at the N-terminus (ATCUN motif), exhibiting a lower g value than the lower-affinity sites.34 Detailed studies of the correlation of CW EPR parameters (in particular A and g values),43 were used to establish the square planar arrangement of the CuN4 site present in the ATCUN motif.34
Upon addition of more than 1 molar equivalent of CuII this feature did not grow any further, suggesting that the corresponding site was already fully occupied. Additional variations in spectral line shapes (for individual CW spectra see Fig. S2†) further demonstrated the simultaneous presence of multiple CuII species, suggesting more than one CuII coordination mode involving nitrogen (histidine/imidazole; Tris buffer) and oxygen atoms (2N2O; 3N1O).31,43,44
Even though the involvement of histidine residues in CuII binding of HSA is beyond doubt,2,34 we did not observe any clear 14N superhyperfine (SHF) couplings in the CW EPR spectra as seen previously for another CuII-binding plasma protein.24 This might suggest close proximity of CuII-binding sites in HSA leading to dipolar line broadening and thus, loss of resolution of SHF coupling, or the involvement of other complicating factors, such as anisotropic hyperfine interactions. A previous CW EPR study on CuII binding of albumin and CuII-histidine complexes could demonstrate the enhanced resolution of the superhyperfine splittings using lower frequency measurements (S-band, 3.87 GHz).34
To further investigate the identity of the high-affinity site yielding the spectral component around 3420 G in CW EPR, a set of HSA histidine mutants (H3A, H9A, H67A, H247A, and H288A, Fig. 1D) was generated and subjected to cryogenic CW EPR measurements in presence of two molar equivalents of CuII. Results clearly demonstrated that the high-affinity binding site was directly related to the histidine residue at position 3 (H3), as only in the H3A mutant the associated spectral component was absent (Fig. 1C and Fig. S3†), confirming previous data.9,34,45 The CuII coordinated by the ATCUN motif has been shown to be square planar,34 with an apical water molecule observed in the DAHK peptide.13 Square planar CuII is expected to show lower g⊥ values (and thus, CW EPR signals at higher field) than tetrahedral, octahedral and Jahn–Teller distorted geometries. This was confirmed by simulations (Fig. S4 and Table S3†).
Additionally, relaxation time measurements were performed on the samples of the pseudo-titration series to investigate the change in relaxation behaviour between 0.5 and 5.0 molar equivalents of CuII. To ensure relevance for the timings of the RIDME experiment, deuterated samples were used for these measurements. While the general trend was consistent, with continuously faster inversion recovery (T1 relaxation) and faster two-pulse electron spin echo decay (T2 relaxation) with increasing amounts of CuII present, substantially different relaxation times were found between the 0.5 molar equivalents of CuII and the samples with higher CuII load, suggesting that the N-terminal high affinity site might be less dynamic even in frozen solution (Fig. S5 and Table S4†). However, the fact that we observe a less dynamic CuII centre when the square planar ATCUN motif is mainly occupied is coincidental, not causal, i.e. the determining factors are currently unclear.
Data from hyperfine spectroscopy provided additional information on the chemical environment of the metal ion binding sites in HSA. Three-pulse ESEEM was used to identify weakly coupled nuclei.33 Directly coordinated ligand atoms to the paramagnetic CuII are commonly too strongly coupled to be observed in ESEEM under our conditions. ESEEM spectra for all samples of the pseudo-titration series revealed very similar frequencies typical for nuclear quadrupole interactions (NQI, narrow lines between ∼0.5 to 2 MHz in Fig. 2A and Fig. S6†) of 14N and the double quantum transition (DQ, a broader line at around 4 MHz in Fig. 2A and Fig. S6†) of remote 14N in the imidazole ring of histidine residues,24,33,44 thereby confirming the involvement of histidine residues in CuII coordination of HSA.
 | ||
| Fig. 2 (A) Stacked plots of three-pulse ESEEM data for the CuII pseudo-titration series, obtained at X-band frequencies (9.5 GHz) and on the maximum of the field-swept spectra. Vertical lines indicate the frequencies expected in the presence of two (or more) histidine residues (NQI: ν0, ν+, ν−; DQ: νdq). (B) Representative (+,+) HYSCORE spectra for HSA with 0.5 (1) and 5.0 (II) molar equivalents of CuII, obtained at X-band frequencies (9.5 GHz) and on the maximum of the field-swept spectra. (C) (+,+) HYSCORE spectrum for HSA H3A with 0.9 molar equivalents of CuII, obtained at X-band frequencies (9.5 GHz) and on the maximum of the field-swept spectrum. | ||
While a distinct DQ peak was exhibited for all samples of the pseudo-titration with CuII, its amplitude, which is directly correlated to the number of histidines involved in the coordination of the metal ion at a particular site,32 was gradually decreasing with increasing CuII load. This suggested that the average number of histidine residues available per site was also decreasing, or in other words, data indicated that weaker binding sites with fewer histidines were bound at higher CuII concentration. HSA has a total of 16 histidine residues, and besides the high affinity N-terminal metal ion binding site around H3 a number of additional lower affinity sites have been postulated for various metal ions, some of them including one or more histidine residues.18
Three-pulse ESEEM for the recombinant H3A mutant confirmed that the NQI and DQ peaks characteristic for histidine residues in the binding environment were still present even if the N-terminal ATCUN motif is disrupted by removing the respective histidine residue, suggesting that additional histidine residues were involved in CuII coordination of the lower affinity binding sites (Fig. S7†). However, we cannot exclude that a second histidine residue is involved in CuII binding at the N-terminal high affinity site which would give rise to a similar ESEEM spectrum. Speculatively, this second histidine residue could be H9, as it is in very close proximity to H3, and a variation of the CoII-binding site B, termed site B′, has been shown to involve besides H9 the aspartate residue at position 1 instead of the one at position 13, meaning H9 can interact directly with residues of the NTS.1 Therefore, joint participation of H3 and H9 in the NTS at low CuII load would explain the larger DQ peak observed for the 0.5 molar equivalents of CuII sample, while at higher CuII load H9 is involved in a different site (vide infra). However, we currently do not have sufficient information to distinguish between this model and other weaker binding sites involving fewer histidine residues.
ESEEM data were further supported by results from the corresponding two-dimensional HYSCORE experiments, with all spectra of the CuII pseudo-titration series exhibiting highly similar combination peaks for weakly coupled 14N (∼0.5–4 MHz, Fig. 2B and Fig. S8†), while the amount of water-coordinated CuII was found to be increasing with CuII load (splitting in the signal at ∼14 MHz).
Interestingly, the HYSCORE spectrum of the H3A mutant with 0.9 molar equivalents of CuII (Fig. 2C and Fig. S9†) showed weaker nitrogen signals compared to the 1 molar equivalent spectrum of the pseudo-titration series, emphasising the role of the H3 for the intensity of the nitrogen peaks. Notably, the H3A also displayed a much stronger signal from water-coordinated CuII strongly indicating the absence of an apical water molecule in HSA, in contrast to the DAHK peptide.13
With the combined results from the CW EPR and hyperfine spectroscopy, we have been able to confirm that the histidine at position 3 in the ATCUN motif is the CuII-binding site of highest affinity in HSA, exhibiting a distinct spectral component with lower g value, in line with a previous EPR study.34 In addition, it could be shown that other histidine residues are involved in further binding sites. However, the actual identity of the residues involved has not yet been resolved. For CoII, for which HSA is the main carrier in the circulation, several other binding sites in HSA have been identified, including site A (involving histidine residues H67 and H247), site B (involving H9), and a secondary site involving H288, which has also been shown to play a role in ZnII binding in ESA.1,18
In the following, single His → Ala HSA constructs for these four candidate histidine residues (H9, H67, H247, H288), in addition to the NTS mutant (H3A), were employed to investigate the effect of the removal of individual histidines on the resulting distance distribution in PDS (RIDME) experiments compared to the HSA WT (Fig. 3 and Table S5†). Two main distances were observed for the recombinantly expressed HSA WT, at ∼2.7 nm and ∼3.5 nm (Fig. 3A). Distance distributions obtained for the HSA H67A and the HSA H247A mutants were highly similar (Fig. 3B and C), exhibiting the nearly identical main distances as the HSA WT. This suggested that these two histidine residues did not majorly contribute to the observed distributions and thus, were not significantly involved in high affinity CuII-binding sites, contrarily to what was proposed previously.15,16 In contrast, elimination of H3 dramatically altered the resulting distance distribution, with the disappearance of the longer distance, while the ∼2.7 nm distance remained (Fig. 3D), again confirming the involvement of the ATCUN motif in HSA high-affinity CuII-binding. Interestingly, a similar effect was observed for the HSA H9A mutant, in this case with the shorter distance becoming drastically reduced in amplitude, while the longer distance remained untouched (Fig. 3E). This suggested the involvement of H9 in CuII-binding, which may be able to coordinate CuII in a similar fashion to the proposed site B for CoII, which involved an additional O-ligand from the aspartate residue 13.1 Another dramatic alteration of the distance distribution was obtained for the H288A mutant, where both, the ∼2.7 and the ∼3.5 nm distances were substantially, if not completely, reduced, with only the indication of a very short distance present remaining (Fig. 3F). Note that all raw RIDME traces are clearly modulated (insets Fig. 3) and were processed with zero-bias.
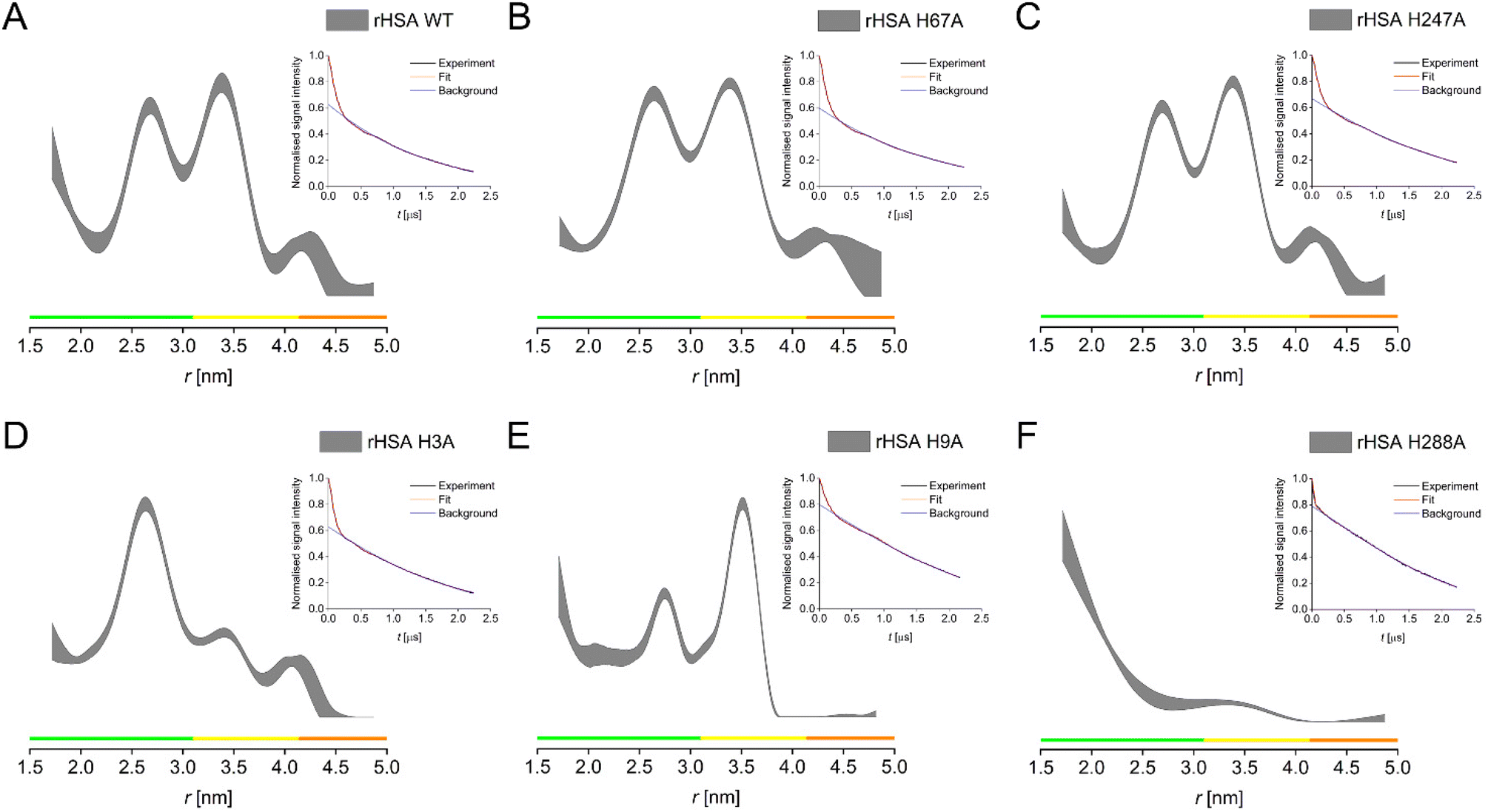 | ||
| Fig. 3 PDS (RIDME) distance distributions for recombinantly produced HSA WT (A), H67A (B), H247A (C), H3A (D), H9A (E), H288A (F). Distance distributions are given as 95% confidence estimate. Colour bars represent reliability ranges (green: shape reliable; yellow: mean and width reliable; orange: mean reliable). Insets show raw RIDME traces (black) with fit (red) and background (blue) for each mutant. | ||
To rationalise these data, we hypothesised the presence of three high-affinity CuII-binding sites in HSA, involving a triangle of three histidine residues, which must involve three distances in between these residues. RIDME data provided evidence that of the two main distance peaks observed in the WT, the longer distance is associated with the H3 residue, while the shorter one is associated with the H9 residue. The third distance of the proposed triangle is supposedly so short that it is at or beyond the limit of PDS capabilities (lower distance limit of ∼1.5 nm), which would likely be the distance between the H3 and the H9 residues. This was supported by the increase in distance probabilities towards the lower distance limit in all distance distributions. It also further supports our speculation that H9 might be involved in CuII binding at the ATCUN site as second histidine at low CuII load, explaining the larger ESEEM DQ peak at low CuII equivalents, while it becomes a lower-affinity binding site on its own when more CuII is present.
RIDME data for H288 confirmed this residue as the third vertex of the triangle. H288 was found to be a secondary binding site for ZnII in ESA, together with H157 and E153.18 While H157 is not conserved in HSA, which has a phenylalanine in position 157, it can be speculated that the lower coordination number is sufficient for CuII binding at this site in HSA. Thus, based on RIDME distance distributions, we confirmed a set of three sites involving histidine residues H3, H9, and H288 (Fig. S10†), as the sites of high-affinity CuII-binding in HSA.
Conclusions
In this study, we investigated CuII-binding of HSA and have demonstrated that in addition to the high-affinity ATCUN motif there are two further CuII-binding sites present in HSA. Using a suite of EPR techniques in combination with site-directed mutagenesis, we were able to identify these sites to involve histidine residues H9 and H288, respectively, which both have, to our knowledge, not been linked to putative HSA CuII-binding sites. Given that both sites were occupied at only two molar equivalents of CuII present in the samples, we conclude that they must have similar affinities, which are, however, much lower compared to the NTS involving H3. While the latter had been demonstrated to have a square planar CuN4 coordination, the characteristic high-field spectral component in CW EPR was not observed for any of the other sites, making H3 the only square planar high affinity site. Further studies will show the effect of fatty acid loading on CuII binding to HSA, and reveal implications for human disease, as has been shown for altered ZnII dependent coagulation in type 2 diabetes.7Author contributions
Katrin Ackermann: conceptualisation (supporting), data curation (lead), formal analysis (lead), funding acquisition (supporting), investigation (equal), methodology (equal), writing – original draft (lead), writing – review & editing (equal). Dongmei Wu: conceptualisation (supporting), formal analysis (supporting), investigation (equal), methodology (equal), writing – original draft (supporting), writing – review & editing (equal). Alan J. Stewart: conceptualisation (equal), investigation (supporting), formal analysis (supporting), funding acquisition (equal), methodology (supporting), supervision (equal), writing – original draft (supporting), writing – review & editing (equal). Bela E. Bode: conceptualisation (equal), investigation (supporting), formal analysis (supporting), funding acquisition (equal), methodology (supporting), supervision (equal), writing – original draft (supporting), writing – review & editing (equal).Data availability
The research data supporting this publication can be accessed at https://doi.org/10.17630/cf121a40-8ee6-491d-b783-5bf65304cd8d.46Conflicts of interest
There are no conflicts to declare.Acknowledgements
For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) license to any Accepted Author Manuscript version arising. They acknowledge support by the Wellcome Trust (204821/Z/16/Z), by the EPSRC (EP/X016455/1), and the British Heart Foundation (PG/15/9/31270 and FS/15/42/31556). B. E. B. acknowledges equipment funding by BBSRC (BB/R013780/1 and BB/T017740/1). The authors thank the StAnD (St Andrews and Dundee) EPR grouping for long-standing support and the St Andrews mass spectrometry and proteomics facility.References
- D. Wu, M. Gucwa, M. P. Czub, D. R. Cooper, I. G. Shabalin, R. Fritzen, S. Arya, U. Schwarz-Linek, C. A. Blindauer, W. Minor and A. J. Stewart, Chem. Sci., 2023, 14, 6244–6258 RSC.
- T. Peters, in All About Albumin, ed. T. Peters, Academic Press, San Diego, 1995, pp. 76–132, DOI:10.1016/B978-012552110-9/50005-2.
- W. Bal, M. Sokołowska, E. Kurowska and P. Faller, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5444–5455 CrossRef CAS PubMed.
- T. Peters and A. J. Stewart, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5351–5353 CrossRef CAS PubMed.
- J. P. C. Coverdale, H. A. van den Berg, S. Khazaipoul, H. E. Bridgewater, A. J. Stewart and C. A. Blindauer, Chem. Commun., 2022, 58, 7384–7387 RSC.
- H. H. Haeri, J. Tomaszewski, B. Phytides, H. Schimm, G. Möslein, M. Niedergethmann, D. Hinderberger and M. Gelos, ACS Pharmacol. Transl. Sci., 2020, 3, 1188–1198 CrossRef CAS PubMed.
- A. I. S. Sobczak, K. G. H. Katundu, F. A. Phoenix, S. Khazaipoul, R. Yu, F. Lampiao, F. Stefanowicz, C. A. Blindauer, S. J. Pitt, T. K. Smith, R. A. Ajjan and A. J. Stewart, Chem. Sci., 2021, 12, 4079–4093 RSC.
- L. W. Gray, T. Z. Kidane, A. Nguyen, S. Akagi, K. Petrasek, Y.-L. Chu, A. Cabrera, K. Kantardjieff, A. Z. Mason and M. C. Linder, Biochem. J., 2009, 419, 237–245 CrossRef CAS PubMed.
- C. Harford and B. Sarkar, Acc. Chem. Res., 1997, 30, 123–130 CrossRef CAS.
- T. Kirsipuu, A. Zadorožnaja, J. Smirnova, M. Friedemann, T. Plitz, V. Tõugu and P. Palumaa, Sci. Rep., 2020, 10, 5686 CrossRef CAS PubMed.
- I. M. Kolthoff and B. R. Willeford Jr., J. Am. Chem. Soc., 1958, 80, 5673–5678 CrossRef.
- M. Rózga, M. Sokołowska, A. M. Protas and W. Bal, JBIC, J. Biol. Inorg. Chem., 2007, 12, 913–918 CrossRef PubMed.
- C. Hureau, H. Eury, R. Guillot, C. Bijani, S. Sayen, P.-L. Solari, E. Guillon, P. Faller and P. Dorlet, Chem. – Eur. J., 2011, 17, 10151–10160 CrossRef CAS PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174–182 CrossRef CAS PubMed.
- P. J. Sadler and J. H. Viles, Inorg. Chem., 1996, 35, 4490–4496 CrossRef CAS PubMed.
- W. Bal, J. Christodoulou, P. J. Sadler and A. Tucker, J. Inorg. Biochem., 1998, 70, 33–39 CrossRef CAS PubMed.
- G. Ferraro, L. Massai, L. Messori and A. Merlino, Chem. Commun., 2015, 51, 9436–9439 RSC.
- K. B. Handing, I. G. Shabalin, O. Kassaar, S. Khazaipoul, C. A. Blindauer, A. J. Stewart, M. Chruszcz and W. Minor, Chem. Sci., 2016, 7, 6635–6648 RSC.
- C. Altenbach, S. L. Flitsch, H. G. Khorana and W. L. Hubbell, Biochemistry, 1989, 28, 7806–7812 CrossRef CAS PubMed.
- D. Abdullin and O. Schiemann, ChemPlusChem, 2020, 85, 353–372 CrossRef PubMed.
- L. Galazzo, M. Teucher and E. Bordignon, in Methods Enzymol, ed. R. D. Britt, Academic Press, 2022, vol. 666, pp. 79–119 Search PubMed.
- A. Giannoulis, Y. Ben-Ishay and D. Goldfarb, in Methods Enzymol, ed. J. A. Cotruvo, Academic Press, 2021, vol. 651, pp. 235–290 Search PubMed.
- T. F. Cunningham, M. R. Putterman, A. Desai, W. S. Horne and S. Saxena, Angew. Chem., Int. Ed., 2015, 54, 6330–6334 CrossRef CAS PubMed.
- K. Ackermann, S. Khazaipoul, J. L. Wort, A. I. S. Sobczak, H. E. Mkami, A. J. Stewart and B. E. Bode, J. Am. Chem. Soc., 2023, 145, 8064–8072 CrossRef CAS PubMed.
- G. R. Eaton, S. S. Eaton, D. P. Barr and R. T. Weber, Quantitative EPR, Springer, Vienna, 2010 Search PubMed.
- W. B. Mims, Phys. Rev. B: Solid State, 1972, 5, 2409–2419 CrossRef.
- W. B. Mims, Phys. Rev. B: Solid State, 1972, 6, 3543–3545 CrossRef.
- L. G. Rowan, E. L. Hahn and W. B. Mims, Phys. Rev., 1965, 137, A61–A71 CrossRef.
- P. Höfer, A. Grupp, H. Nebenführ and M. Mehring, Chem. Phys. Lett., 1986, 132, 279–282 CrossRef.
- L. Fábregas Ibáñez, J. Soetbeer, D. Klose, M. Tinzl, D. Hilvert and G. Jeschke, J. Magn. Reson., 2019, 307, 106576 CrossRef PubMed.
- D. Klose, S. P. B. Vemulapalli, M. Richman, S. Rudnick, V. Aisha, M. Abayev, M. Chemerovski, M. Shviro, D. Zitoun, K. Majer, N. Wili, G. Goobes, C. Griesinger, G. Jeschke and S. Rahimipour, Phys. Chem. Chem. Phys., 2022, 24, 6699–6715 RSC.
- K. I. Silva, B. C. Michael, S. J. Geib and S. Saxena, J. Phys. Chem. B, 2014, 118, 8935–8944 CrossRef CAS PubMed.
- S. Van Doorslaer, eMagRes, 2017, 6, 51–69 CAS.
- M. Valko, H. Morris, M. Mazúr, J. Telser, E. J. L. McInnes and F. E. Mabbs, J. Phys. Chem. B, 1999, 103, 5591–5597 CrossRef CAS.
- L. V. Kulik, S. A. Dzuba, I. A. Grigoryev and Y. D. Tsvetkov, Chem. Phys. Lett., 2001, 343, 315–324 CrossRef CAS.
- S. Milikisyants, F. Scarpelli, M. G. Finiguerra, M. Ubbink and M. Huber, J. Magn. Reson., 2009, 201, 48–56 CrossRef CAS PubMed.
- O. Schiemann and T. F. Prisner, Q. Rev. Biophys., 2007, 40, 1–53 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- A. Volkov, C. Dockter, T. Bund, H. Paulsen and G. Jeschke, Biophys. J., 2009, 96, 1124–1141 CrossRef CAS PubMed.
- L. Fábregas Ibáñez, G. Jeschke and S. Stoll, Magn. Reson., 2020, 1, 209–224 CrossRef PubMed.
- S. G. Worswick, J. A. Spencer, G. Jeschke and I. Kuprov, Sci. Adv., 2018, 4, eaat5218 CrossRef CAS PubMed.
- G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C. R. Timmel, D. Hilger and H. Jung, Appl. Magn. Reson., 2006, 30, 473–498 CrossRef CAS.
- J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 165, 691–708 CrossRef CAS PubMed.
- E. P. Wagner, K. C. Gronborg, S. Ghosh and S. Saxena, J. Chem. Educ., 2019, 96, 1752–1759 CrossRef CAS.
- J. P. Laussac and B. Sarkar, Biochemistry, 1984, 23, 2832–2838 CrossRef CAS PubMed.
- K. Ackermann, D. Wu, A. J. Stewart and B. E. Bode, EPR spectroscopic characterisation of native CuII-binding sites in human serum albumin (dataset), University of St Andrews Research Portal, 2024, https://doi.org/10.17630/cf121a40-8ee6-491d-b783-5bf65304cd8d Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PDF with supplementary figures, tables, and CDA reports. See DOI: https://doi.org/10.1039/d4dt00892h |
| This journal is © The Royal Society of Chemistry 2024 |