
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The coordination of alkali–metal nickelates to organic π-systems: synthetic, structural and spectroscopic insights†
Andryj M.
Borys
 *,
Luca
Vedani
and
Eva
Hevia
*
*,
Luca
Vedani
and
Eva
Hevia
*
Departement für Chemie, Biochemie und Pharmacie, Universität Bern, 3012 Bern, Switzerland. E-mail: andryj.borys-smith@unibe.ch; eva.hevia@unibe.ch
First published on 3rd April 2024
Abstract
Low-valent nickelates have recently been shown to be key intermediates in challenging cross-coupling reactions using aryl ethers as electrophiles. Key for the success of these transformations is the activation of the substrate through π-coordination to the nickelate intermediate, however there is still limited knowledge about the fundamental structure and coordination chemistry of these heterobimetallic complexes. Herein, we report the synthesis, structures, and spectroscopic analysis of a diverse family of alkali–metal nickelates derived from phenyl-alkali–metal reagents and Ni(ttt-CDT), where ttt-CDT = trans,trans,trans-1,5,9-cyclododecatriene. The co-complexation of PhLi with Ni(ttt-CDT) was found to yield 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1 or 4:2 lithium nickelates depending on the stoichiometry and reaction conditions employed. The high lability of the ttt-CDT ligand enables facile ligand exchange with an assorted series of organic π-acceptors, ranging from polyaromatic hydrocarbons to ketones, imines and nitriles. For anthracene and phenanthrene, a homologous series of Li, Na and K nickelates could be obtained, which lead to different structural motifs or degrees of aggregation in the solid-state spanning solvated monomers to complex polymeric arrangements. For π-extended systems such as perylene or coronene, competing single-electron-transfer to give the corresponding radical anions was observed, illustrating the highly reducing nature of the alkali–metal nickelates. X-ray crystallographic analysis and NMR spectroscopy of the phenyl-alkali–metal nickelates reveal extreme back-bonding from Ni(0) to the organic π-acceptors due to strong σ-donation from the carbanionic ligands.
:1, 2:1 or 4:2 lithium nickelates depending on the stoichiometry and reaction conditions employed. The high lability of the ttt-CDT ligand enables facile ligand exchange with an assorted series of organic π-acceptors, ranging from polyaromatic hydrocarbons to ketones, imines and nitriles. For anthracene and phenanthrene, a homologous series of Li, Na and K nickelates could be obtained, which lead to different structural motifs or degrees of aggregation in the solid-state spanning solvated monomers to complex polymeric arrangements. For π-extended systems such as perylene or coronene, competing single-electron-transfer to give the corresponding radical anions was observed, illustrating the highly reducing nature of the alkali–metal nickelates. X-ray crystallographic analysis and NMR spectroscopy of the phenyl-alkali–metal nickelates reveal extreme back-bonding from Ni(0) to the organic π-acceptors due to strong σ-donation from the carbanionic ligands.
Introduction
The coordination chemistry of transition-metals has fascinated chemists for over 150 years1 and a plethora of ligands with varying donor strength, accepting properties, denticity, and countless other features have been reported to date.2,3 One unique class of transition-metal complex is low-valent nickelates,4,5 and these are typically accessed by treating a Ni(0)-olefin6,7 precursor [e.g. Ni(C2H4)3] with a polar organometallic reagent (e.g. organolithiums, Scheme 1a). In these complexes, the carbanion from the polar organometallic reagent can be viewed as a strong σ-donating ligand which coordinates to the Ni(0) centre. Stabilisation of these electron-rich Ni(0) complexes is presented in the form of a complementary π-accepting ligand, which may be the parent olefin from which the nickelate is derived, an external π-acceptor, or from the carbanion itself. Low-valent nickelates were widely investigated in the 1970s and 80s,4 and the high reactivity of these systems is exemplified by their ability to activate small molecules such as dinitrogen.8–10 Nevertheless, they remained dormant in the literature for several decades and it is only in recent years that a renaissance in the field has emerged.5 This has been sparked by mechanistic studies which have demonstrated that low-valent nickelates are overlooked intermediates that facilitate challenging cross-coupling reactions.11,12 Cornella has shown that highly-reduced or simple 16-electron Ni(0)-olefin complexes catalyse the low-temperature C(sp2)–C(sp3) Kumada cross-coupling of vinyl bromides with alkyl Grignard reagents (Scheme 1b),13,14 whilst our group and others have employed Ni(COD)2 for the cross-coupling of aryl ethers with phenyl-lithium (Scheme 1c).15–17 In the latter case, the coordination ability of both nickel and lithium is crucial towards activating the substrate and facilitating smooth Caryl–OMe bond cleavage. Despite these recent catalytic and mechanistic advances however, there is still limited fundamental knowledge into the coordination, structure, and bonding of low-valent nickelates, but these insights may provide essential information towards the rational design of new complexes with untapped catalytic potential. | ||
| Scheme 1 (a) Co-complexation of Ni(0)-olefin complexes with polar organometallics to give low-valent nickelates. (b) Proposed magnesium nickelate intermediate in the low-temperature cross-coupling of vinyl bromides with alkyl Grignard reagents. (c) Proposed lithium nickelate intermediate in the cross-coupling or aryl ethers with phenyl-lithium. (d) This work – coordination of 2:1 phenyl-alkali–metal nickelates to organic π-acceptors. | ||
There are three main variables which influence the synthesis, properties, and reactivity of low-valent nickelates – (i) the donor ligand; (ii) the secondary metal; and (iii) the π-accepting ligand. The donor ligand is derived directly from the polar organometallic reagent and is formally a carbanionic centre, although hydrides (e.g. LiAlH4)18,19 and phosphides (e.g. LiPPh2)20 can also fall under this definition. The hybridisation of the donor ligand will influence the properties and examples of sp3 (alkyl),20–22 sp2 (aryl)15,17,23,24 and sp (acetylide)25,26 derived nickelates have been reported. Additionally, the stoichiometry of the donor ligand employed is crucial and this has been shown to vary from 1:1 up to 4:1, with the ability for many additional molecules of donor ligand to co-complex in the structure without direct coordination to Ni(0).26 The identity of the secondary metal (e.g. Li, Mg, Al) cannot be overlooked, since this will dictate how readily formal transfer of the carbanion centre to Ni(0) occurs depending on the electropositivity and Lewis acidity of the secondary metal. The solvation of the secondary metal can also be manipulated to give contacted or solvent-separated species, or to influence the overall aggregation of the low-valent nickelate. Late-stage exchange of the secondary metal is also possible,24 which may grant access to previously inaccessible heterobimetallic complexes. The final, and less well studied feature of low-valent nickelates is the π-accepting ligand. Most examples to date have employed simple olefin ligands such as ethylene, COD (1,5-cyclooctadiene) or ttt-CDT (trans,trans,trans-1,5,9-cyclododecatriene), but it has been shown that these can often be displaced by other π-accepting ligands such as alkynes, arynes, arenes or even N2.8–10 Herein, we systematically explore and expand the scope of π-accepting ligands using low-valent nickelates derived from Ni(ttt-CDT) and phenyl-alkali–metal reagents (Scheme 1d).
Results and discussion
Co-complexation chemistry of Ni(ttt-CDT) and PhLi
The co-complexation chemistry of Ni(COD)2 and PhLi has been systematically studied and yields a structurally diverse family of lithium nickelates depending on the stoichiometry and reaction conditions employed.15,23 The co-complexation chemistry of Ni(ttt-CDT) with PhLi has also been documented by Jonas and Krüger, but since experimental and analytical details are very limited,4 we decided to first investigate this more systematically. The addition of one equivalent of PhLi to a THF solution of Ni(ttt-CDT) at −30 °C leads to a distinct colour change from red to yellow. The addition of excess 12-crown-4 enables the isolation of the 1:1 lithium nickelate, [(ttt-CDT)NiPh][Li(12-crown-4)2] 1, in 59% crystalline yield (Fig. 1a). The solid-state structure of 1 reveals a solvent-separated ion pair in which the lithium cation (Li1) is sequestered by two molecules of 12-crown-4 (Fig. 1b). The Ni1–C1 distance is 2.024(1) Å which is noticeably longer than Cornella's 1:1 lithium nickelate Li(TMEDA)PhNi(C2H4)2 [1.963(1) Å]13 and all other 2:1 lithium nickelates derived from PhLi.15,17,23,24 Compared to Ni(ttt-CDT) in which the Ni(0) centre sits perfectly co-planar within the cyclododecatriene ligand,27 the Ni-centre in 1 is approximately 0.66 Å above the mean plane defined by the 12 carbon atoms. This leads to longer Ni-olefin distances when compared to Ni(ttt-CDT), but only marginal increase in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C distances [1.384(3)–1.386(3) Å]. The low temperature 13C{1H} NMR spectrum of 1 displays a downfield (deshielded) resonance at δ 195.3 ppm for the ipso-carbon (C1), indicating a highly polarised carbon–nickel bond. Four signals are observed for the coordinated ttt-CDT ligand, which are less shielded when compared to [(ttt-CDT)NiCH3][Li(THF)4].21,28 Whilst complex 1 can be isolated, it is unstable above −20 °C and decomposes within minutes in solution at room temperature. This finding contrasts with phenyl-lithium nickelates derived from Ni(COD)2 where the corresponding 1:1 lithium nickelate can only be spectroscopically detected at high concentrations and readily redistributes back to Ni(COD)2 and a 2:1 lithium nickelate.15
C distances [1.384(3)–1.386(3) Å]. The low temperature 13C{1H} NMR spectrum of 1 displays a downfield (deshielded) resonance at δ 195.3 ppm for the ipso-carbon (C1), indicating a highly polarised carbon–nickel bond. Four signals are observed for the coordinated ttt-CDT ligand, which are less shielded when compared to [(ttt-CDT)NiCH3][Li(THF)4].21,28 Whilst complex 1 can be isolated, it is unstable above −20 °C and decomposes within minutes in solution at room temperature. This finding contrasts with phenyl-lithium nickelates derived from Ni(COD)2 where the corresponding 1:1 lithium nickelate can only be spectroscopically detected at high concentrations and readily redistributes back to Ni(COD)2 and a 2:1 lithium nickelate.15
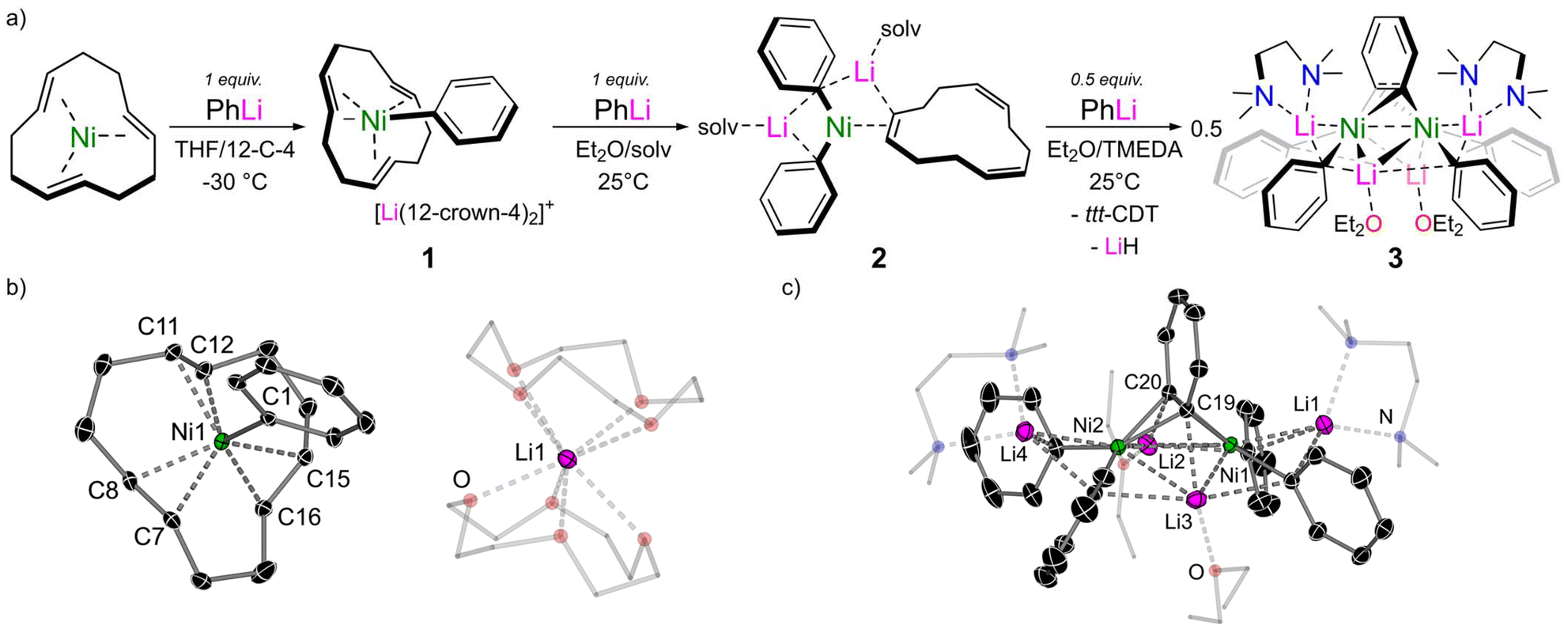 | ||
| Fig. 1 (a) Synthesis of 1:1 lithium nickelate (1), 2:1 lithium nickelate (2), and 4:2 lithium nickelate benzyne-complex (3). (b) Molecular structure of [(ttt-CDT)NiPh][Li(12-crown-4)2] 1. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and 12-crown-4 shown as wireframes for clarity. (c) Molecular structure of Li4(TMEDA)2(Et2O)2Ph4Ni2(μ;η2;η2-C6H4), 3. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated TMEDA and Et2O as wireframes for clarity. | ||
The addition of 2 equivalents of PhLi to a Et2O solution of Ni(ttt-CDT) leads to the clean formation of Li2(solv)nPh2Ni(η2-CDT) (2), however all attempts to grow single crystals suitable for X-ray diffraction were unsuccessful due to its high solubility and instability due to ligand lability (vide infra). The TMEDA solvate could be isolated as a microcrystalline solid in low yields (16%) however and displays similar spectroscopic features to the previously reported COD analogue, Li2(solv)nPh2Ni(η2-COD).15 Complex 2 slowly reacts with an additional 0.5 equivalents of PhLi to give a dinickel-benzyne complex, Li4(TMEDA)2(Et2O)2Ph4Ni2(μ;η2;η2-C6H4) (3). We have previously reported a closely related dinickel-benzyne complex Li6(Et2O)4Ph6Ni2(μ;η2;η2-C6H4),23 which is formed by the addition of a large excess of PhLi to Ni(COD)2. This species was proposed to form by LiH elimination from the third PhLi ligand, preventing the formation of the hypothetical 3:1 lithium nickelate “Li3(solv)nPh3Ni”.29 Formally, this species can be viewed as possessing a neutral benzyne π-ligand, however the highly reducing nature of the lithium nickelate leads to overall reduction to give a {C6H4}2− ligand, as exemplified by the long C–C bond length of 1.449(6) Å.23 In the solid-state structure of 3 (Fig. 1c), no additional PhLi co-complexation is observed and the overall structural and spectroscopic features are comparable to previously reported examples, including its elongated C19–C20 bond length of 1.424(2) Å.
Polyaromatic hydrocarbons as π-ligands to alkali–metal nickelates
Jonas and Krüger have documented that the ttt-CDT ligand in 2:1 lithium nickelate 2 can be displaced by other π-accepting ligands4 and we have recently exploited this strategy to isolate Li2(TMEDA)2Ph2Ni(η2-naphthalene).17 Extending this methodology, the corresponding anthracene analogue Li2(THF)4Ph2Ni(η2-anthracene) 4Li can be readily accessed as its THF solvate in 52% crystalline yield (Fig. 2a). In situ alkali–metal exchange using NaOtBu or KOtBu (2.5 equivalents) in the presence of a suitable polydendate donor gave the corresponding sodium Na2(TMEDA)2Ph2Ni(η2-anthracene) 4Na and potassium K2(PMDETA)2Ph2Ni(η2-anthracene) 4K analogues, as black/green crystalline solids in 42% and 24% yield respectively. The solid-state structure of 4Li is comparable to Li2(TMEDA)2Ph2Ni(η2-naphthalene)17 and Li2(solv)nPh2Ni(η2-COD),15 and features two distinct Li environments; Li2 is coordinated to both ipso-carbons (C1 and C7) whilst Li1 is coordinated to one ipso-carbon [C1⋯Li1 2.252(4) Å] and to one carbon of the coordinated anthracene [C13⋯Li1 2.484(3) Å]. The C13–C26 distance is 1.462(2) Å, which is considerably longer than in free anthracene [1.352(3)–1.356(3) Å]30 and other LnNi(η2-anthracene) complexes bearing trialkylphosphine ligands [1.423–1.427(4) Å],31,32 illustrating strong back-donation from the electron-rich Ni-centre. The solid-state structures of 4Na and 4K display similar bond metrics (CC and Ni–C bond lengths) to 4Li but differ primarily in the coordination of the alkali–metal cations (Fig. 2b and c), a feature that has been observed for alkali–metal main-group metalate complexes.33–35 One sodium (Na1) or potassium (K1) cation is coordinated to two ipso-carbons (C1 and C7) but have additional interactions to ortho-carbons of both phenyl substituents. The second alkali–metal cation (Na2 and K2) coordinates to an ipso-carbon (C7) and ortho-carbon of one phenyl-substituent, and also coordinates in an η3-motif to the central six-membered ring of anthracene. This illustrates the softer character of sodium and potassium which prefer contacts to the arene rings, whilst lithium prefers more localised electrostatic interactions. The 1H NMR spectra of complexes 4Li, 4Na and 4K display upfield shifted resonances for the coordinated anthracene at δ 4.40 (4Li), δ 4.58 and 4.42 (4Na) and δ 4.49 and 4.24 (4K), which are considerably shielded compared to (iPr3P)2Ni(η2-anthracene) [δ 5.32 and 5.55].32
 | ||
| Fig. 2 (a) Synthesis of 2:1 phenyl-alkali–metal nickelate η2-anthracene complexes 4Li, 4Na and 4K. (b) Molecular structure of 4Li. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated THF shown as wireframes for clarity. (c) Molecular structure of 4Na. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated TMEDA shown as wireframes for clarity. (d) Molecular structure of 4K. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated PMDETA shown as wireframes for clarity. | ||
Switching to phenanthrene as the π-accepting ligand, another polyaromatic hydrocarbon consisting of three fused benzene rings which is a non-linear isomer of anthracene, a complete series of 2:1 phenyl-alkali–metal nickelate η2-phenanthrene complexes 5Li, 5Na and 5K could also be obtained, all as their THF solvates (Fig. 3a). The lithium congener Li2(THF)4Ph2Ni(η2-phenanthrene) 5Li was previously documented by Jonas and Krüger4 and the solid-state structure (Fig. 3b) shows a solvated monomer which shares similar features to other 2:1 phenyl-lithium nickelate complexes.15,17,23,24 The sodium analogue 5Na is dimeric in the solid-state (Fig. 3c), akin to other 2:1 phenyl-sodium nickelates that have been reported to date.23,36,37 Finally, the potassium analogue 5K is polymeric in the solid-state (Fig. 3d) and propagates along the crystallographic b axis through numerous K⋯π-arene interactions to the phenyl substituents and coordinated phenanthrene ligand. Unlike 5Li and 5Na in which the phenanthrene ligand is essentially planar, the phenanthrene ligand in 5K is distorted away from planarity [14.6° deviation of mean planes defined by outer six-membered rings] and has considerable torsion [10.9(4)°]. In all complexes, the Ni(0) coordinates to the exposed 9,10-position of phenanthrene (labelled as C13 and C14) which is elongated relative to free phenanthrene [cf. 1.373 Å;385Li 1.453(2) Å; 5Na 1.443(5) or 1.452(3) Å; 5K 1.447(4) Å]. In the 1H NMR spectra, the H13/14 resonances are observed at δ 2.77 (5Li), 3.05 (5Na) and 3.09 (5K), whilst in the 13C{1H} NMR spectra, the C13/14 resonances are observed at δ 39.0 (5Li), 38.9 (5Na) and 40.5 (5K). These resonances are significantly shielded (upfield shifted) relative to free phenanthrene [1H: δ 7.74; 13C{1H}: δ 127.8], indicating very strong back-donation from the Ni(0)-centre into the π-accepting ligand. Despite showing higher degrees of aggregation in the solid-state, 1H DOSY (diffusion ordered spectroscopy) NMR studies indicate that 5Na and 5K are both solvated monomers in THF-d8 solution (see Fig. S3 and S4†).
 | ||
| Fig. 3 (a) Synthesis of 2:1 phenyl-alkali–metal nickelate η2-phenanthrene complexes 5Li, 5Na and 5K. (b) Molecular structure of 5Li. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated THF shown as wireframes for clarity. (c) Molecular structure of 5Na. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated THF shown as wireframes for clarity. (d) Molecular structure of 5K. Thermal ellipsoids shown at 30% probability. Hydrogen atoms and coordinated THF on K2 omitted for clarity. | ||
We next moved on to π-extended polyaromatic hydrocarbons to further assess how the identity of the π-accepting ligand influences the structure and properties of alkali–metal nickelates. Treatment of an in situ prepared solution of Li2(solv)nPh2Ni(η2-CDT) 2 with 1 equivalent of perylene leads to an immediate colour change, from which lustrous black crystals of [Li(THF)2Ph2Ni(η3-perylene)][Li(THF)4] 6Li could be isolated in 71% yield (Fig. 4a). The solid-state structure of 6Li (Fig. 4b) reveals a pseudo-solvent-separated ion pair in which one lithium cation (Li1) is coordinated to both ipso-carbons (C1 and C7) whilst the second lithium cation is coordinated to four molecules of THF and is sequestered away from the lithium nickelate anion. The Ni-centre coordinates to the perylene ligand in a pseudo-η3-motif – stronger binding to C14 and C15 is observed however, as evidenced by shorter Ni–C distances [1.977(2) and 2.088(2) Å vs. 2.171(2) for Ni1–C13] and longer C–C distances [1.413(3) Å for C14–C15 vs. 1.401(3) for C13–C14]. The perylene ligand is slightly distorted away from planarity, with torsion angles across the bay-positions ranging from 2.5(3) to 5.5(3)°. Isolated samples of 6Li were found to be contaminated with a paramagnetic impurity identified as the perylene radical anion (see Fig. S5† for EPR spectrum).39 Moreover, attempts to prepare the sodium and potassium analogues via alkali–metal exchange with AMOtBu led exclusively to the formation of the corresponding perylene radical anions, as confirmed by EPR spectroscopy and single-crystal X-ray diffraction (see Fig. S6, S7 and S18†), and the target alkali–metal nickelates could not be isolated. This suggests that the formation of the radical anion first proceeds through the targeted alkali–metal nickelates and not only illustrates the highly reducing nature of alkali–metal nickelates [cf. one-electron reduction potential of perylene = −1.98 V],40 but also suggests that the identity of the alkali–metal cation influences the reducing strength. Oxidised Ni(I) or Ni(II) species are proposed to be nickel-containing by-products of the reaction, but attempts to identify these by NMR or EPR spectroscopy were unsuccessful. Extending the conjugation of π-accepting ligands even further, the treatment of Li2(solv)nPh2Ni(η2-ttt-CDT) 2 with 1 equivalent of coronene (a.k.a. superbenzene) gave the corresponding lithium nickelate [Li(THF)2Ph2Ni(η2-coronene)][Li(THF)4] 7Li as a black crystalline solid. The solid-state structure of 7Li (Fig. 4c) shows a pseudo-solvent-separated ion pair, akin to 6Li. The observation of this structural motif for perylene and coronene indicates that these π-accepting ligands are better at distributing electron density and delocalising negative charge. In 7Li, the Ni-centre coordinates in a η2-motif to a peripheral “double bond” causing significant elongation for C13–C14 to 1.460(2) Å – in contrast, the five-remaining peripheral “double bonds” have distances ranging from 1.353(3)–1.377(2) Å. At room temperature, only a single very broad resonance is observed around δ 7.00 in the 1H NMR spectrum for the coordinated coronene ligand (Fig. 4d), indicative of exchange processes in which Ni coordinates/decoordinates to all six CC bonds equally. Cooling down to −40 °C however leads to splitting of the resonances into well-defined signals and the appearance of a sharp singlet at δ 3.30 for the position coordinated to Ni (13C resonance for C13/14 at δ 42.3). Similarly, the 7Li NMR spectrum of 7Li at room temperature displays a sharp singlet at δ 0.17, which splits into two broad signals at δ 0.52 and 0.00 upon cooling to −40 °C (see Fig. S2†). Consistent with the lower reduction potential of coronene (−2.36 V),40 only traces of the coronene radical anion41,42 (see Fig. S8†) are observed during the synthesis of potassium nickelate K2(DME)4Ph2Ni(η2-coronene) 7K (see Fig. S20† for molecular structure).
 | ||
| Fig. 4 (a) Synthesis of 2:1 phenyl-lithium nickelate η3-perylene complex 6Li and η2-coronene complex 7Li. (b) Molecular structure of 6Li (anion only). Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated THF shown as wireframes for clarity. (c) Molecular structure of 7Li (anion only). Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated THF shown as wireframes for clarity. (d) Stacked 1H NMR spectra of 7Li at variable temperatures. | ||
The polyaromatic hydrocarbons explored (anthracene, phenanthrene, perylene and coronene) have numerous different sites in which a transition-metal could hypothetically coordinate, and we therefore aimed to justify the coordination modes observed for the diverse series of 2:1 phenyl-alkali–metal nickelate complexes. Polyaromatic hydrocarbons can have several resonance forms which are typically depicted by their Kekulé structures (i.e. containing alternating single and double bonds). An alternate depiction is the Clar structure, which groups six π-electrons into an aromatic π-sextet and is represented by a circle.43 According to Clar's rules, the resonance structure of benzenoid polyaromatic hydrocarbons which contains the most aromatic π-sextets is the most important in characterising its chemical and physical properties. Interestingly, in all nickelate complexes reported herein, the Ni-centre coordinates to “exposed double bonds”44 (shown in red in Scheme 2) that are established in the Clar structure since these possess the greatest π-accepting ability, which is necessary to attenuate the high electron density at the Ni-centre. Supporting this hypothesis, no evidence of coordination is observed with benzene or biphenyl, benzenoid aromatic compounds in which no isolated double bonds are present in their Clar structures.
 | ||
| Scheme 2 Clar structures of anthracene, phenanthrene, perylene and coronene and their respective coordination to Ni(0) in the 2:1 phenyl-alkali–metal nickelate complexes. | ||
Heteroatom-based π-acceptors
We next turned our attention to unsaturated organic compounds containing heteroatoms which have the ability to act as a σ-donor, as well as a π-acceptor and π-donor. Utilising the same synthetic strategy, the treatment of in situ prepared solution of Li2(solv)nPh2Ni(η2-CDT) 2 with 1 equivalent of benzophenone gave an intensely coloured solution from which purple crystals of [Li2(THF)3Ph2Ni(η2-Ph2CO)]2 (8Li) were isolated in 62% yield (Scheme 3, left). The solid-state structure (see Fig. S21† for full structure) reveals a dimeric motif with a Li2O2 core, a feature which is common in alkali–metal alkoxide structures.45–47 The Ni-centre coordinates in a η2-motif to the carbonyl fragment resulting in significant elongation of the C–O bond to 1.389(2)–1.393(2) Å [cf. 1.223(2) Å in free Ph2CO].48 Noticeably, this bond length is considerably longer than other LnNi(η2-benzophenone) complexes [1.331(3)–1.353(6) Å]49–52 and is even approaching that observed in the benzophenone dianion [1.406 Å for (Ph2C–O)Li2].53 Complex 8Li could be treated with KOtBu in the presence of DME (dimethoxyethane) to give [K2(DME)3Ph2Ni(η2-Ph2CO)]2 (8K) as deep blue crystals in 50% yield, and showed the same dimeric aggregation in the solid-state and comparable structural parameters to 8Li (see Fig. S22† for full structure). | ||
| Scheme 3 Synthesis of [Li2(THF)3Ph2Ni(η2-Ph2CO)]2 (8Li), Li2(THF)5Ph2Ni(η2-PhCHNPh) (9Li) and [Li2(THF)3Ph2Ni(η2-Ph2CHCN)]2 (10Li). | ||
The reaction of Li2(solv)nPh2Ni(η2-CDT) 2 with 1 equivalent of N-benzylideneaniline afforded Li2(THF)5Ph2Ni(η2-PhCHNPh) (9Li) as a red crystalline solid in 50% yield (Scheme 3). The solid-state structure (Fig. 5) reveals a monomeric structure in which one lithium cation (Li1) is coordinated to the two ipso-carbons (C14 and C20) whilst the second lithium cation (Li2) is coordinated to N1 and solvated by three molecules of THF. The C1–N1 bond length [1.419(2) Å] is again elongated significantly when compared to free N-benzylideneaniline [1.260(3) Å]54 and other reported LnNi(η2-PhCHNPh) complexes [1.368(3) Å].55 Consistent with strong back-donation from Ni, the C1–H resonance in 9Li is observed at δ 3.81 in the 1H NMR spectrum, considerably shielded (upfield shifted) with respect to free N-benzylideneaniline [cf. δ 8.50 ppm].
 | ||
| Fig. 5 Molecular structure of 9Li. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated THF shown as wireframes for clarity. | ||
Extending the scope of π-acceptors to nitriles, the treatment of Li2(solv)nPh2Ni(η2-ttt-CDT) 2 with 1 equivalent of diphenylacetonitrile afforded yellow crystals of [Li2(THF)3Ph2Ni(η2-Ph2CHCN)]2 (10Li) in a 42% yield (Scheme 3). The solid-state structure of 10Li reveals a dimeric motif (Fig. 6) akin to 8Li, with characteristic elongation of the C13–N1 bond [1.242(2) Å vs. 1.147(2) Å for Ph2CHCN].56 In addition, the C14–C13–N1 unit is significantly distorted away from linearity [128.9(1)° vs. 177.9(1)° for Ph2CHCN],56 a feature that is observed for the coordination of triply bonded acetylenes to alkali–metal nickelates.24,25 The isolation of complexes 8Li, 9Li and 10Li is particularly surprising given that these unsaturated substrates readily react with PhLi via nucleophilic addition,57–59 or α-deprotonation in the case of diphenylacetonitrile.56 No evidence of competitive nucleophilic addition of Ph–M (where M = Li or Ni) or reduction is observed in the synthesis of 8Li and 9Li, and these complexes are stable for several days at room temperature in solution. We propose that coordination of these CO or CN substrates to Ni decreases their electrophilicity and thus tendency towards nucleophilic addition. Contrastingly, 10Li can only be isolated in low yields and is thermally sensitive, indicating that competing pathways or onward decomposition is facile.
 | ||
| Fig. 6 Molecular structure of 10Li. Thermal ellipsoids shown at 30% probability. Hydrogen atoms omitted and coordinated THF shown as wireframes for clarity. | ||
To further investigate the coordination chemistry and quantify the back-donation of alkali–metal nickelates, we next targeted the corresponding 1:1 and 2:1 lithium nickelate carbonyl complexes Li(solv)nPhNi(CO)3 and Li2(solv)nPh2Ni(CO)2. Despite repeated efforts and numerous synthetic routes however, all attempts to isolate or characterise these targets were unsuccessful. Treatment of in situ prepared solutions of [(ttt-CDT)NiPh][Li(solv)n] 1 or Li2(solv)nPh2Ni(η2-ttt-CDT) 2 with 1 atmosphere of carbon monoxide initially leads to a colour change to deep red at −196 °C, but rapid decomposition occurs upon warming to give an intractable mixture of compounds alongside nickel black. Direct synthesis from Ni(CO)4 and 1 or 2 equivalents of PhLi also gave intractable mixtures even at low temperatures, and only traces of Li2(THF)4Ph4Ni(II) could be identified,60 suggesting that CO may be reduced under these conditions. Ligand exchange by treatment of (Ph3P)2Ni(CO)2 with two equivalents of PhLi also gave a mixture of species, from which the mixed phosphine/phosphide complex Li(THF)3(PPh3)2(CO)3Ni2(μ-PPh2) (11Li) was identified as the major species by 31P NMR spectroscopy and single crystal X-ray diffraction (see Fig. S25† for molecular structure). Similar complexes have previously been documented by treatment of (Ph3P)2Ni(CO)2 with potassium metal,61 whilst the formation of 11Li likely originates from Ni-mediated P–C bond cleavage.51,62–64 The lack of selectivity and temperature sensitivity of these reactions is unsurprising given that phenyl-lithium itself reacts unselectively with carbon monoxide to give a mixture of organic species including benzophenone, benzoin and diphenylacetophenone upon acidic workup.65 Examples of nickelate-carbonyl complexes have been documented by Wilke, Pörschke and Jonas, but these are reported to be extremely temperature sensitive and readily react further to give acyl-lithium nickelate complexes.21,28 The synthetic challenge and toxicity risks associated with the preparation of nickel-carbonyl complexes has been addressed through the introduction of computed electronic parameters (CEP) for a series of known and hypothetical L–Ni(CO)3 complexes.66 This demonstrated that carbanionic ligands such as Ph− are much stronger donors compared to neutral ligands such as phosphines and N-heterocyclic carbenes, consistent with the structural and spectroscopic features observed in isolated phenyl-alkali–metal nickelate complexes reported herein.
Conclusions
In conclusion, this work has explored the co-complexation of PhLi with Ni(ttt-CDT), and the rich coordination chemistry of 2:1 phenyl-alkali–metal-nickelates towards a range of organic π-accepting ligands. As exemplified by the homologous series of Li, Na and K nickelates obtained for anthracene and phenanthrene, the overall nickelate core remains very similar whilst significant differences in the coordination environment of the alkali–metal cation are observed, with the π-affinity of the larger and softer potassium cation being most prevalent. The alkali–metal effects also lead to differences in reducing capability, as evidenced by the spectroscopic observation of perylene and coronene radical anions when preparing potassium nickelates. For polyaromatic hydrocarbons, we found that the strong back-donation from the electron-rich Ni centre reveals the Clar structure which contains exposed double bonds with the greatest π-accepting ability. The alkali–metal nickelate coordination chemistry can also be extended to heteroatom based π-acceptors such as ketones, imines, and nitriles, where surprisingly no formal reduction to radical anions or dianions is observed. Given the emerging role of alkali–metal nickelates in catalysis, we believe that these coordination effects will have important mechanistic implications, which can be leveraged towards designing new transformations. We are currently exploiting these coordination and alkali–metal effects for further bond activation chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The X-ray crystal structure determination service unit of DCBP Universität Bern is acknowledged for measuring, solving, and refining new structures. We also thank Dr Ilche Gjuroski (NMR), Andrea Bill (CHN), Claudia Bühr (CHN) and Dr Wowa Stroek (EPR) for analytical services. We are grateful to the Swiss National Science Foundation (SNF) (Project 200021_188573) and Universität Bern for their generous sponsorship of this research.Notes and references
- E. C. Constable and C. E. Housecroft, Chem. Soc. Rev., 2013, 42, 1429–1439 RSC.
- G. A. Lawrance, Introduction to Coordination Chemistry, Wiley, 2010 Search PubMed.
- Ligand Design in Metal Chemistry, ed. M. Stradiotto and R. J. Lundgren, Wiley, 2016 Search PubMed.
- K. Jonas and C. Krüger, Angew. Chem., Int. Ed. Engl., 1980, 19, 520–537 CrossRef.
- A. M. Borys and E. Hevia, Chimia, 2023, 77, 242–245 CrossRef CAS PubMed.
- V. B. Bogdanović, M. Kröner and G. Wilke, Justus Liebigs Ann. Chem., 1966, 669, 1–23 CrossRef PubMed.
- K. Fischer, K. Jonas and G. Wilke, Angew. Chem., Int. Ed. Engl., 1973, 12, 565–566 CrossRef.
- C. Krüger and Y.-H. Tsay, Angew. Chem., Int. Ed. Engl., 1973, 12, 998–999 CrossRef.
- K. Jonas, Angew. Chem., Int. Ed. Engl., 1973, 12, 997–998 CrossRef.
- K. Jonas, D. J. Brauer, C. Krüger, P. J. Roberts and Y.-H. Tsay, J. Am. Chem. Soc., 1976, 98, 74–81 CrossRef CAS.
- H. Ogawa, H. Minami, T. Ozaki, S. Komagawa, C. Wang and M. Uchiyama, Chem. – Eur. J., 2015, 21, 13904–13908 CrossRef CAS PubMed.
- K. Kojima, Z. K. Yang, C. Wang and M. Uchiyama, Chem. Pharm. Bull., 2017, 65, 862–868 CrossRef CAS PubMed.
- L. Nattmann, S. Lutz, P. Ortsack, R. Goddard and J. Cornella, J. Am. Chem. Soc., 2018, 140, 13628–13633 CrossRef CAS PubMed.
- S. Lutz, L. Nattmann, N. Nöthling and J. Cornella, Organometallics, 2021, 40, 2220–2230 CrossRef CAS.
- A. M. Borys and E. Hevia, Angew. Chem., Int. Ed., 2021, 60, 24659–24667 CrossRef CAS PubMed.
- A. M. Borys and E. Hevia, Synthesis, 2022, 2976–2990 CrossRef CAS.
- H. Liang, A. M. Borys, E. Hevia, M. E. L. Perrin and P. A. Payard, J. Am. Chem. Soc., 2023, 145, 19989–19999 CrossRef CAS PubMed.
- K. Pörschke and G. Wilke, Chem. Ber., 1985, 118, 313–322 CrossRef.
- K. R. Pörschke and G. Wilke, J. Organomet. Chem., 1988, 358, 519–524 CrossRef.
- K. Jonas, K. R. Pörschke, C. Krüger and Y.-H. Tsay, Angew. Chem., Int. Ed. Engl., 1976, 15, 621–622 CrossRef.
- K. R. Pörschke, K. Jonas, G. Wilke, R. Benn, R. Mynott and R. Goddard, Chem. Ber., 1985, 118, 275–297 CrossRef.
- K. R. Pörschke, K. Jonas and G. Wilke, Chem. Ber., 1988, 121, 1913–1919 CrossRef.
- R. J. Somerville, A. M. Borys, M. Perez-Jimenez, A. Nova, D. Balcells, L. A. Malaspina, S. Grabowsky, E. Carmona, E. Hevia and J. Campos, Chem. Sci., 2022, 13, 5268–5276 RSC.
- A. M. Borys and E. Hevia, Dalton Trans., 2023, 52, 2098–2105 RSC.
- A. M. Borys, L. A. Malaspina, S. Grabowsky and E. Hevia, Angew. Chem., Int. Ed., 2022, 61, e202209797 CrossRef CAS PubMed.
- A. M. Borys and E. Hevia, Chem. Commun., 2023, 59, 7032–7035 RSC.
- D. J. Brauer and C. Krüger, J. Organomet. Chem., 1972, 44, 397–402 CrossRef CAS.
- K. R. Pörschke and G. Wilke, Chem. Ber., 1984, 117, 56–68 CrossRef.
- R. Taube and N. Stransky, Z. Chem., 1979, 19, 412–413 CrossRef CAS.
- C. P. Brock and J. D. Dunitz, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 795–806 CrossRef.
- D. J. Brauer and C. Krüger, Inorg. Chem., 1977, 16, 884–891 CrossRef CAS.
- J. A. Hatnean, R. Beck, J. D. Borrelli and S. A. Johnson, Organometallics, 2010, 29, 6077–6091 CrossRef CAS.
- R. E. Mulvey, Chem. Commun., 2001, 1049–1056 RSC.
- D. R. Armstrong, E. Brammer, T. Cadenbach, E. Hevia and A. R. Kennedy, Organometallics, 2013, 32, 480–489 CrossRef CAS.
- D. R. Armstrong, H. S. Emerson, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Dalton Trans., 2014, 43, 14229–14238 RSC.
- D. J. Brauer, C. Krüger, P. J. Roberts and Y.-H. Tsay, Angew. Chem., Int. Ed. Engl., 1976, 15, 48–49 CrossRef.
- K. Jonas, Angew. Chem., Int. Ed. Engl., 1976, 15, 47 CrossRef.
- J. Trotter, Acta Crystallogr., 1963, 16, 605–608 CrossRef CAS.
- T. V. Balashova, S. K. Polyakova, V. A. Ilichev, E. V. Baranov, G. K. Fukin, K. A. Kozhanov, G. Y. Zhigulin, S. Y. Ketkov and M. N. Bochkarev, Organometallics, 2023, 42, 3283–3291 CrossRef CAS.
- K. Seto, T. Nakayama and B. Uno, J. Phys. Chem. B, 2013, 117, 10834–10845 CrossRef CAS PubMed.
- M. Sato, K. Yamamoto, H. Sonobe, K. Yano, H. Matsubara, H. Fujita, T. Sugimoto and K. Yamamoto, J. Chem. Soc., Perkin Trans. 2, 1998, 1909–1914 RSC.
- C. Janiak and H. Hemling, Chem. Ber., 1994, 127, 1251–1253 CrossRef CAS.
- M. Solà, Front. Chem., 2013, 1, 4–11 Search PubMed.
- K. P. Vijayalakshmi and C. H. Suresh, New J. Chem., 2010, 34, 2132–2138 RSC.
- C. Frenzel and E. Hey-Hawkins, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 143, 1–17 CrossRef CAS.
- J. Geier, H. Rüegger and H. Grützmacher, Dalton Trans., 2005, 129–136 Search PubMed.
- T. A. Scott, B. A. Ooro, D. J. Collins, M. Shatruk, A. Yakovenko, K. R. Dunbar and H. C. Zhou, Chem. Commun., 2009, 65–67 RSC.
- A. Matsumoto, S. Tsuchiya, Y. Hagiwara, K. Ishikawa, H. Koshima, T. Asahi and K. Soai, Chem. Lett., 2016, 45, 526–528 CrossRef CAS.
- T. T. Tsou, J. C. Huffman and J. K. Kochi, Inorg. Chem., 1979, 18, 2311–2317 CrossRef CAS.
- A. Flores-Gaspar, P. Pinedo-González, M. G. Crestani, M. Muñoz-Hernández, D. Morales-Morales, B. A. Warsop, W. D. Jones and J. J. García, J. Mol. Catal. A: Chem., 2009, 309, 1–11 CrossRef CAS.
- S. Sabater, M. J. Page, M. F. Mahon and M. K. Whittlesey, Organometallics, 2017, 36, 1776–1783 CrossRef CAS.
- A. E. King, S. C. E. Stieber, N. J. Henson, S. A. Kozimor, B. L. Scott, N. C. Smythe, A. D. Sutton and J. C. Gordon, Eur. J. Inorg. Chem., 2016, 1635–1640 CrossRef CAS.
- B. Bogdanović, C. Krüger and B. Wermeckes, Angew. Chem., Int. Ed. Engl., 1980, 19, 817–818 CrossRef.
- R. Basta, A. M. Arif and R. D. Ernst, CSD Commun., DOI:10.5517/ccdc.csd.cc25rylh.
- A. L. Iglesias, M. Muñoz-Hernández and J. J. García, J. Organomet. Chem., 2007, 692, 3498–3507 CrossRef CAS.
- E. Iravani and B. Neumüller, Organometallics, 2003, 22, 4129–4135 CrossRef CAS.
- C. Vidal, J. García-Álvarez, A. Hernán-Gõmez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 53, 5969–5973 CrossRef CAS PubMed.
- C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2016, 55, 16145–16148 CrossRef CAS PubMed.
- G. Dilauro, M. Dell'Aera, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2017, 56, 10200–10203 CrossRef CAS PubMed.
- R. Taube and G. Honymus, Angew. Chem., Int. Ed. Engl., 1975, 14, 261–262 CrossRef.
- B. Kesanli, D. R. Gardner, B. Scott and B. W. Eichhorn, J. Chem. Soc., Dalton Trans., 2000, 1291–1296 RSC.
- D. R. Fahey and J. E. Mahan, J. Am. Chem. Soc., 1976, 98, 4499–4503 CrossRef CAS.
- P. E. Garrou, Chem. Rev., 1985, 85, 171–185 CrossRef CAS.
- S. A. Macgregor, Chem. Soc. Rev., 2007, 36, 67–76 RSC.
- N. S. Nudelman and A. A. Vatale, J. Organomet. Chem., 1983, 241, 143–156 CrossRef CAS.
- L. Perrin, E. Clot, O. Eisenstein, J. Loch and R. H. Crabtree, Inorg. Chem., 2001, 40, 5806–5811 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic details, crystallographic information, and NMR spectra. CCDC 2326086–2326102. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00889h |
| This journal is © The Royal Society of Chemistry 2024 |