
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagneto-structural correlation in lanthanide luminescent [2.2]paracyclophane-based single-molecule magnets†
Hadrien
Flichot
a,
Annika
Sickinger
b,
Jules
Brom
c,
Bertrand
Lefeuvre
a,
Vincent
Dorcet
a,
Thierry
Guizouarn
a,
Olivier
Cador
 a,
Boris
Le Guennic
a,
Laurent
Micouin
c,
Olivier
Maury
b,
Erica
Benedetti
*c and
Fabrice
Pointillart
*a
a,
Boris
Le Guennic
a,
Laurent
Micouin
c,
Olivier
Maury
b,
Erica
Benedetti
*c and
Fabrice
Pointillart
*a
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, 35000 Rennes, France. E-mail: fabrice.pointillart@univ-rennes.fr
bUniv Lyon, ENS de Lyon, Université Claude Bernard Lyon 1, CNRS UMR 5182, Laboratoire de Chimie, Lyon F-69342, France
cUniversité Paris Cité, CNRS, Laboratoire de Chimie et de Biochimie Pharmacologiques et Toxicologiques, F-75006 Paris, France. E-mail: erica.benedetti@u-paris.fr
First published on 22nd April 2024
Abstract
The association of lanthanide ions and paracyclophane derivatives has been very scarcely reported in the literature. In this study, elaboration of five coordination lanthanide complexes involving the 1,4(1,4)-dibenzenacyclohexaphane-12,43-diylbis(diphenylphosphine oxide) ligand (L) was achieved with the determination of single-crystal X-ray diffraction structures of four mononuclear complexes of formula [Ln(hfac)3(L)] (hfac- = 1,1,1,5,5,5-hexafluoroacetylacetonate) (Ln = Dy(III) (1-Dy) and Yb(III) (2-Yb)) and [Ln(tta)3(L)] (tta− = 2-tenoyl-trifluoroacetylacetonate) (Ln = Dy(III) (3-Dy) and Yb(III) (4-Yb)) and one dinuclear complex [Na(Dy2(hfac)6(L)2)](BArF) (BArF− = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) (5-Dy). The compounds were characterized using elemental analysis, IR spectroscopy, DC and AC magnetic measurements and photophysical investigations. L is an efficient organic chromophore for the sensitization of both visible Dy(III) (1-Dy) and near-infrared Yb(III) (2-Yb and 4-Yb) luminescence. The combination of excitation and emission spectra allowed the determination of the crystal field spitting of both the 2F7/2 ground state and 2F5/2 excited state for 2-Yb and 4-Yb. Moreover, 3-Dy and the two Yb(III) derivatives displayed field-induced single-molecule magnet (SMM) behaviour with slow magnetic relaxation occurring through the Raman process only for 2-Yb and 4-Yb, whereas a combination of Orbach and Raman processes was identified for 3-Dy.
Introduction
Since the end of the 40's and the discovery of the [2.2]paracyclophane (pCp) and its derivatives, their unique structural and electronic properties have made them a centre of interest for chemists.1 Indeed, these organic molecules are composed of two ethylene arms linking two distorted benzene, which can be decorated with a plethora of chemical groups.2 [2.2]Paracyclophanes have been successfully exploited in materials science for the development of organic light-emitting diodes (OLEDs) and nonlinear optical materials.3 The most fascinating property of the pCps is probably their planar chirality.4 Since their rigid structure precludes any rotational motion of their aromatic rings, all mono-substituted pCps are optically active compounds, and different planar chiral derivatives can be rapidly generated by increasing the number of substituents on the pCp core. The resulting optically active pCp derivatives were exploited to build families of luminescent compounds displaying electronic circular dichroism (ECD) and circularly polarized luminescence (CPL).5 Moreover, such pCp could be used as ligands for designing coordination complexes or catalysts in asymmetric catalysis and stereoselective synthesis.6,7Focusing on coordination complexes, lanthanide ions are fascinating metal centres due to their magnetic and optical behaviours. On the one hand, their high magnetic moment and strong magnetic anisotropy made it possible to observe a slow magnetic relaxation for a terbium mononuclear complex in 2003,8 called single-molecule magnet (SMM) behavior,9 paving the way for potential applications in high-density data storage,10 quantum computing11 and spintronics.12 Then, chemists developed several approaches to enhance the magnetic performances of SMMs, such as the involvement of organic radicals to favour strong magnetic exchange interaction,13,14 organometallic chemistry to impose strong uniaxial crystal field and high blocking temperatures15–18 and metal–metal bond to increase the coercive field.19 On the other hand, lanthanide ions are well-known for their specific emission line-like spectra and long emission lifetimes,20–22 finding applications in bioimaging,23–25 optical telecommunication devices26–30 and OLEDs.31,32 For more than a decade, several groups demonstrated that magneto-structural correlation could be established between the magnetic and emissive properties in order to gain deeper insight into the physical behaviour of such systems.33–39
The association of lanthanide coordination complexes with chirality paved the way for the observation of new physical properties due to the cross-effect or synergy between two properties. For instance, circularly polarized luminescence and magneto-chiral dichroism (MChD) can be detected with remarkable signal intensities for chiral luminescent magnetic lanthanide complexes.40–44 In this context, some of us exploited chiral ligands with helicoidal (helicene ligands) and axial (binaphtyl ligands) chirality to design multifunctional materials displaying CPL and/or MChD.45–48 The use of pCp with planar chirality could be a suitable approach for designing new CPL/MChD active lanthanide systems, allowing us to go further into the origin of these properties by highlighting the role of the type of chirality. However, examples of pCp-based lanthanide compounds are very scarce and have only been studied in their racemic form for their optical properties (emission and singlet oxygen generation).49,50 To the best of our knowledge, no single-crystal X-ray diffraction structures of these compounds have been reported so far.
In the next lines and as a first step before going to design chiral pCp lanthanide compounds, we proposed to use a racemic [2.2]paracyclophane decorated bisoxophosphine ligand (L) to coordinate the building blocks of formula Ln(β-diketonate)3(H2O)2 (Ln = Dy(III) and Yb(III) (β-diketonate = hfac−, 1,1,1,5,5,5-hexafluoroacetylacetonate or tta−, 2-tenoyl-trifluoroacetylacetonate) and extend the rare reported examples of pCp-based lanthanide coordination complexes. Their association led to the formation of four mononuclear complexes [Ln(hfac)3(L)] (Ln = Dy(III) (1-Dy) and Yb(III) (2-Yb)) and [Ln(tta)3(L)] (Ln = Dy(III) (3-Dy) and Yb(III) (4-Yb)) and one dinuclear complex [Na(Dy2(hfac)6(L)2)](BArF) (5-Dy) in the presence of the bulky tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF−) anion. All molecular structures were elucidated using single-crystal X-ray diffraction, and their magnetic and photophysical properties were investigated. A magneto-structural correlation was performed and rationalized by a computational approach.
Results and discussion
Synthesis of the [2.2]paracyclophane-based bisoxophosphine ligand (L)
The pCp-based ligand L was prepared in two steps, starting from commercially available 4,16-dibromo[2.2]paracyclophane (P1) via an optimization of previously reported procedures. P1 was first submitted to an isomerization reaction occurring in triglyme at 220 °C under microwave irradiation. This transformation allowed us to isolate the desired racemic 4,12-dibromo[2.2]paracyclophane (P2) in 84% yield after purification over silica gel column chromatography. This compound was then submitted to a double bromine-lithium exchange, followed by a reaction with diphenylphosphinyl chloride (2.2 equiv.) to obtain the desired product in 45% isolated yield (Scheme 1). The racemic pCp ligand L was used for the preparation of all mononuclear Ln complexes. The latter were easily obtained by an equimolar reaction of L with Ln(hfac)3(H2O)2 or Ln(tta)3(H2O)2 (Ln = Dy(III) and Yb(III)).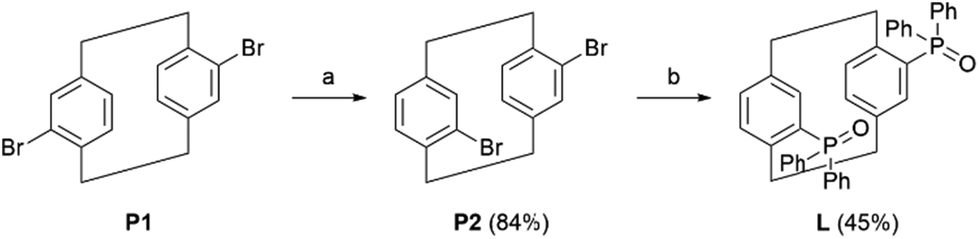 | ||
| Scheme 1 Synthetic route for L with reaction conditions: (a) μW, triglyme, 220 °C, 2 h (three iterations); (b) t-BuLi (1M in THF, 4 equiv.), THF, −78 °C to rt, 15 min, then Ph2POCl (2.2 equiv.), rt, 2 h. | ||
Crystal structure analysis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length (2.249 Å) is shorter than the Dy–Ohfac bond length (2.385 Å). The arrangement of the ligands around the lanthanide ion leads to a dodecahedron (D2d symmetry) as coordination polyhedra. The distortion is visualized by continuous shape measures performed with SHAPE 2.1 (Table S2†).51 The single crystal of 1-Dy contains a 1
O bond length (2.249 Å) is shorter than the Dy–Ohfac bond length (2.385 Å). The arrangement of the ligands around the lanthanide ion leads to a dodecahedron (D2d symmetry) as coordination polyhedra. The distortion is visualized by continuous shape measures performed with SHAPE 2.1 (Table S2†).51 The single crystal of 1-Dy contains a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Rp and Sp enantiomers of L. The torsion angle between the benzene rings of the paracyclophane moiety is ±24.6°. The crystal packing revealed π-interactions between the phenyl rings of the pCp and diphenyl oxo-phosphine groups (Fig. S2†). The shortest Dy⋯Dy distance was measured at 11.618 Å. All the complexes in the (110) plane (Fig. S2†) involved L ligand with the same chirality while the neighbouring plane contains complexes with ligand L of opposite chirality. The phase purity was checked by powder X-ray diffraction comparing the experimental PXRD of 1-Dy with the simulated PXRD obtained from the single crystal X-ray structure (Fig. S3†).
:1 mixture of Rp and Sp enantiomers of L. The torsion angle between the benzene rings of the paracyclophane moiety is ±24.6°. The crystal packing revealed π-interactions between the phenyl rings of the pCp and diphenyl oxo-phosphine groups (Fig. S2†). The shortest Dy⋯Dy distance was measured at 11.618 Å. All the complexes in the (110) plane (Fig. S2†) involved L ligand with the same chirality while the neighbouring plane contains complexes with ligand L of opposite chirality. The phase purity was checked by powder X-ray diffraction comparing the experimental PXRD of 1-Dy with the simulated PXRD obtained from the single crystal X-ray structure (Fig. S3†).
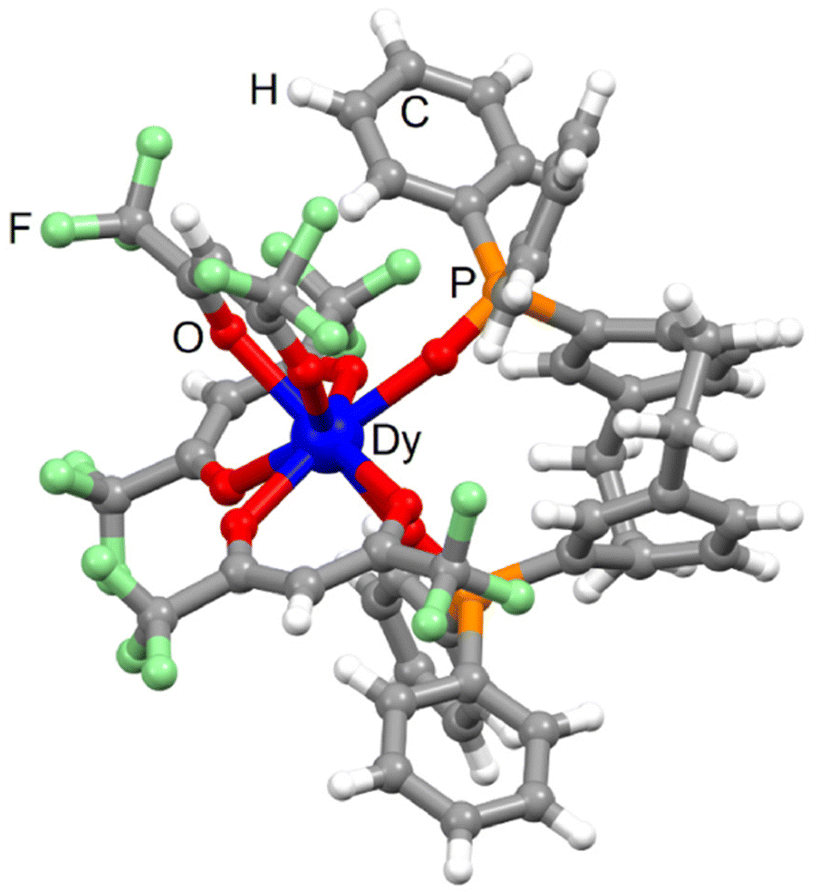 | ||
| Fig. 1 Molecular structure of 1-Dy. Colour code: white, H; grey, C; green, F; red, O; orange, P; blue, Dy. | ||
O and Yb–Ohfac bond lengths are equal to 2.188 Å and 2.336 Å, respectively. The torsion angle is ±22.4° and the shortest Yb⋯Yb intermolecular distance is equal to 12.148 Å. The phase purity was checked by powder X-ray diffraction comparing the experimental PXRD of 2-Yb with the simulated PXRD obtained from the single crystal X-ray structure (Fig. S5†).
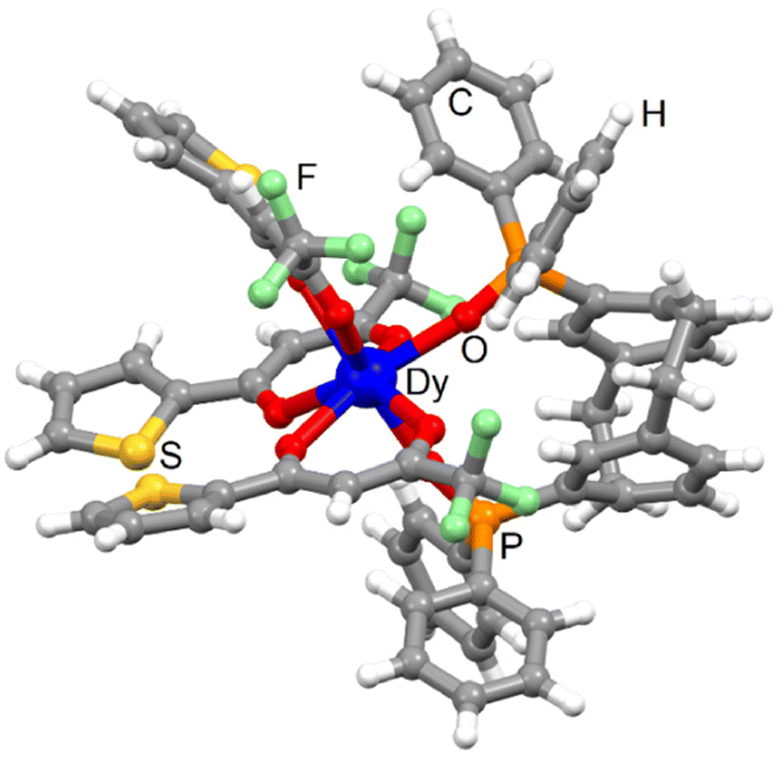 | ||
| Fig. 2 Molecular structure of 3-Dy. Code colour: white, H; grey, C; green, F; red, O; orange, P; yellow, S; blue, Dy. | ||
The Dy(III) centre is surrounded by eight oxygen atoms coming from three tta− anions and one bischelating L. The average Dy–OPO bond length (2.265 Å) [2.222 Å] is shorter than the Dy–Otta one (2.374 Å) [2.344 Å] and they are close to those found for 1-Dy. The replacement of hfac− with tta− did not change the arrangement of the ligands around the lanthanide ions. The coordination polyhedron keeps the dodecahedron prism (D2d symmetry) with a similar distortion from the ideal symmetry than the one calculated for 1-Dy [3-Dy] and 2-Yb [4-Yb] (Table S2†).51 The torsion angle was measured at ±23.6° [22.4°]. Intramolecular S⋯S contacts were identified with the S2⋯S3 short contact (3.631 Å) [3.590 Å]. Crystal packing revealed π-interactions between the phenyl rings of the pCp and diphenyl oxo-phosphine groups and additional intermolecular S2⋯S2 short contact (3.601 Å) [3.678 Å] (Fig. S8†). The shortest Dy⋯Dy distance was measured to be 11.163 Å [11.217 Å]. The phase purity was checked using powder X-ray diffraction comparing the experimental PXRD of 3-Dy with the simulated PXRD obtained from the single crystal X-ray diffraction (Fig. S9†).
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (N°2) (Table S1†). The asymmetric unit is composed of two [Dy(hfac)3(L)] complexes, one Na(I) cation and one BArF− anion (Fig. 3 and Fig. S10†).
(N°2) (Table S1†). The asymmetric unit is composed of two [Dy(hfac)3(L)] complexes, one Na(I) cation and one BArF− anion (Fig. 3 and Fig. S10†).
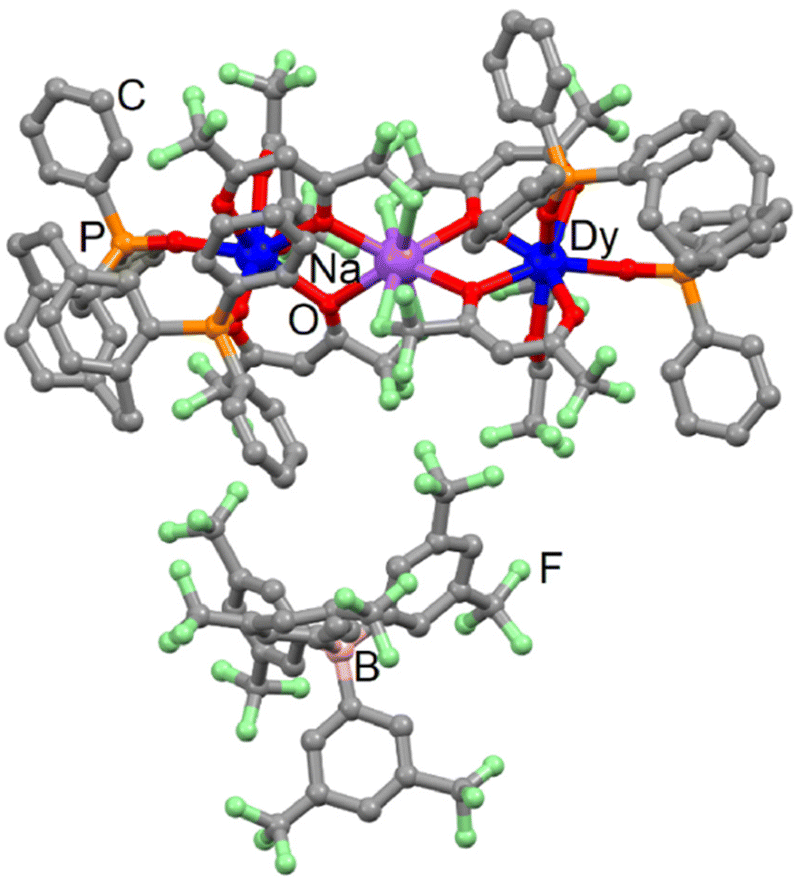 | ||
| Fig. 3 Molecular structure of 5-Dy. Code colour: grey, C; green, F; red, O; orange, P; purple, Na; blue, Dy. | ||
5-Dy can be seen as two 1-Dy complexes linked by a sodium cation. The latter is eight coordinated to four fluorine atoms (Na–F = 2.416 Å) and four oxygen atoms (Na–O = 2.435 Å) from the hfac− ligand. Such an assembly is surprising because some of us showed that the Ln(hfac)3 building block is able to release a β-diketonate in the presence of NaBArF salt.52 The sodium cation links two complexes involving L with opposite chirality. The torsion angles are −23.9° and +15.1° for the pCp, respectively, coordinated to the Dy1 and Dy2 centres. The average Dy⋯Na distance is 3.935 Å while the intramolecular Dy⋯Dy distance is 7.846 Å. Moreover, the shortest intermolecular Dy⋯Dy distance is 11.826 Å. The crystal packing highlighted that the BArF− anion prevents π-interactions between the phenyl rings of paracyclophane and bisphenyl oxophosphine groups (Fig. S11†).
Magnetic investigations
The temperature dependence of χMT for samples 1–5 are represented in Fig. 4. Room temperature values are 13.74 cm3 K mol−1, 2.37 cm3 K mol−1, 13.39 cm3 K mol−1, 2.26 cm3 K mol−1 and 26.56 cm3 K mol−1 for 1–5, respectively. Such values are in agreement with the expected values of 14.17 cm3 K mol−1 (for one Dy(III) with a 6H15/2 ground state (GS) and gJ = 4/3), 2.57 cm3 K mol−1 (for one Yb(III), 2F7/2 GS, gJ = 8/7) and 28.34 cm3 K mol−1 for two Dy(III) ions.53,54 Upon cooling, χMT decreases monotonically down to 10.55 cm3 K mol−1, 1.00 cm3 K mol−1, 11.10 cm3 K mol−1, 0.84 cm3 K mol−1 and 19.12 cm3 K mol−1 for 1–5, respectively. Such a decrease could be attributed to both thermal depopulation of the mJ states of the Dy(III) and Yb(III) ions and antiferromagnetic interactions between the different magnetic centres. Magnetizations for 1–5 at 2 K are depicted in Fig. S12† with experimental values of 4.86Nβ, 1.47Nβ, 5.02Nβ, 1.46Nβ and 9.89Nβ at 50 kOe, respectively. DC magnetic measurements show that the two Yb(III) derivatives have very similar magnetic behaviour while significant differences are observed for the Dy(III) derivatives. One could thus anticipate very similar dynamic magnetic behaviour for the two Yb(III) derivatives while some differences may appear for the two Dy(III) derivatives.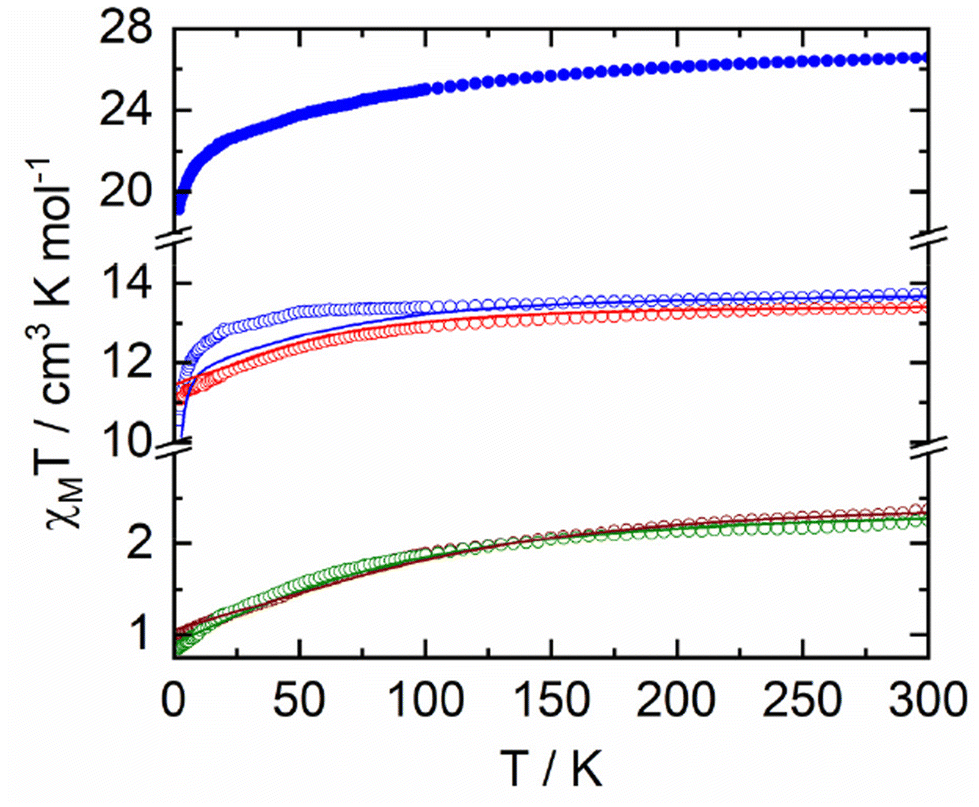 | ||
| Fig. 4 Temperature dependence of χMT for 1-Dy (open blue circles), 2-Yb (open wine circles), 3-Dy (open red circles), 4-Yb (open green circles) and 5-Dy (full blue circles). Full lines correspond to calculated data. | ||
As shown in Fig. 4 and Fig. S12,† DC magnetic data are well corroborated by the results of ab initio calculations (see computational details in Experimental section) on 1-Dy, 2-Yb, 3-Dy and 4-Yb. As already seen in previous works,55,56 while a good agreement is obtained at the CASSCF level for the Dy complexes, adding dynamical correlation through perturbative treatment is mandatory for the Yb systems. Computed energy and wavefunction composition for each MJ state of the ground-state multiplet as well as component values of the Lande g factor are given in Tables S9–S12.† In particular, one may notice the highly mixed composition of the ground state wavefunction of 2-Yb and 4-Yb that precludes any exciting SMM behaviour. For the Dy derivatives, while 1-Dy presents a GS with non-negligible |±13/2〉 contribution and extremely close first excited state, 3-Dy behaves differently with a well-separated ground state mainly composed of Mj = |±15/2〉 giving rise to Ising g-tensor (gx = gy = 0; gz = 19.5), a prerequisite for observing SMM behaviour.
The in-phase (χ′M) and out-of-phase (χ′′M) components of the AC magnetic susceptibility for compounds 1–5Ln were measured using immobilized selected and crunched single crystals. For both compounds 1-Dy and 5-Dy, no out-of-phase signal was detected in zero and applied magnetic field in the 1–10000 Hz frequency range and at 2 K. The three other compounds 2–4Ln did not display any χ′′M contribution in zero DC field due to significant quantum tunnelling of the magnetization (QTM). Nevertheless, QTM could be cancelled by applying an external DC field (Fig. S13–S15†). Field dependence of the magnetic susceptibility for 2–4Ln were measured, and the relaxation times (τ) were extracted with an extended Debye model57–59 (eqn (S1) and Tables S3–S5†) fitting simultaneously the two in-phase (χ′M) and out-of-phase (χ′′M) components of magnetic susceptibility. The best fit of the field dependence of the relaxation time at 2 K was obtained using eqn (1).
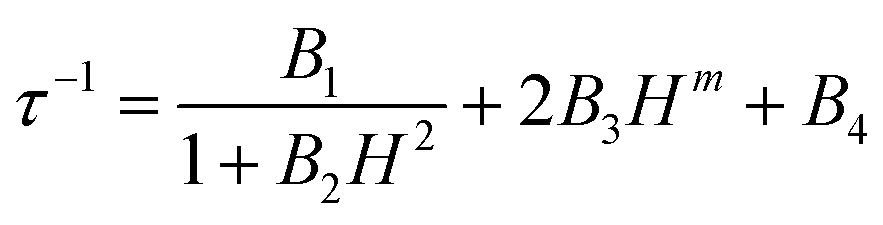 | (1) |
From left to right, the terms are the expressions of QTM, direct and thermally activated (Orbach + Raman) contributions. The best-fitted parameters for m fixed to 4 are: B1 = 3.33(2) × 106 s−1, B2 = 8.85(6) × 10−3 Oe−2, B3 = 8.20(5) × 10−13 s−1 K−1 Oe−4 and B4 = 29.25(6) s−1 for 3-Dy, B1 = 1.32(13) × 103 s−1, B2 = 7.71(15) × 10-6 Oe−2, B3 = 7.67(13) × 10−13 s−1 K−1 Oe−4 and B4 = 7.95(13) × 102 s−1 for 2-Yb and B1 = 9.54(42) × 103 s−1, B2 = 1.83(1) × 10−5 Oe−2, B3 = 8.21(5) × 10−12 s−1 K−1 Oe−4 and B4 = 60.03(51) × 102 s−1 for 4-Yb. It is worth noting that the fits are used in a qualitative manner to evaluate the nature of the magnetic relaxation involved at 2 K under an applied magnetic field. Thus, the fitting procedure highlights that QTM, thermally activated mechanisms (Orbach + Raman) and direct relaxation processes are dominating in weak, moderate and high fields, respectively. The selected field, defined as a compromise between the relaxation time and the amplitude of the out-of-phase component, is 1000 Oe for 2-Yb, 3-Dy and 4-Yb.
Under such an applied field, 2–4Ln show frequency dependence of the out-of-phase signal of the magnetization (Fig. S16–S18†), which can be analysed in the framework of the extended Debye model.57–59 The temperature dependence of the relaxation time is plotted and depicted in Fig. 5d (Tables S6–S8†), and the normalized Argand (Fig. S19–S21†) concluded that more than 90% of the sample was slowly relaxing under the selected applied DC field. The thermal variation of the relaxation time could be fitted using eqn (2) for the three complexes considering a Raman process. For 3, an additional Orbach process was identified for T > 4 K.
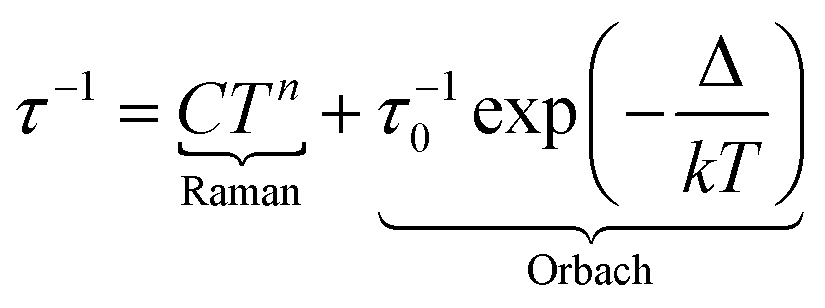 | (2) |
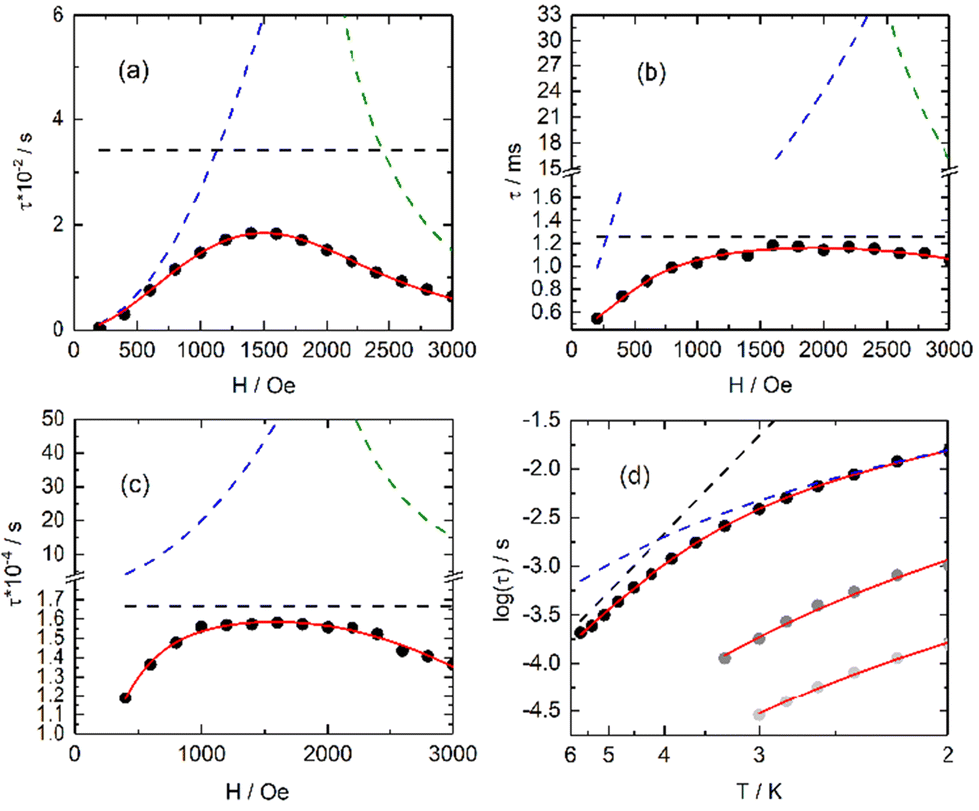 | ||
| Fig. 5 Field dependence of the magnetic relaxation time (full black circles) at 2 K in the field range of 0–3000 Oe for 3-Dy (a), 2-Yb (b) and 4-Yb (c) with the corresponding best fit depicted in the full red line. The separated thermally activated Orbach + Raman, QTM and direct processes are drawn in dashed black, blue and green lines, respectively. (d) Thermal dependence of the magnetic relaxation time for 2-Yb under 1000 Oe applied magnetic field in the 2–3.4 K temperature range (full grey circles), for 3-Dy under 1000 Oe applied magnetic field in the 2–5.7 K temperature range (full black circles) and for 4-Yb under 1000 Oe applied magnetic field in the 2–3 K temperature range (full light grey circles). Full red lines are the best-fitted curves (see text) while the dashed lines are the Orbach (black) and Raman (blue) contributions. | ||
The best fit was obtained with C = 36.7(8) K−n s−1 and n = 4.53(2) for 2-Yb, C = 8.2(3) K−n s−1 with n = 2.96(2) and τ0 = 2.12(11) × 10−6 s and Δ = 27.7(4) K (19 cm−1) for 3-Dy and C = 496.3(26) K−n s−1 with n = 1.83(6) for 4-Yb, where C and n are the constant and exponent factor of the Raman process, τ0 and Δ are the relaxation time and the energy barrier of the Orbach relaxation process. The energy barrier found from the AC data (19 cm−1) and ab initio calculation (31.8 cm−1) are of the same order of magnitude. The expected n value for Kramers ions should be 9,60 but it is well known that for molecular systems, the presence of both acoustic and optical phonons can lead to lower values comprised between 2 and 761–63 or even lower than 2 for some organometallic complexes and when phonon bottleneck process is involved.64
As expected from the DC magnetic measurements, the two Yb(III) derivatives (2-Yb and 4-Yb) behave similarly with field-induced SMM behaviour while the Dy(III) compounds show different magnetic behaviour with no slow magnetic relaxation for 1-Dy and 5-Dy but a field-induced SMM behaviour for 3-Dy. This difference in SMM behaviour within the Dy series is well illustrated computationally by the magnetic transition moments calculated for 1-Dy and 3-Dy (Fig. S22†) and computed energy levels (Tables S9–S12†). Moreover, the main magnetic anisotropy axis appears perpendicular to the PO–Dy–OP direction and along the two hfac− anions, which are localized in the same plane. Such direction corresponds to the most charged orientation of the oblate Dy(III) ion coordination sphere (Fig. S23†) while the less charged orientation is preferred for the main component of the magnetic anisotropy axis in the case of prolate Yb(III) derivatives (2-Yb and 4-Yb) (Fig. S24†).
Photophysical properties
The emission properties of the four mononuclear complexes 1–4Ln were recorded at room temperature and at low temperature (77 K) in the solid state under 340 nm light irradiation. 1-Dy displayed the classical visible emission of the Dy(III) ion attributed to the 4F9/2 → 6Hn/2 transitions (Fig. 6a and Fig. S25†) while the two 2-Yb and 4-Yb complexes displayed the classical NIR Yb(III) centred emission attributed to the 2F5/2 → 2F7/2 transitions (Fig. 6b and Fig. S26†). On the contrary, 3-Dy showed no metal-centred emission, probably because Dy(III) luminescence (475 nm, 21053 cm−1) is quenched by the too-low-energy triplet state of the tta− anion in the Ln(tta)3 moiety (527 nm, 18975 cm−1).65 The emission spectra recorded at room temperature are not resolved enough to define the crystal field splitting of the ground state. Thus, emission measurements were carried out at liquid nitrogen temperature. At 77 K, the emission spectrum of 1-Dy displayed eight contributions centred at 472.7 nm (21155 cm−1), 474.3 nm (21084 cm−1), 476.4 nm (20991 cm−1), 478.3 nm (20907 cm−1), 479 nm (20877 cm−1), 481.1 nm (20786 cm−1), 482.2 nm (20738 cm−1) and 489 nm (20450 cm−1). The confrontation with the excitation spectra (Fig. 6a) allowed us to attribute the two highest-energy lines to hot bands and the emission contribution at 20991 cm−1 to the zero-phonon line. Thus, six of the eight expected contributions for the 4F9/2 → 6H15/2 transition could be identified while the two missing contributions cannot be unambiguously identified. The computational investigation suggested that the energy difference between the two lowest energy Kramers doublets (KDs) is very weak (7 cm−1). Such a small energy gap was not observable on the luminescence spectrum of 1-Dy but it was in agreement with the lack of slow magnetic relaxation. As already observed for similar Dy(III) coordination compounds,66,67 the energy splitting for the 6H15/2 ground state tends to be underestimated by calculation at the CASSCF level (Fig. 6a).
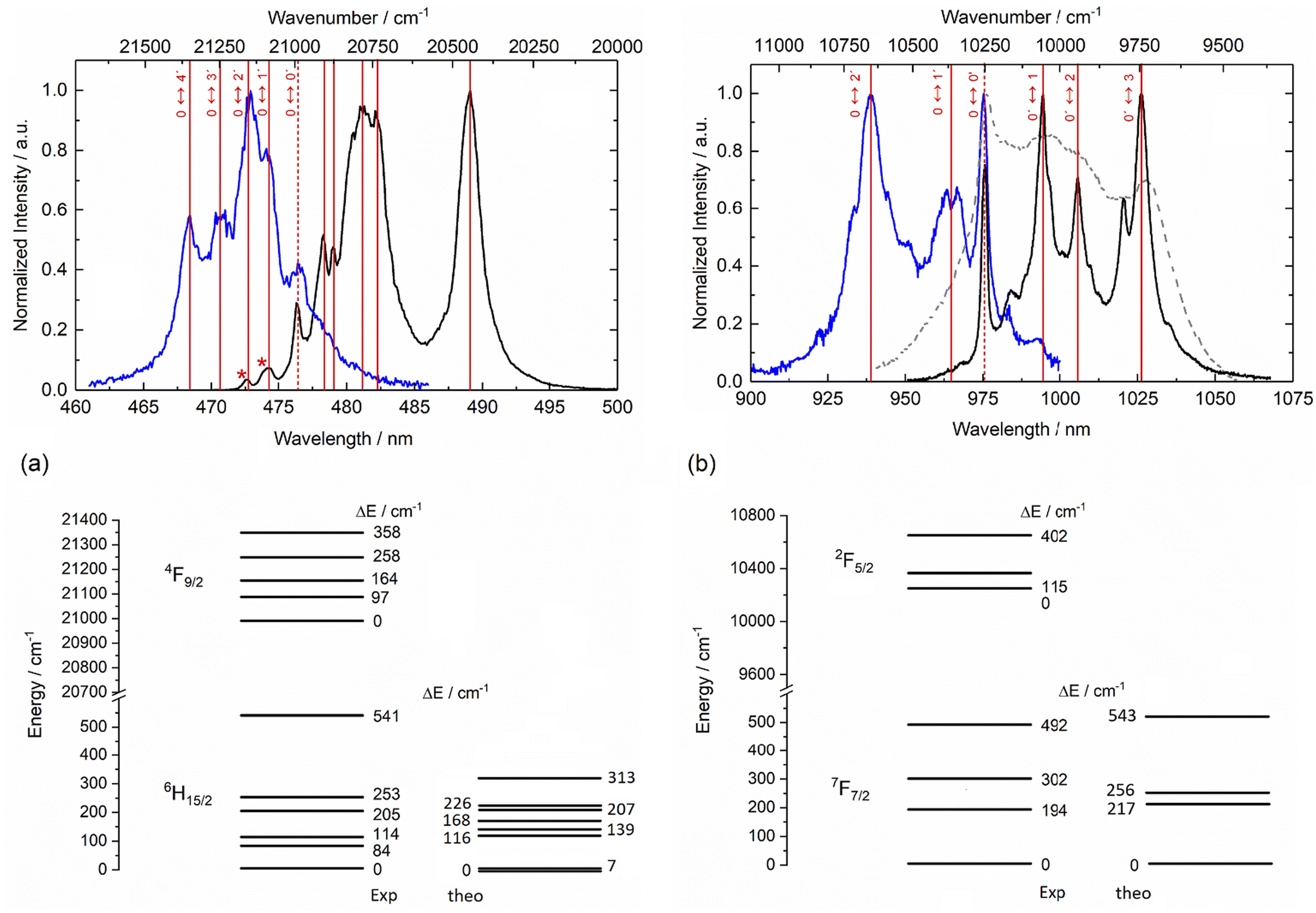 | ||
| Fig. 6 Room (dashed grey line, only for Yb(III) derivatives) and low (full black line) temperature (77 K) solid-state emission spectra for 1-Dy (a) and 2-Yb (b) under irradiation at λexc = 340 nm and their corresponding excitation spectra (full blue line) obtained by detection at 490 nm for 1-Dy and 1025 nm for 2-Yb. The vertical red sticks represent the experimental energy splitting of the ground and first excited levels, with the dashed vertical sticks indicating the zero-phonon line. The corresponding energy diagrams of the ground and first excited state splitting for the two compounds are depicted at the bottom part of the corresponding spectra. | ||
Irradiation at 340 nm of the solid-state samples of 2-Yb and 4-Yb at 77 K led to the observation of resolved Yb(III) centred emission spectra (Fig. 6b and Fig. S26†). Each spectrum displayed the four main signals expected for the 2F5/2 → 2F7/2 transition. The energy differences between each experimentally observed mJ doublet correspond to the crystal field splitting of the 2F7/2 ground state and are represented in the respective energy diagrams (right part of Fig. 6b and Fig. S26†). The measurements of the excitation spectra for both compounds led to the determination of the energy splitting for the 2F5/2 excited state as well as the identification of the zero-phonon line. The energy splitting of both ground and excited states are similar for 2-Yb and 4-Yb, which is in agreement with their structural analysis. The total energy splitting for the ground state is 492 cm−1 for 2-Yb and 529 cm−1 for 4-Yb. Such total splitting values indicate low symmetrical coordination environments of the metal ion,68,69 as already observed in previously reported compounds.46 This splitting is well recovered at the CASPT2 level for 2-Yb, especially the energy gap between the lowest KDs (ΔEexp = 194 cm−1 and ΔEcalc = 217 cm−1) (Fig. 6b) while it remains slightly too low in the case of 4-Yb (ΔEexp = 283 cm−1 and ΔEcalc = 121 cm−1) (Fig. S26†). The poor agreement with the computed energy splitting for 4-Yb (Fig. S26†) could be due to the uncertainties of the experimental emission contributions linked to the lower resolution of the 4-Yb emission spectra compared to that of 2-Yb. Additional emission lines can be identified for the two Yb(III) analogues, which can be attributed to hot bands and vibrational contributions.47,48 The emission decay curves can be fitted by mono-exponential functions (Fig. S27†), confirming the presence of a single spectroscopic Yb(III) site and thus supporting the existence of single monomeric species in solution. The order of magnitude of the observed lifetimes (τobs) is τobs = 21.0 μs for 2-Yb and 13.9 μs for 4-Yb.
Conclusions
The reaction between the [2.2]paracyclophane-based Bisoxophosphine ligand and [Ln(hfac)3(H2O)2] or [Ln(tta)3(H2O)2] (Ln(III) = Dy and Yb) led to the formation of rare examples of lanthanide coordination complexes involving paracyclophane ligand. A dinuclear complex could be obtained in the presence of NaBArF salt. The mononuclear complexes of formula [Yb(hfac)3(L)] (2-Yb), [Dy(tta)3(L)] (3-Dy) and [Yb(tta)3(L)] (4-Yb) displayed field-induced SMM behaviour with magnetic relaxation occurring through the Raman process for 2-Yb and 4-Yb, and a combination of Orbach and Raman processes for 3-Dy. The [2.2]paracyclophane decorated with two oxophosphine groups played the role of an organic chromophore for the efficient sensitization of both visible Dy(III) (for 1-Dy) and near-infrared Yb(III) (for 2-Yb and 4-Yb) emissions. In cases of Yb(III) derivatives, magneto-structural correlation and comparison of both excitation and luminescence spectra led to the determination of the crystal field splitting of the 2F7/2 ground state and 2F5/2 first excited state. 2-Yb and 4-Yb can be described as unprecedented field-induced luminescent lanthanide SMMs involving paracyclophane ligands. The elaboration of the enantiopure complexes for chiroptical investigations is in progress in our laboratory.Experimental section
Materials
The metal building blocks Ln(hfac)3(H2O)2 (hfac− = 1,1,1,5,5,5-hexafluoroacetylacetonate anion)70 and Ln(tta)3(H2O)2 (tta− = 2-thenoyltrifluoroacetylacetonate anion)71 (Ln = Dy(III) and Yb(III)) were synthesized following previously reported methods. Commercially available 4,16-dibromo[2.2]paracyclophane was purchased from Fisher Scientific and used as received. All other reagents were purchased from Merck Co., Inc. and used without further purification.Synthesis
Synthesis of 4,12-dibromo[2.2]paracyclophane (P2). In a 30 mL glass microwave vessel were added commercially available 4,16-dibromo[2.2]paracyclophane (P1, 1 equiv., 5 g, 13.6 mmol) and triglyme (20 mL). The reaction mixture was then heated to 220 °C in a microwave reactor. After 2 h, the reaction was cooled to room temperature, diluted with triglyme and the solid was filtered. The filtrate, which contains the reaction product, was diluted with water and extracted with diethyl ether (3×). The combined organic phases were washed with water and brine, dried over MgSO4, and the solvents were evaporated under reduced pressure. The solid (starting material) was recovered and engaged once again in the isomerization reaction under microwave irradiation. This step was repeated until no more solids were recovered after the process. The combined crude products were purified via silica gel column chromatography using cyclohexane/ethyl acetate 9
:1 as the eluent to obtain the desired product (4.2 g, 11.4 mmol, 84% yield) as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.19 (d, J = 1.6 Hz, 2H), 6.52 (dt, J = 7.8, 4.6 Hz, 4H), 3.45 (ddd, J = 13.3, 9.8, 2.0 Hz, 2H), 3.14–2.97 (m, 4H), 2.81 (ddd, J = 13.4, 10.3, 6.8 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3): δ 141.3, 138.7, 135.0, 132.7, 131.6, 126.6, 35.8, 32.4 ppm. Spectroscopic data are consistent with the literature data for this compound.73
Synthesis of 1,4(1,4)-dibenzenacyclohexaphane-12,43-diylbis(diphenylphosphine oxide) (L). tert-Butyllithium (4 equiv., 13.7 mL, 21.85 mmol) was added dropwise over an hour to a solution of 4,12-dibromo[2.2]paracyclophane (P2, 1 equiv., 2 g, 5.46 mmol) in dry THF (60 mL) at −78 °C. After 15 minutes, the reaction mixture was allowed to warm up to room temperature, and diphenylphosphinic chloride (2.2 equiv., 2.29 mL, 12.02 mmol) was added. The reaction was stirred for 2 hours at rt, then poured into 2 N HCl (150 mL). The resulting aqueous mixture was extracted with CH2Cl2 (3×). The organic layers were combined, dried over MgSO4, and the solvent was evaporated. The resulting solid was heated in an ethyl acetate/pentane mixture (2
:3), then cooled and filtered to afford the desired ligand L (1.3 g, 2.46 mmol, 45% yield) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.75–7.60 (m, 4H), 7.60–7.42 (m, 10H), 7.38 (td, J = 7.4, 1.2 Hz, 2H), 7.34–7.21 (m, 4H), 7.14 (dd, J = 14.6, 1.5 Hz, 2H), 6.76 (d, J = 7.6 Hz, 2H), 6.64 (dd, J = 7.6, 4.1 Hz, 2H), 3.35 (ddd, J = 12.6, 10.4, 6.7 Hz, 2H), 3.27–3.18 (m, 2H), 3.07–2.96 (m, 2H), 2.73 (ddd, J = 13.2, 10.4, 6.7 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ 144.7 (br), 139.6 (d, J = 12.8 Hz), 138.1 (d, J = 11.6 Hz), 135.3 (d, J = 11.2 Hz), 132.14 (d, J = 9.5 Hz), 131.9 (br), 131.7 (d, J = 1.6 Hz), 131.5 (br), 128.2 (d, J = 12.0 Hz), 35.7, 34.4. 31P NMR (202 MHz, CDCl3) δ 24.7 ppm. Spectroscopic data are consistent with the literature data for this compound.73
[Dy(hfac) 3 (L)] ( 1-Dy ). 1 equiv. (16.4 mg, 0.02 mmol) of Dy(hfac)3(H2O)2 was dissolved in 5 mL of CH2Cl2 and 1 equiv. (12.2 mg, 0.02 mmol) of L was dissolved in 6 mL of CH2Cl2. The two resulting CH2Cl2 solutions were mixed and stirred for 30 minutes at room temperature. After this delay, 50 mL of n-hexane was layered. Slow diffusion, followed by slow evaporation, led to colourless single crystals that were suitable for X-ray studies. 16.7 mg, 60% yield. Anal. calcd (%) for C55H37DyF18O8P2: C 47.40, H 2.66; found: C 47.32, H 2.71. I. R. (KBr, range 3200–500 cm−1) 3061, 2946, 2865, 1657, 1554, 1528, 1506, 1256, 1204, 1144, 800, 684, 662 and 585 cm−1.
[Yb(hfac) 3 (L)] ( 2-Yb ). A similar protocol as for 1-Dy was employed using 16.7 mg of Yb(hfac)3(H2O)2 instead of Dy(hfac)3(H2O)2. 14.6 mg, 52% yield. Anal. Calcd (%) for C55H37YbF18O8P2: C 47.08, H 2.64; found: C 47.12, H 2.66. I. R. (KBr, range 3200–500 cm−1) 3060, 2946, 2865, 1655, 1554, 1528, 1503, 1258, 1204, 1144, 798, 685, 660 and 585 cm−1.
[Dy(tta) 3 (L)] ( 3-Dy ). A similar protocol as for 1-Dy was employed using 17.3 mg of Dy(tta)3(H2O)2 instead of Dy(hfac)3(H2O)2. 14.1 mg, 49% yield. Anal. Calcd (%) for C64H46DyF9O8P2S3: C 53.53, H 3.21; found: C 53.44, H 3.19. I. R. (KBr, range 3200–500 cm−1) 3058, 2950, 2865, 1655, 1554, 1529, 1500, 1260, 1207, 1144, 799, 685, 660 and 584 cm−1.
[Yb(tta) 3 (L)] ( 4-Yb ). A similar protocol as for 3-Dy was employed using 18.0 mg of Yb(tta)3(H2O)2 instead of Dy(tta)3(H2O)2. 14.1 mg, 49% yield. Single crystals suitable for X-ray studies were obtained after recrystallization in toluene and diffusion of n-pentane. Anal. Calcd (%) for C64H46YbF9O8P2S3: C 53.14, H 3.18; found: C 53.11, H 3.22. I. R. (KBr, range 3200–500 cm−1) 3061, 2954, 2865, 1650, 1552, 1529, 1499, 1261, 1207, 1144, 803, 685, 659 and 584 cm−1.
[Na(Dy 2 (hfac) 6 (L) 2 )](BArF) ( 5-Dy ). 32.9 mg (0.04 mmol) of Dy(hfac)3(H2O)2 and 35.4 mg (0.04 mmol) of NaBArF were dissolved in 5 mL of CH2Cl2, and 12.2 mg (0.02 mmol) of L was dissolved in 5 mL of CH2Cl2. The two resulting CH2Cl2 solutions were mixed and stirred for 30 minutes at room temperature. After this delay, 20 mL of n-hexane was layered. Slow diffusion, followed by slow evaporation, led to colourless single crystals that were suitable for X-ray studies. 12.5 mg, 17% yield. Anal. calcd (%) for C142H73BDy2F60NaO16P4: C 46.43, H 1.99; found: C 46.39, H 2.01. I. R. (KBr, range 3200–500 cm−1) 3115, 3061, 2946, 2865, 1657, 1554, 1528, 1506, 1465, 1415, 1355, 1256, 1204, 1144, 801, 682, 660 and 583 cm−1.
Material characterization
1H NMR and 13C NMR spectra were recorded on Bruker Avance 500 spectrometers operating at 500 (H value) and 125 MHz (C value) in CDCl3. Spectra were referenced to the residual deuterated solvent (for CHCl3: 7.26 ppm, 1H; 77.16 ppm, 13C). Chemical shifts are reported in ppm, and multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet or overlap of nonequivalent resonances), dd (doublet of doublet), td (triplet of doublet), and br (broad signal). Coupling constants, J, are reported in hertz (Hz). All NMR spectra were obtained at 300 K. Analytical thin layer chromatography (TLC) was performed on silica gel plates (Merck 60F254) visualized with a UV lamp (254 nm). Flash chromatography was performed on silica gel (60–230 mesh) unless otherwise specified. Organic extracts were dried over anhydrous MgSO4. Microwave-mediated reactions were carried out using an Antoon Paar Monowave EXTRA. All solid-state characterization studies (elementary analysis, IR, PXRD, magnetic susceptibility and photophysical measurements) were performed on dried samples and are considered without crystallization solvents. The elemental analyses of the compounds were performed at the Centre Régional de Mesures Physiques de l'Ouest, Rennes.X-ray structure analysis
Single crystals were mounted on a D8 VENTURE Bruker-AXS diffractometer for data collection for compounds 1–5 (MoKα radiation source, λ = 0.71073 Å) from the Centre de Diffractométrie X (CDIFX), Université de Rennes, France. Structures were solved with direct methods using the SHELXT Program74 and refined with a full matrix least-squares method on F2 using the SHELXL-14/7 program.75 Crystallographic data are summarized in Table S1.† Crystallographic data for the structure reported in this study have been deposited with the Cambridge Crystallographic Data Centre (insert CCDC 2297628–2297632† for 4-Yb, 2-Yb, 5-Dy, 1-Dy and 3-Dy, respectively). X-ray diffraction (XRD) patterns were recorded at room temperature in the 2θ range 5–30° with a step size of 0.026° and a scan time per step of 600 s using a PANalytical X'Pert Pro diffractometer (Cu-L2, L3 radiation, λ = 1.5418 Å, 40 kV, 40 mA, PIXcel 1D detector). Data collector and HighScore Plus software were used, respectively, for recording and analysing the patterns.
Spectroscopic analysis
Luminescence spectra were measured using a Horiba-Jobin Yvon Fluorolog-3 spectrofluorimeter equipped with a three-slit double grating monochromator. The solid samples (crystalline powders) were placed in quartz tubes (4 mm diameter) and inserted in a quartz dewar that was filled with liquid nitrogen for low-temperature measurements. The excitation was provided by unpolarized light from a 450 W xenon CW lamp and detected at an angle of 90°. An 830 nm long-pass filter was placed between the sample and the detectors to avoid the presence of stray light due to excitation. Spectra were corrected for the emission spectral response (detector and grating). Near-infrared emission was recorded using a liquid nitrogen-cooled Symphony II CCD detector, and the corresponding excitation spectra were recorded using a liquid nitrogen-cooled solid indium/gallium/arsenic detector (850–1600 nm). Both emission and excitation in the UV/Vis region were recorded using a Hamamatsu R928 photomultiplier tube. For luminescence lifetimes, the sample was excited using a pulsed Nd:YAG laser (SpectraPhysics) operating at 10 Hz. Light emitted at right angles to the excitation beam was focused onto the slits of a monochromator (PTI120), which was used to select the appropriate wavelength. The growth and decay of the luminescence at selected wavelengths were detected using a Ge photodiode (Edinburgh Instruments, EI-P) and recorded using a digital oscilloscope (Tektronix TDS320) before being transferred for analysis. Luminescence lifetimes were obtained by iterative reconvolution of the detector response (obtained by using a scatterer) with exponential components for the growth and decay of the metal-centred luminescence.Magnetic analysis
The DC magnetic susceptibility measurements were performed on a solid polycrystalline sample with a Quantum Design MPMS-XL SQUID magnetometer between 2 and 300 K in an applied magnetic field of 0.02 T for temperatures of 2–20 K, 0.2 T for temperatures of 20–80 K and 1 T for temperatures of 80–300 K. AC magnetic susceptibility measurements were performed using a Quantum Design MPMS-XL SQUID for frequencies between 1 and 1000 Hz and a Quantum Design PPMS magnetometers for frequencies between 50 and 10000 Hz. These measurements were all corrected for diamagnetic contribution, as calculated with Pascal's constants.
Computational details
Wavefunction-based calculations were carried out on molecular structures of 1-Dy, 2-Yb, 3-Dy and 4-Yb complexes by using the SA-CASSCF/(MS-CASPT2)/RASSI-SO approach, as implemented in the OpenMolcas quantum chemistry package (versions 22.10).76 In this approach, the relativistic effects are treated in two steps on the basis of the Douglas–Kroll Hamiltonian. First, the scalar terms were included in the basis-set generation and were used to determine the spin-free wavefunctions and energies in the complete active space self-consistent field (CASSCF) method.77 For 1-Dy and 3-Dy, the active space consisted of the nine 4f electrons of the DyIII ion spanning the seven 4f orbitals, i.e., CAS(9,7)SCF while for 2-Yb and 4-Yb, the active space consisted of thirteen 4f electrons of the YbIII ion spanning the seven 4f orbitals, i.e. CAS(13,7)SCF. In order to obtain a better description of the electronic structure of the Ytterbium systems (2-Yb and 4-Yb), calculations were computed using perturbation theory at the second order,78i.e., MS-CAS(13,7)PT2, based on the previous CAS(13,7)SCF results. Next, spin–orbit coupling was added within the restricted-active-space-state-interaction (RASSI-SO) method, which uses spin-free wavefunctions as basis states.79,80 Spin–orbit (SO) integrals are calculated using the AMFI (atomic mean-field integrals) approximation.81 The resulting wavefunctions and energies are used to compute the magnetic properties and g-tensors of the lowest states from the energy spectrum using the pseudo-spin S = 1/2 formalism in the SINGLE-ANISO routine.82,83 Cholesky decomposition of the bielectronic integrals was employed to save disk space and speed up the calculations.84 For 1-Dy and 3-Dy, state-averaged CASSCF calculations were performed for all of the sextets (21 roots), all of the quadruplets (224 roots), and 300 out of the 490 doublets of the DyIII ion. 21 sextets, 128 quadruplets, and 107 doublets were mixed through spin−orbit coupling in RASSI-SO. For 2-Yb and 4-Yb, state-averaged CASSCF calculations were performed for the 7 doublets. All atoms were described by ANO-RCC basis sets.85,86 The following contractions were used: [8s7p4d3f2g1h] for Dy and Yb, [4s3p2d] for the O atoms, [4s3p] for the P and S atoms, [3s2p] for the C and F atoms and [2s] for the H atoms. The atomic positions were extracted from the X-ray crystal structures. Only the positions of the H atoms were optimized with the AMS program suite (revision 2022.103).87,88 These calculations utilized the scalar all-electron zeroth-order regular approximation (ZORA)S15 along with the revPBE (Perdew–Burke–Ernzerhof) functional.89,90 For all atoms, the atomic basis set corresponded to the triple-ζ polarized (TZP) Slater-type orbital (STO) all-electron basis set.91Author contributions
J. B. and E. B. performed the organic syntheses, H. F. performed the coordination chemistry and crystallisations; V. D. realised the single crystal X-ray diffraction experiments and refined the X-ray structures; T. G. and O. C. performed and analysed the magnetic measurements. B. L. G. performed the computational investigation. A. S. and O.M. investigated the absorption and emission properties. L. M. and E. B. discussed the idea and the results and commented on the manuscript. F. P. conceived and designed the experiments and drove the writing of the article. All authors have read and agreed on the published version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by CNRS, Université de Rennes, the European Research Council through the ERC-CoG 725184 MULTIPROSMM (project no. 725184) and the Agence Nationale de la Recherche (SMMCPL ANR-19-CE29-0012-02; PhotoChiraPhane ANR-19-CE07-0001). B. L. G. thanks the French GENCI/IDRIS-CINES centres for high-performance computing resources. Yannick Guyot is acknowledged for the luminescence lifetime measurements.References
- C. J. Brown and A. C. Farthing, Nature, 1949, 164, 915–916 CrossRef CAS.
- H. Hope, J. Bernstein and K. N. Trueblood, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 1733–1743 CrossRef CAS.
- G. P. Bartholomew, I. Ledoux, S. Mukamel, G. C. Bazan and J. Zyss, J. Am. Chem. Soc., 2002, 124, 13480–13485 CrossRef CAS PubMed.
- D. J. Cram and N. L. Allinger, J. Am. Chem. Soc., 1955, 77, 6289 CrossRef CAS.
- S. Felder, M.-L. Delcourt, R. Rodríguez, L. Favereau, J. Crassous, L. Micouin and E. Benedetti, J. Mater. Chem. C, 2023, 11, 2053–2062 RSC.
- Z. Hassan, E. Spuling, D. M. Knoll, J. Lahann and S. Bräse, Chem. Soc. Rev., 2018, 47, 6947–6963 RSC.
- S. E. Gibson and J. D. Knight, Org. Biomol. Chem., 2003, 1, 1256–1269 RSC.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194–197 CrossRef CAS PubMed.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- K. S. Pedersen, A.-M. Ariciu, S. McAdams, H. Weihe, J. Bendix, F. Tuna and S. Piligkos, J. Am. Chem. Soc., 2016, 138, 5801–5804 CrossRef CAS PubMed.
- J. D. Rinehart, M. Fang, W. K. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed.
- S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546–18549 CrossRef CAS PubMed.
- R. A. Layfield, M.-L. Tong, B. M. Day, Y.-C. Chen, F.-S. Guo and A. Mansikkamaki, Science, 2018, 362, 1400–1403 CrossRef PubMed.
- K. R. McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, Chem. Sci., 2018, 9, 8492–8503 RSC.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- A. H. Vincent, Y. L. Whyatt, N. F. Chilton and J. R. Long, J. Am. Chem. Soc., 2023, 145, 1572–1579 CrossRef CAS PubMed.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E.-S. Choi, J.-G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS PubMed.
- J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- S. V. Eliseeva and J.-C. G. Bünzli, in Lanthanide spectroscopy, Materials, and Bioapplications, Springer Series on Fluorescence, ed. P. Hänninen and H. Härmä, Springer, Verlag, Berlin, 2010, vol. 7 Search PubMed; Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, ed. A. De Bettencourt-Diaz, Wiley, New York, 2014 Search PubMed.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS PubMed.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- A. D'Aleo, A. Bourdolle, S. Bulstein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet and O. Maury, Angew. Chem., Int. Ed., 2012, 51, 6622–6625 CrossRef PubMed.
- S. Banerjee, L. Huebner, M. D. Romanelli, G. A. Kumar, R. E. Riman, T. J. Emge and J. G. Brennan, J. Am. Chem. Soc., 2005, 127, 15900–15906 CrossRef CAS PubMed.
- G. A. Kumar, R. E. Riman, L. A. Diaz Torres, S. Banerjee, M. D. Romanelli, T. J. Emge and J. G. Brennan, Chem. Mater., 2007, 19, 2937–2946 CrossRef CAS.
- S. Banerjee, G. A. Kumar, R. E. Riman, T. J. Emge and J. G. Brennan, J. Am. Chem. Soc., 2007, 129, 5926–5931 CrossRef CAS PubMed.
- M. Romanelli, G. A. Kumar, T. J. Emge, R. E. Riman and J. G. Brennan, Angew. Chem., Int. Ed., 2008, 47, 6049–6051 CrossRef CAS PubMed.
- L. Song, X. Liu, Z. Zhen, C. Chen and D. Zhang, J. Mater. Chem., 2007, 17, 4586–4590 RSC.
- M. D. McGehee, T. Bergstedt, C. Zhang, A. P. Saab, M. B. O'Regan, G. C. Bazan, V. I. Srdanov and A. J. Heeger, Adv. Mater., 1999, 11, 1349–1354 CrossRef CAS.
- A. de Bettencourt-Dias, Dalton Trans., 2007, 2229–2241 RSC.
- F. Pointillart, B. Le Guennic, O. Cador, O. Maury and L. Ouahab, Acc. Chem. Res., 2015, 48, 2834–2842 CrossRef CAS PubMed.
- J. H. Jia, Q.-W. Li, Y.-C. Chen, J.-L. Liu and M.-L. Tong, Coord. Chem. Rev., 2019, 378, 365–381 CrossRef CAS.
- R. Marin, G. Brunet and M. Murugesu, Angew. Chem., Int. Ed., 2021, 60, 1728–1746 CrossRef CAS PubMed.
- G. Brunet, R. Marin, M.-J. Monk, U. Resch-Genger, D. A. Gálico, F. A. Sigoli, E. A. Suturina, E. Hemmer and M. Murugesu, Chem. Sci., 2019, 10, 6799–6808 RSC.
- J. Wang, J. J. Zakrzewski, M. Heczko, M. Zychowicz, K. Nakagawa, K. Nakabayashi, B. Sieklucka, S. Chorazy and S. Ohkoshi, J. Am. Chem. Soc., 2020, 142, 3970–3979 CrossRef CAS PubMed.
- D. Errulat, R. Marin, D. A. Gálico, K. L. M. Harriman, A. Pialat, B. Gabidullin, F. Iikawa, O. D. D. Couto Jr, J. O. Moilanen, E. Hemmer, F. A. Sigoli and M. Murugesu, ACS Cent. Sci., 2019, 5, 1187–1198 CrossRef CAS PubMed.
- P.-Y. Liao, Y. Liu, Z.-Y. Ruan, H.-L. Wang, C.-G. Shi, W. Deng, S.-G. Wu, J.-H. Jia and M.-L. Tong, Inorg. Chem., 2023, 62, 1075–1085 CrossRef CAS PubMed.
- Y. B. Tan, Y. Okayasu, S. Katao, Y. Nishikawa, F. Asanoma, M. Yamada, J. Yuasa and T. Kawai, J. Am. Chem. Soc., 2020, 142, 17653–17661 CrossRef CAS PubMed.
- J. L. Lunkley, D. Shirotani, K. Yamanari, S. Kaizaki and G. Muller, J. Am. Chem. Soc., 2008, 130, 13814–13815 CrossRef CAS PubMed.
- O. G. Willis, F. Zinna, G. Pescitelli, C. Micheletti and L. Di Bari, Dalton Trans., 2022, 51, 518–523 RSC.
- N. F. M. Mukthar, N. D. Schley and G. Ung, J. Am. Chem. Soc., 2022, 144, 6148–6153 CrossRef CAS PubMed.
- B.-A. N. Willis, D. Schnable, N. D. Schley and G. Ung, J. Am. Chem. Soc., 2022, 144, 22421–22425 CrossRef CAS PubMed.
- C. A. Mattei, K. Dhbaibi, B. Lefeuvre, V. Dorcet, G. Argouarch, O. Cador, B. Le Guennic, O. Maury, C. Lalli, S. Guy, A. Bensalah-Ledoux, F. Riobé, B. Baguenard and F. Pointillart, Chirality, 2021, 1–14 Search PubMed.
- C. A. Mattei, V. Montigaud, B. Lefeuvre, V. Dorcet, G. Argouarch, O. Cador, B. Le Guennic, O. Maury, C. Lalli, Y. Guyot, S. Guy, C. Gindre, A. Bensalah-Ledoux, F. Riobé, B. Baguenard and F. Pointillart, J. Mater. Chem., 2023, 11, 7299–7310 RSC.
- M. Atzori, K. Dhbaibi, H. Douib, M. Grasser, V. Dorcet, I. Breslavetz, K. Paillot, O. Cador, G. L. J. A. Rikken, B. Le Guennic, J. Crassous, F. Pointillart and C. Train, J. Am. Chem. Soc., 2021, 143, 2671–2675 CrossRef CAS PubMed.
- K. Dhbaibi, M. Grasser, H. Douib, V. Dorcet, O. Cador, N. Vanthuyne, F. Riobé, O. Maury, S. Guy, A. Bensalah-Ledoux, B. Baguenanrd, G. L. J. A. Rikken, C. Train, B. Le Guennic, M. Atzori, F. Pointillart and J. Crassous, Angew. Chem., Int. Ed., 2023, e202215558 CAS.
- R. Grykien, B. Luszczynska, I. Glowacki, L. Puntus, I. Pekareva, K. Lyssenko, F. Kajzar, I. Rau and C. A. Lazar, Opt. Mater., 2016, 57, 114–119 CrossRef CAS.
- S. Wu, L. Abad Galan, M. Roux, F. Riobé, B. Le Guennic, Y. Guyot, T. Le Bahers, L. Micouin, O. Maury and E. Benedetti, Inorg. Chem., 2021, 60, 16194–16203 CrossRef CAS PubMed.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE Program for the Stereochemical Analysis of Molecular Fragments by Means of Continuous Shape Measures and Associated Tools, Departament de Quimica Fisica, Departament de Quimica Inorganica, and Institut de Quimica Teorica i Computacional – Universitat dè Barcelona, Barcelona, Spain Search PubMed.
- H. Douib, K. Dhbaibi, B. Lefeuvre, V. Dorcet, T. Guizouarn and F. Pointillart, Chirality, 2022, 1–10 Search PubMed.
- C. Benelli and D. Gatteschi, Introduction to Molecular Magnetism: From Transition Metals to Lanthanides, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015 Search PubMed.
- O. Kahn, Molecular Magnetism, VCH, Weinhem, Germany, 1993 Search PubMed.
- J. Jung, T. T. da Cunha, B. Le Guennic, F. Pointillart, C. L. M. Pereira, J. Luzon, S. Golhen, O. Cador, O. Maury and L. Ouahab, Eur. J. Inorg. Chem., 2014, 3888–3894 CrossRef CAS.
- F. Gendron, S. Di Pietro, L. Abad Galan, F. Riobé, V. Placide, L. Guy, F. Zinna, L. Di Bari, A. Bensalah-Ledoux, Y. Guyot, G. Pilet, F. Pointillart, B. Baguenard, S. Guy, O. Cador, O. Maury and B. Le Guennic, Inorg. Chem. Front., 2021, 8, 914–926 RSC.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS.
- R. Orbach, Proc. R. Soc. London, Ser. A, 1961, 264, 458–484 CAS.
- R. Orbach, Proc. Phys. Soc., 1961, 77, 821–826 CrossRef CAS.
- C. Dekker, A. F. M. Arts, H. W. de Wijn, A. J. van Duyneveldt and J. A. Mydosh, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 11243–11251 CrossRef CAS PubMed.
- J. Tang and P. Zhang, Lanthanide Single Molecule Magnets, Springer, Berlin, Germany, 2015 Search PubMed.
- P. Evans, D. Reta, G. F. S. Whitehead, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2019, 141, 19935–19940 CrossRef CAS PubMed.
- D. Reta and N. F. Chilton, Phys. Chem. Chem. Phys., 2019, 21, 23567–23575 RSC.
- K. N. Shrivastava, Phys. Status Solidi B, 1983, 117, 437–458 CrossRef CAS.
- H. Xu, L.-H. Wang, X.-H. Zhu, K. Yin, G.-Y. Zhong, X.-Y. Hou and W. Huang, J. Phys. Chem. B, 2006, 110, 3023–3029 CrossRef CAS PubMed.
- C. A. Mattei, V. Montigaud, F. Gendron, S. Denis-Quantin, V. Dorcet, N. Giraud, F. Riobé, G. Argouarch, O. Maury, B. Le Guennic, O. Cador, C. Lalli and F. Pointillart, Inorg. Chem. Front., 2021, 8, 947–962 RSC.
- C. A. Mattei, V. Montigaud, V. Dorcet, F. Riobé, G. Argouarch, O. Maury, B. Le Guennic, O. Cador, C. Lalli and F. Pointillart, Inorg. Chem. Front., 2021, 8, 963–976 RSC.
- Y. Kishi, L. Cornet, F. Pointillart, F. Riobé, B. Lefeuvre, O. Cador, B. Le Guennic, O. Maury, H. Fujiwara and L. Ouahab, Eur. J. Inorg. Chem., 2018, 458–468 CrossRef CAS.
- F. Guégan, J. Jung, B. Le Guennic, F. Riobé, O. Maury, B. Gillon, J.-F. Jacquot, Y. Guyot, C. Morel and D. Luneau, Inorg. Chem. Front., 2019, 6, 3152–3157 RSC.
- M. F. Richardson, W. F. Wagner and D. E. Sands, J. Inorg. Nucl. Chem., 1968, 30, 1275–1289 CrossRef CAS.
- A. I. Vooshin, N. M. Shavaleev and V. P. Kazakov, J. Lumin., 2000, 91, 49–58 CrossRef.
- S. Felder, S. Wu, J. Brom, L. Micouin and E. Benedetti, Chirality, 2021, 33, 506–527 CrossRef CAS PubMed.
- P. J. Pye, K. Rossen, R. A. Reamer, N. N. Tsou, R. P. Volante and P. J. Reider, J. Am. Chem. Soc., 1997, 119, 6207–6208 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- I. F. Galván, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov, R. K. Carlson, L. F. Chibotaru, J. Creutzberg, N. Dattani, M. G. Delcey, S. S. Dong, A. Dreuw, L. Freitag, L. M. Frutos, L. Gagliardi, F. Gendron, A. Giussani, L. González, G. Grell, M. Guo, C. E. Hoyer, M. Johansson, S. Keller, S. Knecht, G. Kovačević, E. Källman, G. Li Manni, M. Lundberg, Y. Ma, S. Mai, J. P. Malhado, P. Å. Malmqvist, P. Marquetand, S. A. Mewes, J. Norell, M. Olivucci, M. Oppel, Q. M. Phung, K. Pierloot, F. Plasser, M. Reiher, A. M. Sand, I. Schapiro, P. Sharma, C. J. Stein, L. K. Sørensen, D. G. Truhlar, M. Ugandi, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, O. Weser, T. A. Wesołowski, P.-O. Widmark, S. Wouters, A. Zech, J. P. Zobel and R. Lindh, J. Chem. Theory Comput., 2019, 15, 5925–5964 CrossRef PubMed.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- K. Andersson, P.-Å. Malmqvist, B. O. Roos, A. J. Sadlev and K. Wolinski, J. Phys. Chem., 1990, 94, 5483–5488 CrossRef CAS.
- P.-A. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- P.-A. Malmqvist and B. O. Roos, Chem. Phys. Lett., 1989, 155, 189–194 CrossRef CAS.
- B. A. Heß, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef.
- L. F. Chibotaru and L. Ungur, J. Chem. Phys., 2012, 137, 064112 CrossRef CAS PubMed.
- L. F. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., Int. Ed., 2008, 47, 4126–4129 CrossRef CAS PubMed.
- F. Aquilante, P.-A. Malmqvist, T. B. Pedersen, A. Ghosh and B. O. Roos, J. Chem. Theory Comput., 2008, 4, 694–702 CrossRef CAS PubMed.
- B. O. Roos, R. Lindh, P.-A. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2004, 108, 2851–2858 CrossRef CAS.
- B. O. Roos, R. Lindh, P.-A. Malmqvist, V. Veryazov, P.-O. Widmark and A.-C. Borin, J. Phys. Chem. A, 2008, 112, 11431–11435 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- AMS 2022.103, Amsterdam, The Netherlands, https://www.scm.com.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SCXRD and PXRD data, computational details, DC and AC magnetic data, emission data. CCDC 2297628–2297632. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00536h |
| This journal is © The Royal Society of Chemistry 2024 |