
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A homochiral tartrate-bridged dinuclear chromium(III) complex anion with a resonance-assisted hydrogen bond for proton conduction†
Marko
Dunatov
 a,
Zhibo
Zhao
b,
Dijana
Žilić
a and
Lidija
Androš Dubraja
*a
a,
Zhibo
Zhao
b,
Dijana
Žilić
a and
Lidija
Androš Dubraja
*a
aRuđer Bošković Institute, Bijenička cesta 54, 10000 Zagreb, Croatia. E-mail: lidija.andros@irb.hr; Tel: +385 1 4561184
bInstitute of Nanotechnology, Karlsruhe Institute of Technology, Kaiserstraße 12, 76131 Karlsruhe, Germany
First published on 7th May 2024
Abstract
The synthesis of a homochiral building block based on L-tartrate-chromium(III) complex anions is reported. The dinuclear complex anion, which contains two bridging L-tartrate ligands and one aromatic N-donor ligand coordinated to chromium(III) ions, exhibits a boat conformation in which intramolecular resonance-assisted hydrogen bonding is present. The sodium L-tartrate-chromium(III) compound with the formula Na[Cr2(bpy)2(L-tart)2H]·9H2O (1) crystallizes from a methanol–water solution as a high water content material in the monoclinic space group P2. The as-synthesized compound is only stable at high relative humidity and undergoes structural transformations during drying, which are accompanied by water loss. However, these transformations are reversible and upon wetting, the material returns to its high water content structure. Based on a combination of experimental techniques (PXRD, in situ ATR-FTIR and EPR spectroscopy), the structure of the complex anions appears to be insensitive to the humidity variable processes (wetting and drying). Due to the presence of several hydrogen acceptor and donor groups in the L-tartrate-chromium(III) complex anion, we investigated the proton transport properties of a sodium L-tartrate-chromium(III) compound by impedance spectroscopy under dry and wet conditions at different temperatures. Since the relative humidity affects the structural transformations in this system, it also has a large influence on the proton conductivity, which varies by up to four orders of magnitude depending on the degree of hydration. These results confirm that the proton conductivity can be tuned in flexible structures in which non-covalent interactions determine the crystal packing.
Introduction
It has been experimentally and theoretically proved that structural water molecules have a major influence on the crystal structures of certain synthetic and biological materials by adding additional bonding interactions to the polar functional groups that help stabilize the structure but also provide more flexibility due to their reversible non-covalent bonding.1–4 For structures with a high water content, a certain degree of flexibility can be correlated with that of biological membranes and natural tissues, providing an excellent platform for solid-state research with regard to transportation applications.5 Due to the above-mentioned flexibility, the structures of such materials with a high water content are highly dependent on the degree of hydration, i.e. air humidity.6,7 Since atmospheric humidity is a large reservoir of energy that is continuously available, it seems reasonable to try to exploit it to generate electricity.8 One way to utilize humidity is to develop proton conductors in the solid state, either in purely organic and inorganic forms or as hybrid metal–organic systems that could be used in fuel cells for clean and efficient energy generation.9,10 For the design of highly proton-conducting materials, specific functional groups, such as carboxylates, phosphonates, and sulphonates, contribute to the increase of proton conductivity and have been extensively used to synthesize proton conductors.11–13The flexibility of structures containing naturally occurring carboxylic acids, such as L-tartaric acid, has been demonstrated in several transition metal complexes that undergo reversible solid-state transformation upon exposure to moisture.14–19L-Tartaric acid serves several purposes as a ligand in coordination chemistry: it coordinates, chelates or bridges the metal centres, can be pH-adjusted to form different ionic species (−4 ≤ ionic charge ≤ 0) and contains sufficient functional groups to participate in non-covalent interactions that stabilize the structure. In most of the structures found in the Cambridge Structural Database (CSD),20 the L-tartrate ligand bridges the transition metal centres, leading either to extended polymeric structures, as in a series of isomorphous compounds {[M(NN)(L-tart-H2)]·xH2O}n (M2+ = Mn, Co, Cu, and Zn; NN = 2,2′-bipyridine, 1,10-phenanthroline; L-tart-H2 = dianion of L-tartaric acid; x = 5, 6),21–26 or to isolated dinuclear complexes of the formula [{M(NN)}2(L-tart-H2)] (M2+ = Pd and Pt).27,28 Complexes containing two tartrate bridging groups of the same enantiomeric configuration are considered to be energetically more stable than those with bridges of opposite chirality.19 For this reason, these complexes are also very stable toward intramolecular racemization, making them suitable building blocks for the construction of chiral extended inorganic–organic frameworks with a polar crystal structure. This type of structural arrangement is particularly important for achieving nonlinear optical properties and ferroelectricity.
In this article, we report the synthesis of a homochiral dinuclear L-tartrate-chromium(III) complex anion, which crystallizes with sodium cations and a large number of water of crystallization molecules in a compound Na[Cr2(bpy)2(L-tart)2H]·9H2O (1), [bpy = 2,2′-bipyridine (C10H8N2); L-tart = completely deprotonated L-tartaric acid, (C4O6H2)4−]. In contrast to divalent transition metal cations,21–28 chromium(III) ions form bridged dinuclear complexes with meso-, D- and L-tartaric acid, in which one proton per two tetranegative tartrate ligands remains undissociated (as shown in Scheme 1).19,29 The structure of the meso-tartrate-chromium(III) complex anion with an aromatic N-donor ligand was described by Ortega et al.30 and the structure of the L-tartrate-chromium(III) complex anion is topologically the same. Very short intramolecular hydrogen bonds, also known as resonance-assisted hydrogen bonds,31–33 exist in these complex anions. Compound 1 undergoes a reversible structural transformation depending on the relative humidity. The proton conducting properties were investigated under dry and humid conditions by electrochemical impedance spectroscopy to establish the direct connection between the structure and activity. In addition the magnetic properties were determined by magnetization and electron paramagnetic resonance measurements.
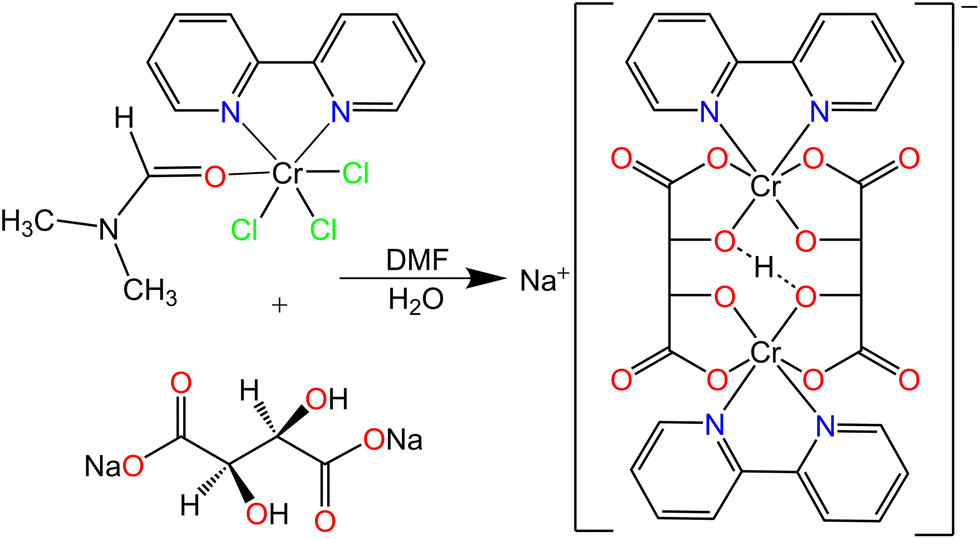 | ||
| Scheme 1 Solution based synthesis of compound Na[Cr2(bpy)2(L-tart)2H]·9H2O (1) through the reaction of [CrCl3(DMF)(bpy)] and sodium L-tartrate. | ||
Experimental
Starting materials
The chromium(III) precursor [CrCl3(DMF)(bpy)] was prepared according to a slightly modified literature procedure,34 and the other reagents were obtained from commercial sources. Chromium(III) chloride hexahydrate, CrCl3·6H2O, (1.066 g, 4 mmol) was dissolved in DMF (6 ml) and 2,2′-bipyridine (0.625 g, 4 mmol) was added to the hot solution, which was stirred and refluxed at 383 K. After 10 minutes of stirring at 383 K, the reaction mixture was cooled to room temperature, and the crystalline product [CrCl3(DMF)(bpy)] was separated by filtration, washed with cold DMF and dry ether, and used as such for further synthesis. ATR-FTIR bands (4000–400 cm−1): 3109 (m), 3056 (m), 2956 (m), 2856 (w), 1642 (vs), 1601 (s), 1564 (m), 1495 (m), 1469 (m), 1444 (s), 1432 (s), 1420 (s), 1406 (m), 1362 (vs), 1312 (m), 1279 (w), 1245 (m), 1225 (w), 1117 (w), 1155 (m), 1116 (m), 1102 (m), 1071 (w), 1061 (m), 1045 (w), 1033 (m), 863 (w), 813 (w), 771 (vs), 744 (w), 731 (s), 706 (vs), 664 (m), 650 (s), 453 (w), 430 (m), 417 (m).Synthetic procedure of Na[Cr2(bpy)2(L-tart)2H]·9H2O (1)
A DMF–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) solution (4 mL) containing sodium L-tartrate dihydrate (0.345 g, 1.50 mmol) and [CrCl3(DMF)(bpy)] (0.581 g, 1.50 mmol) was stirred and heated at 393 K to give a red solution. After 10 min, the reaction mixture was allowed to cool to room temperature. After about 30 min, the product was obtained in the form of a red crystalline powder with the formula Na[Cr2(bpy)2(L-tart)2H]·9H2O (1). The crystalline powder was separated by filtration, washed with absolute ethanol and dried in air (yield: 75%). Single crystals of 1, suitable for X-ray analysis, were obtained by recrystallization of the red powder from a methanol–water (2:1) solution. ATR-FTIR bands (4000–400 cm−1): 3412 (m, br), 3110 (m), 3081 (m), 2856 (w), 1622 (vs), 1600 (vs), 1494 (w), 1471 (m), 1445 (s), 1353 (vs), 1310 (s), 1278 (m), 1248 (w), 1173 (w), 1158 (w), 1129 (m), 1106 (s), 1055 (m), 1033 (m), 925 (w), 897 (m), 841 (m), 826 (m), 774 (s), 743 (s), 734 (vs), 661 (s), 652 (s), 642 (s), 631 (s), 607 (m), 571 (s), 540 (m), 519 (m), 454 (s), 423 (m), 403 (w).
:3) solution (4 mL) containing sodium L-tartrate dihydrate (0.345 g, 1.50 mmol) and [CrCl3(DMF)(bpy)] (0.581 g, 1.50 mmol) was stirred and heated at 393 K to give a red solution. After 10 min, the reaction mixture was allowed to cool to room temperature. After about 30 min, the product was obtained in the form of a red crystalline powder with the formula Na[Cr2(bpy)2(L-tart)2H]·9H2O (1). The crystalline powder was separated by filtration, washed with absolute ethanol and dried in air (yield: 75%). Single crystals of 1, suitable for X-ray analysis, were obtained by recrystallization of the red powder from a methanol–water (2:1) solution. ATR-FTIR bands (4000–400 cm−1): 3412 (m, br), 3110 (m), 3081 (m), 2856 (w), 1622 (vs), 1600 (vs), 1494 (w), 1471 (m), 1445 (s), 1353 (vs), 1310 (s), 1278 (m), 1248 (w), 1173 (w), 1158 (w), 1129 (m), 1106 (s), 1055 (m), 1033 (m), 925 (w), 897 (m), 841 (m), 826 (m), 774 (s), 743 (s), 734 (vs), 661 (s), 652 (s), 642 (s), 631 (s), 607 (m), 571 (s), 540 (m), 519 (m), 454 (s), 423 (m), 403 (w).
Infrared spectroscopy
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were recorded in the 4000–400 cm−1 range using a PerkinElmer Frontier FT-IR spectrometer. Humidity-dependent ATR-FTIR measurements were recorded in situ in a chamber with a manual device (HH414, Omega Engineering, Inc.) for monitoring the relative humidity (RH) that was attached to the ATR module of the spectrometer. The RH value was adjusted in two ways: (i) in dynamic experiments by mixing dry N2 and wet air; (ii) in static experiments by exposure to saturated aqueous solutions of NaBr (RH 60%) and NaCl (RH 75%) salts, as well as normal room humidity (RH 28 and 35%).Electrical study
The electrical properties were studied by impedance spectroscopy using an impedance analyser (PalmSens4) in the frequency range from 10 Hz to 1 MHz in a sample cell whose specific RH value was achieved by exposure to saturated aqueous solutions of different salts (NaBr and NaCl) and at room humidity. For measurements the polycrystalline sample was pressed (estimated by ca. 8 GPa) into a cylindrical pellet with a diameter of 5 mm and a thickness of 0.5 mm having 3.5 mm diameter sputter coated Au electrodes on the opposite surfaces of the pellet. The impedance spectra were analysed by equivalent circuit modelling using the non-linear least-squares fitting procedure.Magnetic study
The temperature dependence of the magnetic susceptibility in the range of 2–293 K in a field of 1000 Oe was measured with an MPMS-3 magnetometer equipped with a superconducting quantum interferometer device. Electron paramagnetic resonance (EPR) measurements were performed using a Bruker Elexsys 580 FT/CW spectrometer. The spectra were recorded in the temperature range 78–297 K with a microwave frequency of about 9.7 GHz, a magnetic field modulation amplitude of 0.5 mT and a modulation frequency of 100 kHz.Crystallography
Single-crystal X-ray diffraction data were collected by ω-scans using Cu-Kα radiation (λ = 1.54179 Å, a microfocus tube, and a mirror monochromator) on a Rigaku XtaLAB Synergy diffractometer at 200 and 100 K. The crystal data, experimental conditions and the final refinement parameters are summarized in Table 1 and Table S1.† Data reduction, including multiscan absorption correction, was performed with the CrysAlisPRO software package. Molecular and crystal structures were solved by direct methods using the program SIR201935 and spherical refinement was performed using the full-matrix least-squares method based on F2 with anisotropic displacement parameters for all non-hydrogen atoms (SHELXL-2014/7).36 Both programs were operated under the WinGX program package.37 In the structure at 200 K, one sodium atom and water molecules with the labels O38–O48 located near a crystallographic 2-fold rotation axis were modelled with an occupancy of 0.5. The positions of hydrogen atoms attached to carbon were found in the electron density map, but due to the complex structure with many disordered water molecules, all hydrogen atoms in the structural model were placed in idealized positions, with a C–H distance of 0.93 Å and displacement parameters were assigned as Uiso(H) = 1.2Ueq(C). The hydrogen atom that is shared by two L-tartrate ligands was also recognized in a difference map. Other hydrogen atoms of the water molecules (72 per unit cell) were not determined due to orientational disorder of some water molecules.
| Temperature/K | 200 |
| Crystal colour, habit | Red, stick |
| Empirical formula | C28H39Cr2N4NaO21 |
| M r/g mol−1 | 894.62 |
| Crystal system | Monoclinic |
| Space group | P2 |
| a/Å | 19.9810(5) |
| b/Å | 8.7003(2) |
| c/Å | 22.9085(5) |
| α/° | 90 |
| β/° | 99.220(2) |
| γ/° | 90 |
| V/Å3 | 3930.98(16) |
| Z′ | 4 |
| ρ calcd/g cm−3 | 1.512 |
| μ/mm−1 | 5.430 |
| F(000) | 1848 |
| θ range/° | 3.21–77.82 |
| Measured reflections | 29681 |
| Independent reflections | 11359 |
| Observed reflections | 10097 |
| No. of parameters, restraints | 1061, 3 |
| R int | 0.0453 |
| R, wR [I > 2σ(I)] | 0.0753, 0.2164 |
| R, wR [all data] | 0.0840, 0.2244 |
| Goodness of fit | 1.089 |
| Flack parameter | 0.006(6) |
| Δρmax, Δρmin/e Å−3 | 1.138, −0.718 |
The data collected at 100 K show that lowering the temperature has no significant effect on the thermal motion of the water molecules, which are located together with the sodium atoms in layer-like voids (35% of the unit cell, 1400 Å3). Due to the difficulties in modelling the water molecules, we additionally used the PLATON SQUEEZE tool.38 The refinement results for squeezed and un-squeezed data at 100 and 200 K are shown in Table S1.† Geometrical calculations were carried out with PLATON38 and the figures were generated using ORTEP37 and CCDC-Mercury.39 The powder X-ray diffraction (PXRD) data were collected in reflection mode with Cu-Kα1 radiation (λ = 1.54060 Å) on a Malvern PANalytical Empyrean diffractometer using a step size of 0.007° in the 2θ range between 5° and 50°.
Results and discussion
Synthesis
The chromium(III) species used as precursor for the synthesis of the new L-tartrate compound formed in almost stoichiometric yield after refluxing a DMF solution of CrCl3·6H2O and the N-donor ligand (bpy).34 Heating the as-synthesized precursor [CrCl3(DMF)(bpy)] with sodium L-tartrate in DMF–water solution leads to the dissociation of DMF and chloride anions from chromium(III) to give the title compound Na[Cr2(bpy)2(L-tart)2H]·9H2O (1), as shown in Scheme 1. The use of DMF is crucial for this reaction to occur, since it dissolves the chromium(III) precursor and is employed as a reagent for the deprotonation of sodium L-tartrate.Crystal structure
The compound Na[Cr2(bpy)2(L-tart)2H]·9H2O (1) crystallizes in the monoclinic space group P2 with four [Cr2(bpy)2(L-tart)2H]− anions in the unit cell, two of which are symmetry independent (Fig. 1). The [Cr2(bpy)2(L-tart)2H]− complex anion is composed of two chromium(III) ions that are bridged with two tartrate anions and chelated with 2,2-bipyridine molecules. The coordination sphere around chromium(III) is octahedral and the complex anion has a boat configuration. The Cr–N and Cr–O bond lengths are in the ranges 2.055–2.082 Å and 1.905–1.989 Å, which are typical of bis(bidentate) chromium(III) complexes with N and O donor atoms.30,40–42 The distances between two CrIII ions bridged by tartrate ligands in complex anions are 4.928(2) and 4.978(2) Å. The tartrate anions are almost fully deprotonated: only one hydrogen atom remains per two tartrate anions, which is located between the two hydroxyl groups of the opposite tartrates. The specific bis(bidentate) coordination mode of the tartrate anions to chromium(III) atoms enables such a configuration in which short O⋯O distances of 2.482(8) and 2.491(9) Å (two symmetrically independent anions) between the half-ionized hydroxyl groups are accomplished and further stabilized by the presence of a bridging hydrogen atom. Similar structures with an –O–H–O– bridge are reported in the CSD,20 where the O⋯O distance varies between 2.231 and 2.773 Å, with an average value of 2.442 Å. | ||
| Fig. 1 Molecular structures of two symmetrically independent dinuclear [Cr2(bpy)2(L-tart)2H]− anions present in the structure of Na[Cr2(bpy)2(L-tart)2H]·9H2O (1). Displacement ellipsoids are drawn at a probability level of 50% and hydrogen atoms are shown as spheres of arbitrary radii. | ||
Charge balance in compound 1 is accomplished with the presence of hydrated sodium ions. One sodium atom is located in a special position surrounded by two oxo-oxygen atoms of the symmetry-related [Cr2(bpy)2(L-tart)2H]− anions (Cr1, Cr2) and four water molecules. The other two symmetry-related [Cr2(bpy)2(L-tart)2H]− anions (Cr3 and Cr4) are also connected via a sodium atom, which is in orientational disorder due to its position near the crystallographic 2-fold rotation axis. One additional sodium atom is located close to the [Cr2(bpy)2(L-tart)2H]− anion (Cr3 and Cr4) stabilizing the deprotonated tartrate ligands.
The crystal structure is stabilized by stacking interactions (Fig. 2a and Table S2†). Each aryl group of the complex anion is connected to the nearest aryl group of the neighbouring complex anion, forming infinite intermolecular π-stacking across the (100) plane. The distance between two CrIII ions connected by π-stacks is 7.677 and 7.751 Å. Between the layers of π-stacked complex anions, there are a large number of water molecules (36 per unit cell) and four sodium atoms (Fig. 2b). Some water molecules located near special positions are refined with half occupancy. The water molecules together with the hydroxyl and carboxyl groups of the tartrate ligands form a rather complex hydrogen bonding network that extends in three dimensions (geometrical parameters of hydrogen bonding are given in Table S3†).
 | ||
| Fig. 2 Crystal packing feature in compound 1: (a) stacking interactions formed by [Cr2(bpy)2(L-tart)2H]− anions in the (100) plane (chromium ions are shown as green spheres); (b) blue areas represent the cavities in which water molecules and sodium ions are located (octahedra around chromium ions are shown in green). | ||
Crystal transformation
Compound 1 with a large number of water molecules is not stable at room temperature when the relative humidity is low. The TGA/DSC analysis showed that all water molecules are already eliminated at 365 K (see Fig. S3 and Table S4 in the ESI†). The complete decomposition of the organic part of the molecule ends at 680 K, leaving behind the oxide residue. In situ ATR-FTIR spectra recorded during exposure to a dry nitrogen atmosphere reveal that water molecules exit the crystal structure upon drying. This is detectable as a decrease in the intensity of ν(O–H) stretching vibrations appearing as a broad band in the range 3650–3150 cm−1 (Fig. 3a). Less pronounced but observable changes in the spectra are associated with vibrations originating from the tartrate ligands as they form hydrogen bonds with the surrounding water molecules. The positions of the bands related to the asymmetric stretching vibrations of the carbonyl group [νas(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)] in the initial phase appear at 1622 and 1600 cm−1, and in the dry phase, they shift to 1634 and 1608 cm−1, respectively.18,19,43 The symmetric stretching vibration of the carbonyl group [νs(CO)] shifts from 1353 cm−1 in the initial phase to 1348 cm−1 in the dry phase. The vibrations in the bpy molecule are less sensitive to intermolecular hydrogen bonding and remain at their original frequencies; e.g. bands at 1471, 1445 and 734 cm−1 which are related to aromatic ring deformations.43 When the dry phase is exposed to RH 60% (achieved by the presence of saturated NaBr solution) the spectrum recovers to the initial one (green line in Fig. 3a).
O)] in the initial phase appear at 1622 and 1600 cm−1, and in the dry phase, they shift to 1634 and 1608 cm−1, respectively.18,19,43 The symmetric stretching vibration of the carbonyl group [νs(CO)] shifts from 1353 cm−1 in the initial phase to 1348 cm−1 in the dry phase. The vibrations in the bpy molecule are less sensitive to intermolecular hydrogen bonding and remain at their original frequencies; e.g. bands at 1471, 1445 and 734 cm−1 which are related to aromatic ring deformations.43 When the dry phase is exposed to RH 60% (achieved by the presence of saturated NaBr solution) the spectrum recovers to the initial one (green line in Fig. 3a).
 | ||
| Fig. 3 (a) In situ humidity-dependent ATR-FTIR spectra following water loss during exposure to a dry nitrogen atmosphere; (b) PXRD patterns following the structural transformation from the initial hydrate phase 1 to the dry phase and the recovered hydrate phase. A simulated diffractogram from the single-crystal XRD data of 1 is given for comparison. | ||
The structural model obtained from the single crystal XRD data of 1 gave a satisfactory fit for Rietveld refinement of powder data (Fig. S2, ESI†). The crystals of 1 show a strong preferential orientation in the [100] direction, which is due to the mild grinding conditions under which the material is prepared for the PXRD measurement to minimize the structural transformation that occurs as soon as the material is exposed to a relative humidity of less than 60%. According to PXRD the initial high water content structure of 1 undergoes complete structural transformation during exposure to a dry nitrogen atmosphere, which is associated with water release (Fig. 3b). The transformation is reversible and the initial PXRD pattern of the hydrated phase recovers after exposure to high relative humidity levels (RH > 60%).
Proton-conducting studies
The proton conductivities of compound 1 were investigated at different RH values by AC impedance spectroscopy using pressed pellets of the powder sample with two gold electrodes sputtered onto opposite surfaces. The dependence of conductivity (σ) on RH (Fig. 4a) clearly indicates that there are two regimes of conduction. At low humidity (below RH 30%) the conductivity is rather small at 298 K and reaches 2.86 × 10−10 S cm−1. A significant increase, by four orders of magnitude to a value of 4.23 × 10−6 S cm−1, is observed under humid conditions at RH 75%. This behaviour is related to the structural transformation that occurs in this system, where two distinct phases appear to exist based on PXRD and in situ ATR-FTIR (Fig. 3), the fully hydrated phase at high RH levels and the dry phase at low RH levels. Fig. 4b and c show Nyquist plots for the dry phase measured at a RH of 28% and the hydrated phase at a RH of 75%, respectively. The corresponding Nyquist plots can be described using an equivalent circuit consisting of a resistor (R) and a constant phase element (CPE) connected in parallel. Under humid conditions the complex impedance spectrum for compound 1 can be described by two parallel equivalent circuits (R–CPE) connected in series. The semicircle at higher frequencies is usually considered as an intrinsic property of the material (grain), whereas the semicircle at low frequencies is associated with the grain boundaries as a result of the polycrystalline form of compound 1 pressed in a pellet. The reason for the more obvious contribution of the grain boundaries under humid conditions could be related to the presence of so-called ice-like and liquid-like water layers that form on the surface and grain boundaries when RH is high enough. The presence of molecular adsorbed water layers has been experimentally confirmed by spectroscopic and impedance methods on silicon oxide and ceramic materials,44,45 and recently in a metal–organic system composed of ammonium cations and a vanadium(IV)-L-tartrate complex.18 In a metal–organic system under humid conditions (RH 75%), the generation of so-called liquid-like layers of adsorbed water leads to a one order of magnitude increase in proton conductivity.18 In compound 1, due to the very large number of water molecules in the structure, it is difficult to distinguish the contributions of ice-like and liquid-like water layers by ATR-FTIR spectroscopy, but impedance spectroscopy clearly indicates their formation, which is reflected in the difference in conductivity measured at RH 60 and 75% (Fig. 4a). Similar to the vanadium(IV)-L-tartrate compound,18 in compound 1 water molecules also serve a dual purpose by both enhancing proton conduction and facilitating the structural transformation from dry to fully hydrated phases.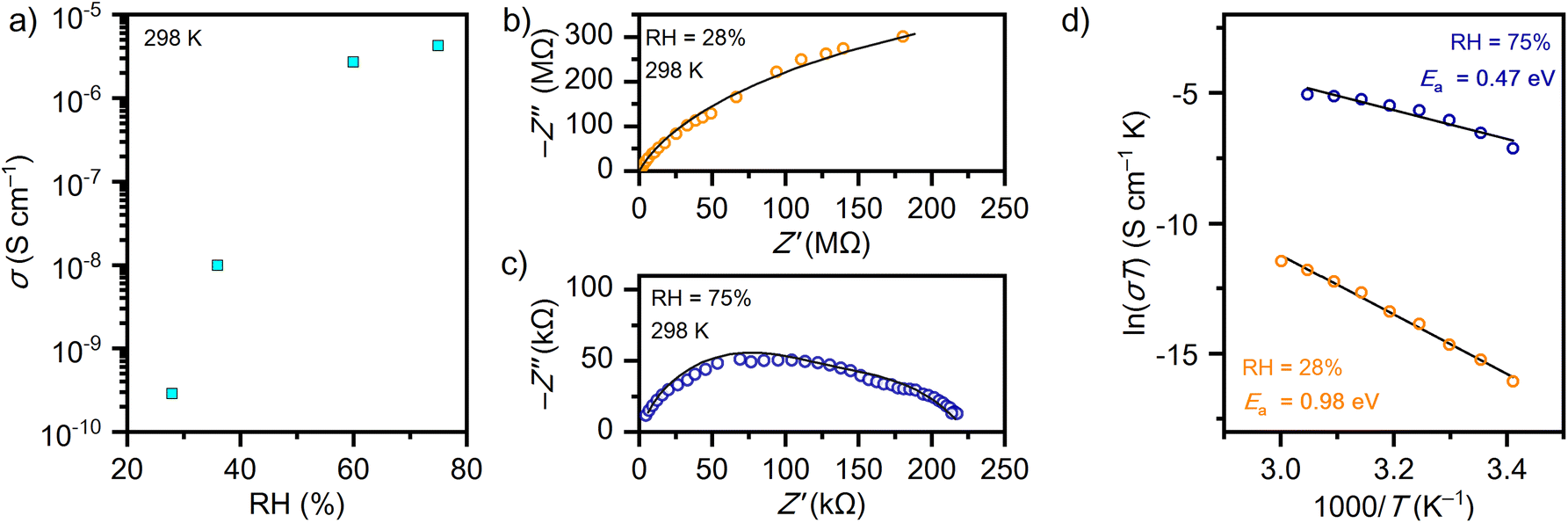 | ||
| Fig. 4 (a) Plot of conductivity vs. RH at 298 K; (b and c) complex impedance plane (Nyquist plot) recorded at different RH values at 298 K with fitted arcs (solid lines); (d) ln(σT) vs. T−1 plot with the Arrhenius fit as a solid line. | ||
The thermal dependences of conductivity were investigated under dry and wet conditions in the temperature range of 293–333 K. The activation energies (Ea) were calculated based on the Arrhenius fit for the linear part of the ln(σT) vs. T−1 plot (Fig. 4d). The calculated Ea values are 0.98 and 0.47 eV at 35 and 75% RH, respectively. The difference in the activation energies under dry and wet conditions indicates that different types of mechanisms of conduction exist. With the elimination of a considerable amount of water molecules in the dry phase, the structure of hydrogen bonds is lost and the conduction pathways are probably based on the diffusion of protonated charge carriers via the so-called vehicle mechanism.46 In contrast, when the high water level is restored in hydrated form these water molecules mediate proton transport via pathways arranged on the network of hydrogen bonds through the Grotthuss mechanism.47
Magnetic study
Compound 1 contains four octahedral chromium(III) ions in the form of two dinuclear tartrate bridged complex anions per asymmetric unit. Magnetization measurements as a function of temperature were performed to reveal the magnetic properties of the chiral dinuclear L-tartrate chromium(III) complex anion.The magnetic susceptibility calculated in the field of 1000 Oe (see Fig. 5) indicates a paramagnetic-like behaviour. According to the oxidation state of chromium and the ligand field around it the magnetic susceptibility χ was modelled supposing that the spin S = 3/2 of CrIII ions has the zero-field splitting type of single ion anisotropy with a neglected transversal term. Program PHI was used to fit the data.48 The value of 6.44 emu mol−1 Oe−1 K observed for χMT at 293 K is in good agreement with the Curie contribution of four chromium(III) ions with S = 3/2 (value calculated from the spin only formula 6.48 emu mol−1 Oe−1 K). This value decreases slowly as the temperature is lowered, reaching 3.89 cm3 mol−1 Oe−1 K at 2 K. The values obtained for the g-factor and the zero-field splitting term D, g = 1.844(1) and |D| = 3.39(5) cm−1, are in good agreement with the EPR spectra and also in very good correlation with previously determined D for chromium(III) in similar environments.49,50 Similar to the L-tartrate bridged vanadium and lanthanide complexes no exchange interactions are involved despite the short ligand metal–metal pathways present in the tartrate-bridged structures (of about 4.2 Å in the vanadium complex, 5.8 Å in the lanthanide complexes, and 4.9 Å in 1).15,51
 | ||
| Fig. 5 Temperature dependence of molar magnetic susceptibility (χM) of 1 (calculated per four Cr atoms) measured in the field of 1000 Oe. The inset shows the temperature dependence of the susceptibility and temperature product. The solid red lines represent the corresponding fitting curves. | ||
EPR measurements were performed to reveal fine changes in the local environment of CrIII ions in compound 1 under dry and wet conditions concerning RH. The X-band EPR spectra of the investigated samples, recorded at three selected temperatures of 78 K, 200 K, and 297 K, are shown in Fig. 6. The spectra obtained are characteristic of CrIII ions with spin S = 3/2. It can be seen that the spectra of 1 under dry and wet conditions are very similar, indicating that the chromium coordination spheres are not much affected by the water content in the structure. To obtain spin-Hamiltonian parameters, it is necessary to perform EPR spectroscopy in a stronger magnetic field and at a higher frequency (HF-EPR). Namely, the spectra shown in Fig. 6 are related to the allowed central transition Ms = −1/2 ↔ +1/2. To detect the other two allowed transitions −3/2 ↔ −1/2 and +1/2 ↔ +3/2, HF-EPR spectroscopy is required, as we reported previously.40,50 However, if we use the values of the g-factor and the zero-field splitting parameter D obtained from magnetization measurements, the obtained simulated spectrum (with a Lorentzian linewidth of 160 mT) is in approximately good agreement with the experimental spectra, as can be seen in Fig. 6.52
 | ||
| Fig. 6 The temperature dependence of normalized X-band EPR spectra of the initial hydrated phase 1 and its dry phase. The simulated spectrum is obtained using the values of the g-factor and the zero-field splitting parameter D obtained from magnetization measurements. | ||
Conclusions
In summary, we report the synthesis of a homochiral building block based on L-tartrate-bridged chromium(III) ions of the formula [Cr2(bpy)2(L-tart)2H]−, which can potentially be used for the design of polar crystal structures of interest for applications in molecular magnetism, nonlinear optics, ferroelectrics and other switchable dielectrics. The anion adopts a boat conformation in which a short distance between two deprotonated hydroxyl groups is stabilized by the presence of a hydrogen atom in a resonance-assisted type of intramolecular hydrogen bond. The sodium salt crystallizes in the monoclinic space group P2 as a highly hydrated compound, as is frequently observed in tartrate complexes. The water molecules of crystallization give rise to increased proton conductivity, but also limit the stability of the as-synthesized compound under humid conditions. Under dry conditions, the structure of the initial (as-synthesized) phase changes driven by water loss, and as a result, proton transfer pathways are lost, affecting the decrease in proton conductivity by four orders of magnitude. The fact that the structure of the initial phase with a high water content can be restored when exposed to humid conditions at room temperature and pressure clearly demonstrates the importance of water from the air in solid-state transformations of metal–organic compounds. According to spectroscopic (ATR-FTIR) and magnetic (EPR) measurements, the geometry and bonding topology of the anion are not significantly affected by water-assisted transformations, indicating the stability of the [Cr2(bpy)2(L-tart)2H]− anion under various conditions. The observed electrical responsiveness of these transformations to external triggers such as humidity, on the other hand, is a valuable property that can be used to develop new sensors and switchable devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Croatian Science Foundation (UIP-2019-04-7433 and IP-2022-10-9292) is gratefully acknowledged.Notes and references
- J. Bae, S. H. Park, D. Moon and N. C. Jeong, Commun. Chem., 2022, 5, 51 CrossRef CAS PubMed.
- P. Yin, Z. M. Zhang, H. Lv, T. Li, F. Haso, L. Hu, B. Zhang, J. Bacsa, Y. Wei, Y. Gao, Y. Hou, Y.-G. Li, C. L. Hill, E.-B. Wang and T. Liu, Nat. Commun., 2015, 6, 6475 CrossRef CAS PubMed.
- S. Dong, J. Leng, Y. Feng, M. Liu, C. J. Stachouse, A. Schönhals, L. Chiappisi, L. Gao, W. Chen, J. Shang, L. Jin, Z. Qi and C. A. Schalley, Sci. Adv., 2017, 3, eaao0900 CrossRef PubMed.
- S. Han, Q. Wu, J. Zhu, J. Zhang, A. Chen, S. Su, J. Liu, J. Huang, X. Yang and L. Guan, Mater. Horiz., 2023, 10, 1012–1019 RSC.
- A. Ramanthrikkovil Variyam, M. Stolov, J. Feng and N. Amdursky, ACS Nano, 2024, 18(6), 5101–5112 CrossRef CAS PubMed.
- K. Otake, K. Otsubo, T. Komatsu, S. Dekura, J. M. Taylor, R. Ikeda, K. Sugimoto, A. Fujiwara, C.-P. Chou, A. Wibawa Sakti, Y. Nishimura, H. Nakai and H. Kitagawa, Nat. Commun., 2020, 11, 843 CrossRef CAS PubMed.
- J. Aríñez-Soriano, J. Albalad, C. Vila-Parrondo, J. Pérez-Carvajal, S. Rodríguez-Hermida, A. Cabeza, J. Juanhuix, I. Imaz and D. Maspoch, Chem. Commun., 2016, 52, 7229–7232 RSC.
- X. Liu, H. Gao, L. Sun and J. Yao, Adv. Mater., 2023, 2300748 Search PubMed.
- Y. Gao, R. Broersen, W. Hageman, N. Yan, M. C. Mittelmeijer-Hazeleger, G. Rothenberg and S. Tanase, J. Mater. Chem. A, 2015, 3, 22347–22352 RSC.
- A. Martinelli, J. M. Otero-Mato, M. N. Garaga, K. Elamin, S. M. Habibur Rahman, J. W. Zwanziger, U. Werner-Zwanziger and L. Varela, J. Am. Chem. Soc., 2021, 143, 13895–13907 CrossRef CAS PubMed.
- M.-J. Wei, J.-Q. Fu, Y.-D. Wang, Y. Zhang, H.-Y. Zang, K.-Z. Shao, Y.-G. Li and Z.-M. Su, CrystEngComm, 2017, 19, 7050–7056 RSC.
- D.-W. Lim, M. Sadakiyo and H. Kitagawa, Chem. Sci., 2019, 10, 16 RSC.
- S. Liu, M. Liu, X. Li, Q. Xu, Y. Sun and G. Zeng, J. Mater. Chem. A, 2023, 11, 13965–13970 RSC.
- Y.-H. Liu, S.-H. Lee, J.-C. Chiang, P.-C. Chen, P.-H. Chien and C.-I. Yang, Dalton Trans., 2013, 42, 16857–16867 RSC.
- U. Huizi-Rayo, A. Zabala-Lekuona, A. Terenzi, C. M. Cruz, J. M. Cuerva, A. Rodríguez-Diéguez, J. Angel García, J. M. Seco, E. San Sebastian and J. Cepeda, J. Mater. Chem. C, 2020, 8, 8243–8256 RSC.
- K. Ohno, Y. Kusano, S. Kaizaki, A. Nagasawa and T. Fujihara, Inorg. Chem., 2018, 57, 14159–14169 CrossRef CAS PubMed.
- M. Dunatov, A. Puškarić and L. Androš Dubraja, J. Mater. Chem. C, 2023, 11, 2880–2888 RSC.
- M. Dunatov, K. Molčanov, Z. Štefanić, R. Kruk and L. Androš Dubraja, Inorg. Chem., 2024, 63, 163–172 CrossRef CAS PubMed.
- G. L. Robbins and R. E. Tapscott, Inorg. Chem., 1976, 15, 155–159 Search PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, B72, 171–179 CrossRef PubMed.
- X.-F. Zhang, D.-G. Huang, C. Feng, C.-N. Chen, Q.-T. Liu and L.-C. Sun, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, C59, m402–m404 CrossRef CAS PubMed.
- S. C. Manna, E. Zangrando, J. Ribas and N. R. Chaudhuri, Eur. J. Inorg. Chem., 2007, 4592–4595 CrossRef CAS.
- M. McCann, F. Humphreys and V. McKee, Polyhedron, 1997, 16, 3655–3661 CrossRef CAS.
- G.-Y. Dong, C.-H. He, T.-F. Liu, G.-H. Cuia and X.-C. Deng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, E67, m1005–m1006 CrossRef PubMed.
- J. Lu, J.-H. Yu, X.-Y. Chen, P. Cheng, X. Zhang and J.-Q. Xu, Inorg. Chem., 2005, 44(17), 5978–5980 CrossRef CAS PubMed.
- R. Saha, S. Biswasa and G. Mostafa, CrystEngComm, 2011, 13, 1018–1028 RSC.
- K. Ohno, H. Tanuma, Y. Kusano, S. Kaizaki, A. Nagasawaa and T. Fujihara, Dalton Trans., 2017, 46, 7612–7618 RSC.
- K. Ohno, T. Sugaya, M. Kato, N. Matsumoto, R. Fukano, Y. Ogino, S. Kaizaki, T. Fujihara and A. Nagasawa, Cryst. Growth Des., 2014, 14(8), 3675–3679 CrossRef CAS.
- S. Kaizaki, J. Hidaka and Y. Shimura, Bull. Chem. Soc. Jpn., 1969, 42, 988–994 CrossRef CAS.
- R. B. Ortega, R. E. Tapscott and C. F. Campana, Inorg. Chem., 1982, 21, 2517–2519 CrossRef CAS.
- L. A. Malaspina, A. A. Hoser, A. J. Edwards, M. Woińska, M. J. Turner, J. R. Price, K. Sugimoto, E. Nishibori, H.-B. Bürgi, D. Jayatilaka and S. Grabowsky, CrystEngComm, 2020, 22, 4778–4789 RSC.
- L. A. Malaspina, A. J. Edwards, M. Woińska, D. Jayatilaka, M. J. Turner, J. R. Price, R. Herbst-Irmer, K. Sugimoto, E. Nishibori and S. Grabowsky, Cryst. Growth Des., 2017, 17, 3812–3825 CrossRef CAS.
- B. Dereka, Q. Yu, N. H. C. Lewis, W. B. Carpenter, J. M. Bowman and A. Tokmakoff, Science, 2021, 371, 160–164 CrossRef CAS PubMed.
- J. Broomhead and F. Dwyer, Aust. J. Chem., 1961, 14(2), 250–252 CAS.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef PubMed.
- F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef.
- L. Androš Dubraja, M. Jurić, J. Popović, D. Pajić, Y. Krupskaya, V. Kataev, B. Büchner and D. Žilić, Dalton Trans., 2018, 47, 3992–4000 RSC.
- L. Androš Dubraja, M. Jurić, F. Torić and D. Pajić, Dalton Trans., 2017, 46, 11748–11756 RSC.
- L. Androš, M. Jurić, K. Molčanov and P. Planinić, Dalton Trans., 2012, 41, 14611–14624 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley, New York, 6th edn, 2009 Search PubMed.
- D. B. Asay and S. H. Kim, J. Phys. Chem. B, 2005, 109, 16760–16763 CrossRef CAS PubMed.
- S. Ø. Stub, E. Vøllestad and T. Norby, J. Phys. Chem. C, 2017, 121, 12817–12825 CrossRef CAS.
- K. Hirata, K. Akasaka, O. Dopfer, S. Ishiuchi and M. Fujii, Chem. Sci., 2024, 15, 2725–2730 RSC.
- I. Popov, Z. Zhu, A. R. Young-Gonzales, R. L. Sacci, E. Mamontov, C. Gainaru, S. J. Paddison and A. P. Sokolov, Commun. Chem., 2023, 6, 77 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- L. Androš Dubraja, M. Jurić, W. Lafargue-Dit-Hauret, D. Pajić, A. Zorko, A. Ozarowski and X. Rocquefelte, New J. Chem., 2018, 42, 10912–10921 RSC.
- D. Žilić, L. Androš, Y. Krupskaya, V. Kataev and B. Büchner, Appl. Magn. Reson., 2015, 46, 309–321 CrossRef.
- J. Garcia-Jaca, M. Insausti, R. Cortes, T. Rojo, J. L. Pizarro and M. I. Arriortua, Polyhedron, 1994, 13, 357–364 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Qualitative elemental EDX analysis (Fig. S1), PXRD data (Fig. S2), crystallographic details and geometry (Tables S1–S3), and TG/DTA results (Fig. S3 and Table S4). CCDC 2336524, 2351123 and 2351124. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00783b |
| This journal is © The Royal Society of Chemistry 2024 |