
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of Schiff base-[C,N,S] pincer palladacycles: hydrolysis renders singular trinuclear, tetranuclear, and heteropentanuclear Pd3W2 coordinated complexes†
Francisco
Reigosa‡
 ,
Paula M.
Polo‡
,
M. Teresa
Pereira
and
José M.
Vila
*
,
Paula M.
Polo‡
,
M. Teresa
Pereira
and
José M.
Vila
*
Departamento de Química Inorgánica, Universidad de Santiago de Compostela, E-15782 Santiago de Compostela, Spain. E-mail: josemanuel.vila@usc.es
First published on 19th April 2024
Abstract
Treatment of the Schiff base ligands a–f with Li2[PdCl4]/NaAcO in methanol under reflux gave the single nuclear palladacycles 1a–1f, with the metal atom bonded to a terdentate monoanionic [C,N,S] iminic ligand and to a chloride ligand that completes the palladium coordination sphere. Reaction of 1a–1c with silver perchlorate/triphenylphosphine in acetone at room temperature yielded the single nuclear complexes 2a–2c as the perchlorate salts, after substitution of the chloride ligand by a triphenylphosphine. However, reaction of a–c with Na2[PdCl4]/NaAcO in methanol at room temperature also gave compounds 1a–1c albeit contaminated with small amounts of the corresponding free aldehyde (mixture A). Reaction of mixture A with silver perchlorate/triphenylphosphine in acetone at room temperature gave analogously 2a–2c with some of the corresponding free aldehyde (mixture B). Attempts to purify mixtures A and B via recrystallization produced single crystals of 5 and 6 respectively: two serendipitously formed complexes, bearing thiomethyl aniline and/or acetate ligands, and void of aldehyde or iminic residue; the structures contain eight- and six-membered rings of alternating palladium and nitrogen atoms, respectively. To clarify this situation the aniline itself was reacted with palladium(II) acetate or with Na2[PdCl4]; in the latter case after recrystallization a unique behavior is revealed, giving rise to a tetranuclear complex containing a Pd4N4 ring with three differing coordination environments on the palladium atoms. Treatment of 1d with Ph2PCH2PPh2 (dppm)/AgClO4 or with Ph2PCH2(PPh2)W(CO)5/AgClO4 gave 3d, with a mono-coordinated dppm ligand, and 4d, respectively; complex 3d could not be converted into 4d by reaction with W(CO)5(THF). Recrystallization of 4d gave a still further noticeable species, complex 8: a pentanuclear trans-configured heterometallic mixed valent Pd(II)/W(0) linear complex with the palladium atoms supported by two acetate and two thiomethyl aniline bridging ligands. The complexes were fully characterized by microanalysis, IR, 1H, and 31P NMR spectroscopies, as appropriate. The X-ray single-crystal analyses for compounds 1b, 5, 6, 7 and 8 are described.
Introduction
The chemistry of palladacycles,1,2 first reported by Cope and Siekman,3 constitutes a flourishing part of organometallics that has attracted much research interest in past years attributable to a great extent to their versatile structural and reactivity features, mainly derived from the fact that their properties can be easily tuned, for example by modification of the cyclometallated ligand or the ancillary ligands at the metal. They are also well known for their broad applications4 in numerous fields such as in organic synthesis,5,6 photochemistry,7,8 optical resolution processes,9 and catalysis, which after the pioneering work by Herrmann et al.,10,11 continues to be researched,12–21 as potential biologically active materials,22–31 and liquid crystals.32–34In the past we have shown that thiosemicarbazones yield palladacycles with the ligand in a terdentate [C,N,S] fashion35 with formation of tetranuclear compounds possessing two distinct palladium–sulfur bonds, i.e., Pd–Schelating and Pd–Sbridging, with the ligand in a pincer mode; Kawamoto et al.36 have reported similar structures for Schiff base [C,N,S] Pd(II) and Pt(II) complexes. This results in three strong bonds at the metal, namely the Pd–C, Pd–N and Pd–Schelating bonds pertaining to two fused five-membered rings, that maintain the metal tightly bonded so that only one of the four coordination sites in the square-planar palladium environment is capable of further reaction. Earlier results by this laboratory also put forward that the said complexes react with small bite strong chelating diphosphines to give species with an uncoordinated phosphorus donor atom,37 thus performing as metalloligands, which were able to coordinate to a second metal atom, providing homo- and heterobimetallics. We then sought out to look for analogous systems that would retain the excellent pincer properties of the terdentate [C,N,S] ligands and for this purpose Schiff bases derived from 2-thiomethylaniline were chosen; this changes the thiosemicarbazone sequence –C(Me)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–N(H)–C(NHR)–S to the corresponding Schiff base one –C(H)N–CC–SMe. We reasoned that these related systems bearing analogous backbones of nitrogen and sulfur donors would behave likewise and even in the absence of the thiolate sulfur the non-thiolate thiomethyl group would bind well in agreement with Pearson's concept.38 The preparation of [C,N,S] metallacycles can be achieved by quite numerous routes such as reactions with lithium derivatives,39 oxidative addition and photooxidation,40 and direct orthometallation for which Pd(DMSO)2Cl2,41 Pd(OAc)2, Li2[PdCl4] or Na2[PdCl4] are amongst the more common reagents. The lithium salt has proven to be a quite useful palladium source for the synthesis of palladacycles, and was used in the present preparations, albeit requires prior synthesis of the salt using moisture-sensitive reagents; therefore, the commercially available sodium salt was also used to compare results and to reduce the synthetic steps. Thus, the latter two were chosen and although the initial steps seemed to agree with the expectations the ensuing iminic metalloligands and the corresponding bimetallics similar to those mentioned above, could not be prepared from the sodium salt, drifting the process to a different route: we tentatively attribute this to partial hydrolysis of the iminic ligand. The spontaneous decay of the products yielded unprecedented rearrangements producing tri- and tetranuclear homometallic complexes, with the methylthioaniline precursor simultaneously acting as a chelating and bridging ligand, leading to new insights into their chemistry. This led us to study the reactivity of the 2-thiomethylaniline itself to test our hypothesis. Furthermore, the bimetallic palladium/tungsten also decayed on recrystallization to yield a pentanuclear heterometallic complex. A more recent example related to spontaneous palladacycle disruption has been reported by us related to the Schiff base palladacycles: an innovate structural rearrangement from single-nuclear to double-nuclear pseudo-pentacoordinated complexes, that we dare say is even more surprising since it could be termed as a special case of self-cyclopalladation.42 We tentatively coin these processes as typical cases of serendipity in palladacycle chemistry, which on the other hand put forward novel and exciting reactivity patterns yet to be accounted for, developing new substances that may be of use in any of the fields of application of these compounds.
N–N(H)–C(NHR)–S to the corresponding Schiff base one –C(H)N–CC–SMe. We reasoned that these related systems bearing analogous backbones of nitrogen and sulfur donors would behave likewise and even in the absence of the thiolate sulfur the non-thiolate thiomethyl group would bind well in agreement with Pearson's concept.38 The preparation of [C,N,S] metallacycles can be achieved by quite numerous routes such as reactions with lithium derivatives,39 oxidative addition and photooxidation,40 and direct orthometallation for which Pd(DMSO)2Cl2,41 Pd(OAc)2, Li2[PdCl4] or Na2[PdCl4] are amongst the more common reagents. The lithium salt has proven to be a quite useful palladium source for the synthesis of palladacycles, and was used in the present preparations, albeit requires prior synthesis of the salt using moisture-sensitive reagents; therefore, the commercially available sodium salt was also used to compare results and to reduce the synthetic steps. Thus, the latter two were chosen and although the initial steps seemed to agree with the expectations the ensuing iminic metalloligands and the corresponding bimetallics similar to those mentioned above, could not be prepared from the sodium salt, drifting the process to a different route: we tentatively attribute this to partial hydrolysis of the iminic ligand. The spontaneous decay of the products yielded unprecedented rearrangements producing tri- and tetranuclear homometallic complexes, with the methylthioaniline precursor simultaneously acting as a chelating and bridging ligand, leading to new insights into their chemistry. This led us to study the reactivity of the 2-thiomethylaniline itself to test our hypothesis. Furthermore, the bimetallic palladium/tungsten also decayed on recrystallization to yield a pentanuclear heterometallic complex. A more recent example related to spontaneous palladacycle disruption has been reported by us related to the Schiff base palladacycles: an innovate structural rearrangement from single-nuclear to double-nuclear pseudo-pentacoordinated complexes, that we dare say is even more surprising since it could be termed as a special case of self-cyclopalladation.42 We tentatively coin these processes as typical cases of serendipity in palladacycle chemistry, which on the other hand put forward novel and exciting reactivity patterns yet to be accounted for, developing new substances that may be of use in any of the fields of application of these compounds.
In this work we report palladacycles bearing tridentate [C,N,S] Schiff base ligands and differing palladium salts, together with the corresponding reactions with mono- and diphosphines, leading to the discovery of new multinuclear complexes.
Experimental section
General procedures
Solvents were purified by standard methods. Chemicals (lithium chloride, palladium(II) chloride), the phosphines PPh3 and Ph2PCH2PPh2 (dppm), aldehydes and 2-thiomethyl aniline were used as supplied from commercial sources. Lithium tetrachloropalladate was made in situ by treating palladium(II) chloride with lithium chloride in methanol. Ph2PCH2P(Ph2)W(CO)5 was prepared by literature synthesis. Microanalyses were carried out at the Servicio de Análisis Elemental at the University of Santiago using a FISONS elemental analyzer, Model 1108. IR spectra were recorded as KBr pellets or polythene discs on BRUKER Model IFS-66v and IR-FT Mattson Model Cygnus-100 spectrophotometers, and on a JASCO FT/IR-4600 spectrometer equipped with an ATR, model ATR-PRO ONE. NMR spectra were obtained as CDCl3, DMSO-d6 or Me2CO-d6 solutions as appropriate and referenced to SiMe4 (1H) or 85% H3PO4 (31P–{1H}) and were recorded on BRUKER DPX 250 and Varian Inova 400 spectrometers. All chemical shifts, in ppm, were reported downfield from the standards.Syntheses
Preparation of the ligands
The corresponding aldehyde and 2-thiomethyl aniline were added together in chloroform (ca. 30 cm3) in a round-bottomed flask to give a pale-yellow solution which was refluxed in a modified Dean–Stark apparatus under dry nitrogen for 24 h, after which the resulting yellow to dark-yellow solution was cooled to room temperature and the solvent removed under reduced pressure.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) N[2-(SMe)C6H4] a.
Yellow solid. Yield: 87%. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H, HCN), 7.66 (s, 1H, H2), 7.47 (dd, 3J = 8.3 Hz, 4J = 1.9 Hz, 1H, H6), 7.24 (td, 3J = 7.3 Hz, 4J = 1.7 Hz, 1H, H10), 7.19 (d, 3J = 7.3 Hz, 1H. H11), 7.15 (td, 3J = 7.3 Hz, 4J = 1.7 Hz, 1H, H9), 7.08 (dd, 3J = 8.3 Hz, 4J = 2.0 Hz, 1H, H5), 7.04 (dd, 3J = 7.6 Hz, 4J = 1.7 Hz, 1H, H8), 3.89 (s, 6H, OMe), 2.42 (s, 3H, SMe). IR cm−1ν(CN) 1618. Anal. found: C, 67.1; H, 5.9; N, 5.0; S, 10.9%, C16H17NO2S (287.38 g mol−1) requires C, 66.9; H, 6.0; N, 4.9; S, 11.2%.
N[2-(SMe)C6H4] b.
Yellow solid. Yield: 92%. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H, HCN), 7.82 (d, 4J = 2.1 Hz, 1H, H2), 7.77 (dd, 3J = 8.4 Hz, 4J = 2.3 Hz, 1H, H6), 7.23 (td, 3J = 7.4 Hz, 4J = 1.7 Hz, 1H, H10), 7.18 (dd, 3J = 7.7 Hz, 4J = 1.7 Hz, 1H, H11), 7.15 (td, 3J = 7.4 Hz, 4J = 1.8 Hz, 1H), 7.06 (d, 3J = 8.4 Hz, 1H, H5), 7.03 (dd, 3J = 7.5 Hz, 4J = 1.6 Hz, 1H, H8), 3.93 (s, 3H, OMe), 2.42 (s, 3H, SMe), 2.25 (s, 3H, Me). IR cm−1ν(CN) 1625. Anal. found: C, 70.5; H, 6.2; N, 5.2; S, 11.9%, C16H17NOS (271.38 g mol−1) requires C, 70.8; H, 6.3; N, 5.2; S, 11.8%.
N[2-(SMe)C6H4] c.
Orange oil. Yield: 88%. 1H NMR (400 MHz, acetone-d6) δ 8.38 (s, 1H, HCN), 7.52 (s, 1H, H2), 7.46 (d, 3J = 8.7 Hz, 1H, H6), 7.23 (m, 2H, H9/H11), 7.18–7.13 (m, 1H), 7.04 (d, 3J = 8.2 Hz, 1H, H8), 6.96 (d, 3J = 8.3 Hz, 1H, H5), 4.34 (d, 3J = 5.4 Hz, 4H, CH2), 2.42 (s, 3H, SMe). IR cm−1ν(CN) 1621. Anal. found: C, 67.1; H, 5.3; N, 4.7; S, 11.5%, C16H15NO2S (285.36 g mol−1) requires C, 67.3; H, 5.3; N, 4.9; S, 11.2%.
N[2-(SMe)C6H4] d.
Yellow oil. Yield 85%. 1H NMR (250 MHz, DMSO-d6) δ 8.44 (s, 1H, HCN), 7.88 (vd, N = 8.0 Hz, 2H, H2, H6), 7.10–7.30 (m, 4H, H9, H10, H11, H12), 7.07 (vd, N = 8.0 Hz, 2H, H3, H5), 3.82 (s, 3H, OMe), 2.37 (s, 3H, SMe). IR cm−1ν(CN) 1626. Anal. found: C, 70.1; H, 6.0; N, 5.7; S, 12.3%, C15H15NOS (257.35 g mol−1) requires C, 70.0; H, 5.9; N, 5.4; S, 12.5%.
N[2-(SMe)C6H4] e.
Yellow oil. Yield 82%. 1H NMR (250 MHz, CDCl3) δ 8.73 (s, 1H, HCN), 8.19 (d, 3J(H6H5) = 8.7 Hz, 1H, H6), 6.93–7.17 (m, 4H, H9, H10, H11, H12), 6.56 (dd, 3J(H5H6) = 8.7 Hz, 4J(H5H3) = 2.3 Hz, 1H, H5), 6.43 (d, 4J(H3H5) = 2.3 Hz, 1H, H3), 3.84 (s, 3H, OMe), 3.82 (s, 3H, OMe), 2.42 (s, 3H, SMe). IR cm−1ν(CN) 1601. Anal. found: C, 67.0; H, 5.9; N, 5.0; S, 11.0%, C16H17NO2S (287.38 g mol−1) requires C, 66.8; H, 6.0; N, 4.9; S, 11.2%.
N[2-(SMe)C6H4] f.
Yellow oil. Yield 98%. 1H NMR (250 MHz, CDCl3): δ 8.65 (s, 1H, HCN), 7.97 (d, 3J(H6H5) = 8.8 Hz, 1H, H6), 6.70–7.18 (m, 4H, H9, H10, H11, H12), 6.96 (d, 3J(H5H6) = 8.8 Hz, 1H, H5), 3.94 (s, 3H, OMe), 3.90 (s, 3H, OMe), 3.87 (s, 3H, OMe), 2.43 (s, 3H, SMe). IR cm−1ν(CN) 1623. Anal. found: C, 64.1; H, 6.0; N, 4.5; S, 10.2%, C17H19NO3S (317.40 g mol−1) requires C, 64.3; H, 6.0; N, 4.4; S, 10.1%.
N[2-(SMe)C6H4] a.
Yellow solid. Yield: 87%. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H, HCN), 7.66 (s, 1H, H2), 7.47 (dd, 3J = 8.3 Hz, 4J = 1.9 Hz, 1H, H6), 7.24 (td, 3J = 7.3 Hz, 4J = 1.7 Hz, 1H, H10), 7.19 (d, 3J = 7.3 Hz, 1H. H11), 7.15 (td, 3J = 7.3 Hz, 4J = 1.7 Hz, 1H, H9), 7.08 (dd, 3J = 8.3 Hz, 4J = 2.0 Hz, 1H, H5), 7.04 (dd, 3J = 7.6 Hz, 4J = 1.7 Hz, 1H, H8), 3.89 (s, 6H, OMe), 2.42 (s, 3H, SMe). IR cm−1ν(CN) 1618. Anal. found: C, 67.1; H, 5.9; N, 5.0; S, 10.9%, C16H17NO2S (287.38 g mol−1) requires C, 66.9; H, 6.0; N, 4.9; S, 11.2%.
N[2-(SMe)C6H4] b.
Yellow solid. Yield: 92%. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H, HCN), 7.82 (d, 4J = 2.1 Hz, 1H, H2), 7.77 (dd, 3J = 8.4 Hz, 4J = 2.3 Hz, 1H, H6), 7.23 (td, 3J = 7.4 Hz, 4J = 1.7 Hz, 1H, H10), 7.18 (dd, 3J = 7.7 Hz, 4J = 1.7 Hz, 1H, H11), 7.15 (td, 3J = 7.4 Hz, 4J = 1.8 Hz, 1H), 7.06 (d, 3J = 8.4 Hz, 1H, H5), 7.03 (dd, 3J = 7.5 Hz, 4J = 1.6 Hz, 1H, H8), 3.93 (s, 3H, OMe), 2.42 (s, 3H, SMe), 2.25 (s, 3H, Me). IR cm−1ν(CN) 1625. Anal. found: C, 70.5; H, 6.2; N, 5.2; S, 11.9%, C16H17NOS (271.38 g mol−1) requires C, 70.8; H, 6.3; N, 5.2; S, 11.8%.
N[2-(SMe)C6H4] c.
Orange oil. Yield: 88%. 1H NMR (400 MHz, acetone-d6) δ 8.38 (s, 1H, HCN), 7.52 (s, 1H, H2), 7.46 (d, 3J = 8.7 Hz, 1H, H6), 7.23 (m, 2H, H9/H11), 7.18–7.13 (m, 1H), 7.04 (d, 3J = 8.2 Hz, 1H, H8), 6.96 (d, 3J = 8.3 Hz, 1H, H5), 4.34 (d, 3J = 5.4 Hz, 4H, CH2), 2.42 (s, 3H, SMe). IR cm−1ν(CN) 1621. Anal. found: C, 67.1; H, 5.3; N, 4.7; S, 11.5%, C16H15NO2S (285.36 g mol−1) requires C, 67.3; H, 5.3; N, 4.9; S, 11.2%.
N[2-(SMe)C6H4] d.
Yellow oil. Yield 85%. 1H NMR (250 MHz, DMSO-d6) δ 8.44 (s, 1H, HCN), 7.88 (vd, N = 8.0 Hz, 2H, H2, H6), 7.10–7.30 (m, 4H, H9, H10, H11, H12), 7.07 (vd, N = 8.0 Hz, 2H, H3, H5), 3.82 (s, 3H, OMe), 2.37 (s, 3H, SMe). IR cm−1ν(CN) 1626. Anal. found: C, 70.1; H, 6.0; N, 5.7; S, 12.3%, C15H15NOS (257.35 g mol−1) requires C, 70.0; H, 5.9; N, 5.4; S, 12.5%.
N[2-(SMe)C6H4] e.
Yellow oil. Yield 82%. 1H NMR (250 MHz, CDCl3) δ 8.73 (s, 1H, HCN), 8.19 (d, 3J(H6H5) = 8.7 Hz, 1H, H6), 6.93–7.17 (m, 4H, H9, H10, H11, H12), 6.56 (dd, 3J(H5H6) = 8.7 Hz, 4J(H5H3) = 2.3 Hz, 1H, H5), 6.43 (d, 4J(H3H5) = 2.3 Hz, 1H, H3), 3.84 (s, 3H, OMe), 3.82 (s, 3H, OMe), 2.42 (s, 3H, SMe). IR cm−1ν(CN) 1601. Anal. found: C, 67.0; H, 5.9; N, 5.0; S, 11.0%, C16H17NO2S (287.38 g mol−1) requires C, 66.8; H, 6.0; N, 4.9; S, 11.2%.
N[2-(SMe)C6H4] f.
Yellow oil. Yield 98%. 1H NMR (250 MHz, CDCl3): δ 8.65 (s, 1H, HCN), 7.97 (d, 3J(H6H5) = 8.8 Hz, 1H, H6), 6.70–7.18 (m, 4H, H9, H10, H11, H12), 6.96 (d, 3J(H5H6) = 8.8 Hz, 1H, H5), 3.94 (s, 3H, OMe), 3.90 (s, 3H, OMe), 3.87 (s, 3H, OMe), 2.43 (s, 3H, SMe). IR cm−1ν(CN) 1623. Anal. found: C, 64.1; H, 6.0; N, 4.5; S, 10.2%, C17H19NO3S (317.40 g mol−1) requires C, 64.3; H, 6.0; N, 4.4; S, 10.1%.
Preparation of the chloride compounds
Route a: Palladium chloride and lithium chloride were added together in oxygen-free methanol in a carousel flask under argon. The mixture was stirred at room temperature until a reddish color appeared. Then, the ligand was added and a color change was observed, followed by turbidity and by the formation of a solid within the solution. The reaction mixture was heated to 70 °C for 4 h and one equivalent of sodium acetate was added, which produced the instantaneous formation of an orange solid that was separated by centrifugation.Route b: In a 100 mL round bottom flask the ligand was dissolved in methanol with stirring and at room temperature. Then, sodium tetrachloropalladate was added, followed by sodium acetate, and upon further stirring the formation of an orange precipitate was observed. The solid was separated by filtration, washed and dried under vacuum.
Route c (for 1d, 1e, 1f): To a stirred dark-red solution of lithium tetrachloropalladate(II) in methanol the ligand, d, e, f, was added and the mixture was refluxed for 1 h under dry dinitrogen. After cooling to room temperature sodium acetate was added to the resulting solution. The solid formed was filtered off, washed with ethanol and air-dried.
N[2-(SMe)C6H4]}(Cl)] 1a.
Orange solid. Yield: 58%. 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H, HCN), 7.60 (d, 3J = 8.0 Hz, 1H, H11), 7.57 (t, 3J = 8.0 Hz, 1H, H10), 7.46 (s, 1H, H2), 7.45 (td, 3J = 8.4 Hz, 4J = 1.4 Hz, 1H, H9), 7.40–7.33 (m, 1H), 7.02 (s, 1H, H5), 4.01 (s, 3H, OMe), 3.86 (s, 3H, OMe), 2.81 (s, 3H, SMe). IR cm−1ν(CN) 1579; ν(Pd–Cl) 322. Anal. found: C, 45.1; H, 3.8; N, 3.5; S, 7.3%, C16H16ClNO2PdS (428.24 g mol−1) requires C, 44.9; H, 3.8; N, 3.3; S, 7.5%.
N[2-(SMe)C6H4]}(Cl)] 1b.
Orange solid. Yield: 63%. 1H NMR (400 MHz, CDCl3) δ 8.47 (s, 1H, HCN), 7.60 (dd, 3J = 8.5 Hz, 4J = 1.2 Hz, 1H), 7.57 (dd, 3J = 7.8 Hz, 4J = 1.5 Hz, 1H), 7.44 (td, 3J = 8.4 Hz, 4J = 1.5 Hz, 1H), 7.39 (s, 1H, H2), 7.38–7.29 (m, 2H), 3.96 (s, 3H, OMe), 2.81 (s, 3H, SMe), 2.14 (s, 3H, Me). IR cm−1ν(CN) 1581; ν(Pd–Cl) 329. Anal. found: C, 46.7; H, 3.9; N, 3.6; S, 7.9%, C16H16ClNOPdS (412.24 g mol−1) requires C, 46.6; H, 3.9; N, 3.4; S, 7.8%.
N[2-(SMe)C6H4]}(Cl)] 1c.
Orange solid. Yield: 74%. 1H NMR (400 MHz, acetone-d6) δ 9.01 (s, 1H, HCN), 7.99 (d, 3J = 8.4 Hz, 1H, H11), 7.81 (d, 3J = 7.8 Hz, 1H, H8), 7.58 (t, 3J = 7.4 Hz, 3H, H10), 7.48 (t, 3J = 8.0 Hz, 1H, H9), 7.21 (s, 1H, H2), 7.14 (s, 1H, H5), 7.34 (d, J = 4.1 Hz, 2H, CH2), 4.27 (s, 2H, CH2), 2.82 (d, J = 1.6 Hz, 3H, SMe). IR cm−1ν(CN) 1581; ν(Pd–Cl) 327. Anal. found: C, 44.8; H, 3.2; N, 3.1; S, 7.2%, C16H14ClNO2PdS (426.22 g mol−1) requires C, 45.1; H, 3.3; N, 3.3; S, 7.5%.
N[2-(SMe)C6H4]}(Cl)] 1d.
Yellow solid. Yield 83%. 1H NMR (250 MHz, DMSO-d6): δ 9.12 (s, 1H, HCN), 7.98 (d, 3J(H11H12) = 8.4 Hz, 1H, H12), 7.83 (d, 3J(H9H10) = 7.9 Hz, 1H, H9), 7.55 (t, 3J(H10H11) = 8.0 Hz, 1H, H10), 7.53 (d, 3J(H2H3) = 8.8 Hz, 1H, H2), 7.44 (t, 1H, H11), 7.11 (d, 4J(H5H3) = 2.7 Hz, 1H, H5), 6.71 (dd, 1H, H3), 3.80 (s, 3H, OCH3), 2.75 (s, 3H, SCH3). IR 1603 ν(CN), 315 ν(Pd–Cltrans-N) cm−1. Anal. found: C, 45.0; H, 3.4; N, 3.4; S, 8.0%, C15H14ClNOPdS (398.22 g mol−1) requires C, 45.2; H, 3.5; N, 3.5; S, 8.1%.
N[2-(SMe)C6H4]}(Cl)] 1e.
Orange solid. Yield 77%. 1H NMR (250 MHz, CDCl3)δ 8.77 (s, 1H, HCN), 7.61 (d, 3J(H11H12) = 8.2 Hz, 1H, H12), 7.51 (d, 3J(H9H10) = 7.8 Hz, 1H, H9), 7.39 (t, 1H, H10), 7.27 (t, 1H, H11), 7.03 (d, 4J(H5H3) = 2.1 Hz, 1H, H5), 6.06 (d, 4J(H3H5) = 2.1 Hz, 1H, H3), 3.89 (s, 3H, OMe), 3.80 (s, 3H, OMe), 2.77 (s, 3H, SMe). IR 1585 ν(CN), 332 ν(Pd–Cltrans-N) cm−1. Anal. found: C, 45.1; H, 3.8; N, 3.2; S, 7.4%, C16H16ClNO2PdS (428.24 g mol−1) requires C, 44.9; H, 3.8; N, 3.3; S, 7.5%.
N[2-(SMe)C6H4]}(Cl)] 1f.
Yellow solid. Yield 82%. 1H NMR (250 MHz, CDCl3) δ 8.73 (s, 1H, HCN), 7.64 (d, 3J(H12H11) = 8.3 Hz, 1H, H12), 7.54 (d, 3J(H9H10) = 7.7 Hz, 1H, H9), 7.42 (t, 3J(H10H11) = 7.2 Hz, 1H, H10), 7.15 (s, 1H, H5); 7.30 (t, 1H, H11), 4.02 (s, 3H, OMe), 3.95 (s, 3H, OMe), 3.77 (s, 3H, OMe), 2.76 (s, 3H, SMe). IR 1606 ν(CN), 333 ν(Pd–Cltrans-N) cm−1. Anal. found: C, 44.5; H, 3.9; N, 3.2; S, 7.1%, C17H18ClNO3PdS (458.27 g mol−1) requires C, 44.6; H, 4.0; N, 3.1; S, 7.0%.
Preparation of the triphenylphosphine palladacycles
The appropriate palladacycle and silver perchlorate were added in acetone (15 mL) in a carousel tube to give a light orange solution, which was stirred for 10 min. Then, the solution becomes dark orange and the resulting white solid (AgCl) was removed by centrifugation prior to addition of triphenylphosphine. Removal of the solvent under reduced pressure gave the expected complex as an orange solid, which was filtered off and dried.N[2-(SMe)C6H4]}(PPh3)][ClO4] 2a.
Orange solid. Yield: 78%. 1H NMR (400 MHz, acetone-d6) δ 9.29 (d, 4JP–H = 9.3 Hz, 1H, HCN), 8.05 (d, 3J = 8.4 Hz, 1H, H11), 7.89–7.81 (m, 7H), 7.74–7.51 (m, 9H), 7.43 (s, 1H, H2), 6.83 (t, 3J = 7.8 Hz, 1H, H9), 6.61 (t, 3J = 7.8 Hz, 1H, H10), 6.05 (d, 4JP–H = 5.3 Hz, 1H, H5), 3.76 (s, 3H, OMe), 2.93 (s, 3H, OMe), 2.12 (s, 3H, SMe). 31P-NMR (400 MHz, acetone-d6) δ 38.89. IR ν(CN) 1582; ν(ClO4) 1094 cm−1. Anal. found: C, 53.8; H, 3.9; N, 1.8; S, 4.1%, C34H31ClNO6PPdS (754.53 g mol−1) requires C, 54.1; H, 4.1; N, 1.9; S, 4.3%.
N[2-(SMe)C6H4]}(PPh3)][ClO4] 2b.
Orange solid. Yield: 80%. 1H NMR (400 MHz, acetone-d6) δ 9.25 (d, 4JP–H = 9.1 Hz, 1H, HCN), 8.06 (d, 3J = 8.6 Hz, 1H, H11), 7.85 (dd, J = 12.4, 8.0 Hz, 6H), 7.74–7.56 (m, 9H), 7.51 (d, J = 6.8 Hz, 11H), 7.43 (s, 1H, H2), 6.84 (t, J = 7.9 Hz, 1H, H9), 6.62 (t, J = 7.9 Hz, 1H, H10), 6.05 (d, 4JP–H = 4.3 Hz, 1H, H5), 2.95 (s, 3H, OMe), 2.26 (s, 3H, Me), 2.12 (s, 3H, SMe). 31P-NMR (400 MHz, acetone-d6) δ 37.49. IR cm−1ν(CN) 1582; ν(ClO4) 1091. Anal. found: C, 55.1; H, 4.1; N, 1.9; S, 4.4%, C34H31ClNO5PPdS (738.53 g mol−1) requires C, 55.3; H, 4.2; N, 1.9; S, 4.3%.
N[2-(SMe)C6H4]}(PPh3)][ClO4] 2c.
Orange solid. Yield: 85%. 1H NMR (400 MHz, acetone-d6) δ 9.30 (d, 4JP–H = 9.0 Hz, 1H, HCN), 8.10 (d, 3J = 8.3 Hz, 1H, H11), 8.06–7.97 (m, 6H), 7.78–7.49 (m, 9H), 7.46 (t, 3J = 7.6 Hz, 1H, H10), 7.36 (m, 1H, H9), 7.09 (s, 1H, H2), 7.02 (s, 1H, H2), 5.95 (d, 4JP–H = 5.0 Hz, 1H, H5), 4.17–4.10 (m, 4H, OCH2CH2O), 2.14 (s, 3H, SMe). 31P-NMR (400 MHz, acetone-d6) δ 38.54. IR cm−1ν(CN) 1576; ν(ClO4) 1094. Anal. found: C, 54.5; H, 3.9; N, 2.1; S, 4.1%, C34H29ClNO6PPdS (752.51 g mol−1) requires C, 54.3; H, 3.9; N, 1.9; S, 4.3%.
Reactivity of 2-methylthio aniline
Route a: Palladium acetate was introduced into a carousel tube fitted with a stirring bar. 10 mL of toluene are added, and a partial dissolution of palladium salt is observed. Methylthioaniline is then introduced, and it is observed that the solution becomes orange. It is allowed to react with stirring at 50 °C for 24 h, after which the formation of a yellow solid is observed within the solution that is separated by centrifugation. The supernatant is brought to dryness, resulting in an orange solid. 1H NMR (400 MHz, acetone-d6) δ 7.27 (d, 3J = 7.6 Hz, 1H), 7.02 (t, 3J = 7.7 Hz, 1H), 6.77 (d, 3J = 8.1 Hz, 1H), 6.59 (t, 3J = 7.5 Hz, 1H), 4.91 (d, 3J = 10.2 Hz, 2H), 2.31 (s, 3H).Route b: In a carousel tube fitted with a stirring bar sodium tetrachloropalladate was added in methanol (10 mL) and a reddish solution was formed; which quickly changes to yellow upon addition of methylthioaniline. The reaction mixture was stirred for 24 h and a white solid formed was separated by centrifugation, after which the resulting solution was reduced to low volume to give an orange solid. 1H NMR (400 MHz, DMSO-d6) δ 7.84 (d, 3J = 7.7 Hz, 1H), 7.48 (t, 3J = 7.5 Hz, 1H), 7.42 (t, 3J = 7.5 Hz, 1H), 7.33 (d, 3J = 7.9 Hz, 1H), 7.18 (d, 3J = 7.7 Hz, 1H), 6.97 (t, 3J = 7.7 Hz, 1H), 6.68 (d, 3J = 8.0 Hz, 1H), 6.53 (t, 3J = 7.5 Hz, 1H), 5.16 (s, 2H, NH), 2.84 (s, 3H, SMe), 2.29 (s, 3H, SMe).
Preparation of the diphosphine palladacycles
N[2-(SMe)C6H4]}(PPh2CH2PPh2-P)](ClO4) (3d).
Silver perchlorate (10.4 mg, 0.05 mmol) was added to a suspension of the cyclometallated complex 1a (20 mg, 0.05 mmol) in acetone (15 cm3). The mixture was stirred for 4 h at r.t. and filtered over Celite to remove the silver chloride precipitate. PPh2CH2PPh2 (19 mg, 0.05 mmol) was added to the filtrate and the solution stirred for 24 h at r.t., the solvent was removed and the residue was recrystallized from dichloromethane/hexane. Orange solid. Yield 78%. 1H NMR (400 MHz, CDCl3) δ 8.88 (d, 1H, HCN, 4JP–H = 8.4 Hz), 7.30–7.80 (m, 20H, PPh2); H2, 7.70–7.00 (H10–H12 hidden by the resonance of aromatic phosphine system), 7.15 (d, 1H, H9), 6.61 (d, 1H, H3, 3J(H2H3) = 7.5 Hz), 5.84 (s, 1H, H5), 4.19 (ta, 2H, PCH2P, 2J(HP) = 2.4 Hz), 3.22 (s, 3H, OCH3), 2.07 (s, 3H, SCH3). 31P NMR (400 MHz, CDCl3) δ −23.60 (d, 2J(PP) = 51.5 Hz); 31.30 (d, 2J(PP) = 51.5 Hz). IR cm−1ν(CN) 1598; ν(ClO4) 1093. Anal. found: C, 56.8; H, 4.3; N, 1.7; S, 4.3%, C40H36ClNO5P2PdS (846.60 g mol−1) requires C, 57.0; H, 4.2; N, 1.6; S, 3.8%.
N[2-(SMe)C6H4]}(PPh2CH2P(Ph2)W(CO)5-P)](ClO4) (4d).
To a suspension of 1e (20 mg, 0.05 mmol) in acetone (15 cm3). AgClO4 was added. The mixture was stirred for 4 h, after which time the silver chloride formed was filtered off through Celite. PPh2CH2P(Ph2)W(CO)5 (11 mg, 0.025 mmol) was added to the filtrate and the solution stirred for 4 h; reducing to low volume gave a solid which was filtered off and dried. Yellow solid. Yield 53% (1200.52). Calc. found. 1H NMR (400 MHz, CDCl3) δ 8.90 (d, 1H, HCN, 4JP–H = 7.6 Hz), 7.70–7.00 (H9–H12 hidden by the resonance of aromatic phosphine system), 6.42 (d, 2J(PP) = 72.0 Hz, 1J(PW) = 247.9 Hz), 6.02 (d, 1H, H3, 4J(H3H5) = 3.2 Hz), 5.55 (dd, 1H, H5, 4J(H5H3) = 3.2 Hz, 4J(H5P) = 7.1 Hz), 4.93 (t, 1H, CH2, 2J(HP) = 9.3 Hz, 2J(HP) = 11.5 Hz), 3.98 (dd, 1H, CH2, 2J(HP) = 6.3 Hz, 2J(HP) = 11.7 Hz), 3.82 (s, 3H, OCH3), 3.40 (s, 3H, OCH3), 1.70 (s, 3H, SCH3). 31P NMR (400 MHz, CDCl3) δ 26.6 (d, 2J(PP) = 72.0 Hz); IR cm−1ν(CN) 1583; ν(ClO4) 1095. Anal. found: C, 45.8; H, 3.3; N, 1.7; S, 3.2%, C46H38ClNO11P2PdSW (1200.52 g mol−1) requires C, 46.0; H, 3.2; N, 1.2; S, 2.7%.
Results and discussion
For the convenience of the reader the compounds and reactions are shown in Schemes 1–3. The compounds described in this paper were characterized by elemental analysis (C, H, N, S), and by IR spectroscopy, and by 1H, 31P–{1H} spectroscopy and, in part, crystal structure analysis (see Experimental, Table 1 and Fig. 1–5). | ||
| Fig. 1 ORTEP drawing of compound 1b with thermal ellipsoid plot shown at 50% probability level. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond distances (Å) and angles (°): Pd(1)–C(1) 1.9936(11), Pd(1)–N(1) 2.0039(9), Pd(1)–S(1) 2.3987(3), Pd(1)–Cl(1) 2.2963(3), S(1)–Pd(1)–Cl(1) 98.815(10), N(1)–Pd(1)–S(1) 84.63(3), N(1)–Pd(1)–Cl(1) 175.43(3), C(1)–Pd(1)–S(1) 166.53(3), C(1)–Pd(1)–Cl(1) 94.65(3), C(1)–Pd(1)–N(1) 81.91(4). | ||
 | ||
| Fig. 2 ORTEP drawing of compound 5 with thermal ellipsoid plot shown at 50% probability level. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond distances (Å) and angles (°): Pd(01)–S(005) 2.262(3), Pd(01)–N(00N) 2.065(8), Pd(01)–N(00M) 2.032(9), Pd(01)–O(00I) 2.042(8), N(00N)–Pd(01)–S(005) 99.5(3), N(00N)–Pd(01)–O(00I) 88.1(3), N(00M)–Pd(01)–O(00I) 87.0(3), N(00M)–Pd(01)–S(005) 85.7(3); Pd(02)–O(00G) 2.044(8), Pd(02)–N(00N) 2.014(9), Pd(02)–N(00M) 2.063(9), Pd(02)–S(006), 2.257(3), N(00N)–Pd(02)–S(006) 86.2(3), N(00N)–Pd(02)–O(00G) 91.2(4), N(00M)–Pd(02)–O(00G) 88.4(3), N(00M)–Pd(02)–S(006) 94.4(3). | ||
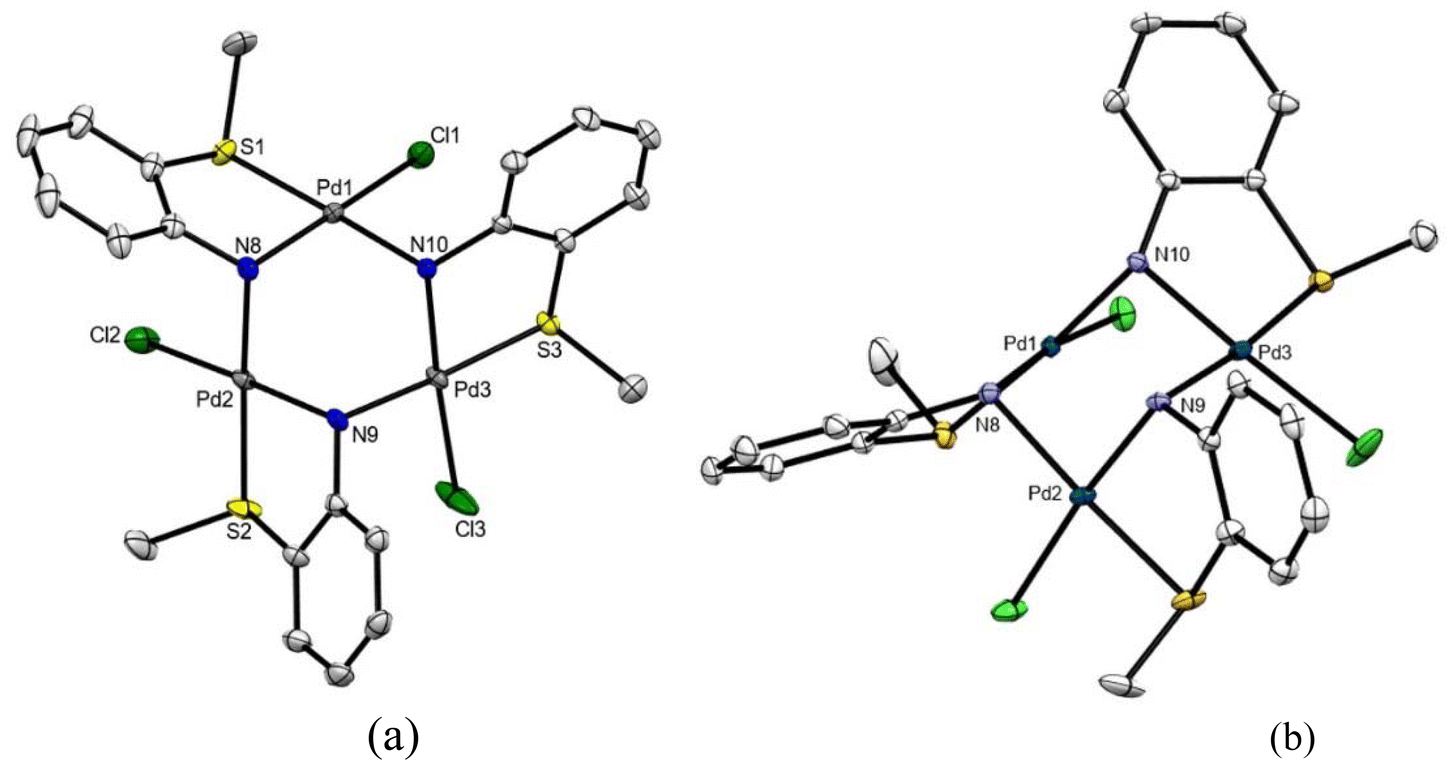 | ||
| Fig. 3 (a) ORTEP drawing of compound 6 with thermal ellipsoid plot shown at 50% probability level. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond distances (Å) and angles (°): Pd(1)–Cl(1) 2.3248(9), Pd(1)–N(8) 2.025(3), Pd(1)–N(10) 2.096(3), Pd(1)–S(1) 2.2803(8), N(10)–Pd(1)–Cl(1) 94.61(8), N(8)–Pd(1)–S(1) 84.21(8), N(8)–Pd(1)–N(10) 90.77(11), Cl(1)–Pd(1)–S(1) 90.72(3); Pd(2)–Cl(2) 2.3286(9), Pd(2)–N(8) 2.079(3), Pd(2)–N(9) 2.026(3), Pd(2)–S(2) 2.2863(9), N(8)–Pd(2)–Cl(2) 95.26(8), Cl(2)–Pd(2)–S(2) 90.51(8), N(8)–Pd(2)–N(9) 89.19(11), N(9)–Pd(2)–S(2) 85.25(8); Pd(3)–Cl(3) 2.3284(10), Pd(3)–N(9) 2.092(3), Pd(3)–N(10) 2.023(3), Pd(3)–S(3) 2.2791(8), N(9)–Pd(3)–Cl(3) 95.65(8), N(10)–Pd(3)–S(3) 85.87(8), N(9)–Pd(3)–N(10) 89.07(11), Cl(3)–Pd(3)–S(3) 89.44(3). (b) Detailed view of the chair conformation for the Pd3N3 ring. | ||
 | ||
| Fig. 4 ORTEP drawing of compound 7 with thermal ellipsoid plot shown at 50% probability level. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond distances (Å) and angles (°): Pd(1)–N(1) 2.067(6), Pd(1)–N(4) 2.053(6), Pd(1)–S(1) 2.280(2), Pd(1)–S(4) 2.272(2), N(1)–Pd(1)–S(1) 85.95(16), N(4)–Pd(1)–S(4) 86.22(18), N(1)–Pd(1)–N(4) 95.2(2), S(1)–Pd(1)–S(4) 92.70(8); Pd(2)–N(1) 2.092(6), Pd(2)–N(2) 2.033(7), Pd(2)–S(2) 2.256(2), Pd(2)–Cl(1) 2.330(2), N(1)–Pd(2)–Cl(1) 92.02(17), Cl(1)–Pd(2)–S(2) 88.61(7), N(1)–Pd(2)–N(2) 92.4(2), N(2)–Pd(2)–S(2) 86.92(19); Pd(3)–N(2) 2.046(6), Pd(3)–N(3) 2.055(6), Pd(3)–Cl(2) 2.3156(19), Pd(3)–Cl(3) 2.3261(19), N(2)–Pd(3)–Cl(3) 86.86(19), N(3)–Pd(3)–Cl(2) 87.50(18), N(2)–Pd(3)–Cl(2) 92.85(19), Cl(3)–Pd(3)–N(3) 93.17(18); Pd(4)–N(3) 2.030(6), Pd(4)–N(4) 2.087(6), Pd(4)–S(3) 2.253(2), Pd(4)–Cl(4) 2.3373(19), N(4)–Pd(4)–Cl(4) 91.97(18), Cl(4)–Pd(4)–S(3) 88.60(7), N(3)–Pd(4)–N(4) 92.9(2), N(3)–Pd(4)–S(3) 86.62(19). | ||
 | ||
| Fig. 5 ORTEP drawing of compound 8 with thermal ellipsoid plot shown at 50% probability level. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond distances (Å) and angles (°): Pd(1)–N(1) 2.044(8), Pd(1)–P(1) 2.287(3), Pd(1)–S(1) 2.254(3), Pd(1)–O(7) 2.076(7), Pd(2)–N(1) 2.041(8), Pd(2)–O(8) 1.998(7), N(1)–Pd(1)–S(1) 85.95(16), S(1)–Pd(1)–P(1) 86.22,(18), P(1)–Pd(1)–O(7) 95.2(2), O(7)–Pd(1)–N(1) 92.70(8); N(2)–Pd(1)–P(1) 174.0(3), S(1)–Pd(1)–O(7) 177.4(2), N(1)–Pd(2)–O(8) 89.8(3). | ||
 | ||
| Scheme 1 Reaction sequence leading to the synthesis of the compounds. | ||
 | ||
| Scheme 2 Reactivity of 2-thiomethyl aniline with differing palladium reagents. | ||
 | ||
| Scheme 3 Reaction sequence leading to the compounds with diphosphine. | ||
| Compound | 1b | 5 | 6 | 7 | 8 |
| Empirical formula | C16H16ClNOPdS | C17H20Cl4N2O6Pd2S2 | C25H36Cl3N3O2Pd3S5 | C31H35Cl13N4Pd4S4 | C78H66Cl2N2O22PdPd3S2W2 |
| Formula weight | 412.245 | 767.131 | 996.518 | 1479.647 | 2329.12 |
| Temperature/K | 100.0 | 100.0 | 100.0 | 100.00 | 100.0 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Orthorhombic | Triclinic |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | Pbca |
P |
| a/Å | 8.1340(2) | 11.6987(6) | 9.6057(5) | 10.6947(8) | 10.8665(13) |
| b/Å | 11.6466(3) | 15.0875(8) | 19.0596(10) | 22.0850(16) | 12.0797(14) |
| c/Å | 17.0711(4) | 18.3377(9) | 19.8471(11) | 40.285(3) | 19.070(2) |
| α/° | 90 | 105.262(3) | 90 | 90 | 72.291(7) |
| β/° | 101.9138(10) | 104.871(3) | 91.4728(17) | 90 | 89.037(7) |
| γ/° | 90 | 107.653(4) | 90 | 90 | 78.172(7) |
| Volume/Å3 | 1582.37(7) | 2769.0(3) | 3632.4(3) | 9515.0(12) | 2331.2(5) |
| Z | 4 | 4 | 4 | 8 | 1 |
| ρ calc./g cm−3 | 1.730 | 1.840 | 1.822 | 2.066 | 1.659 |
| μ/mm−1 | 1.471 | 15.748 | 2.006 | 2.425 | 3.262 |
| F(000) | 821.5 | 1518.5 | 1961.1 | 5733.3 | 1136 |
| Crystal size/mm3 | 0.156 × 0.127 × 0.092 | 0.08 × 0.03 × 0.02 | 0.14 × 0.09 × 0.05 | 0.09 × 0.07 × 0.03 | 0.200 × 0.090 × 0.030 |
| Radiation | Mo Kα (λ = 0.71073) | Cu Kα (λ = 1.54178) | Mo Kα (λ = 0.71073) | Mo Kα (λ = 0.71073) | Mo Kα (λ = 0.71073) |
| 2θ/° | 4.88 to 77.14 | 6.62 to 140.14 | 4.74 to 56.76 | 4.32 to 56.68 | 1.810 to 26.445 |
| Index ranges | −14 ≤ h ≤ 14, −20 ≤ k ≤ 19, −29 ≤ l ≤ 29 | −14 ≤ h ≤ 14, −18 ≤ k ≤ 18, −22 ≤ l ≤ 22 | −12 ≤ h ≤ 12, −25 ≤ k ≤ 25, −25 ≤ l ≤ 26 | −14 ≤ h ≤ 14, −29 ≤ k ≤ 29, −33 ≤ l ≤ 53 | −13 ≤ h ≤ 13, −15 ≤ k ≤ 15, −23 ≤ l ≤ 23 |
| Reflections collected | 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 757 757 |
68334 |
86053 |
98993 |
70681 |
| Independent reflections | 8909 [Rint = 0.0353, Rsigma = 0.0233] | 10523 [Rint = 0.1983, Rsigma = 0.1049] |
9079 [Rint = 0.0378, Rsigma = 0.0198] | 11826 [Rint = 0.0686, Rsigma = 0.0392] |
9506 [Rint = 0.1012] |
| Data/restraints/parameters | 8909/0/193 | 10523/0/602 |
9079/24/459 | 11826/24/574 |
9506/439/550 |
| Goodness-of-fit on F2 | 1.037 | 1.020 | 1.050 | 1.045 | 1.087 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0232, wR2 = 0.0510 | R 1 = 0.0676, wR2 = 0.1638 | R 1 = 0.0328, wR2 = 0.0737 | R 1 = 0.0727, wR2 = 0.1316 | R 1 = 0.0762, wR2 = 0.1540 |
| Final R indexes [all data] | R 1 = 0.0312, wR2 = 0.0547 | R 1 = 0.1213, wR2 = 0.2051 | R 1 = 0.0370, wR2 = 0.0764 | R 1 = 0.0832, wR2 = 0.1368 | R 1 = 0.1130, wR2 = 0.1649 |
| Largest diff. peak/hole/e Å−3 | 0.73/−1.16 | 1.59/−1.82 | 3.16/−2.83 | 1.63/−1.91 | 2.303/−2.307 |
The Schiff base ligands a–f were prepared by reaction of 2-thiomethylaniline with the corresponding aldehyde to give pure air-stable solids which were fully characterized. Distinctive of the spectrum of d were the two virtual doublets stemming from the aromatic AA′XX′ spin system with N = 8.0 ppm. Treatment of the ligands with Li2[PdCl4]/NaAcO in methanol gave the palladacycles 1a–1c (Scheme 1) and 1d–1f (Scheme 3) that were isolated as air-stable solids with the ligand in a tridentate [C,N,S] coordination mode; preparative details and characteristic microanalytical and spectroscopic data are given in the Experimental section. Noteworthy to mention are the shift of the ν(CN) stretch to lower wavenumbers by ca. 40 cm−1 in the IR spectra, and absence of the C(6)–H resonance in the NMR spectra;43 also, the SMe resonance was downfield shifted in the 1H NMR spectra, ca. 0.4 ppm, in agreement with Pd–S coordination, and the metallated ligand of 1d showed the absence of the AA′XX′ system consequent on Pd–C bond formation.
Crystal structure of 1b
Suitable crystals were grown by slow evaporation of a chloroform solution. The crystal structure of 1b (Fig. 1 and Table 1) consists of molecules with the palladium(II) atom bonded in a slightly distorted square-planar environment to four different donor atoms, the aryl C(6) carbon, the iminic N(1) nitrogen, and the S(1) sulfur atoms, of the tridentate iminic and to the chlorine atom Cl(1). The sulfur atom in SMe becomes a chiral center upon metalation, and in the structure there is a racemic mixture of both enantiomers. The angles between adjacent atoms in the palladium coordination sphere are ca. 90° with the most noticeable distortion in the N(1)2Pd(1)2C(6) angle of 81.7(2)°, a consequence of chelation; the sum of angles around the palladium atom is 360.03°. All bond lengths are within the expected range, with allowance for Pd(1)2C(1) length, of 2.044(4) Å, shorter than the expected value of 2.081 Å44 suggesting some degree of multiple bond character in the Pd2C(aryl)linkage.45
Reaction of 1a–1c with PPh3/AgClO4 gave complexes 2a–2c, after abstraction of the chloride ion as AgCl, which were fully characterized (see Experimental). Attempts to produce species with simultaneous linkage of the phosphine and chloride ligands to the metal were to no avail; this is in keeping with a rather strong Pd–S bond. The IR spectra showed absence and of the ν(Pd–Cl) band; whilst the 1H NMR spectra the H(5) was upfield shifted and coupled to the phosphorus resonance.
To study the suitability of other metallating agents a slightly different synthetic approach was tested with ligands a–c; this allowed disclosure of a new performance of the ligands and complexes. Thus, treatment of ligands a–c with Na2[PdCl4]/NaAcO in methanol gave a solution containing the 1a–1c complexes, with analogous spectroscopic features as those described above, plus the starting aldehyde and amine, for which the 1H NMR spectra showed the signals for the HCO, NH2 and SMe resonances ca. 10, 6.7 and 2.3 ppm, respectively, together with other aromatic resonances (mixtures type A). This did not seem to be unusual because in the previous reactions using Li2[PdCl4] as metal salt, prior to addition of the base, aliquots of the reaction mixture were taken and some free aldehyde was observed due to partial hydrolysis; however, in those cases after addition of NaAcO and ensuing work up the final compounds were obtained pure (vide supra). Then, treatment of the said mixture A with PPh3/AgClO4 yielded compounds 2a–2c impurified with free aldehyde and amine (mixtures type B). To separate the palladacycles and confirm the alternative use of the sodium salt as a metallating agent, recrystallization was performed for both types of mixtures.
Crystal structure of 5
Suitable crystals of mixture A were grown from a chloroform solution, labelled 5. The crystal structure of 5 (Fig. 2 and Table 1) consists of molecules with the palladium(II) atom bonded in a slightly distorted square-planar environment to two mutually perpendicular pairs of [O,O] and [N,S] donors, pertaining to bridging acetate and chelating 2-thiomethylanilide ligands, respectively; the nitrogen atoms in turn act as bridging donors between two palladiums. The nitrogen atoms are arranged in a square-planar geometry and in turn act as bridging donors holding the palladium atoms at a midway distance between each pair of nitrogens to give an eight-membered ring of alternating Pd/N centers. Thus, the palladium coordination planes are set as two pairs of mutually facing parallel planes ca. 90° to the Pd4N4 ring.We reasoned that mixture B should produce either crystals of 2a–2c, as appropriate, or a structure analogous to 5 depending on whether or not the acetate ligands were lost in the corresponding preparation. Again, the experimental facts show that an absolutely distinctive structure was obtained: a trinuclear crystal void of any acetate ligands, labelled 6.
Crystal structure of 6
Suitable crystals were grown from mixture B from a DMSO-d6 solution, labelled 6. The crystal structure (Fig. 3 and Table 1) consists of a trinuclear molecule with a central Pd3N3 six-membered ring of alternating palladium and nitrogen atoms in a chair conformation, with the Pd(2) and N(10) atoms pointing to opposite directions from the Pd(1)N(8)N(9)Pd(3) plane (Fig. 5b). Each palladium atom is bonded to a chelating bidentate [N,S] 2-thiomethylanilide, that in turn also coordinates as a bridging ligand between two metal atoms via the nitrogen donor. The fourth coordination site at each palladium is occupied by a chloride ligand trans to nitrogen for the three metal centers. The palladium coordination planes [S(1)N(8)Cl(1)Pd(1)N(10)] (plane 1), [S(2)N(8)Cl(2)Pd(2)N(9)] (plane 2) and [S(3)N(9)Cl(3)Pd(3)N(10)] (plane 3) are set as follows: 1^2 83.44°; 1^3 77.08°; 2^3 79.91°.In view of this unexpected behavior of the amine ligand stemming from partial hydrolysis of the parent Schiff base, we sought out to study the behavior of free 2-methylthioaniline itself in order to determine if compounds 5 and 6 were produced only from the corresponding palladacycles, or if it was solely dependent on the presence of the free amine. For such a purpose 2-thiomethylaniline was treated independently with Pd(OAc)2 in toluene or with Na2[PdCl4] in methanol (see Scheme 2 and Experimental for preparative details).
The former process gave only the free amine, as was determined by 1H NMR spectroscopy. However, in the latter case the 1H NMR spectrum shows there are two singlets assignable to the SMe groups and two sets of aromatic peaks containing four doublets and four triplets in agreement with a four-spin system of an ortho-substituted phenyl ring. This is a differing situation from that found for compounds 5 and 6, where the 2-thiomethylanilide ligands were in equivalent chemical environments, pointing to two types of non-equivalent amine molecules that are present in the final compound. Fortunately, single-crystals could be obtained to clarify this finding and the result is even more fascinating than for structures 5 and 6 (vide infra).
Crystal structure of 7
Suitable crystals were grown from a chloroform solution, labelled 7. The crystal structure (Fig. 4 and Table 1) consists of four palladium atoms, four chloride and four 2-thiomethylanilide ligands displayed such that there are three types of palladium coordination environments: 2 + 1 + 1. The aniline moieties are linked to the Pd(1), Pd(2) and Pd(4) atoms in a bidentate [N,S] fashion and the four palladium atoms are bonded together via the nitrogen from bridging 2-MeC6H4NH2-N units; the chlorine atoms complete the coordination sphere at the metal as terminal ligands. Thus, Pd(1) is bonded to two chelating [N,S] aniline ligands with the donor atoms in a cis geometry; Pd(2) and Pd(4) to one chelating amine, a terminal chlorine atom and a nitrogen atom from an adjacent organic ligand; Pd(3) to two mutually trans terminal chlorine atoms and two nitrogen donors form nearby amine ligands also in a trans arrangement. All of which results in a Pd4N4 eight-membered ring of alternating palladium and nitrogen atoms. The palladium coordination planes [Pd(1)N(1)N(4)S(1)S(4)] (1), [Pd(2)N(1)N(2)S(2)Cl(1)] (2), [Pd(3)N(2)N(3)Cl(2)Cl(3)] (3) [Pd(4)N(3)N(4)S(3)Cl(4)] (4) [1^2 89.59°; 1^3 87.60°, 1^4 86.55°, 2^3 85.92°; 3^4 85.81°; 2^4 8.99°] are angled ca. 90°, save for planes 2 and 4 which are close to a parallel disposition.Next, we attempted to study the behavior of the 1a–1f class complexes with diphosphines to determine whether they would give species analogous to the thiosemicarbazone-[C,N,S] palladacycles we have described in the past, i.e., complexes with a mono-coordinated diphosphine and also the ensuing dinuclear compounds with the diphosphine ligand bridging two metal centers; or if alternatively, and given the results depicted above, they would evolve to unprecedented multinuclear compounds; for which purpose we chose compound 1d and the diphosphine dppm, Ph2PCH2PPh2.
Hence, treatment of 1d with dppm in 1:1 molar ratio and NaClO4 gave the hoped for complex 3d as a pure air-stable solid, which was fully characterized. The 31P NMR spectrum displayed two doublets at −23.6 and 31.3 ppm, for the two inequivalent phosphorus nuclei. The resonance at lower frequency was assigned to the non-coordinated phosphorus nucleus; whereas the one at higher frequency was assigned to the phosphorus nucleus bonded to palladium. The HCN resonance was a doublet coupled to the trans phosphorus nucleus (4J(HiP) = 8.4 Hz). Both the MeO and SMe resonances were shifted to lower frequency ca. 0.6 and 0.7 ppm respectively, due to shielding by the phosphine phenyl rings. The proton signal was an apparent triplet from the AA′XX′ four spin system P(CH2)P with an N value of 2.4 Hz. Attempts to prepare the heterobimetallic complex 4d by treatment of 3d with [W(CO)5(THF)] gave an untreatable mixture which was not further investigated. Likewise, reaction of 3d with other substrates bearing labile ligands such as [Fe(CO)4(THF)] and [Pd(Cl)2(PhCN)2], to yield the corresponding dinuclear species were also deemed unsuccessful. Alternatively, reaction of 1d with [W(CO)5{Ph2PCH2PPh2–P}] did produce the expected compound 4d as a yellow solid. The 31P NMR spectrum displayed two doublets at 26.6 and 6.42 ppm, for the two inequivalent phosphorus nuclei, with the latter one showing the tungsten satellites (1JPW = 247.9 Hz). The NMR tube solution of 4d was set aside and left to stand whereupon crystals appeared; the resulting X-ray crystallographic analysis showed them to be quite different from the expected complex, rendering an altogether new compound. The beauty of this serendipitous contingency is that it seems not to be an occasional issue, but rather a repeatable event, i.e., three-fold recrystallization attempts rendered the same structure.
Crystal structure of 8
Suitable crystals were grown for a chloroform solution of 4d (Fig. 5 and Table 1). The unit cell consists of one molecule of the compound and two perchlorate anions. The structure depicts a pentanuclear palladium/tungsten trans-configured heterometallic linear complex with the metal atoms connected by three linkers: a dppm diphosphine, two acetate anions, and two sulfur-containing aniline molecules stemming from the pincer ligand after hydrolysis of the CN double bond; no reduction of palladium was observed. The palladium atoms themselves are bridged by two nitrogen atoms: Pd(1)N(1)Pd(2)N(2)Pd(3), from two 2-MeC6H4NH2-N units, and also by two μ-MeCOO-O,O ligands.46 The outer palladium atoms show similar coordination environments each bonded to a chelating [N,S] aniline ligand with the donor atoms in a cis geometry; an oxygen donor from the bridging acetate ligand a tungsten metalloligand Ph2PCH2P(Ph)2W(CO)5, with the nitrogen and phosphorus atoms, on the one hand and the sulfur and oxygen donors, on the other, in trans arrangements. The coordination planes at palladium are essentially planar, with slight deviations consequent upon chelation of the aniline moieties. The angles on palladium are ca. 90° in the range 89.78–90.22° [Pd(2)], 83.78–95.35° [Pd(1), Pd(3)]. The coordination planes on Pd(1) [Pd(1)N(1)O(7)S(1)P(1)] and Pd(2) [Pd(2)O(8)N(1)O(8)N(1)] are at an angle of 83.68°.
Conclusions
In this work we have shown that the reaction conditions for the preparation of cyclometallated compounds from terdentate-[C,N,S] Schiff base ligands have a meaningful effect on the purity of the resulting products. In the case of the lithium metallating agent, the product is obtained in good yield and pure, whereas and in the case of the alternative sodium salt route, needing less restricted reaction conditions, the target compounds are formed but side reactions occur leading to presence of the parent aldehyde after partial decomposition of the organic ligand, as was detected by NMR spectroscopy. Therefore, cleavage of the Schiff base ligand occurs, and the released amine is in turn transformed into a chelating and/or bridging ligand to the metal. Thus, structures with eight- and six-membered rings of alternating palladium and nitrogen atoms are assembled. In either case it was possible to obtain the corresponding triphenylphosphine palladacycles prior abstraction of the chloride ligand. Moreover, thiomethyl aniline itself also shows a unique behavior when reacted with the appropriate palladium(II) reagent yielding a tetranuclear complex containing a Pd4N4 eight-membered ring that displays three different varieties of palladium coordination. This finding leads to the conclusion that the behavior previously described stems from a decomposition process which is unique to these iminic palladacycles and not from reaction of the hydrolyzed ligand with unreacted metallating agent. Last, but not least, this more than singular chemical performance of the coordinated Schiff base ligand is evident in the case of the dinuclear Pd/W metallacycle which evolves upon recrystallization to give a trans-configured heterometallic mixed valent Pd(II)/W(0) linear complex.Author contributions
Francisco Reigosa: formal analysis, methodology, conceptualization. Paula Polo: validation, methodology, investigation. Teresa Pereira: conceptualization, methodology. José M. Vila: conceptualization, writing – original draft, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was also made possible thanks to the financial support received from the Xunta de Galicia (Galicia, Spain) under the Grupos de Referencia Competitiva Programme (project GRC2019/14). F. R. thanks the Spanish Ministry of Education (grant FPU15/07145).References
- M. T. Pereira and J. M. Vila, The P-C Building Block of Palladacycles: A Cornerstone for Stoichiometric C-C and C-X Bond Assemblage, in Palladacycles. Synthesis, Characterization and Applications, ed. J. Dupont and M. Pfeffer, Wiley-VCH, 2008, pp. 87–108 Search PubMed.
- J. M. Vila, M. T. Pereira, F. Lucio-Martínez and F. Reigosa, Palladacycles as Efficient Precatalysts for Suzuki-Miyaura Cross-Coupling Reactions, Palladacycles. Catalysis and Beyond, 2019, pp. 1–20 Search PubMed.
- A. C. Cope and R. W. Siekman, Formation of Covalent Bonds from Platinum or Palladium to Carbon by Direct Substitution, J. Am. Chem. Soc., 1965, 87(14), 3272–3273, DOI:10.1021/ja01092a063.
- I. Omae, Intramolecular Five-Membered Ring Compounds and Their Applications, Coord. Chem. Rev., 2004, 248(11–12), 995–1023, DOI:10.1016/j.ccr.2004.05.011.
- J. Tsuji, Palladium Reagents and Catalysts, 2004, vol. 9, DOI:10.1002/0470021209.
- M. Albrecht, Cyclometalation Using D-Block Transition Metals: Fundamental Aspects and Recent Trends, Chem. Rev., 2010, 110(2), 576–623, DOI:10.1021/cr900279a.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Light-Emitting Devices Based on Organometallic Platinum Complexes as Emitters, Coord. Chem. Rev., 2011, 255(21–22), 2401–2425, DOI:10.1016/j.ccr.2011.01.049.
- Á. Díez, E. Lalinde and M. T. Moreno, Heteropolynuclear Cycloplatinated Complexes: Structural and Photophysical Properties, Coord. Chem. Rev., 2011, 255(21–22), 2426–2447, DOI:10.1016/j.ccr.2010.12.024.
- S. B. Wild, Resolutions of Tertiary Phosphines and Arsines with Orthometallated Palladium(II) – Amine Complexes, Coord. Chem. Rev., 1997, 166, 291–311, DOI:10.1016/s0010-8545(97)00046-5.
- W. A. Herrmann, C. Brossmer, K. Öfele, C.-P. Reisinger, T. Priermeier, M. Beller and H. Fischer, Palladacycles as Structurally Defined Catalysts for the Heck Olefination of Chloro- and Bromoarenes, Angew. Chem., Int. Ed. Engl., 1995, 34(17), 1844–1848, DOI:10.1002/anie.199518441.
- M. Beller, H. Fischer, W. A. Herrmann, K. Öfele and C. Brossmer, Palladacycles as Efficient Catalysts for Aryl Coupling Reactions, Angew. Chem., Int. Ed. Engl., 1995, 34(17), 1848–1849 CrossRef CAS.
- B. Bermudez, L. A. Adrio, F. Lucio-Martinez, F. Reigosa, J. M. Vila and J. M. Ortigueira, Imine Palladacycles: Synthesis, Structural Analysis and Applications in Suzuki-Miyaura Cross Coupling in Semi-Aqueous Media, Molecules, 2022, 27, 3146, DOI:10.3390/molecules27103146.
- F. Lucio-Martínez, L. A. Adrio, P. Polo-Ces, J. M. Ortigueira, J. J. Fernández, H. Adams, M. T. Pereira and J. M. Vila, Palladacycle Catalysis: An Innovation to the Suzuki-Miyaura Cross-Coupling Reaction, Dalton Trans., 2016, 45(44), 17598–17601, 10.1039/c6dt03542f.
- J. Dupont, M. Pfeffer and J. Spencer, Palladacycles – An Old Organometallic Family Revisited: New, Simple, and Efficient Catalyst Precursors for Homogeneous Catalysis, Eur. J. Inorg. Chem., 2001, 1917–1927, DOI:10.1002/1099-0682(200108)2001:8<1917::AID-EJIC1917>3.0.CO;2-M.
- R. B. Bedford, Palladacyclic Catalysts in C–C and C–Heteroatom Bond-Forming Reactions, Chem. Commun., 2003, 1787–1796 RSC.
- I. P. Beletskaya and A. V. Cheprakov, Palladacycles in Catalysis – A Critical Survey, J. Organomet. Chem., 2004, 689(24 SPEC. ISS.), 4055–4082, DOI:10.1016/j.jorganchem.2004.07.054.
- A. F. Littke and G. C. Fu, Palladium-Catalyzed Coupling Reactions of Aryl Chlorides, Angew. Chem., Int. Ed., 2002, 41(22), 4176–4211, DOI:10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U.
- J. Dupont, C. S. Consorti and J. Spencer, The Potential of Palladacycles: More than Just Precatalysts, Chem. Rev., 2005, 105(6), 2527–2571, DOI:10.1021/cr030681r.
- M. T. Chen, C. A. Huang and C. T. Chen, Synthesis, Characterization, and Catalytic Applications of Palladacyclic Complexes Bearing C,N,S-Donor Ligands, Eur. J. Inorg. Chem., 2006, 4642–4648, DOI:10.1002/ejic.200600601.
- S. Doherty, J. G. Knight, N. A. B. Ward, D. M. Bittner, C. Wills, W. McFarlane, W. Clegg and R. W. Harrington, Electron-Rich Trialkyl-Type Dihydro-KITPHOS Monophosphines: Efficient Ligands for Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling. Comparison with Their Biaryl-like KITPHOS Monophosphine Counterparts, Organometallics, 2013, 32(6), 1773–1788, DOI:10.1021/om3011992.
- L. A. Bulygina, N. S. Khrushcheva, K. A. Lyssenko and A. S. Peregudov, Synthesis of Unsymmetrical Pincer CNN Palladium Complex of 8-Dimethylamino-3-Ferrocenylmethyl-3-Azabicyclo[3.2.1]Octane and Its Catalytic Activity in the Suzuki Reaction, Mendeleev Commun., 2022, 32(3), 331–333, DOI:10.1016/j.mencom.2022.05.012.
- A. Habtemariam, B. Watchman, B. S. Potter, R. Palmer, S. Parsons, A. Parkin and P. J. Sadler, Control of Aminophosphine Chelate Ring-Opening in Pt(II) and Pd(II) Complexes: Potential Dual-Mode Anticancer Agents, J. Chem. Soc., Dalton Trans., 2001, 1306–1318, 10.1039/b009117k.
- C. Navarro-Ranninger, I. López-Solera, V. M. González, J. M. Pérez, A. Alvarez-Valdés, A. Martín, P. R. Raithby, J. R. Masaguer and C. Alonso, Cyclometalated Complexes of Platinum and Palladium with N-(4-Chlorophenyl)-α-Benzoylbenzylideneamine. in Vitro Cytostatic Activity, DNA Modification, and Interstrand Cross-Link Studies, Inorg. Chem., 1996, 35(18), 5181–5187, DOI:10.1021/ic960050y.
- A. G. Quiroga and C. N. Ranninger, Contribution to the SAR Field of Metallated and Coordination Complexes: Studies of the Palladium and Platinum Derivatives with Selected Thiosemicarbazones as Antitumoral Drugs, Coord. Chem. Rev., 2004, 248(1–2), 119–133, DOI:10.1016/j.cct.2003.11.004.
- N. Cutillas, G. S. Yellol, C. De Haro, C. Vicente, V. Rodríguez and J. Ruiz, Anticancer Cyclometalated Complexes of Platinum Group Metals and Gold, Coord. Chem. Rev., 2013, 257(19–20), 2784–2797, DOI:10.1016/j.ccr.2013.03.024.
- F. Reigosa-Chamorro, L. R. Raposo, P. Munín-Cruz, M. T. Pereira, C. Roma-Rodrigues, P. V. Baptista, A. R. Fernandes and J. M. Vila, In Vitro and in Vivo Effect of Palladacycles: Targeting A2780 Ovarian Carcinoma Cells and Modulation of Angiogenesis, Inorg. Chem., 2021, 60(6), 3939–3951, DOI:10.1021/acs.inorgchem.0c03763.
- J. Albert, R. Bosque, M. Cadena, L. D'Andrea, J. Granell, A. González, J. Quirante, C. Calvis, R. Messeguer, J. Badía, L. Baldomà, T. Calvet and M. Font-Bardia, A New Family of Doubly Cyclopalladated Diimines. A Remarkable Effect of the Linker between the Metalated Units on Their Cytotoxicity, Organometallics, 2014, 33(11), 2862–2873, DOI:10.1021/om500382f.
- R. Gigli, G. J. S. Pereira, F. Antunes, A. Bechara, D. M. Garcia, D. G. Spindola, M. G. Jasiulionis, A. C. F. Caires, S. S. Smaili and C. Bincoletto, The Biphosphinic Paladacycle Complex Induces Melanoma Cell Death through Lysosomal-Mitochondrial Axis Modulation and Impaired Autophagy, Eur. J. Med. Chem., 2016, 107, 245–254, DOI:10.1016/j.ejmech.2015.11.008.
- S. Aliwaini, A. J. Swarts, A. Blanckenberg, S. Mapolie and S. Prince, A Novel Binuclear Palladacycle Complex Inhibits Melanoma Growth in Vitro and in Vivo through Apoptosis and Autophagy, Biochem. Pharmacol., 2013, 86(12), 1650–1663, DOI:10.1016/j.bcp.2013.09.020.
- A. L. de Andrade Querino, J. T. da Silva, J. T. Silva, G. M. Alvarenga, C. H. da Silveira, M. T. Q. de Magalhães, O. A. Chaves, B. A. Iglesias, R. Diniz and H. Silva, Mono and Dinuclear Platinum and Palladium Complexes Containing Adamantane–Azole Ligands: DNA and BSA Interaction and Cytotoxicity, J. Biol. Inorg. Chem., 2019, 24(7), 1087–1103, DOI:10.1007/s00775-019-01719-5.
- D. Ćoćić, S. Jovanović, M. Nišavić, D. Baskić, D. Todorović, S. Popović, Ž. D. Bugarčić and B. Petrović, New Dinuclear Palladium(II) Complexes: Studies of the Nucleophilic Substitution Reactions, DNA/BSA Interactions and Cytotoxic Activity, J. Inorg. Biochem., 2017, 175, 67–79, DOI:10.1016/j.jinorgbio.2017.07.009.
- M. Marcos, in Metallomesogens. Synthesis, Properties and Applications, ed. J. L. Serrano, 1996 Search PubMed.
- D. Pucci, G. Barberio, A. Bellusci, A. Crispini and M. Ghedini, Tailoring “Non Conventional” Ionic Metallomesogens around an Ortho-Palladated Fragment, J. Organomet. Chem., 2006, 691(6), 1138–1142, DOI:10.1016/j.jorganchem.2005.11.039.
- A. Ionescu, N. Godbert, A. Crispini, R. Termine, A. Golemme and M. Ghedini, Photoconductive Nile Red Cyclopalladated Metallomesogens, J. Mater. Chem., 2012, 22(44), 23617–23626, 10.1039/c2jm34946a.
- J. M. Vila, M. T. Pereira, J. M. Ortigueira, M. Graña, A. Suárez, J. J. Fernández, A. Fernández, M. López-torres and H. Adams, Formation, Characterization, and Structural Studies of Novel Thiosemicarbazone Palladium(II) Complexes. Crystal Structures of [{Pd[C6H4C(Et)NNC(S)NH2]}4], [Pd{C6H4C(Et)NNC(S)NH2}(PMePh2)] and [{Pd[C6H4C(Et)NNC(S)NH2]}2(μ-Ph2PCH2PPh2)], Dalton Trans., 1999, 4193–4201 RSC.
- T. Kawamoto, I. Nagasawa, H. Kuma and Y. Kushi, Pd⋯H–C Interactions. Preparation and Structure of Orthometalated Tetranuclear Complexes of Palladium(II) and Platinum(II), Inorg. Chem., 1996, 35(9), 2427–2432, DOI:10.1021/ic950769q.
- J. Martinez, M. T. Pereira, I. Buceta, G. Alberdi, A. Amoedo, J. J. Fernandez, M. Lopez-Torres, J. M. Vila, J. M. Ortigueira and J. M. Vila, Palladium(II) Cyclometallated Thiosemicarbazone Compounds: A New Class of Bidentate P,S Metalloligands, Organometallics, 2003, 22, 5581–5584 CrossRef CAS.
- R. G. Pearson, Hard and Soft Acids and Bases, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- Y. Qiao and G. Jin, Nickel(II) and Palladium(II) Complexes with Tridentate [C,N,S] and [C,N,P] Ligands: Syntheses, Characterization, and Catalytic Norbornene Polymerization, Organometallics, 2013, 32, 1932–1937, DOI:10.1021/om400033n.
- J. Yasuda, K. Inoue, K. Mizuno, S. Arai, K. Uehara, A. Kikuchi, Y. N. Yan, K. Yamanishi, Y. Kataoka, M. Kato, A. Kawai and T. Kawamoto, Photooxidation Reactions of Cyclometalated Palladium(II) and Platinum(II) Complexes, Inorg. Chem., 2019, 58(23), 15720–15725, DOI:10.1021/acs.inorgchem.9b01492.
- S. Mondal, S. Maity and P. Ghosh, Orthopalladated 1,4-Iminonaphthoquinone Derivative: Syntheses, Redox Series, Molecular and Electronic Structures, Inorg. Chim. Acta, 2019, 487, 240–246, DOI:10.1016/j.ica.2018.12.003.
- B. al Janabi, F. Reigosa, G. Alberdi, J. M. Ortigueira and J. M. Vila, An Innovative Structural Rearrangement in Imine Palladacycle Metaloligand Chemistry: From Single-Nuclear to Double-Nuclear Pseudo-Pentacoordinated Complexes, Molecules, 2023, 28, 2328, DOI:10.3390/molecules28052328.
- H. Onoue and I. Moritani, Ortho-Metalation Reactions of N-Phenylbenzaldimine and Its Related Compounds by Palladium(II) Acetate, J. Organomet. Chem., 1972, 43(2), 431–436, DOI:10.1016/S0022-328X(00)81621-6.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, New York, 3rd edn, 1960 Search PubMed.
- C. Navarro-Ranninger, I. López-Solera, A. Alvarez-Valdés, J. H. Rodríguez-Ramos, J. R. Masaguer, J. L. García-Ruano and X. Solans, Cyclometalated Complexes of Palladium(II) and Platinum(II) with N-Benzyl- and N-(Phenylethyl)-α-Benzoylbenzylideneamine. Delocalization in the Cyclometalated Ring as a Driving Force for the Orthometalation, Organometallics, 1993, 12(10), 4104–4111, DOI:10.1021/om00034a052.
- The ligands come from the starting salt and certainly a small amount is probably not removed in the purification process. Their presence is not apparent in the elemental analysis or in the NMR spectrum. The small amount of acetate does not suffice to significantly distort the microanalysis. The resonance of the MeCOO group is overlapped by the impurity of commercial deuterated chloroform ca. 2.1 ppm.
Footnotes |
| † CCDC 2333528 (1b), 2333529 (5), 2333530 (7), 2333531 (6) and 2333532 (8). For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00756e |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |