
A reduced polyoxometalate-encapsulated organo cobalt modified phosphate framework for improving photocatalytic reduction of CO2†
Yinhua
Zhu‡
,
Pinfang
Yan‡
,
Lingtong
Xu
,
Zeyu
Du
,
Hua
Mei
* and
Yan
Xu
 *
*
College of Chemical Engineering, State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, Nanjing 211800, P. R. China. E-mail: lingtongxu@njtech.edu.cn
First published on 11th April 2024
Abstract
A reduced polyoxometalate-based organo-metallophosphate (MOPO) framework formulated as [Co4(PO4)(C7H8N4)6](PWVI10WV2O40) (Co-PO4-PW12) with an ultra-high CO production rate of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 676 μmol g−1 h−1 has been presented through photocatalytic CO2 reduction investigations.
676 μmol g−1 h−1 has been presented through photocatalytic CO2 reduction investigations.
What is getting more and more urgent is that humans are under higher pressure to discover effective and useful solutions to issues on account of the use of limited fossil fuels, which results in massive CO2 emissions and then leads to global warming.1,2 Under this background, carbon capture, use, and storage, often known as CCUS, is seen as an essential and advantageous method for reducing CO2 emissions and converting CO2 to other resources.3 Among all kinds of CO2 utilization technologies, constructing manufactured photosynthetic systems that employ direct sunlight to transform CO2 into various energy-related substances (such as CO, CH4, HCOOH, etc.) could serve as a “killing two birds with one stone” way to solve this predicament.4–6 Moreover, as we know, chemically inert CO2 molecules must go through multiple electron transfers with a proton-assisted process, in order to achieve these photocatalytic CO2 reduction reaction (CO2RR) processes.7 Hence, designing economical and efficient photocatalysts to assist the CO2RR process is particularly significant.8,9
Polyoxometalates (POMs), which constitute a subfamily of metal oxide cluster materials composed of earth-abundant elements, possess vital value in various fields due to their well-defined structures and diverse components.10–15 Because of the semiconductor characteristics, strong light absorption, and excellent redox capabilities, it is viable for POMs to undergo reversible and continuous electron transport rapidly in catalytic reactions including water splitting and the photocatalytic CO2RR without structural changes. Furthermore, another significant characteristic is that POMs have a rich surface with oxygen atoms and a high number of negative charges, which always display diverse coordination modes that can coordinate with different types of transition metals forming ligand-to-metal charge transfer bands so as to play a role in regulating the absorbance and energy levels.16 Nevertheless, the facile solubility of pure POMs poses a tough threat to their recyclability, which prevents them from complying with industry requirements.17,18 Therefore, the development of novel organic–inorganic hybrid materials with attractive architectures and superior properties has been an intriguing and challenging topic.19–21
Metal–organic frameworks (MOFs), which may be employed as functional platforms to unhomogenise POMs, are compounds with the ability to expand through repeating coordination of organic ligands and metal ions.22–27 One promising solution is to synthesize POM-based MOF (POMOF) materials by combining POMs as the guest and a porous MOF as the host matrix, which can be used to overcome pure POMs’ solubility.28,29 Generally speaking, a POMOF provides the following advantages: (i) it benefits from strong thermal stability and recyclability; (ii) the crystal form of a POMOF with a defined structure and chemical composition is advantageous for researching catalytic procedures; (iii) MOFs are splendid platforms for the catalytic realization of hierarchical solar energy conversion centers, which is because of their enormous surface area, abundant metal content, and active flexibility in the design of a web framework. The organo-metallophosphate (MOPO), with inorganic PO4 tetrahedra as its basic structural component, is a member of the MOF family.30 Such porous materials are favored due to the flexible structure and suitable porosity of conventional MOFs in addition to having an arranged phosphate skeleton in the structure, which is beneficial for facilitating multiple electron transport events occurring during photosynthesis.31,32 Enlightened by these discoveries, integrating POMs with MOPO frameworks could accelerate CO2 photoreduction catalysis while also boosting the diversity of POMOFs. Moreover, reduced polyoxometalate-encapsulated POMOFs may improve photocatalytic performance due to better electron transport.
Keeping the above factors in mind, reduced [Co4(PO4)(C7H8N4)6](PWVI10WV2O40) (Co-PO4-PW12) was synthesized by encasing H5PWVI10WV2O40 inside the interior cavity of an MOPO framework utilizing a hydrothermal procedure. In Co-PO4-PW12, PO43− anions build an MOPO framework by combining with the bidentate N-containing Bim ligands and Co2+ ions, with the reduced Keggin-type PWVI10WV2O40 ingeniously encased in the framework as its size is suitable. Simultaneously, Co-PO4-PW12 and the [Ru(bpy)3]Cl2 complex might combine to provide a controllable system for CO2 photoreduction. The [Ru(bpy)3]Cl2 complex is in charge of collecting visible light, while the MOPO frameworks act as active sites for CO2 adsorption and activation. Co-PO4-PW12 demonstrated a phenomenal CO production rate of 13676 μmol g−1 h−1 and selectivity of 94.5% in CO2 photoreduction on account of reduced-POM and MOPO frameworks operating together synergistically.
The Co-PO4-PW12 crystals with a smooth appearance and no cracks were selected under an optical microscope and fixed on the top of a capillary. Also, the crystal data under 296 K Mo-Kα (λ = 0.71073 A) irradiation were collected on a Bruker Apex II CCD diffractometer. The SHELXL-2018/3 package was used for optimization using direct methods and the full matrix least squares method. The selected crystallographic data for Co-PO4-PW12 are presented in Table S3.†
One effective and innovative approach for creating a MOPO is hydrothermal one-step synthesis. However, parameters including the pH of the solution, reaction temperature, and reaction duration may have an effect on the crystal nucleation and growth process in hydrothermal environments. It is also shown that the pH and reaction temperature had a significant impact on the creation of Co-PO4-PW12 based on a series of parallel tests. It should be noted that while the ideal reaction temperature was 140 °C, high-quality target compounds could not be synthesized when pH values were higher than 4.5 or lower than 2.5.
The trigonal system with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group is where Co-PO4-PW12 crystallizes, with regard to single-crystal X-ray studies. The asymmetric unit of the MOPO is composed of the C7H8N4 (Bim) organic ligand, phosphate anions (PO43−), and Co ions (Fig. S3†). Interestingly, reduced Keggin type (PWVI10WV2O40)5− polyanions (PW12) are encased in the MOPO framework. The valences of W atoms were measured by X-ray photoelectron spectroscopy (XPS) and are shown in Fig. 2d. The XPS analysis of Co-PO4-PW12, as shown in Fig. 2d, explicitly determined four partially overlapped peaks and the ratio of WV to WVI is 2:10. In addition, each independent Co ion in Co-PO4-PW12 displays four-coordinated modes. The coordination environment of Co ions is linked by one oxygen atom from the PO43− anion and three Bim ligands’ N atoms. Table S2† displays that the distance of Co–N ranges between 1.969(14) Å and 2.002(13) Å, whereas that of N–Co–N ranges between 105.1(6) Å and 110.3(4) Å. As shown in Fig. 1, the Co ions are linked by ligands to form a [Co2(Bim)2]4+ cationic sixteen-membered ring. Subsequently, six PO43− anions then act as bridge linkers to join with six macrocycles with 16 members, resulting in rings with dimensions of 6.62 × 5.42 Å, shaped like a “Star of David” (Fig. 1a). An ingenious coordination between the organophosphate ring and Co ions occurs through Bim ligands leading to a MOPO cage (Fig. 1b). The MOPO cage has the size of 23.69 × 20.14 × 17.25 Å, while the Keggin-type PW12 exactly fits the diameters of the cage in Co-PO4-PW12. Thus, the MOPO cage provides sufficient space for in situ encapsulation of Keggin-type POMs. Also, adjacent MOPO cages are edge-shared to form a 2D layer (Fig. 1c). These 2D layers are further expanded into a 3D structure through the PO43− anion linkage (Fig. 1d). From a topological perspective, the MOPO framework as a whole can be represented as sql topology with the point symbol {6;82}3{63;83}2 if PO43− anions and cobalt ions are successively regarded as 4- and 3-connected nodes, respectively (Fig. 1e).
space group is where Co-PO4-PW12 crystallizes, with regard to single-crystal X-ray studies. The asymmetric unit of the MOPO is composed of the C7H8N4 (Bim) organic ligand, phosphate anions (PO43−), and Co ions (Fig. S3†). Interestingly, reduced Keggin type (PWVI10WV2O40)5− polyanions (PW12) are encased in the MOPO framework. The valences of W atoms were measured by X-ray photoelectron spectroscopy (XPS) and are shown in Fig. 2d. The XPS analysis of Co-PO4-PW12, as shown in Fig. 2d, explicitly determined four partially overlapped peaks and the ratio of WV to WVI is 2:10. In addition, each independent Co ion in Co-PO4-PW12 displays four-coordinated modes. The coordination environment of Co ions is linked by one oxygen atom from the PO43− anion and three Bim ligands’ N atoms. Table S2† displays that the distance of Co–N ranges between 1.969(14) Å and 2.002(13) Å, whereas that of N–Co–N ranges between 105.1(6) Å and 110.3(4) Å. As shown in Fig. 1, the Co ions are linked by ligands to form a [Co2(Bim)2]4+ cationic sixteen-membered ring. Subsequently, six PO43− anions then act as bridge linkers to join with six macrocycles with 16 members, resulting in rings with dimensions of 6.62 × 5.42 Å, shaped like a “Star of David” (Fig. 1a). An ingenious coordination between the organophosphate ring and Co ions occurs through Bim ligands leading to a MOPO cage (Fig. 1b). The MOPO cage has the size of 23.69 × 20.14 × 17.25 Å, while the Keggin-type PW12 exactly fits the diameters of the cage in Co-PO4-PW12. Thus, the MOPO cage provides sufficient space for in situ encapsulation of Keggin-type POMs. Also, adjacent MOPO cages are edge-shared to form a 2D layer (Fig. 1c). These 2D layers are further expanded into a 3D structure through the PO43− anion linkage (Fig. 1d). From a topological perspective, the MOPO framework as a whole can be represented as sql topology with the point symbol {6;82}3{63;83}2 if PO43− anions and cobalt ions are successively regarded as 4- and 3-connected nodes, respectively (Fig. 1e).
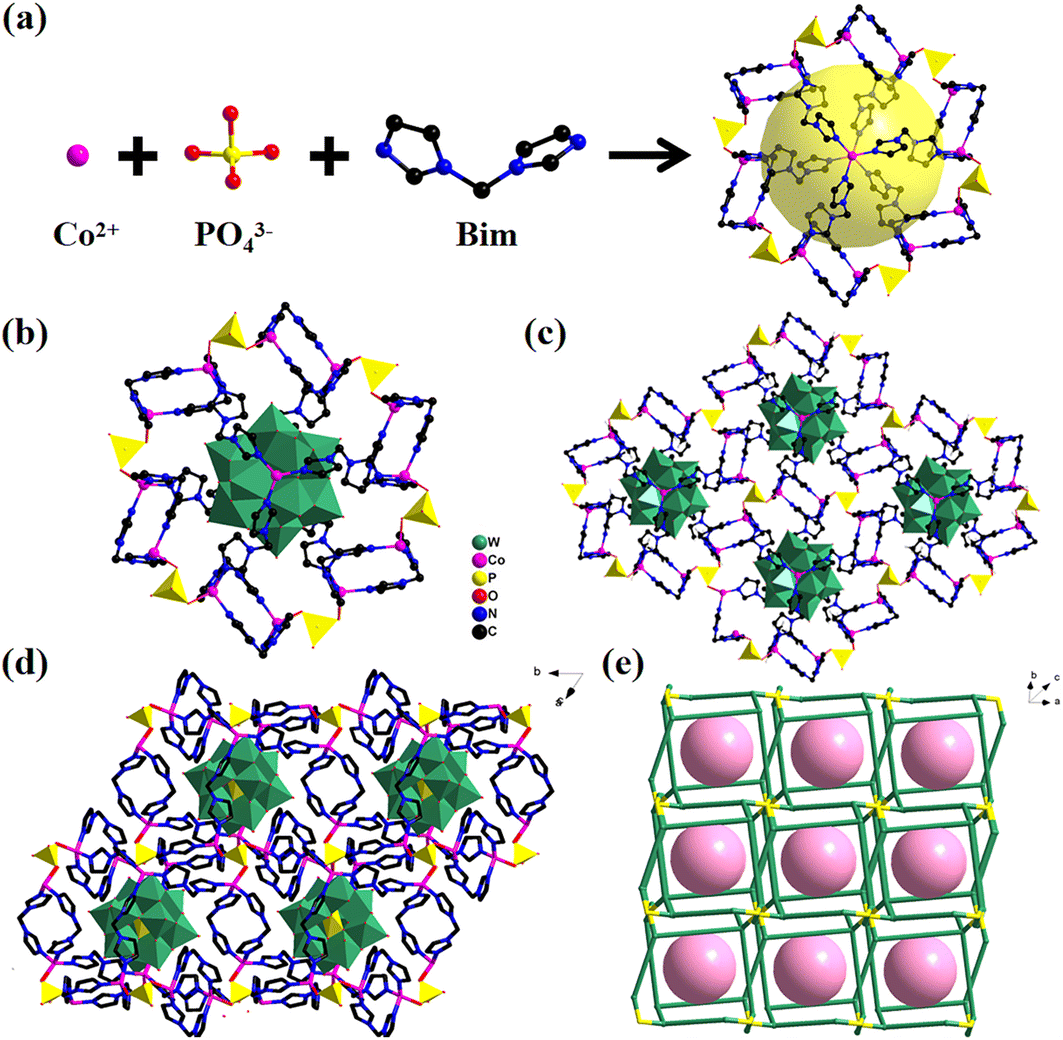 | ||
| Fig. 1 (a) Formation of the MOPO cage; (b) a single POM-encapsulated MOPO cage; (c) 2D layer of Co-PO4-PW12; (d) 3D structure of Co-PO4-PW12; (e) simplified structure of Co-PO4-PW12. Rose balls represent POMs. H atoms are omitted for clarity. | ||
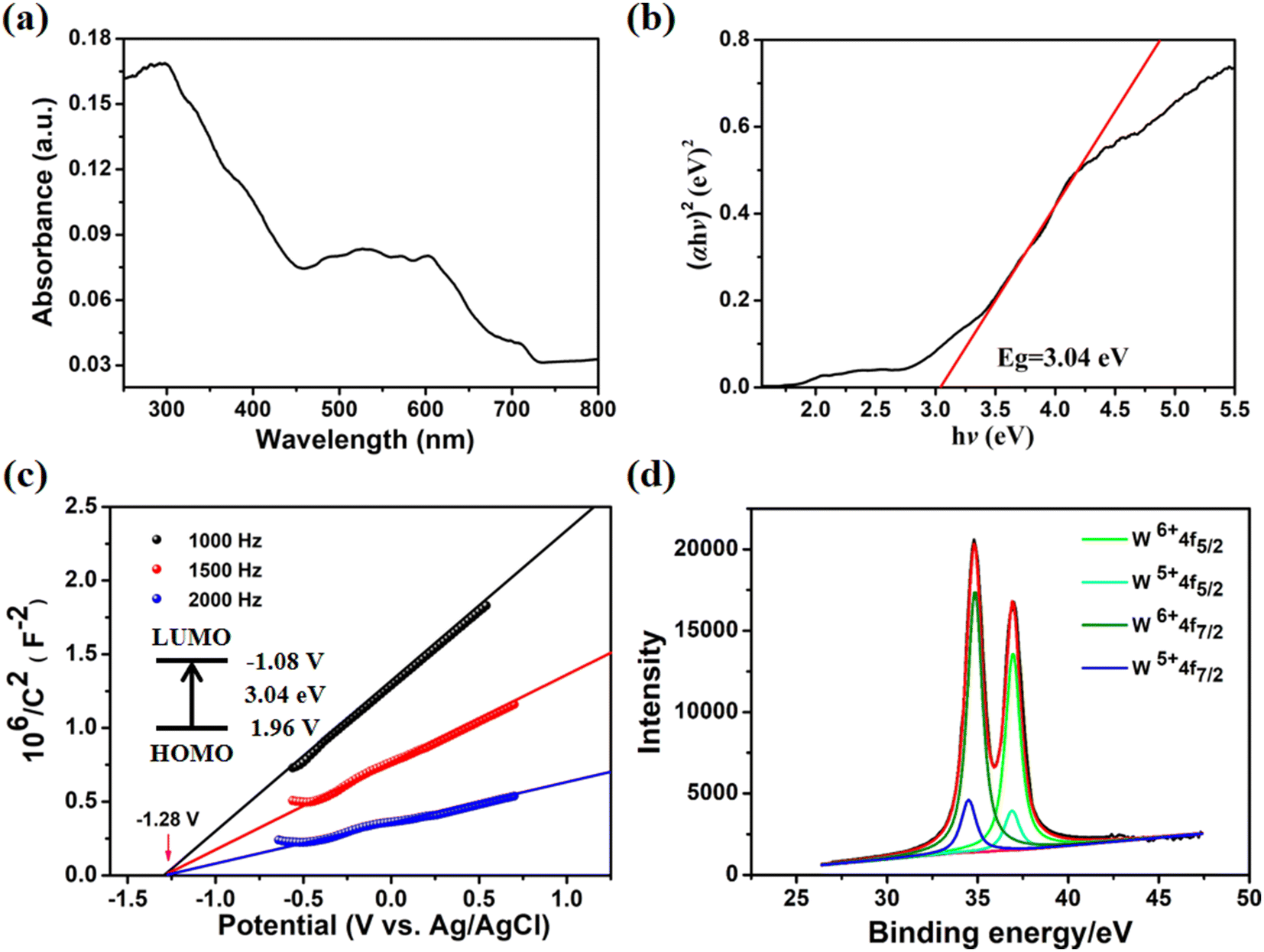 | ||
| Fig. 2 (a) UV–visible diffuse reflectance spectra of Co-PO4-PW12, (b) (αhν) 2vs. hν curve, (c) Mott–Schottky plots, (d) fit of the curve providing positions of the WV 4f7/2, WVI 4f7/2, WV 4f5/2 and WVI 4f5/2 peaks at 34.48 eV, 34.86 eV, 36.90 eV and 36.93 eV, respectively, in the XPS spectrum of W4f peaks for Co-PO4-PW12. | ||
Powder X-ray diffraction (PXRD) study provided the proof that Co-PO4-PW12 is a pure phase. A study was made on Co-PO4-PW12's chemical stability as well. For 24 hours, the crystals (20 mg) were submerged in a mixed solvent of 50 mL of several organic solvents, with CH3CN and TEOA. Its great stability in these organic solvents provided the necessary conditions for using it as a heterogeneous photocatalyst (Fig. S6†). The compound's thermal stability was also measured by thermogravimetric (TG) examination (Fig. S9†). The TG curve may be divided into one major stage, in essence. If the temperature climbs over 172 °C, the TG curve degrades by 23.16% due to the breakdown of the organic ligands and a portion of the metal framework. The FT-IR spectra, shown in Fig. S8,† of Co-PO4-PW12 were further acquired (400–4000 cm−1). The strong and broad absorption bands around 3483 cm−1 can be considered as the –OH stretching vibration. The obvious peaks at 1645 and 1415 cm−1 could prove that the C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of Bim ligands exist in Co-PO4-PW12. The P–O vibration signature is represented by the peaks at 1097 cm−1, while the Keggin POM anions’ WO and W–O–W vibrations are located at 956, 811, and 751 cm−1.
C of Bim ligands exist in Co-PO4-PW12. The P–O vibration signature is represented by the peaks at 1097 cm−1, while the Keggin POM anions’ WO and W–O–W vibrations are located at 956, 811, and 751 cm−1.
A variety of tests were conducted to assess the photocatalytic activity of Co-PO4-PW12 in addition to the structural properties listed above, including UV-vis diffuse reflectance spectra and Mott–Schottky plots (Fig. 2). It is very obvious that a wide absorption band is displayed between 450 nm and 700 nm (Fig. 2a), which may be caused by the effective charge migration between the Co metal ion of Co-PO4-PW12 and the Bim ligands.33 Additionally, as shown in Fig. 2b, the Tauc plot approach showed the band gap energy (Eg) of Co-PO4-PW12 to be 3.04 eV. All of these results clearly suggest that the Co-PO4-PW12 photocatalyst possesses a lot of promise. Co-PO4-PW12 showed n-type semiconductor properties, as indicated by the positive slope of the curve in Mott–Schottky plots measured at 1000, 1500, and 2000 Hz (Fig. 2c). The LUMO position value of Co-PO4-PW12 is −1.08 V more negative than the reduction potential value of E (CO/CO2) which is −0.53 V vs. NHE, offering a theoretical prospect for its utilization in photocatalytic CO2 reduction.
As a consequence of the inspiring characterization findings mentioned above, we performed the photocatalytic CO2RR employing Co-PO4-PW12 in a pure CO2 atmosphere with V(MeCN:TEOA) = 4:1 as the solvent and [Ru(bpy)3]Cl2 as the photosensitizer.34 When 3 mg of Co-PO4-PW12 was primarily used to drive CO production under visible light, the overall increasing trend of CO production as a function of time was similar to a linear increase according to Fig. 3a. The main gaseous product detected by chromatography was CO with trace amounts of CH4 and H2. After 5 h of reaction, the amount of CO produced, catalyzed by Co-PO4-PW12, reached 205.15 μmol, that is, the reaction rate was 13676 μmol g−1 h−1, also showing 94.5% CO selectivity. Then, the effects of 1 mg, 5 mg, and 7 mg of Co-PO4-PW12 on the photocatalytic CO2RR were further investigated (Fig. 3b). When 1 mg of Co-PO4-PW12 was added, the CO yield of the reaction system also reached 46.3 μmol which was considerably lower than that for 3 mg. Once the catalyst addition was upped to 5 mg and 7 mg, the CO production of Co-PO4-PW12 reached 261.05 μmol and 293.59 μmol after 5 h, respectively. Nonetheless, it is worth noting that the CO production rate instead decreased, reaching only 10441.9 μmol g−1 h−1 and 8388 μmol g−1 h−1 owing to insufficient photoelectron transfer on [Ru(bpy)3]Cl2 to Co-PO4-PW12.35
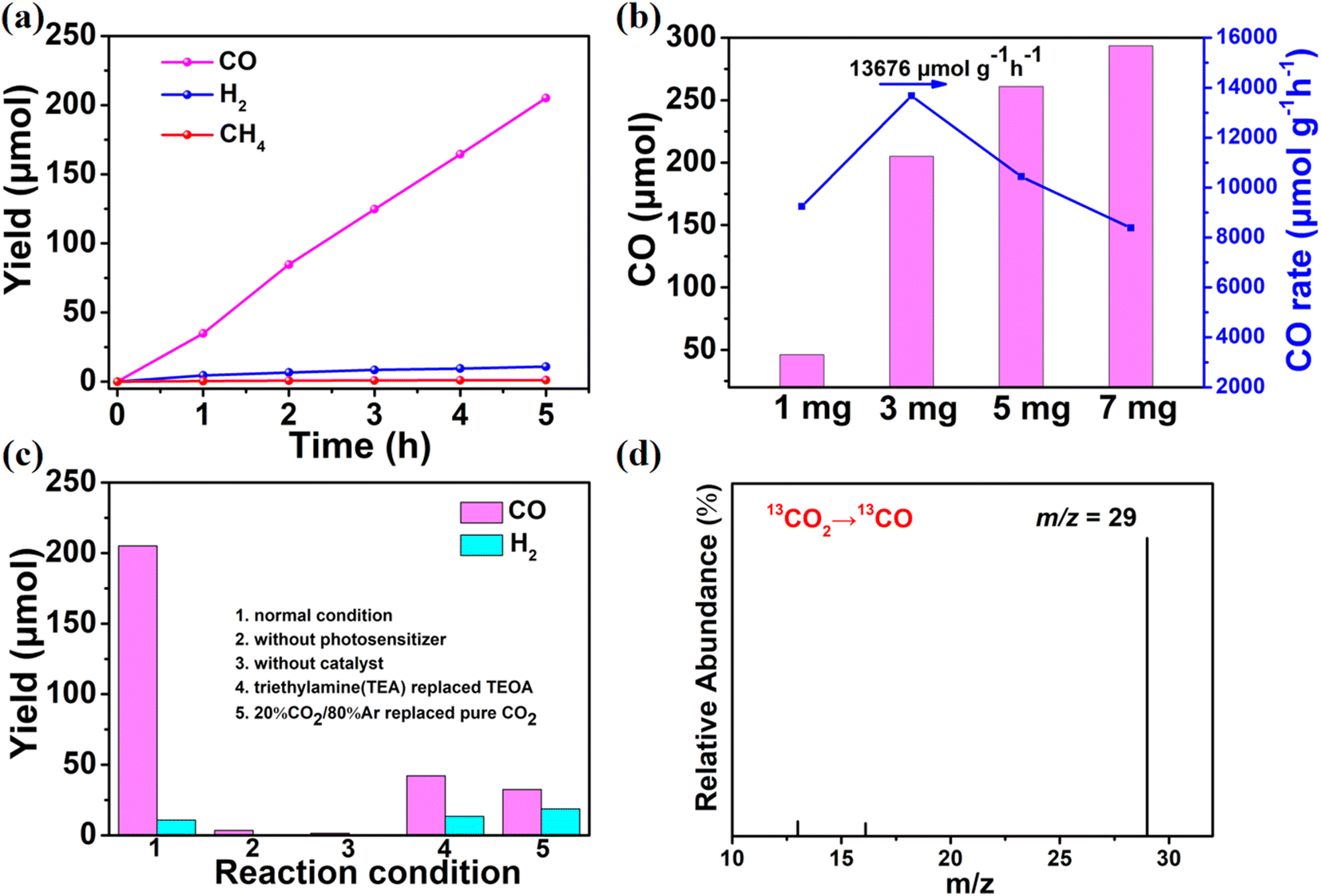 | ||
| Fig. 3 (a) Production of CO over time is depicted on a trend line, (b) the effect of quantity on the production of CO, (c) the study of the CO2RR under varied situations, and (d) the mass spectra in the 13CO2 isotopic detection experiment over Co-PO4-PW12 as a photocatalyst. | ||
In order to further verify the important role of other components in the CO2 photoreduction system, multiple control experiments were performed. The results are presented in Fig. 3c. When the experiment was conducted in the dark, no product was observed, indicating that this is a light-driven catalytic reaction. In the absence of a Ru-photosensitizer or the catalyst Co-PO4-PW12, almost no CO and H2 were detected, indicating that this is a photodriven catalytic reaction enabled by the synergism of the photosensitizer and catalyst. Similarly, a certain amount of CO was obtained once the photocatalytic CO2RR experiment was executed in a low concentration of 20%CO2/80%Ar surrounding, revealing that a practical application value was created for the reaction to take place. In addition, the production of CO requires a suitable sacrificial electron donor as the CO yield was drastically and significantly reduced when TEOA was replaced with TEA. Only a trace amount of CO was generated when TEOA was not added, suggesting that TEOA could help retain and activate CO2 molecules, act as an electron donor, and be indispensable for promoting CO2 conversion. On top of that, the trial also supported the product's carbon source. As shown in Fig. 3d, the carbon tracing experiment enabled the detection of the peak at m/z = 29 that belonged to 13CO. These studies provided persuasive evidence that CO originated from CO2.
By analyzing the photoluminescence (PL) spectra of acetonitrile solutions of the photosensitizer [Ru(bpy)3]Cl2 in the presence of the sacrificial agent TEOA or catalyst Co-PO4-PW12, and by preparing the experimental solutions in equal portions in accordance with the concentration used for the photocatalytic reaction, we were able to better understand the process of electron migration. Due to the interaction of its holes and electrons, the excited state of [Ru(bpy)3]2+* exhibits a powerful emission spectrum at around 580 nm. The intensity of the luminescence will diminish as a result of the reduction or oxidation of the photosensitizer, depending on whether it is quenched by the TEOA electron donor or the catalyst. The photoluminescence intensity of [Ru(bpy)3]2+ was altered a little when various TEOA concentrations were added (Fig. 4b and c). The catalyst Co-PO4-PW12 had a considerable quenching effect when we applied it at the same molar amount. Consequently, the rapid complexation between holes and electrons on [Ru(bpy)3]2+ is effectively suppressed. These findings have shown that the charge separation between [Ru(bpy)3]2+ and the catalyst Co-PO4-PW12 is more effective. As the catalyst concentration increased, the fluorescence intensity of the excited state of [Ru(bpy)3]2+* steadily dropped, which may be attributed to the catalyst's ongoing electron transfer from the excited state.
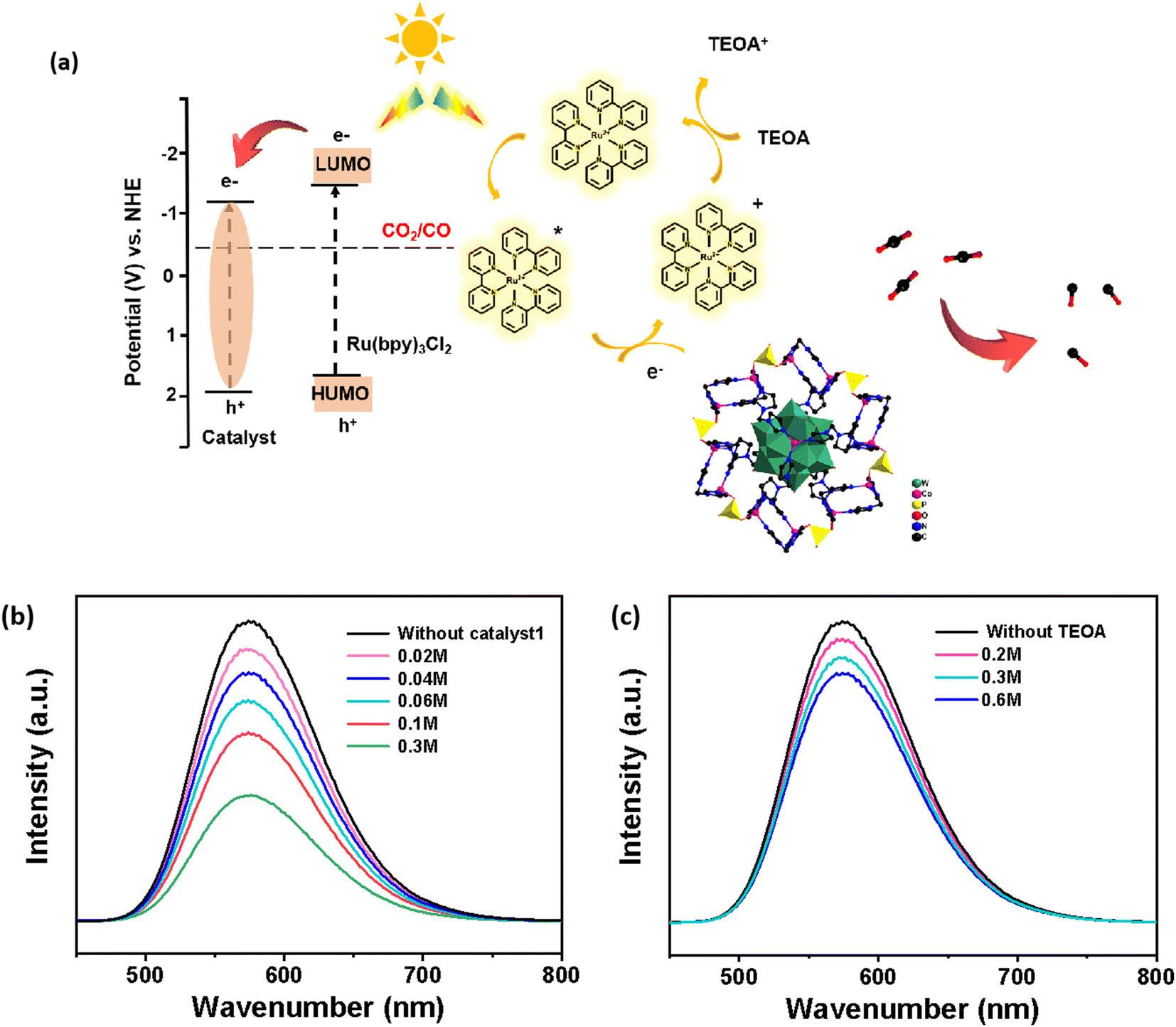 | ||
| Fig. 4 (a) The speculative mechanism for the photocatalytic CO2 reduction by Co-PO4-PW12. (b and c) The photoluminescence spectra of MeCN solution in the presence of [Ru(bpy)3]Cl2 obtained with different doses of Co-PO4-PW12 (0.02 M–0.3 M) and TEOA (0.2 M–0.6 M). | ||
As shown in Fig. 4a, the reaction mechanism and reaction path of the whole photocatalytic CO2RR process catalyzed by Co-PO4-PW12 are deduced according to the above experimental results and experimental phenomena: for starters, under the stimulation of visible light, the electrons of [Ru(bpy)3]Cl2 are transferred from the HOMO to the LUMO to form the Ru(bpy)32+* excited state. Afterwards, the photogenerated electrons are transferred to the catalyst to form Ru(bpy)33+ in the oxidized state and Co-PO4-PW12 in the reduced state due to the matching of the LUMO position. Ultimately, Ru(bpy)33+ in the oxidized state is converted to Ru(bpy)32+ through the sacrificial agent TEOA, that is, the photogenerated holes combine with TEOA to form TEOA+, and CO2 molecules adsorbed on the catalyst's surface are reduced to CO and separated, completing the whole catalytic cycle accompanied by the catalyst switching back to its original form.36–40
To sum up, we have carefully synthesized the catalyst Co-PO4-PW12 with a MOPO linked by PO43− and Co2+ anions, and the reduced PW12 polyanions are encapsulated into the MOPO framework. The CO production rate of Co-PO4-PW12 reached 13676 μmol g−1 h−1 with a selectivity of over 94.5% due to the synergistic impact of POMs and MOPOs. This work creatively combines MOPO frameworks with reduced POMs to provide new insights into the design of efficient photocatalytic materials for CO2 reduction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (92161109).References
- W. Kim, B. A. McClure, E. Edri and H. Frei, Chem. Soc. Rev., 2016, 45, 3221–3243 RSC
.
- X. Yue, L. Cheng, F. Li, J. Fan and Q. Xiang, Angew. Chem., Int. Ed., 2022, 40, e202208414 Search PubMed
- Z. Y. Du, Y. Z. Yu, Y. L. Hong, N. F. Li, Y. M. Han, J. P. Cao, Q. Sun, H. Mei and Y. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 57174–57181 CrossRef CAS PubMed
- J. Zhang, X. Cui, Y. Zhou, T. Kong, Y. Wang, X. Wei and Y. Xiong, Chem. Commun., 2023, 59, 2299–2302 RSC
- J. Zhou, H. Wu, C. Y. Sun, C. Y. Hu, X. L. Wang, Z. H. Kang and Z. M. Su, J. Mater. Chem. A, 2018, 6, 21596–21604 RSC
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed
- H. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S. Yu and H. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed
- D. C. Hong, T. K. Y. Kawanishi, Y. T. Tsukakoshi, K. T. Hiroaki, T. Y. Ishizuka and T. K. H. K. Kojima, J. Am. Chem. Soc., 2019, 141, 20309–20317 CrossRef CAS PubMed
- P. Madhusudan, R. Shi, S. L. Xiang, M. T. Jin, B. N. Chandrashekar, J. W. Wang, W. J. Wang, O. W. Peng, A. Amini and C. Cheng, Appl. Catal., B, 2021, 282, 119600 CrossRef CAS
- Y. H. Zhu, J. B. Yang, X.-M. Liu, J. L. Wang, Q. D. Ping, Z. Y. Du, J. N. Li, T. T. Zang, H. Mei and Y. Xu, Dalton Trans., 2022, 51, 3502–3511 RSC
- J. Becerra, D.-T. Nguyen, V.-N. Gopalakrishnan and T.-O. Do, ACS Appl. Energy Mater., 2020, 3, 7659–7665 CrossRef CAS
- S. Rong, M. Wang and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202310018 CrossRef CAS PubMed
- C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2012, 131, 1883–1888 CrossRef PubMed
- Y. Chen, H. An, S. Chang, T. Xu, Y. Li, Q. Zhu, Y. Huang and Y. Wei, Inorg. Chem., 2023, 26, 10120–10130 CrossRef PubMed
- W. Jiang, X. M. Liu, J. Liu, J. Shi, J. P. Cao, X. M. Luo, W. S. You and Y. Xu, Chem. Commun., 2019, 55, 9299–9302 RSC
- B. N. Li, X. J. Yu, H. J. Pang, Q. B. Shen, Y. Hou, J. Ma and H. Y. Xin, Chem. Commun., 2020, 56, 7199–7202 RSC
- X.-X. Li, J. Liu, L. Zhan, L.-Z. Dong, Z.-F. Xin, S.-L. Li, Q. Xue, H. Fu, K. Huang and Y.-Q. Lan, ACS Appl. Mater. Interfaces, 2019, 11, 25790–25795 CrossRef CAS PubMed
- J.-S. Qin, D.-Y. Du, W. Guan, X.-J. Bo, Y.-F. Li, L.-P. Guo, Z.-M. Su, Y.-Y. Wang, Y.-Q. Lan and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 7169–7177 CrossRef CAS PubMed
- R. Das, R. Paul, A. Parui, A. Shrotri, C. Atzori, K. A. Lomachenko, A. K. Singh, J. Mondal and S. C. Peter, J. Am. Chem. Soc., 2022, 145, 422–435 CrossRef PubMed
- R. Paul, A. Boruah, R. Das, S. Chakraborty, K. Chahal, D. J. Deka, S. C. Peter, B. K. Mai and J. Mondal, Small, 2024, 20, 2305307 CrossRef CAS PubMed
- R. Paul, R. Das, N. Das, S. Chakraborty, C. W. Pao, Q. Thang Trinh, G. T. K. Kalhara Gunasooriya, J. Mondal and S. C. Peter, Angew. Chem., Int. Ed., 2023, 62, e202311304 CrossRef CAS PubMed
- S. Suan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. F. Zou, Y. M. Zhang, L. L. Zhang, Y. Fang, J. L. Li and H. C. Zhou, Adv. Mater., 2018, 30, 170430 Search PubMed
- Y. Dong, J. Zhang, W. Wang, B. Hu, D. Xia, K. Lin, L. Geng and Y. Yang, Small, 2023, 29, 2301824 CrossRef PubMed
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126 RSC
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364 CrossRef CAS PubMed
- S.-Q. You, Y.-J. Dong, B.-S. Hou, M. Dong, J.-L. Tong, L.-X. Wang, X.-L. Wang, C.-Y. Sun, W. Guan and Z.-M. Su, J. Mater. Chem. C, 2023, 11, 7389–7396 RSC
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735 RSC
- Z.-M. Zhang, T. Zhang, C. Wang, Z. Lin, L.-S. Long and W. Lin, J. Am. Chem. Soc., 2015, 137, 3197–3200 CrossRef CAS PubMed
- X.-J. Kong, Z. K. Lin, Z.-M. Zhang, T. Zhang and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 6411–6416 CrossRef CAS PubMed
- L. Shao, Y. Li, J. H. Yu and R. R. Xu, Inorg. Chem., 2012, 51, 225–229 CrossRef CAS PubMed
- E. Gianotti, M. Manzoli, M. E. Potter, V. N. Shetti, D. Sun, J. Paterson, T. M. Mezza, A. Levy and R. Raja, Chem. Sci., 2014, 5, 1810–1819 RSC
- Z. Y. Du, Y. N. Xue, X. M. Liu, N. F. Li, J. L. Wang, H. Mei and Y. Xu, J. Mater. Chem. A, 2022, 10, 3469–3477 RSC
- B. Li, X. Yu, H. Pang, Q. Shen, Y. Hou, H. Ma and J. Xin, Chem. Commun., 2020, 56, 7199–7202 RSC
- J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217–234 CrossRef CAS
- J. N. Qin, S. B. Wang and X. C. Wang, Appl. Catal., B, 2017, 209, 476–482 CrossRef CAS
- N. Li, J. Liu, B. X. Dong and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 20779–20793 CrossRef CAS PubMed
- X. X. Li, L. Zhang, J. Liu, L. Yuan, T. Wang, J. Y. Wang, L. Z. Dong, K. Huang and Y. Q. Lan, JACS Au, 2021, 1, 1288–1295 CrossRef CAS PubMed
- L. Zhang, R. H. Li, X. X. Li, J. Liu, W. Guan, L. Z. Dong, S. L. Li and Y. Q. Lan, Proc. Natl. Acad. Sci., 2022, 119, e2210550119 CrossRef CAS PubMed
- L. Qiao, M. Song, A. Geng and S. Yao, Chin. Chem. Lett., 2019, 30, 1273–1276 CrossRef CAS
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: PXRD patterns, IR spectra, and TGA characterization. CCDC 2339653. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00812j |
| ‡ These authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2024 |