
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal complexes of the (2,2,2-trifluoroethyl)phosphinate NOTA analogue as potential contrast agents for 19F magnetic resonance imaging†
Filip
Koucký
 a,
Tereza
Dobrovolná
a,
Jan
Kotek
*a,
Ivana
Císařová
a,
Jana
Havlíčková
a,
Alan
Liška
b,
Vojtěch
Kubíček
a and
Petr
Hermann
a
a,
Tereza
Dobrovolná
a,
Jan
Kotek
*a,
Ivana
Císařová
a,
Jana
Havlíčková
a,
Alan
Liška
b,
Vojtěch
Kubíček
a and
Petr
Hermann
a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 8, 128 42 Prague 2, Czech Republic. E-mail: modrej@natur.cuni.cz
bDepartment of Molecular Electrochemistry and Catalysis, J. Heyrovský Institute of Physical Chemistry AS CR, Dolejškova 2155/3, 182 23 Prague 8, Czech Republic
First published on 26th March 2024
Abstract
A new hexadentate 1,4,7-triazacyclononane-based ligand bearing three coordinating methylene-(2,2,2-trifluoroethyl)phosphinate pendant arms was synthesized and its coordination behaviour towards selected divalent (Mg2+, Ca2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+) and trivalent (Cr3+, Fe3+, Co3+) transition metal ions was studied. The ligand forms stable complexes with late divalent transition metal ions (from Co2+ to Zn2+) and the complexes of these metal ions are formed above pH ∼3. A number of complexes with divalent metal ions were structurally characterized by means of single-crystal X-ray diffraction. The complex of the larger Mn2+ ion adopts a twisted trigonally antiprismatic geometry with a larger coordination cavity and smaller torsion of the pendant arms, whereas the smaller ions Ni2+, Cu2+ and Zn2+ form octahedral species with a smaller cavity and larger pendant arm torsion. In the case of the Co2+ complexes, both coordination arrangements were observed. The complexes with paramagnetic metal ions were studied from the point of view of potential utilization in 19F magnetic resonance imaging. A significant shortening of the 19F NMR longitudinal relaxation times was observed: a sub-millisecond range for complexes of Cr3+, Mn2+ and Fe3+ with symmetric electronic states (t2g3 and HS-d5), the millisecond range for the Ni2+ and Cu2+ complexes and tens of milliseconds for the Co2+ complex. Such short relaxation times are consistent with a short distance between the paramagnetic metal ion and the fluorine atoms (∼5.5–6.5 Å). Among the redox-active complexes (Mn3+/Mn2+, Fe3+/Fe2+, Co3+/Co2+, Cu2+/Cu+), the cobalt complexes show sufficient stability and a paramagnetic–diamagnetic changeover with the redox potential lying in a physiologically relevant range. Thus, the Co3+/Co2+ complex pair can be potentially used as a smart redox-responsive contrast agent for 19F MRI.
Introduction
Magnetic Resonance Imaging (MRI) is one of the most powerful imaging methods in current medicine. Compared to the classical radiological methods utilizing ionizing radiation such as Computed Tomography (CT), Single-Photon Emission Computed Tomography (SPECT) or Positron Emission Tomography (PET), MRI utilizes the nuclear magnetic resonance effect and uses non-ionizing radiation for detection which brings a strong benefit to the patients. Classical MRI utilizes the detection of the NMR effect of water 1H nuclei and distinguishes the different tissues according to the water content and/or signal relaxation times (longitudinal, T1, and transversal, T2). The image contrast can be further improved by the application of contrast agents (CAs) which influence the water proton relaxation times, and a suitable experimental setup selectively increases or decreases the water 1H signal according to the CA distribution in the tissue.1 Other MRI techniques have been developed, including the detection of 1H signals of compounds other than water (e.g. fat) or utilization of responsive (“smart”) contrast agents which change their properties according to the surrounding conditions.2 In addition, other NMR active nuclei such as 13C, 19F or 31P can also be detected by MRI.3 Among them, fluorine is of special interest, as it is a monoisotopic element with a high gyromagnetic ratio which provides a very high sensitivity towards the NMR effect. The gyromagnetic ratio of 19F is very close to that of 1H which enables the detection of the 19F NMR signal with the use of standard MRI scanners with only small hardware and software modifications. Furthermore, fluorine has almost no abundance in living organisms and, thus, it has no natural background in the imaging experiments. This fact predisposes the 19F nuclei for “hot-spot” imaging where only the externally supplied xenobiotic contrast agent molecules provide the 19F MRI signal.4 As both 1H and 19F MRI experiments can be performed on the same hardware, the need to overlay 19F “hot-spots” with the anatomical image can be elegantly solved by 1H + 19F tandem MRI.5 Such an imaging method could be especially useful for example for following the fate of labelled cells after transplantation.6The compounds first tested as 19F NMR contrast agents were usually highly fluorinated hydrocarbons and their derivatives (for simplicity called perfluorocarbons, PFCs). The design of fluorinated contrast agents takes advantage of the generally high stability of the C–F bond towards hydrolysis which makes these compounds inert and non-biodegradable.7 Highly fluorinated organic compounds therefore usually exhibit a low acute toxicity, and some of the PFCs have been already studied for a long time as potential blood substitutes due to the high solubility of molecular oxygen8–11 or as materials in vitreo-retinal surgery due to their optical properties.12,13 Although some adverse health effects have been reported in the latter application,14 they have been attributed mainly to the presence of trace toxic impurities like non-fully substituted hydrocarbons and other derivatives.15,16 However, although PFCs show high stability, it was recently found that they cause some environmental risks.17
The compounds originally used for 19F MRI are not water-soluble and have been used in the form of nano- or microemulsions which complicates some applications (e.g. cellular labelling). Further complication arises from a generally long T1 relaxation time of the 19F nucleus in the PFC (order of seconds). Therefore, a signal is acquired for several seconds, a long delay between excitation pulses is needed and it prolongs the whole experiment. To solve this problem, complexes of fluorine-containing ligands with some paramagnetic metal ions were introduced, as the presence of a paramagnetic metal ion strongly influences the NMR relaxation times of nuclei in the vicinity.18 The concentration of the contrast agents must be relatively high (on a millimolar scale) to achieve a reasonable signal-to-noise ratio. Therefore, the complexes must exhibit exceptional kinetic inertness as the free metal ions are frequently toxic and, in addition, the contrast agent loses convenient relaxation properties after the dissociation of the complexes. Therefore, the ligands are often based on a macrocyclic scaffold, mostly on 1,4,7-triazacyclononane (tacn), 1,4,7,10-tetraazacyclododecane (cyclen) or 1,4,8,11-tetraazacyclotetradecane (cyclam), Fig. 1, with suitably chosen coordinating pendant arms or substituents which contain fluorine atoms. The first explored groups were derivatives and analogues of cyclen-based H4dota (Fig. 1),19–31 the octadentate ligand family very suitable for complexation of the lanthanide(III) ions. The Ln3+ complexes of such ligands are generally kinetically inert (the Gd3+ complexes are used as ordinary 1H MRI CAs),32 and a range of magnetic properties of individual Ln3+ ions brings a possibility of tuning the properties of the final CAs.21,25,31 More recently, complexes of transition metal ions have also been found to effectively shorten the NMR relaxation times of close 19F nuclei.33 For complexation of transition metal ions, H4dota derivatives have also been used,28,34,35 but hexadentate derivatives of tacn and cyclam are generally more suitable for this purpose.36 Therefore, several tacn derivatives with three coordinating pendant arms containing fluorine were prepared and studied.37–40 Similarly, cyclam-based ligands with two coordinating groups have also been studied.41–47
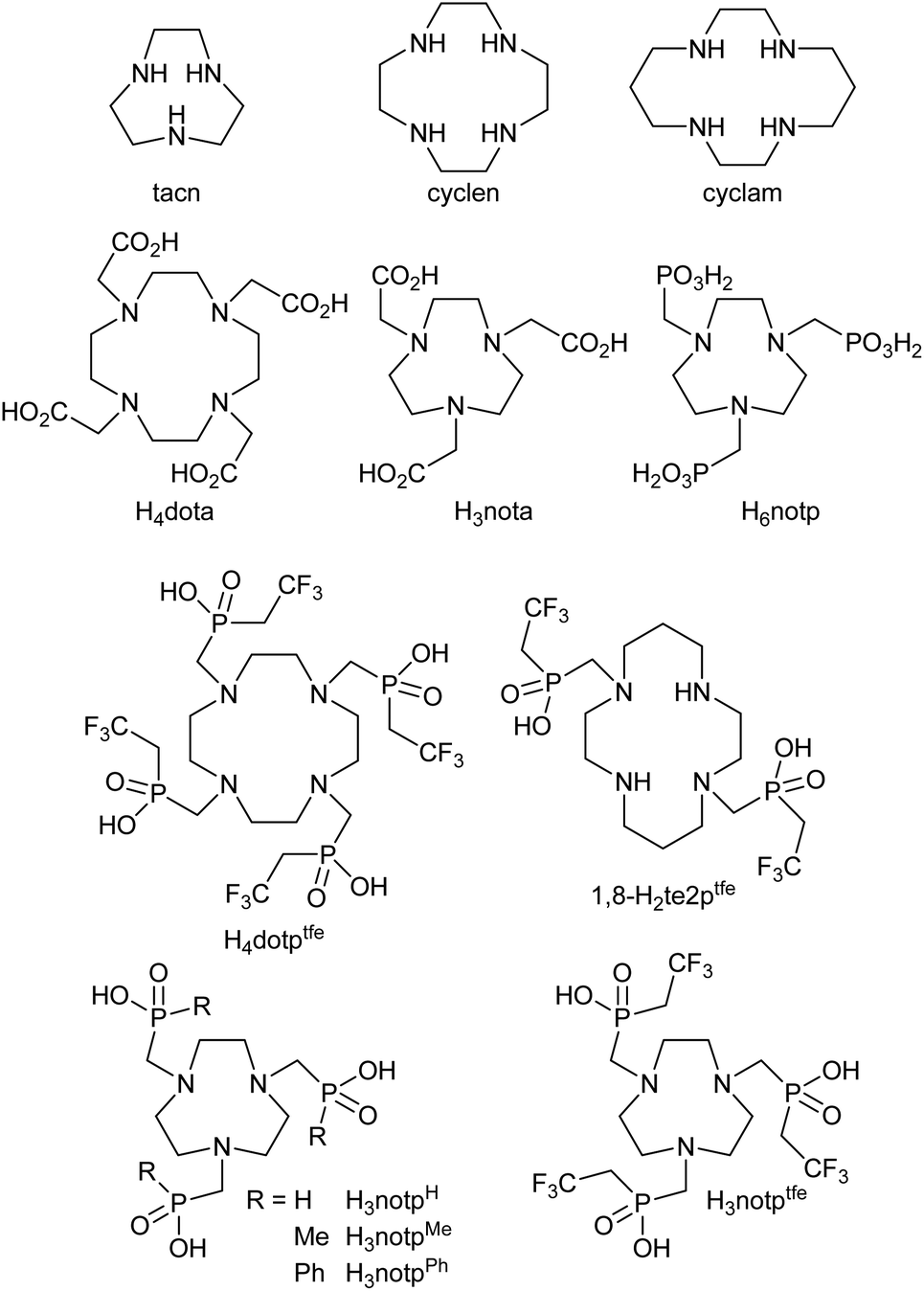 | ||
| Fig. 1 Ligands mentioned in the text. | ||
In our previous contributions, we showed that (2,2,2-trifluoroethyl)phosphinic acid is a suitable synthon for the introduction of the fluorine atoms into a ligand molecule. We used this coordinating pendant arm in the synthesis of the cyclen-based ligand H4dotptfe (Fig. 1) which contains twelve fluorine atoms and we studied its Ln3+ complexes.31 Recently, the same pendant arm has been utilized in the cyclam-based ligand 1,8-H2te2ptfe (Fig. 1) containing six fluorine atoms and a study of its transition metal complexes was reported.47 The results showed that such a concept is viable and brought promising data especially for the Co2+ complex.47
To extend our work in this field, we designed a new tacn-based ligand containing three [methylene-(2,2,2-trifluoroethyl)phosphinic acid] pendant arms, H3notptfe (Fig. 1). It increases the number of fluorine atoms in one small molecule to nine. It would potentially increase the visibility of the contrast agents during the imaging experiment. In this manuscript, we report on the synthesis of the designed ligand and the results of its coordination study with selected first-row transition metal ions.
Experimental
General
Commercial chemicals (Fluka, Aldrich, CheMatech, Lachema, Fluorochem) were used as obtained. Anhydrous solvents were obtained by established procedures.48 2,2,2-Trifluoroethyl-tosylate49 and 2,2,2-trifluoroethylbromide50 were prepared according to published procedures with slight modifications. (2,2,2-Trifluoroethyl)phosphinic acid was prepared analogously as reported earlier47 but 2,2,2-trifluoroethylbromide was used instead of iodide and the workup was modified. Analytical HPLC-MS experiments were carried out on a Waters ACQUITY QDa system (detection: MS – electrospray ionization under atmospheric pressure, m/z 100–1250; UV-Vis – λ 210–800 nm, diode array) equipped with an RP column (Cortecs C18 2.7 μm, 4.6 × 50 mm) using a mixture of water with 0.1% trifluoroacetic acid (TFA) and MeCN with 0.1% TFA; for details on the chromatographic method, see Fig. S1.† Mass spectra were obtained on a Waters ACQUITY QDa with a direct loading using H2O, H2O with 0.1% TFA or MeOH for the sample dilution. Data were processed by Empower 3 software. Flash chromatography was performed on an ECOM ECS28P0X (UV-Vis diode-array detector, λ 200–800 nm). Thin-layer chromatography (TLC) was performed on silica-coated aluminium sheets Silica gel 60 F254 (Merck). Spots were visualized using UV light (254 and 366 nm), dipping in 5% aq. solution of CuSO4 or in aq. solution containing 1% KMnO4 and 2% Na2CO3. UV-Vis spectra were recorded on spectrometer Specord 50 Plus (Analytic Jena) in a quartz–glass cell with an optical path of 1 cm. NMR spectra were recorded on a Varian VNMRS300 (frequencies 299.9 MHz for 1H, 282.2 MHz for 19F and 121.4 MHz for 31P), Varian Inova 400 MHz (frequencies 400.0 MHz for 1H, 100.6 MHz for 13C, 376.3 MHz for 19F and 161.9 MHz for 31P) or Bruker Avance III 600 MHz (frequencies 600.2 MHz for 1H, 150.9 MHz for 13C and 564.7 MHz for 19F). The spectra were acquired at 25 °C unless stated otherwise. Internal references for 1H and 13C NMR spectra were t-BuOH for D2O solutions and CHD2OD residual peak for MeOH-d4 solutions. Aq. H3PO4 (3%) was used as the external reference for 31P NMR (0.5 ppm) and ca. 1% triflic acid for 19F NMR (−79.0 ppm). These secondary references were referenced to 85% H3PO4 (0.0 ppm) and to Freon-11 (0.0 ppm), respectively. Chemical shifts are given in ppm and coupling constants in Hz. Multiplicities of the signals are expressed as follows: s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Chemical shifts of paramagnetic compounds were corrected for a bulk magnetic susceptibility effect as described previously (difference of the 19F signals of 2,2,2-trifluoroethanol between the paramagnetic sample and the insert, Table S1†);47 the corrected values are presented throughout the text. The T1 relaxation times of the nucleus were measured using the Inversion Recovery sequence. The 19F T2 relaxation times of diamagnetic compounds were measured from the CPMG sequence on a Varian Inova 400 MHz and Bruker III 600 MHz; the measurement is not accessible on VNMRS300 as the probe used does not provide accurate 180° pulses for the 19F nuclei. For all paramagnetic complexes, T2* relaxation times were calculated from half-widths of their NMR signals.Ligand synthesis
Elem. anal.: found (calc. for C9H9F3O3S, Mr 254.22) C: 42.44 (42.52), H: 3.43 (3.57), F: 21.27 (22.42), S: 12.10 (12.61).
NMR (CDCl3): 1H: 2.47 (s, 3H, CH3); 4.34 (q, 2H, 3JHF 8.0, CH2);7.38 (d, 2H, 3JHH 8.2, arom.); 7.81 (d, 2H, 3JHH 8.2, arom.). 13C{1H}: 21.8 (CH3); 64.7 (q, 2JCF 38.1, CH2); 122.7 (q, 1JCF 277, CF3); 128.2, 130.3, 132.0, 146.1 (4 × arom.). 19F: −74.2 (t, 3JFH 8.0, CF3).
Recrystallization from boiling 96% aq. EtOH afforded single-crystals suitable for determination of the crystal structure by X-ray diffraction. The crystal structure is the same as that already reported,51 but with significantly better parameters of refinement (see the ESI, Table S4 and Fig. S34†).
NMR (20% v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v solution in CH2Cl2, 20 °C): 1H: 3.72 (q, 2H, 3JHF 8.9, CH2). 13C{1H}: 26.4 (q, 2JCF 37.8, CH2); 123.7 (q, 1JCF 274, CF3). 19F: −69.6 (t, 3JFH 8.9, CF3).
:v solution in CH2Cl2, 20 °C): 1H: 3.72 (q, 2H, 3JHF 8.9, CH2). 13C{1H}: 26.4 (q, 2JCF 37.8, CH2); 123.7 (q, 1JCF 274, CF3). 19F: −69.6 (t, 3JFH 8.9, CF3).
Then, 2,2,2-trifluoroethylbromide (13.6 g, 83.5 mmol, 1.1 equiv.) dissolved in anhydrous DCM (20 ml) was added dropwise to the reaction mixture; the rate of the addition was slow enough to prevent haze formation. The mixture was stirred at room temperature overnight. Then, the reaction mixture (0.1 ml) was hydrolysed in 96% EtOH (1 ml) and 31P NMR of the hydrolysed sample revealed ca. 72% conversion; the main impurity was H3PO3. The whole reaction mixture was hydrolysed by dropwise addition of 96% EtOH (60 ml). The hydrolysed mixture was evaporated on a rotary evaporator at the bath temperature of 50 °C. The residue was dissolved in water (30 ml) and the solution was poured onto a column of a strong cation exchange resin (Dowex 50, H+-cycle, 400 ml of aq. suspension) and a crude product was eluted off with water. During this procedure, only a part of DIPEA was kept on the column and some DIPEA was still present in the product (1H NMR). Therefore, chromatography on the same cation exchange column (re-converted to the H+-cycle) was repeated three more times (until the signals of DIPEA disappeared). The aqueous solution of the purified product was concentrated on a rotary evaporator at a maximum temperature of 40 °C in the bath.
The crude product free from DIPEA was purified on a silica column (100 g of dry SiO2) using conc. aq. NH3:EtOH at 1:25 as an eluent. Fractions containing the pure product were combined and evaporated in a vacuum affording ammonium (2,2,2-trifluoroethyl)phosphinate as a white solid. Yield 7.1 g (57%). Spectroscopic characteristics were identical to those reported previously.47 The single crystals of the product were prepared by the slow evaporation of the aqueous solution to near dryness. Besides the simple ammonium salt, the single crystals of 1-adamantylammonium salt were prepared analogously by evaporation of the aqueous solution of (2,2,2-trifluoroethyl)phosphinic acid obtained from the ammonium salt by elution through a strong cation exchanger (Dowex 50, H+-form, elution with water) neutralized with 1-adamantylamine. For data of these crystal structures, see the ESI (Table S4 and Fig. S35, S36†).
The oily residue was diluted in aq. TFA (50% v/v, 50 ml) and tacn (0.52 g, 4.0 mmol, 1.0 equiv.) was added. After its dissolution, paraformaldehyde (0.48 g, 16 mmol, 4.0 equiv.) was added in one portion. The flask was tightly closed and the suspension was stirred at 40 °C for 1 d. Then, an additional amount of paraformaldehyde was added (0.12 g, 4 mmol, 1 equiv.) and the reaction mixture was stirred at 40 °C for the next 1 d. After cooling, volatiles were removed in vacuum affording a brownish oil. It was diluted with water (10 ml) and loaded onto a column of a strong cation exchanger (Dowex 50, H+-form, 400 ml). Elution with water afforded the required product in a mixture with simple acids [starting with (2,2,2-trifluoroethyl)phosphinic acid, hydroxymethyl-(2,2,2-trifluoroethyl)phosphinic acid, (2,2,2-trifluoroethyl)phosphonic acid and traces of TFA]; the doubly substituted macrocycle derivatives remained on the column. The acid fraction was evaporated on a vacuum rotary evaporator. The crude oily product was diluted with water (10 ml) and purified in two portions by flash chromatography using a C18-AQ stationary phase (PuriFlash, 120 g). A gradient of MeCN in H2O was used (both solvents with 0.1% of TFA) with a flow of 50 ml min−1: 0.0 min 0% MeCN, 3.0 min 0% MeCN, 12.0 min 20% MeCN, 15.0 min 20% MeCN, 15.5 min 100% MeCN, and 18.5 min 100% MeCN. The product retention time was 9.8 min. The pure product-containing fractions were evaporated to dryness and the product was isolated as a non-stoichiometric trifluoroacetate (ca. 0.3 molar equiv. according to 19F NMR). The yield was 1.26 g (ca. 49%) of colourless glassy oil.
NMR (H3notptfe·0.3TFA, D2O, pD 0.5): 1H: 2.88 (dq, 6H, 2JHP 14.6, 3JHF 11.8, PCH2CF3); 3.44 (d, 6H, 2JHP 6.4, NCH2P); 3.61 (s, 12H, NCH2CH2N). 13C{1H}: 36.5 (dq, 1JCP 86.4, 2JCF 28.4, PCH2CF3); 52.4 (s, NCH2CH2N); 56.3 (d, 1JCP 99.0, NCH2P); 125.4 (qd, 1JCF 275, 2JCP 3.1, CF3). 19F: −57.5 (td, 3JFH 11.75, 3JFP 8.7), −75.8 (s, TFA). 31P: 23.6 (broad s). MS-ESI: (+): 610.4 ([M + H]+, calc. 610.1); (−): 608.3 ([M − H]−, calc. 608.1), 722.3 ([M + TFA − H]−, calc. 722.1). TLC: iPrOH:conc. aq. NH3:water 10:3:3; Rf 0.85 (5% aq. CuSO4, blue spot). Purity was checked by HPLC (Fig. S1 and S3†) and 1H, 13C, 19F and 31P NMR spectroscopy (Fig. S4–S7†).
The ligand H3notptfe for some experiments was further purified (removal of any remaining TFA) by chromatography on the strong cationic exchanger (Dowex 50, H+-form) with water as an eluent; TFA is eluted off slightly earlier than H3notptfe which is thus enriched in the later fractions. The fractions were analysed by 19F NMR spectroscopy and combined according to their purity. After repeated procedures, some amount of pure H3notptfe was isolated as a colourless thick oil containing only some small amount of water as an impurity.
Preparation of solutions of the complexes for NMR studies
The ligand stock solution was prepared by dissolution of H3notptfe (600 mg, TFA-free) in water into a 25 ml volumetric flask. The exact ligand concentration (38.3 mM) was determined by 19F qNMR using standardized aq. solution of TFA similarly as described previously.47For determination of magnetic moments, the samples were prepared by mixing exactly the measured volume (200–300 μl) of the aq. ligand stock solution, the appropriate amount of the aq. stock solution of the metal ion salts (MnCl2, (NH4)2Fe(SO4)2, CoCl2, NiCl2, CuCl2; 0.95 equiv., concentration determined by chelatometry), appropriate amount of 1.50 M aq. NaOH (2.90 equiv.) and exactly measured volume (200–300 μl) of aq. HEPES buffer (0.1 M) with pH 7.4. The solutions of the Mn2+, Fe2+ and Cu2+ complexes were measured immediately (a fast complex formation) and the solutions of the Co2+ and Ni2+ complexes were equilibrated at 50 °C in a tightly closed vial overnight. To prepare the Cr3+ complex, the mixture of the weighed solid CrCl3·6H2O and the ligand stock solution (1.05 equiv.) with pH adjusted to ca. 6 with diluted aq. NH3 was heated in a tightly closed vial at 90 °C for 5 d. The Fe3+ complex was prepared by mixing the stock solution of Fe(NO3)3 (concentration determined by iodometry) and the ligand stock solution (1.05 equiv.), pH adjustment to ca. 5 with diluted aq. NH3 and heating at 90 °C in a tightly closed vial overnight. Before measurement, t-BuOH (10 μl) was added to 350 μl of the buffered (pH 7.4) complex solution into an NMR tube, an insert tube containing 0.1% t-BuOH in D2O was inserted and the 1H NMR spectra were acquired. In the case of the Fe2+ complex, a fast formation of a colloidal precipitate was observed when handling the solution on air and, therefore, the complex was prepared under an Ar atmosphere, the solution was filtered through a microfilter and measured immediately.
Samples of the metal complexes of H3notptfe for T1 and T2 NMR experiments were prepared in 1 ml vials by mixing the ligand stock solution (500 μl) with the stock solution (10 μl) of the appropriate metal salt containing 0.9 equiv. of the metal ion (i.e. Mg(ClO4)2, CrCl3, MnCl2, FeCl3, CoCl2, NiCl2, CuCl2 and ZnCl2). These stock solutions were prepared by dissolution of 9 equiv. of the respective metal salts in water (100 μl). The pH of the mixtures was carefully adjusted by stepwise addition of diluted aq. NaOH to ca. 7 (in the case of the Fe3+ complex, diluted aq. NH3 was used). Then, the vials were closed and the mixtures were heated overnight at 50 °C (in the case of the Cr3+ and Fe3+ complexes, the mixtures were heated at 90 °C for 5 d). After cooling, pH was adjusted to 7.4 by diluted aq. NH3 (7.5 in the case of the Mg2+–H3notptfe system).
Successful formation of the complexes was seen from the colour change (Cr3+, Fe3+, Co2+, Ni2+ and Cu2+) and was confirmed by NMR spectroscopy (Tables S1 and S2†) and mass spectrometry. The NMR and UV-Vis spectra of the complexes are shown in Fig. S8–S25.†
[MgII(notptfe)]−: colourless. NMR (H2O, pH 7.5): 19F: −57.2 (35%, broad q, 3JFH = 3JFP ∼10); −57.0 (65%, free ligand, dt). MS-ESI (−): m/z 630.1 ([M]−, calc. 630.1).
[CrIII(notptfe)]: deep purple. NMR (H2O, pH 7.4): 19F: −46.0 (extremely broad). MS-ESI (+): m/z 659.1 ([M + H]+, calc. 659.0).
[MnII(notptfe)]−: colourless. NMR (H2O, pH 7.4): 19F: −41.8 (extremely broad). MS-ESI (−): m/z 661.2 ([M]−, calc. 661.0).
[FeIII(notptfe)]: yellow. NMR (H2O, pH 7.4): 19F: −31.0 (very broad). MS-ESI (+): m/z 663.1 ([M + H]+, calc. 663.0).
[CoII(notptfe)]−: purple-pink. NMR (D2O, pD 7.4, evaporated to dryness and re-dissolved in D2O): 1H: −51.5, −12.8, 24.1, 83.4, 113.9, 115.2 and 157.0 (broad singlets). 13C{1H}: −461, −201, −115, 158 and 190 (all broad s). 19F: −50.5 (broad). 31P{1H}: 204 (broad). MS-ESI (+): m/z 667.2 ([M + 2H]+, calc. 667.0).
[NiII(notptfe)]−: light blue. NMR (H2O, pH 7.4): 19F: −48.1 (91%, broad); −45.4 (9%, broad). MS-ESI (−): m/z 664.9 ([M]−, calc. 664.0).
[CuII(notptfe)]−: deep blue. NMR (H2O, pH 7.4): 19F: −54.2 (broad). MS-ESI (−): m/z 669.1 ([M]−, calc. 669.0).
[ZnII(notptfe)]−: colourless. NMR (D2O, pD 7.4, evaporated to dryness and re-dissolved in D2O): 1H: 2.87 (broad s, 10H); 3.12 (broad s, 14H). 13C{1H}: 36.61 (dq, 1JCP 89.8, 2JCF 28.5, PCH2CF3); 53.11 and 57.05 (2 × broad s, NCH2CH2N); 60.0 (d, 1JCP 102, NCH2P); 126.2 (q, 1JCF 274, 2JCP 1.5, CF3). 19F: −57.21 (pseudo-q, 3JFH = 3JFP ∼10.5). 31P: 29.02 (m, 3JPH = 3JPF ∼10.5). 31P{1H}: 29.02 (q, 3JPF 10.5). MS-ESI (+): m/z 670.1 ([M]−, calc. 670.0).
Electrochemical study
The stock solutions of the H3notptfe complexes for electrochemical experiments were prepared analogously as solutions for NMR studies but starting from the ligand stock solution (exactly 1.000 ml) and the corresponding solid metal salt (exactly 0.9 equiv.; CrCl3·6H2O, MnCl2·4H2O, FeCl3·6H2O, CoCl2·6H2O, NiCl2·6H2O, CuCl2·2H2O); The pH of the solutions was adjusted to 7.4.The H3nota complexes for the electrochemical experiments with the same metal ions were prepared and isolated in the solid form as described in the literature (or by analogous procedures).52 The solids were dissolved in water before the measurement.
Cyclic voltammetry was performed using a potentiostat Polarographic analyzer PA 3 equipped with an XY writer (Laboratorní přístroje Praha). A three-electrode setup was used. Working electrodes were a hanging mercury drop electrode (HMDE) or platinum disc electrode; a saturated calomel electrode (SCE) was used as the reference electrode. Platinum wire with a plate was used as the auxiliary (counter) electrode. Among the several aqueous supporting electrolytes tested (0.1 M NaH2PO4/Na2HPO4 buffer with pH 7.4, 0.05 M LiClO4, 0.05 M (NH4)ClO4, 0.05 M (NEt4)ClO4), 0.05 M aq. LiClO4 was chosen due to the widest measurement window [+0.4−(−2.2) V and +1.5−(−0.9) V for the HMDE and Pt electrode, respectively, vs. SCE]. For each measurement, 0.05 M aq. LiClO4 (10 ml) was deoxygenated by bubbling argon through the solution in the measuring cell for several minutes; the absence of dissolved oxygen was confirmed by the measurement of a blank scan. Then, an appropriate volume of the stock solution of the studied H3notptfe complex or weighed amount of the solid H3nota complex was added to reach a concentration of 1–4 mM for the complexes, and the solution was shortly deoxygenated again before the start of the electrochemical experiment.
Spectro-electrochemical experiments were performed using an optically transparent thin-layer electrochemical cell (OTTLE cell) assembled from a Pt mesh working electrode, Pt mesh auxiliary electrode and Ag wire as the reference electrode placed in a thin layer (optical path 0.2 mm) between two quartz windows.53 Electrodes were connected to a potentiostat WaveDriver 10 (Pine research) driven by Aftermath 1.6 software. The OTTLE cell was placed into a UV-Vis spectrophotometer UV-18010 (Shimadzu).
Electrosynthesis of the [CoIII(notptfe)] complex was performed in an H-shaped electrochemical cell with a fine frit placed in the middle of the horizontal part connecting two vertical tubes. One tube was equipped with the auxiliary electrode (Pt wire with a plate) and was filled with 0.05 M LiClO4. The second tube contained the working electrode (Pt wire with a plate). The experiment was performed starting from 5 mM solution of the [CoII(notptfe)]− complex in 0.05 M LiClO4.
Potentiometric study
The methodology of potentiometric titrations and processing of the experimental data were analogous to those previously reported.47,54 The ligand stock solution was prepared by dissolution of TFA-free H3notptfe (600 mg) in a 50 ml volumetric flask. The exact ligand concentration (19.2 mM) was determined by 19F qNMR using standardized aq. solution of TFA similarly as it was used previously.47 This value agreed well with the value calculated during the fitting of the protonation constants (19.38 mM, difference <1%) which was used for the calculations of the constants.Potentiometric titrations were carried out in a vessel thermostatted to 25 ± 0.1 °C. The titrations of the free ligand and systems containing Mg2+, Ca2+, Mn2+, Cu2+ and Zn2+ were performed in the −log[H+] range 1.6–12.1. The concentration of the ligand in the titration vessel was ca. 0.004 M, ligand-to-metal ratio 1:0.95, ionic strength 0.1 M (NMe4)Cl and starting volume ca. 5 ml. An equilibrium in the systems Co2+–H3notptfe and Ni2+–H3notptfe was established slowly and, therefore, the out-of-cell method was used in these cases. Each solution corresponding to one titration point was prepared in a tube with a ground joint (pH 1.6–6.5, three titrations with 15 points, starting volume ca. 1 ml, the same concentrations as used in a common titration) and the solutions were left to equilibrate at room temperature for one week. The overall protonation constants βh are concentration constants and are defined by βh = [HhL]/([H]h·[L]) (stepwise protonation constants are defined as logK1 = logβ1; logKh = logβh − logβh−1). The overall stability constants are defined by the general equation βhml = [MmHhLl]/([M]m·[H]h·[L]l). Here, the formation of only M:L 1:1 complexes (m = l = 1) was considered. The water ion product, pKw 13.81, and stability constants of M2+ − OH− systems were taken from the literature.55 The constants (with their standard deviations) were calculated with program package OPIUM (Table S3†).56
Single-crystal X-ray diffraction study
:10 mixture as an eluent; under these conditions, the excess of free M2+ was kept at the top of the column. In the case of [Mg(H2O)6][Ni(notptfe)]2·12H2O, the ammonium salt obtained by chromatography was crystallized by concentration of the aq. solution in the presence of an excess of MgCl2 (10 equiv.). The single crystals of [Co(H2O)6][Co(notptfe)]2·14.25H2O·0.75MeOH were prepared by saturation of the ligand solution with an excess of freshly precipitated Co(OH)2, filtration, concentration and a slow vapour diffusion of MeOH into the complex solution. The single crystals of Na3[Co(notptfe)]2Br·3Me2CO were prepared by neutralization of an equimolar aq. solution of the ligand and CoBr2 with NaOH to pH ca. 7, concentration and a slow vapour diffusion of acetone. The single crystals of [ZnCl(H2O)3][Zn(notptfe)]·2H2O were obtained by mixing the ligand and an excess of ZnCl2, neutralization with diluted aq. NH3 to pH ca. 5. and a slow concentration in air.
Results and discussion
Synthesis of the ligand H3notptfe
Synthesis of the studied ligand H3notptfe (H3L) was performed as shown in Scheme 1.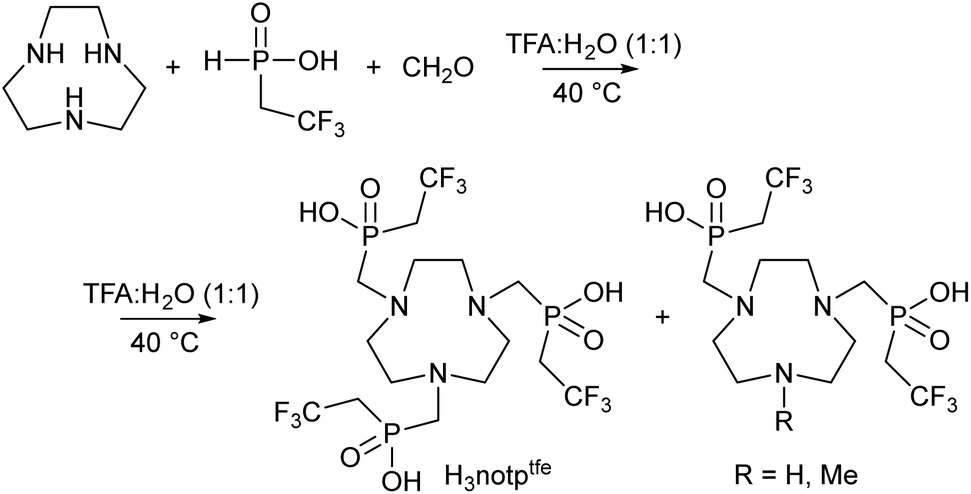 | ||
| Scheme 1 Synthesis of the studied ligand H3notptfe. | ||
The synthesis of the starting (2,2,2-trifluoroethyl)phosphinic acid was reported previously;47 however, the use of commercial 2,2,2-trifluoroethyliodide for an Arbuzov-type reaction with bis(trimethylsilyl)hypophosphite resulted in the presence of hydroiodic acid in the final mixture that complicated further purifications.47 Therefore, we optimized the synthesis using 2,2,2-trifluoroethylbromide. This compound was prepared by reaction of 2,2,2-trifluoroethyl-tosylate49 with KBr in boiling anhydrous diethyleneglycol (boiling point 244 °C).50 However, the synthesis needs non-trivial apparatus as the product is volatile (boiling point 26 °C) (see Experimental and Fig. S2†).
A Mannich-type reaction between tacn, (2,2,2-trifluoroethyl)phosphinic acid and paraformaldehyde was performed in a strong acid solution (50% v/v aq. TFA) at 40 °C. It was found that only a slight excess of (2,2,2-trifluoroethyl)phosphinic acid (3.1 equiv.) and paraformaldehyde (3.5 equiv.) is sufficient to reach almost quantitative conversion according to the 19F and 31P NMR spectra. Only a small extent of oxidation of (2,2,2-trifluoroethyl)phosphinic acid to (2,2,2-trifluoroethyl)phosphonic acid and formation of hydroxymethyl-(2,2,2-trifluoroethyl)phosphinic acid were observed under these conditions. The ligand H3notptfe behaves as a strong acid and can be eluted from a strong cation exchange resin using water. However, its elution is only slightly slowed down compared to the simple acids present in the mixture [TFA, (2,2,2-trifluoroethyl)phosphonic acid, hydroxymethyl-(2,2,2-trifluoroethyl)phosphinic acid, an excess of starting (2,2,2-trifluoroethyl)phosphinic acid]. Traces of macrocyclic by-products with two phosphinic acid pendant arms (a twice-substituted intermediate and its N-methylated derivative, Scheme 1) were kept on the resin and could be eluted using 5% aq. ammonia solution. The ligand H3notptfe was further purified by repeated chromatography on silica with conc. aq. NH3:EtOH 1:25 as the mobile phase to get a non-stoichiometric ammonium salt or by flash chromatography on a reverse phase column using a water:acetonitrile gradient containing 0.1% TFA as a modifier to obtain non-stoichiometric trifluoroacetate. The ammonium salt of the ligand can be converted to the free ligand H3notptfe on a strong cation exchange resin; ammonium cations stay on the column and the acidic form of H3notptfe is eluted with water. In the case of the H3notptfe–TFA adduct, a similar procedure can be used; however here, TFA is eluted only slightly earlier than the free H3notptfe ligand and, thus, the procedure must be repeated to remove TFA completely. The ligand was isolated as a hygroscopic thick semi-solid glassy material, which complicates further analytical work. Therefore, concentrations of its stock solutions had to be determined independently by 19F qNMR using the addition of a known amount of standardized TFA solution.
Equilibrium studies
To study the acid–base behaviour of H3notptfe and the stability of its complexes, potentiometric titrations were employed. Two protonation constants were calculated from the titration data acquired in the pH range 1.6–12.1. Tables 1 and S3† list the results together with data reported for related ligands. The observed protonation steps correspond to the protonations of the amino groups of the tacn skeleton. Going from the fully deprotonated species, the first protonation proceeds in the alkaline region (logK1 > 10) as it is common for other tacn-based ligands; the observed value falls into a range of values for other tacn-tris(phosphinic acid) derivatives (Table 1).60–62 Compared to the acetate (H3nota) and phosphonate (H6notp) derivatives (Fig. 1), the basicity of H3notptfe is lower due to the electron-withdrawing characteristic of the phosphinate groups.63 The protonation constant describing the second protonation step also corresponds well to those of other tacn-tris(phosphinic acid) derivatives (Table 1). Further protonations lay below the pH range accessible by potentiometry pointing to the strong acidity of the phosphinic acid groups. The calculated distribution diagram of differently protonated H3notptfe species is shown in Fig. S26.†
Kh), stability constants of the studied metal complexes (logβ011) and protonation constants of hydroxidocomplexes (logβ011 − logβ−111) of H3notptfe, and comparison with related ligands. Charges of the species are omitted for clarity; “L” means fully deprotonated form of the ligand
| Equilibrium | H3notptfea | H3notpH | H3notpMe | H3nota | H6notp |
|---|---|---|---|---|---|
| a This work, 0.1 M (NMe4)Cl. b 0.1 M KNO3, ref. 60. c 0.1 M (NMe4)Cl, ref. 61. d 0.1 M KCl, ref. 62. e 0.1 M (NMe4)Cl, ref. 64. f 0.1 M (NMe4)Cl, ref. 65. g 0.1 M (NMe4)Cl, ref. 66. h 0.1 M KNO3, ref. 67. i 1 M KNO3, ref. 68. | |||||
| H+ + L = HL | 10.23 | 10.16b | 10.92d | 13.17e | 12.1g |
| 10.48c | 11.9h | ||||
| 11.79i | |||||
| H+ + HL = H2L | 2.86 | 3.13b | 3.97d | 5.74e | 9.4g |
| 3.28c | 9.3h | ||||
| 8.65i | |||||
| H+ + H2L = H3L | — | 1.11b | 2.09d | 3.22e | 7.5g |
| 7.6h | |||||
| 7.09i | |||||
| H+ + H3L = H4L | — | — | — | 1.96e | 5.9g |
| 5.4h | |||||
| 5.38i | |||||
| H+ + H4L = H5L | — | — | — | 0.70f | 2.9g |
| 2.7h | |||||
| 2.53i | |||||
| Mg2+ + L = [Mg(L)] | 5.04 | 5.32b | 6.66d | 10.97c | 11.01i |
| [Mg(L)(OH)] + H+ = [Mg(L)] | 11.71 | — | — | — | — |
| Ca2+ + L = [Ca(L)] | 3.83 | 4.29b | 4.45d | 10.32c | 6.38i |
| [Ca(L)(OH)] + H+ = [Ca(L)] | 12.87 | 11.70b | — | — | — |
| Mn2+ + L = [Mn(L)] | 10.61 | — | — | 16.30e | 16.6i |
| [Mn(L)(OH)] + H+ = [Mn(L)] | 10.17 | — | — | — | — |
| Co2+ + L = [Co(L)] | 13.04 | 12.97b | — | 20.13f | 19.7i |
| Ni2+ + L = [Ni(L)] | 13.18 | 13.40b | — | 19.24f | 19.4i |
| Cu2+ + L = [Cu(L)] | 13.50 | 13.43b | — | 23.33f | 21.3i |
| [Cu(L)(OH)] + H+ = [Cu(L)] | 11.57 | — | — | — | — |
| Zn2+ + L = [Zn(L)] | 13.40 | 13.04b | — | 22.32f | 24.9i |
| [Zn(L)(OH)] + H+ = [Zn(L)] | 12.26 | — | — | — | — |
The thermodynamic stabilities of the complexes with selected alkaline earth (Mg2+, Ca2+) and transition (Mn2+, Co2+, Ni2+, Cu2+, Zn2+) metal ions were studied in the M2+–H3notptfe systems using a slight ligand excess. Except for Co2+ and Ni2+, complexation of all metal ions was fast enough for conventional potentiometry. The systems containing Co2+/Ni2+ ions were studied by the out-of-cell method. The system containing Fe2+ cannot be studied by this method as some precipitate is irreversibly formed during the titration. In general, besides formation of the [M(notptfe)]− complexes, the formation of hydroxido-complexes [M(notptfe)(OH)]2− was observed in strongly alkaline solutions pointing to the weak nucleophilicity of the pendant arm oxygen atoms which are replaced by the hydroxido ligand in a strongly alkaline solution (Table 1). It should be noted that potential formation of the hydroxido complexes of Co2+ and Ni2+ ions cannot be confirmed as the out-of-cell titration used to study the systems cannot be reliably performed up to the alkaline region. Stability constants of [M(notptfe)]− complexes are similar to those found for other phosphinic acid derivatives and are significantly lower in comparison with those of H3nota and H6notp as a result of a generally lower orverall basicity of the donor sites in the phosphinate ligands.63 The ligand H3notptfe shows a slight selectivity for Mg2+ over Ca2+ analogously to other related derivatives; however, these biogenic ions are not fully complexed even in the strongly alkaline region (see Fig. 2 and S27†).
 | ||
| Fig. 2 Distribution diagrams of the M2+–H3notptfe (=M2+–H3L) systems; c(M2+) = c(H3notptfe) = 0.004 M, M = Mg, Mn and Cu. The abundances of the species formed in the Mg2+–H3notptfe system are shown in red, those present in the Mn2+–H3notptfe system are shown in green and those present in the Cu2+–H3notptfe system are shown in blue. Dashed lines represent abundances of the free M2+ ions, full lines represent the [MII(notptfe)]− species, and the dotted lines represent the [MII(notptfe)(OH)]2− species. | ||
The complexes with the transition metal ions are much more stable than those of alkaline earth ions; the Mn2+ complex has the lowest stability among the studied transition metal ions and is fully formed above pH 7 (Fig. 2 and S28†). Complexes with Co2+–Zn2+ have comparable stability and the ligand shows almost no selectivity for the Cu2+ ion; so, the effect of Williams–Irving ordering is negligible. These metal ions are fully complexed at pH > 3; an illustrative distribution diagram is shown in Fig. 2 and S29.†
The observed non-selectivity for the Cu2+ ion is consistent with the non-selectivity of other phosphorus-containing tacn-based ligands but it is in contrast to the selectivity observed for H3nota (Table 1). Overall, the ligand H3notptfe binds paramagnetic transition metal ions into stable complexes with a high selectivity over biogenic alkaline earths ions which is promising for potential in vitro/in vivo use.
Magnetic properties of the complexes in solution
As the value of magnetic moment plays a significant role in the paramagnetic relaxation enhancement mechanism, we determined the magnetic moments of the studied paramagnetic complexes in solution. As the Mn2+/Cu2+–H3notptfe complexes are formed quickly (see Potentiometric study section), they were prepared by mixing the metal salt and ligand (in a slight excess) stock solutions, and adjusting the pH to 7.4. Solutions of the Co2+ and Ni2+ complexes were prepared analogously but the solutions were heated to 50 °C overnight to ensure full complexation. In the system containing Fe2+, some colloidal precipitate was continuously formed; therefore, measurement of the magnetic moment was performed immediately after preparation of the solution under argon and filtration through a microfilter.Solutions of the Cr3+ and Fe3+ complexes were prepared by the reaction of CrCl3·6H2O (solid) or Fe(NO3)3 (stock solution), respectively, with a slight excess of H3notptfe after neutralization to pH 5–6 and heating at 90 °C for 5 d (Cr3+) or overnight (Fe3+). Surprisingly, attempts to prepare a solution of the Co3+ complex by reaction of the ligand with Na3[Co(CO3)3] (a kinetically labile precursor commonly used for a direct preparation of trivalent cobalt complexes)69 failed; the Co2+ complex was formed instead as identified by UV-Vis and 19F NMR spectroscopies.
Magnetic moments were determined using Evans’ method70,71 from the chemical shift difference of t-BuOH present in the solution of the complex with a known concentration and aq. solution in the insert cuvette and are listed in Table 2.
| Complex | μ eff (B.M.) |
|---|---|
| [CrIII(notptfe)] | 3.51 |
| [MnII(notptfe)]− | 5.75 |
| [FeII(notptfe)]− | 5.29 |
| [FeIII(notptfe)] | 6.16 |
| [CoII(notptfe)]− | 4.98 |
| [NiII(notptfe)]− | 2.77 |
| [CuII(notptfe)]− | 1.86 |
The values of μeff clearly show that the complexes of Mn2+, Fe2+/Fe3+ and Co2+ are high-spin and, thus, an overall ligand field induced by the (notptfe)3− anion is relatively small.
Redox properties of the complexes
The redox behaviour of transition metal complexes of the “parent” H3nota is well documented in the literature.52,72 Redox potentials of the M3+/2+–H3nota systems are reported for M = Cr, Mn, Fe, Co and Ni, and values of some of them fall into a biologically relevant range (Table 3). The change of oxidation state governs electronic and magnetic properties of the ions, and can significantly alter the chemical shift of the 19F NMR signal and its relaxation properties. If the redox change can occur under in vivo conditions, the complex can be utilized as a redox-responsive probe.18 Therefore, electrochemical studies of H3notptfe complexes and analogous H3nota complexes were performed to obtain directly comparable data as data reported in the literature were sometimes acquired under different or unspecified conditions (electrode material, supporting electrolyte). Besides the metal ions mentioned above, we also studied the Cu2+ complex as the Cu2+/Cu+ pair can also be employed as a redox probe.18| Redox pair | M–H3notptfe | M–H3nota | |
|---|---|---|---|
| This work | This work | Literature52 | |
| a Hanging mercury drop electrode. b Platinum electrode. c No electrochemical process was observed. | |||
| Cr3+/Cr2+ | −1.32a | −1.39a | −1.41 |
| Mn3+/Mn2+ | 0.67b | 0.56b | 0.56 |
| Mn4+/Mn3+ | 0.96b | 1.02b | — |
| Fe3+/Fe2+ | −0.02a,b | −0.035a | −0.045b |
| −0.045 | |||
| Co3+/Co2+ | 0.95b | −0.23a | −0.24 |
| Ni3+/Ni2+ | —c | 0.95b | 0.92 |
0.05 M aq. LiClO4 solution was chosen as the supporting electrolyte due to a wide measurement window [+0.4−(−2.2) and 1.5−(−0.9) V using the HMDE and Pt electrode, respectively, vs. SCE], and the complexes were studied by cyclic voltammetry (CV). The reversible behaviour of the M2+/M3+–H3notptfe and M2+/M3+–H3nota pairs was found for M = Cr, Mn, Fe and Co (Table 3). In contrast to the [NiII(nota)]− complex, [NiII(notptfe)]− cannot be oxidized to the [NiIII(notptfe)] complex in the accessible potential range.
In the case of the Mn-systems with both ligands, a further reversible process was observed which is attributable to the [MnIV(L)]+/[MnIII(L)] redox pair. This behaviour was reported for the Mn2+–H3nota complex previously72 but the reported potentials of 0.30 and 0.68 V for the [MnIII(nota)]/[MnII(nota)]− and [MnIV(nota)]+/[MnIII(nota)] pairs, respectively, are not consistent with other literature data52 and with our value; probably, the reported potentials were wrongly corrected for the SCE potential. To identify the species and to study their stabilities, a spectro-electrochemical study was performed. If the potential was increased in the range from 0.5 to 1.0 V, a d–d transition band centred at 470 nm gradually appeared (Fig. S30†), attributable to Mn3+ due to the one-electron oxidation of [MnII(L)]− and the formation of [MnIII(L)], consistent with the results obtained by the CV study. Further oxidation (applied potential up to 1.8 V) led to a gradual increase in absorbance over the whole spectral range, with no distinguished absorption band (Fi S31†). It was probably caused by the formation of a colloidal precipitate. Thus, one can conclude that the oxidation of [MnIII(L)] to [MnIV(L)]+ is pseudo-reversible on the short CV timescale but the formed MnIV-species decomposes and colloidal MnO2·nH2O is formed during the longer spectro-electrochemical timescale.
The spectro-electrochemical reduction (applied potential from 0.5 to −0.8 V) of the [FeIII(notptfe)] complex led to the formation of [FeII(notptfe)]− as documented by the gradual spectral change (Fig. S32†). The [FeII(notptfe)]− species was found to be stable on the given timescale (order of seconds); however as observed earlier, it decomposes on longer standing (see the Magnetic properties section).
The electro-synthetic oxidation (applied potential 1.1 V) of the [CoII(notptfe)]− complex (characterized by three close low-intensity absorption bands centred around 520 nm) revealed the formation of the [CoIII(notptfe)] complex (the appearance of two new intense d–d bands at 394 and 557 nm, as shown in Fig. S33†) which is stable in solution for at least several days.
The Cu2+ complexes of both H3notptfe and H3nota underwent irreversible two-electron reduction on HMDE as proved by the anodic peak corresponding to the oxidation of metallic copper; no electrochemical changes were observed using the Pt electrode. According to the measured values, the [CrII(notptfe)]− complex is a very strong reduction agent similar to [CrII(nota)]−, and its potential lies close to the reduction edge of HDME; such a value cannot be practically utilized in biological systems. However, the potentials of [MIII(L)]/[MII(L)]− pairs with M = Mn, Fe and Co lie in the biologically relevant range. Among them, the stability of the Mn2+ complex is not sufficient for utilization in biological systems (full complexation at pH > 7, see Potentiometric study section) and the [FeII(notptfe)]− species was found to be kinetically labile, so only the Co3+/Co2+–H3notptfe redox pair is suitable for in vitro/in vivo utilization. Furthermore, the [CoII(notptfe)]− complex is a high-spin paramagnetic species but the oxidized form [CoIII(notptfe)] is a low-spin diamagnetic complex, which induces a very significant change in the 19F NMR spectra (see below). Therefore, we studied the corresponding redox process using chemical oxidizing/reducing agents. The [CoIII(notptfe)] species can be prepared by chemical oxidation of [CoII(notptfe)]− with K2S2O8 or H2O2 (Fig. S33†). The [CoIII(notptfe)] species can be reduced back to the [CoII(notptfe)]− complex using NaBH4; other reducing agents tested – N2H4, NH2OH, Na2S2O4 – caused no reaction. It should be further highlighted that the reaction of H3notptfe with Na3[CoIII(CO3)3] afforded [CoII(notptfe)]− and not the expected [CoIII(notptfe)] complex. Therefore, although the [CoIII(notptfe)] species is a relatively strong oxidation agent (E½ + 0.95 V vs. SCE, Table 3), some kinetic barrier plays a role in the tested chemical reductions, probably due to a full wrapping of the central metal ion with the ligand which prevents a close contact with a reduction agent. A comparison of the scaled UV-Vis spectra of [CoIII(notptfe)] and [CoII(notptfe)]− is shown in Fig. 3.
 | ||
| Fig. 3 Comparison of the UV-Vis spectra of [CoIII(notptfe)] and [CoII(notptfe)]− complexes. Spectra are arbitrarily scaled. Extinction coefficients in dm3 mol−1 cm−1: [CoII(notptfe)]−: ε(502 nm) 16, ε(520 nm) 17, ε(541 nm) 16; [CoIII(notptfe)]: ε(394 nm) 68, ε(557 nm) 92. | ||
Solid-state structures of the complexes
The H3nota-like ligands usually wrap the metal ions in a hexadentate fashion. Three nitrogen atoms of the tacn skeleton form a basal N3-plane making one face of the coordination polyhedron, and the oxygen atoms of three pendant arms form a parallel upper O3-plane. Formation of the chelate rings upon a coordination of the tacn unit results in a clock-wise or anti-clock-wise torsion on the ethylene groups connecting the coordinated nitrogen atoms (i.e., in the Newman projection of the formal ethylene-diamine fragment, it depends on mutual positions of the C–N bonds of the front and rear carbon atoms, respectively); such conformations are denoted as δ or λ. All three chelate rings formed in the macrocyclic part have to adopt the same geometry due to steric hindrances, i.e. one of two possible macrocycle conformations δδδ or λλλ is formed. The pendant arm coordination leads to a torsion of the pendant arm with respect to the (pseudo)trigonal axis. Here, stereodescriptors Δ and Λ are used for clockwise and anti-clockwise arrangements, respectively [i.e., differing in mutual positions of the O and N atoms belonging to the individual pendant arms in a view along the (pseudo)trigonal axis from the top to the bottom]. If the complex molecule has (pseudo)trigonal symmetry, all pendants are twisted in the same direction. A combination of both rotations leads to the formation of two diastereomeric pairs of enantiomers, ΔΔΔλλλ + ΛΛΛδδδ and ΔΔΔδδδ + ΛΛΛλλλ (hereafter for simplicity denoted as Δλ + Λδ and Δδ + Λλ). Analogous diastereoisomerism is well-documented in the complexes of H4dota-like ligands (Fig. 1) where it leads to the square-antiprismatic (SA isomer, Δλ + Λδ, real pendant torsion >35°) and the twisted-square-antiprismatic (TSA isomer, Δδ + Λλ, real pendant torsion <30°) geometries.63,73–80 Similarly to the complexes of H4dota-like ligands, the diastereoisomers of the complexes of H3nota-like ligands also differ in a mutual twist of the N3- and O3-planes. The larger twist found in the Δλ+Λδ isomer leads to a shorter separation of the N3- and O3-planes and to a geometry closer to an octahedron (OC). The Δδ+Λλ conformations lead to the twisted-trigonal-antiprismatic (TTA) arrangement with a somewhat larger distance between the N3- and O3-planes. In the case of the H3notptfe ligand, a further source of isomerism comes also from the absolute (R/S) configuration of the phosphorus atom upon coordination of the phosphinate group. Three pendant arms with potentially independent R/S-isomerism give rise to a number of isomers (Fig. 4), similarly as observed for the phosphinate analogues of H4dota.81–83 | ||
| Fig. 4 Possible diastereoisomers of H3notptfe complexes; only one of the enantiomers of each diastereoisomer is shown. All shown enantiomers have the λλλ conformations of the macrocycle, the other enantiomers have the δδδ conformations. Left column: the OC species; right column: the TTA species. Colour code: metal ion – magenta, phosphorus – orange, oxygen – red, nitrogen – light blue, carbon – dark grey. | ||
Two Mn2+ phases were obtained where the central Mn2+ ion has the TTA coordination sphere. The absolute configuration on a phosphorus atom of all coordinated pendant phosphinate groups in the complex molecule is the same (in the latter compound forced due to the crystallographic trigonal symmetry of the space group, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ), resulting in the presence of the enantiomeric pair Δδ-SSS and Λλ-RRR in both structures. Selected relevant geometric parameters are listed in Tables 4 and S5† and molecular structures of the complex species are shown in Fig. 5 and 6.
), resulting in the presence of the enantiomeric pair Δδ-SSS and Λλ-RRR in both structures. Selected relevant geometric parameters are listed in Tables 4 and S5† and molecular structures of the complex species are shown in Fig. 5 and 6.
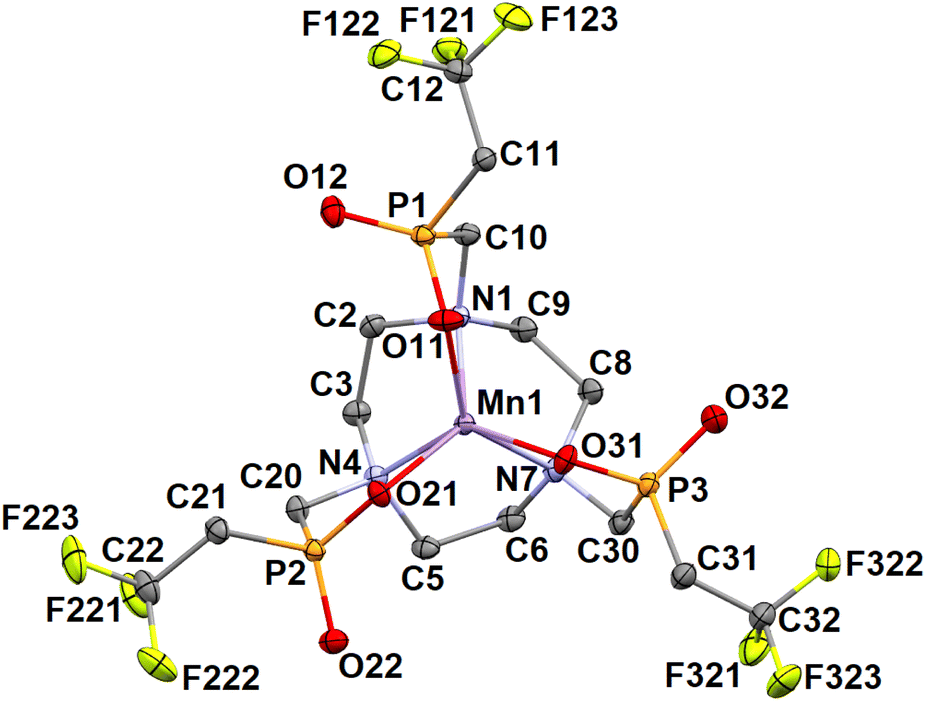 | ||
| Fig. 5 Molecular structure of the [Mn(notptfe)]− anion with the Δδ-SSS TTA geometry found in the crystal structure of (NH4)3[Mn(notptfe)]Cl2·3H2O. Hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 6 Molecular structure of [Mn(H2O)6][Mn(notptfe)]2 fragment found in the crystal structure of [Mn(H2O)6][Mn(notptfe)]2·18H2O. Intermolecular hydrogen bonds connecting two [Mn(notptfe)]− anions to the central [Mn(H2O)6]2+ cation are shown in turquoise. Individual [Mn(notptfe)]− species adopt the Δδ-SSS and Λλ-RRR TTA geometries. Carbon-bound hydrogen atoms are omitted for clarity. | ||
| (NH4)3[Mn(L)]Cl2·3H2O | [Mn(H2O)6][Mn(L)]2·18H2O | (NH4)[Co(L)]·3.5H2O | [Co(H2O)6][Co(L)]2·14.25H2O·0.75MeOH | Na3[Co(L)]2Br·3Me2CO | (NH4)[Cu(L)]·3.5H2O | (NH4)2[Cu(L)]Cl·3H2O | [Mg(H2O)6][Ni(L)]2·12H2O | [ZnCl(H2O)3][Zn(L)]·2H2O | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Geometry | TTA | TTAa | OC | TTA | TTA | TTAa | TTAa | TTA | TTA | OC | square pyramidal | OCa | OCa |
| Δδ-SSS | Δδ-SSS | Δλ-SSS | Λλ-RRR | Δδ-SSS | Λλ-RRR | Δδ-SSS | Λλ-SSS | Λλ-SSS | Λδ-RRR | ΔΛ0-λλλ-SR0 | Λδ-RRR | Λδ-RRR | |
| a The complex molecule possesses trigonal symmetry; F1 = F4 = F7, F2 = F5 = F8, F3 = F6 = F9. b NQ and OQ are centroids of the N3- and O3- planes, respectively. c Formal numbering: fluorine atoms F1–3 belong to the pendant arm attached to N1, fluorine atoms F4–6 to the pendant arm attached to N4, and fluorine atoms F7–9 to the pendant arm attached to N7. d More abundant position of the disordered trifluoroethyl group. | |||||||||||||
| Distances (Å) | |||||||||||||
| M–N1 | 2.360(1) | 2.335(2) | 2.160(2) | 2.168(2) | 2.168(2) | 2.187(2) | 2.170(2) | 2.154(8) | 2.149(8) | 2.251(1) | 2.040(2) | 2.103(1) | 2.191(3) |
| M–N4 | 2.377(1) | 2.335(2) | 2.144(2) | 2.171(2) | 2.178(2) | 2.187(2) | 2.170(2) | 2.163(8) | 2.156(8) | 2.035(1) | 2.021(2) | 2.103(1) | 2.191(3) |
| M–N7 | 2.341(1) | 2.335(2) | 2.166(2) | 2.189(2) | 2.184(2) | 2.187(2) | 2.170(2) | 2.162(8) | 2.141(8) | 2.116(1) | 2.254(2) | 2.103(1) | 2.191(3) |
| M–011 | 2.123(1) | 2.113(2) | 2.062(2) | 2.081(1) | 2.092(1) | 2.084(1) | 2.098(2) | 2.108(6) | 2.127(6) | 1.989(1) | 1.972(1) | 2.059(1) | 2.063(3) |
| M–O21 | 2.144(1) | 2.113(2) | 2.060(2) | 2.100(1) | 2.098(1) | 2.084(1) | 2.098(2) | 2.104(6) | 2.133(6) | 2.006(1) | 1.940(1) | 2.059(1) | 2.063(3) |
| M–O31 | 2.117(1) | 2.113(2) | 2.125(2) | 2.072(1) | 2.075(1) | 2.084(1) | 2.098(2) | 2.093(7) | 2.086(7) | 2.360(1) | — | 2.059(1) | 2.063(3) |
| M⋯NQb | 1.662(1) | 1.633(2) | 1.398(2) | 1.437(1) | 1.436(1) | 1.455(2) | 1.435(2) | 1.411(5) | 1.396(5) | 1.358(1) | 1.315(1) | 1.323(1) | 1.436(1) |
| M⋯OQb | 1.242(1) | 1.140(2) | 1.091(1) | 1.217(1) | 1.209(1) | 1.256(2) | 1.202(2) | 1.234(4) | 1.223(4) | 1.127(1) | — | 1.123(1) | 1.138(1) |
| NQ⋯OQb | 2.9042 | 2.7726 | 2.4912 | 2.6529 | 2.6443 | 2.7110 | 2.6369 | 2.6445 | 2.6189 | 2.4991 | — | 2.4458 | 2.5739 |
| M⋯F1c | 6.005(1) | 5.869(2) | 5.640(2) | 5.545(1) | 5.540(2) | 5.644(1) | 5.544(2) | 5.893(6) | 5.887(7) | 5.634(1) | 5.697(1) | 5.574(1) | 5.662(3) |
| M⋯F2c | 6.415(1) | 6.388(2) | 6.275(2) | 6.277(1) | 6.310(2) | 6.341(1) | 6.294(2) | 6.222(6) | 6.242(6) | 6.248(1) | 6.168(1) | 6.233(1) | 6.309(3) |
| M⋯F3c | 6.877(1) | 6.649(2) | 6.583(2) | 6.339(1) | 6.380(1) | 6.470(1) | 6.406(2) | 6.803(6) | 6.823(6) | 6.545(1) | 6.582(1) | 6.414(1) | 6.556(3) |
| M⋯F4c | 5.958(1) | 5.869(2) | 5.651(2) | 5.613(1) | 5.514(2) | 5.644(1) | 5.544(2) | 5.391(8) | 4.559(7) | 5.556(1) | 5.505(1) | 5.574(1) | 5.662(3) |
| M⋯F5c | 6.468(1) | 6.388(2) | 6.333(2) | 6.284(1) | 6.206(2) | 6.341(1) | 6.294(2) | 5.910(8) | 5.097(7) | 6.271(1) | 6.125(1) | 6.233(1) | 6.309(3) |
| M⋯F6c | 6.831(1) | 6.649(2) | 6.466(2) | 6.428(1) | 6.256(2) | 6.470(1) | 6.406(2) | 6.637(8) | 6.290(7) | 6.409(1) | 6.278(1) | 6.414(1) | 6.556(3) |
| M⋯F7c | 5.880(1) | 5.869(2) | 5.005(3)d | 5.638(1) | 5.654(1) | 5.644(1) | 5.544(2) | 5.765(6) | 5.744(7) | 5.237(2) | 5.264(1) | 5.574(1) | 5.662(3) |
| M⋯F8c | 6.347(1) | 6.388(2) | 5.956(3)d | 6.305(1) | 6.328(1) | 6.341(1) | 6.294(2) | 6.346(6) | 6.349(6) | 6.094(2) | 6.888(2) | 6.233(1) | 6.309(3) |
| M⋯F9c | 6.836(1) | 6.649(2) | 6.455(3)d | 6.438(1) | 6.476(1) | 6.470(1) | 6.406(2) | 6.699(6) | 6.723(6) | 6.664(2) | 7.169(1) | 6.414(1) | 6.556(3) |
| mean(M⋯F) | 6.40 | 6.30 | 6.04 | 6.10 | 6.08 | 6.07 | 6.19 | 6.07 | 6.18 | ||||
| Dihedral angles (°) | |||||||||||||
| N1–NQ–OQ–O11b | 6.76(7) | 24.38(9) | 46.9(1) | 33.43(8) | 33.03(8) | 29.08(8) | 35.99(9) | 34.7(4) | 33.5(3) | 44.82(6) | — | 50.39(4) | 43.0(2) |
| N4–NQ–OQ–O21b | 6.65(6) | 24.38(9) | 49.6(1) | 32.27(8) | 33.45(8) | 29.08(8) | 35.99(9) | 33.2(3) | 33.2(3) | 53.70(6) | — | 50.39(4) | 43.0(2) |
| N7–NQ–OQ–O31b | 8.79(6) | 24.38(9) | 46.0(1) | 31.28(8) | 32.00(8) | 29.08(8) | 35.99(9) | 34.2(4) | 34.7(4) | 45.56(6) | — | 50.39(4) | 43.0(2) |
The coordination cages are relatively large compared to other complexes (see below) as evidenced by the distance between the N3- and O3-planes (2.9 and 2.8 Å, respectively). It is caused by the long length of the N–Mn coordination bonds. The large separation of the planes leads also to a relatively small twist angle between the planes (i.e. pendant arm rotation, ca. 8° and 24° for the complex anions in (NH4)3[Mn(notptfe)]Cl2·3H2O and [Mn(H2O)6][Mn(notptfe)]2·18H2O phases, respectively). In the crystal structure of [Mn(H2O)6][Mn(notptfe)]2·18H2O, two [Mn(notptfe)]− anions are head-to-head connected to the central [Mn(H2O)6]2+ cation through hydrogen bonds (Fig. 6); a similar structural motif was also found in some other crystal structures (see below).
Several Co2+-containing phases were obtained and, surprisingly, different diastereoisomers of the [Co(notptfe)]− anion were found. In the crystal structure of (NH4)[Co(notptfe)]·3.5H2O, the Δλ-SSS and Λδ-RRR enantiomers with the OC geometry were found (Fig. 7 and S37†). In the structure of [Co(H2O)6][Co(notptfe)]2·14.25H2O·0.75MeOH the Δδ-SSS and Λλ-RRR enantiomeric pairs with TTA geometries were found; for an example, see Fig. 8. These complex anions are connected to the [Co(H2O)6]2+ cation through the hydrogen bonds (Fig. S38†) similarly as it was observed in the crystal structure of [Mn(H2O)6][Mn(notptfe)]2·18H2O discussed above, although the compounds are not isostructural. In contrast to the structures of the other Co2+ phases, the sodium salt Na3[Co(notptfe)]2Br·3Me2CO presents a TTA enantiomeric Δδ-RRR/Λλ-SSS pair (Fig. 8 and S39†). Selected relevant geometric parameters for the structures are listed in Tables 4 and S5.†
 | ||
| Fig. 7 Molecular structure of the [Co(notptfe)]− anion with the Δλ-SSS OC geometry found in the crystal structure of (NH4)[Co(notptfe)]·3.5H2O. More abundant part of the disordered pendant arm (P3A and related atoms) is shown; for the disorder, see Fig. S37.† Hydrogen atoms are omitted for clarity. | ||
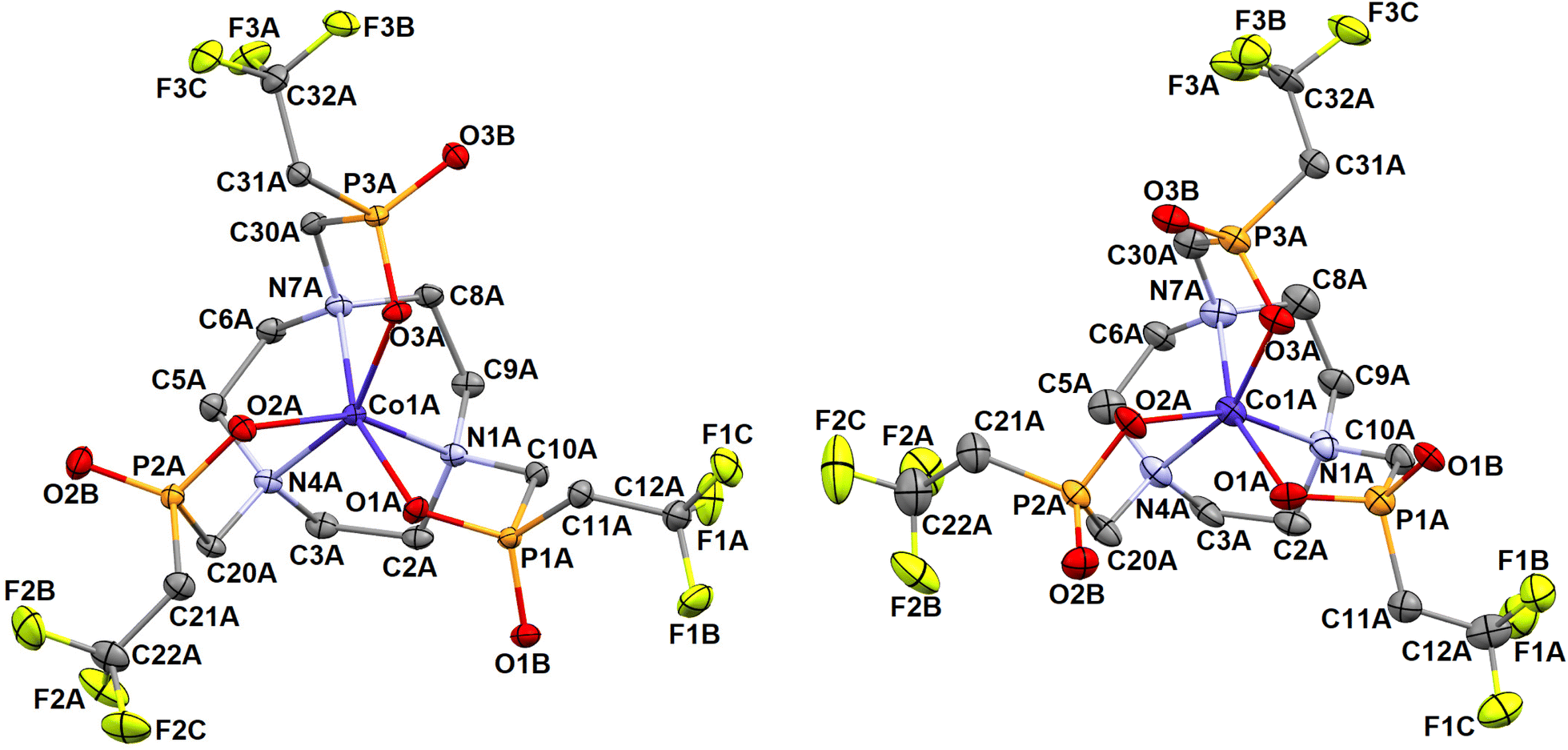 | ||
| Fig. 8 Molecular structures of the [Co(notptfe)]− anion with the Λλ-RRR TTA geometry found in the crystal structure of [Co(H2O)6][Co(notptfe)]2·14.25H2O·0.75MeOH (left) and that with the Λλ-SSS TTA geometry found in the crystal structure of Na3[Co(notptfe)]2Br·3Me2CO (right). Hydrogen atoms are omitted for clarity. | ||
The Ni2+ complex was successfully crystallized as [Mg(H2O)6][Ni(notptfe)]2·12H2O in the space group R and with the lattice parameters close to those of [Mn(H2O)6][Mn(notptfe)]2·18H2O; however, the compounds are not isostructural. As in the [Mn(H2O)6][Mn(notptfe)]2·18H2O and [Co(H2O)6][Co(notptfe)]2·14.25H2O·0.75MeOH phases, two [Ni(notptfe)]− anions are head-to-head connected to the central hexaaqua unit via hydrogen bonds (Fig. S40†). The [Ni(notptfe)]− species form an octahedral Λδ-RRR/Δλ-SSS pair (Fig. 9). For geometric parameters, see Tables 4 and S5.†
 | ||
| Fig. 9 Molecular structure of the [Ni(notptfe)]− anion with the Λδ-RRR OC geometry found in the crystal structure of [Mg(H2O)6][Ni(notptfe)]2·12H2O. Hydrogen atoms are omitted for clarity. | ||
In the case of the Cu2+ complex, two different phases were isolated in the solid state. The compound (NH4)[Cu(notptfe)]·3.5H2O is isostructural with (NH4)[Co(notptfe)]·3.5H2O and even an analogous disorder was found (see the ESI†). The Δλ-SSS/Λδ-RRR enantiomers with the octahedral geometry were found (Fig. S41 and S42†). In the other phase of composition (NH4)2[Cu(notptfe)]Cl·3H2O, only two pendant arms are coordinated and they are turned in mutually opposite directions (Fig. 10). Centrosymmetry of the space group (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) leads formally to ΔΛ-λλλ-SR/ΛΔ-δδδ-RS enantiomers. Very similar pentacoordinated geometry was previously reported for the Cu2+ complexes of H6notp and H3notpPh (Fig. 1).84,85 Selected geometric parameters are listed in Tables 4 and S5.†
) leads formally to ΔΛ-λλλ-SR/ΛΔ-δδδ-RS enantiomers. Very similar pentacoordinated geometry was previously reported for the Cu2+ complexes of H6notp and H3notpPh (Fig. 1).84,85 Selected geometric parameters are listed in Tables 4 and S5.†
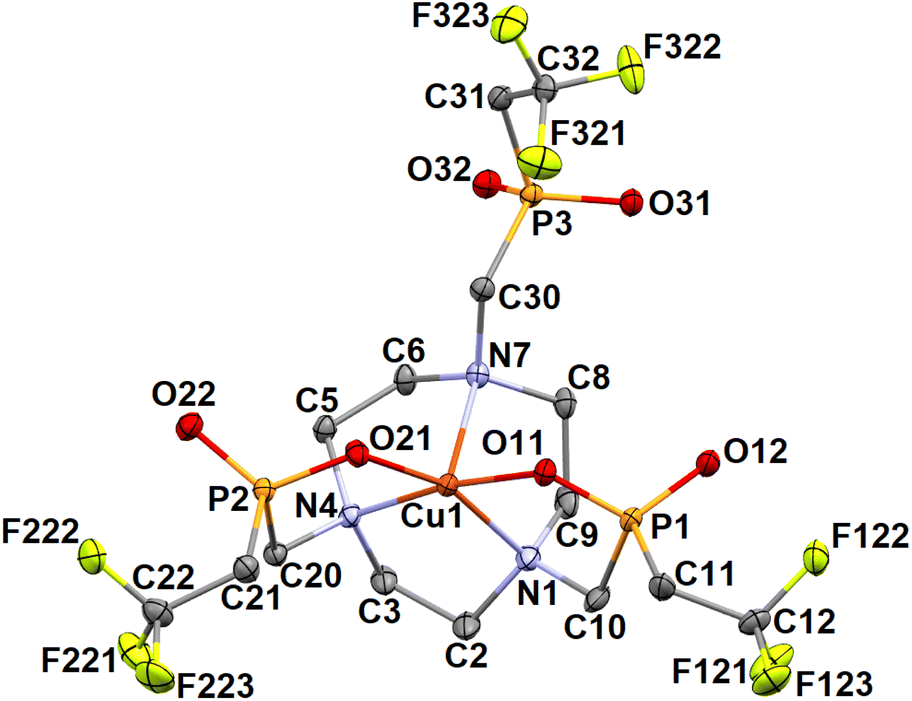 | ||
| Fig. 10 Molecular structure of the pentacoordinated [Cu(notptfe)]− anion adopting the ΔΛ0-λλλ-SR geometry found in the crystal structure of (NH4)2[Cu(notptfe)]Cl·3H2O. Hydrogen atoms are omitted for clarity. | ||
Octahedral species with Λδ-RRR and Δλ-SSS geometry were also observed in the crystal structure of the Zn2+ complex, [ZnCl(H2O)3][Zn(notptfe)]·2H2O (Fig. S43†). Here, the counter-cation [ZnCl(H2O)3]+ is formed from the excess of ZnCl2 used in the reaction. For geometric parameters, see Tables 4 and S5.†
Overall, the twisted trigonally antiprismatic (TTA, Δδ/Λλ) geometry is preferred for the largest ion (Mn2+) as this geometry leads to a larger coordination cage (dNQ⋯OQ 2.62–2.90 Å, Tables 4 and S5†). Smaller Ni2+, Cu2+ and Zn2+ ions form octahedral species (OC, Δλ/Λδ) with a shorter separation of the N3- and O3-planes (2.45–2.57 Å). The Co2+ ion of an intermediate size can adopt both environments, differing in this parameter. The torsion angles of the pendant arms are consistently smaller for the TTA (7–36°, ideal value 30°) than for the OC (43–54°, ideal value 60°) arrangements. Similar structural features were also found for divalent transition metal complexes of the related ligands. All reported divalent complexes of H3notpPh (Co2+, Ni2+, Cu2+ and Zn2+) adopt an OC geometry with the N3–O3 twist in the range 51–52°.86,87 However, the Mn2+ complex of H6notp also adopt the OC geometry but very distorted [twist 38°, distances from the centroids of the N3- and O3-planes, NQ and OQ, respectively, are d(Mn⋯NQ) 1.64 Å, d(Mn⋯NQ) 1.02 Å].88
The critical parameter influencing the relaxation rates of the 19F nuclei is the distance between the paramagnetic ion and the fluorine atoms. Therefore, this parameter is compiled in Table 4 together with selected geometric parameters of the coordination cages. It can be seen that the exact geometry of the coordination sphere influences the M⋯F distance only negligibly – in the TTA species, the distances are only slightly longer (ca. 5.6–6.7 Å) when compared to the OC isomers (ca. 5.5–6.5 Å). Such distances fall in the range suggested to be relevant for a significant influence on the 19F NMR relaxation times.89 Very similar mean M⋯F lengths were observed also in the complexes of 1,8-H2te2ptfe (cyclam-based ligand with the same pendant arms, Fig. 1) which were studied previously.47
NMR spectral properties of the complexes
Measured NMR characteristics of the studied complexes are compiled in Tables 5, 6, S1 and S2.† A comparison of the 19F NMR spectra of transition metal complexes of H3notptfe is shown in Fig. 11. | ||
| Fig. 11 Visual comparison of 19F NMR spectra of the studied transition metal complexes of H3notptfe and free ligand. The inset shows spectra of diamagnetic H3notptfe and [Zn(notptfe)]−. | ||
| 19F Larmor frequency/MHz | 565 | 376 | 282 | ||||
|---|---|---|---|---|---|---|---|
| Temperature/°C | 25 | 37 | 25 | 37 | 25 | 37 | |
| Sample | δ(corr.)/ppm | T 1/ms | |||||
| a Only a rough estimate due to the very fast relaxation. b Spectra cannot be successfully phased. | |||||||
| H3notptfe | −57.12 | 0.66 × 103 | 0.85 × 103 | 0.97 × 103 | 1.2 × 103 | 1.1 × 103 | 1.5 × 103 |
| [MgII(notptfe)]− | −57.30 | 0.61 × 103 | 0.75 × 103 | 0.88 × 103 | 1.0 × 103 | 1.1 × 103 | 1.3 × 103 |
| [CrIII(notptfe)] | −46.0 | 1.0a | 2.0a | —b | —b | —b | —b |
| [MnII(notptfe)]− | −41.8 | 0.5a | 0.8a | —b | —b | —b | —b |
| [FeIII(notptfe)] | −31.0 | 0.4a | 0.5a | 0.5a | 0.5a | 0.5a | 0.8a |
| [CoII(notptfe)]− | −50.5 | 44 | 57 | 64 | 71 | 78 | 83 |
| [NiII(notptfe)]− | −48.5 | 3.2 | 3.9 | 3.3 | 4.0 | 3.6 | 4.8 |
| [CuII(notptfe)]− | −54.2 | 4.7 | 5.7 | 4.1 | 5.0 | 3.8 | 5.2 |
| [ZnII(notptfe)]− | −57.21 | 0.61 × 103 | 0.76 × 103 | 0.95 × 103 | 1.1 × 103 | 1.0 × 103 | 1.4 × 103 |
 (ms, paramagnetic compounds, calculated from the half-widths) of the 19F NMR signals of H3notptfe and its studied complexes. Estimated deviation (based on repeated measurements) is generally < 5%
(ms, paramagnetic compounds, calculated from the half-widths) of the 19F NMR signals of H3notptfe and its studied complexes. Estimated deviation (based on repeated measurements) is generally < 5%
| 19F Larmor frequency/MHz | 565 | 376 | 282 | ||||
|---|---|---|---|---|---|---|---|
| Temperature/°C | 25 | 37 | 25 | 37 | 25 | 37 | |
| Sample | δ(corr.)/ppm | T 2/ms | |||||
| a Measurement of T2 was not possible on a Varian VNMRS300 with an accessible probe. b Only a rough estimate due to the extreme broadness of the signal. c Spectra cannot be successfully phased. | |||||||
| H3notptfe | −57.12 | 0.52 × 103 | 0.69 × 103 | 0.80 × 103 | 0.95 × 103 | —a | —a |
| [MgII(notptfe)]− | −57.30 | 0.22 × 103 | 0.17 × 103 | 0.48 × 103 | 0.44 × 103 | —a | —a |
| [ZnII(notptfe)]− | −57.21 | 0.23 × 103 | 0.35 × 103 | 0.41 × 103 | 0.52 × 103 | —a | —a |
| T 2*/ms | |||||||
| [CrIII(notptfe)] | −46.0 | 0.22b | 0.17b | —c | —c | —c | —c |
| [MnII(notptfe)]− | −41.8 | 0.08b | 0.07b | —c | —c | —c | —c |
| [FeIII(notptfe)] | −31.0 | 0.21b | 0.26b | 0.22b | 0.27b | 0.24b | 0.30b |
| [CoII(notptfe)]− | −50.5 | 1.8 | 1.0 | 2.1 | 1.3 | 2.3 | 1.6 |
| [NiII(notptfe)]− | −48.5 | 1.9 | 2. | 2.1 | 2.6 | 2.3 | 2.7 |
| [CuII(notptfe)]− | −54.2 | 2.1 | 2.6 | 1.8 | 2.2 | 1.7 | 2.2 |
To study the solution structures of the complexes, the NMR spectra of diamagnetic (Mg2+, Zn2+) complexes also were acquired. The 19F NMR spectrum of the Mg2+–H3notptfe system at pH 7.5 fully agrees with the results of potentiometry – the spectrum revealed the presence of the well resolved signals of the free ligand and the complex in ca. 65:35 ratio (Fig. S8†). Besides these two species, some very minor signals were also observed, probably belonging to the complex species with different R/S configurations of the phosphinate pendant groups or to some species in which some of the pendant arms are uncoordinated. It indicates that the ligand and complex species are not in chemical exchange with respect to the NMR time scale, and, very probably, the major complex species has the C3-symmetry. In the case of the Zn2+–H3notptfe system, a full complexation of the metal ion was expected on the basis of potentiometry. It was confirmed by the 19F and 31P NMR spectra (Fig. S23–S25†) where observation of only one symmetric signal points to the presence of only one enantiomeric pair of a single diastereoisomer (one combination of Δ/Λ + δ/λ + R/S) in the solution; it is very probably the octahedral Δλ-SSS/Λδ-RRR species found in the solid state (see above). However, the 1H and 13C{1H} NMR spectra showed a fast fluxionality of the complex species: only two unresolved broad signals were found in the 1H NMR spectrum at 25 °C (Fig. S21†). The signals remained broad even at 5 °C which precludes further study. In the 13C{1H} NMR spectrum, two very broad signals of the macrocyclic carbon atoms were found but other carbon atoms showed well-resolved sharp signals (Fig. S22†).
For the Cr3+, Mn2+ and Fe3+ complexes, only 19F NMR signals can be detected; signals of the other nuclei fully vanished, probably due to the relatively slow electronic relaxation caused by the symmetric electronic state of the metal ions (t2g3 for Cr3+, HS-d5 for the others). The 19F NMR signals of [CrIII(notptfe)] and [MnII(notptfe)]− are extremely broad (ν½ ≈ 2.0 and ≈4.6 kHz at 565 MHz, respectively) whereas the 19F NMR signal of the [FeIII(notptfe)] complex is more narrow (ν½ ≈1.1 kHz at 565 MHz) and can be easily measured. However, the longitudinal relaxation of the 19F NMR signals of all these complexes is extremely fast (T1 < 1 ms, Tables 5 and 6).
In contrast to the paramagnetic complexes discussed above, all 1H, 13C, 19F and 31P NMR spectra could be observed for the [CoII(notptfe)]− complex. As several geometries of the complex were found in the solid state (see above), some dynamic equilibrium between the arrangements can be expected in the solution. However, there is only one symmetric (although broad) peak in the 19F/31P NMR spectra and only one set of a 1H/13C{1H} signal. Thus, fluxionality of the [CoII(notptfe)]− complex species is probably very fast. In the 1H NMR spectra spreading over 200 ppm, 7 of the expected 8 signals were found (Fig. 12 and S13†) as the last signal was very close to the signal of the solvent (water/HDO) and overlapped; it was found by measuring the temperature dependence of the spectra (Fig. S13†). In the 13C{1H} NMR spectra, all of the expected 5 signals were observed in the range from −500 to +200 ppm (Fig. 12 and S14†). A similar very large spectral 13C NMR range was observed previously for the Co2+ complex of the N-(2,2,2-trifluoroethyl) cyclam derivative.45
 | ||
| Fig. 12 1H (left) and 13C{1H} (right) NMR spectra of the [CoII(notptfe)]− complex (D2O, pD 7.4, 37 °C). | ||
In the 19F NMR spectrum of [CoII(notptfe)]−, one broad signal centred at −50.5 ppm is present (Fig. S15†) which shows optimally fast longitudinal relaxation in order tens of milliseconds (T1 40–80 ms, dependent on the external magnetic field strength and temperature, Table 5), although its transversal relaxation is very fast (Table 6). This compound also shows a broad 31P{1H} NMR signal at ca. 200 ppm (Fig. S16†). When the [CoII(notptfe)]− complex is oxidized with K2S2O8 or H2O2, a set of multiplets (pseudo-quartets due to similar values of JFH and JFP) of [CoIII(notptfe)] gradually appears in the 19F NMR spectrum (Fig. 13). It is consistent with the data from spectro-electrochemical experiments (Fig. S33†). The signals are narrow and their relaxation is slow. Thus, the formed [CoIII(notptfe)] complex is obviously diamagnetic with a low-spin d6 arrangement. Several close and hardly separable 19F NMR signals of [CoIII(notptfe)] appear which can be explained by the presence of isomers with different R/S configurations on the phosphinate groups. These isomers are not exchanged due to kinetic inertness and non-fluxionality of the Co3+ complexes, and each affords individual signal(s).
 | ||
| Fig. 13 19F NMR spectra of the reaction mixtures showing gradual oxidation of [CoII(notptfe)]− to [CoIII(notptfe)] with K2S2O8. | ||
The 19F NMR spectrum of the [NiII(notptfe)]− complex contains two broad 19F NMR signals separated by ca. 3 ppm, with a relative intensity ca. 10:90% (Fig. S18†). In the case of [CuII(notptfe)]−, only one broad signal was observed (Fig. S20†). The signals of both complexes relax very fast with T1 in the range of ca. 3–5 ms and T2ca. 2 ms (Tables 5 and 6).
Overall, the observed relaxation times are consistent with those found previously for the complexes of the related cyclam-based ligand 1,8-H2te2ptfe (Fig. 1) with the same pendant arm.47 Fast movement of the 2,2,2-trifluoroethyl group effectively averages the distances; however, the mean value cannot be reliably calculated as it strongly correlates with electronic relaxation times whose exact values are not known and, for individual metal ions, can cover a wide range.47 However, the [CrIII(notptfe)], [MnII(notptfe)]− and [FeIII(notptfe)] complexes cannot be utilised in 19F MRI as they show very broad signals which relax too fast and, therefore, are not reliably detectable. The relaxation characteristics of the [NiII(notptfe)]− and [CuII(notptfe)]− complexes are more suitable but their potential use in the imaging experiments would need special ultrafast measurement techniques90,91 due to too short relaxation times (few milliseconds). In this respect, the most promising is the [CoII(notptfe)]− complex showing optimally fast longitudinal relaxation (T1 in order of tens of milliseconds) which is suitable for standard MRI hardware. Furthermore, the redox potential of the [CoIII(notptfe)]/[CoII(notptfe)]− pair has physiologically relevant value making the compound potentially employable as a redox probe.
Conclusions
A new 1,4,7-triazacyclononane-based ligand substituted with three (2,2,2-trifluoroethyl)phosphinate pendant arms was studied with respect to its potential use in contrast agents for 19F Magnetic Resonance Imaging (19F MRI). With a large metal ion Mn2+, the ligand forms complexes with twisted-trigonal-antiprismatic geometry, whereas with the smaller ions Ni2+, Cu2+ and Zn2+, complex species with an octahedral geometry are formed. With the Co2+ ion, both environments were observed. Complexes of Co2+, Ni2+, Cu2+ and Zn2+ are very stable and are fully formed at pH > ∼3. Paramagnetic metal ion complexes show very short relaxation times of the 19F NMR signal due to a short distance between the paramagnetic metal ion and the fluorine atoms (values found in the crystal structures are ∼5.5–6.5 Å). Most of the studied complexes show too fast longitudinal relaxation for the application (T1 of the Cr3+, Mn2+ and Fe3+ complexes in the sub-millisecond range; the Ni2+ and Cu2+ complexes ∼4 ms), but T1 of [CoII(notptfe)]− (40–80 ms) falls in a range suitable for easy exploitation. Electrochemical studies revealed the formation of a stable diamagnetic Co3+ complex (E½ 0.95 V vs. SCE). Together with a suitable relaxation time, it makes the [CoIII(notptfe)]/[CoII(notptfe)]− pair potentially useful as a smart redox-responsive contrast agent in 19F MRI.Author contributions
FK – investigation, methodology, validation, visualization, and writing – original draft; TD – investigation and writing – original draft, JK – conceptualization, investigation, methodology, validation, visualization, and writing – original draft, review and editing, IC – investigation, JH – investigation, AL – investigation, VK – investigation, PH – conceptualization, funding acquisition, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the Czech Science Foundation (GAČR) 22-34083S is acknowledged.References
- The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. E. Merbach, L. Helm and É. Tóth, John Wiley & Sons, 2nd edn, 2013 Search PubMed.
- C. S. Bonnet and É. Tóth, Chimia, 2016, 70, 102–108 CrossRef CAS PubMed.
- Y. A. Pirogov, Phys. Procedia, 2016, 82, 3–7 CrossRef CAS.
- I. Tirotta, V. Dichiarante, C. Pigliacelli, G. Cavallo, G. Terraneo, F. B. Bombelli, P. Metrangolo and G. Resnati, Chem. Rev., 2015, 115, 1106–1129 CrossRef CAS PubMed.
- J. Ruiz-Cabello, B. P. Barnetta, P. A. Bottomley and J. W. M. Bulte, NMR Biomed., 2011, 24, 114–129 CrossRef CAS PubMed.
- E. T. Ahrens, R. Flores, H. Xu and P. A. Morel, Nat. Biotechnol., 2005, 23, 983–987 CrossRef CAS PubMed.
- T. Zhang, Q. Zhang, J.-H. Tian, J.-F. Xing, W. Guo and X.-J. Liang, MRS Commun., 2018, 8, 303–313 CrossRef CAS.
- K. Waxman, Ann. Emerg. Med., 1986, 15, 1423–1424 CrossRef CAS PubMed.
- A. R. Burgan, W. C. Herrick, D. M. Long and D. C. Long, Biomater., Artif. Cells, Artif. Organs, 1988, 16, 681–682 CrossRef.
- K. C. Lowe, Blood Rev., 1999, 13, 171–184 CrossRef CAS PubMed.
- B. Remy, G. Deby-Dupont and M. Lamy, Br. Med. Bull., 1999, 55, 277–298 CrossRef CAS PubMed.
- G. A. Peyman, J. A. Schulman and B. Sullivan, Surv. Ophthalmol., 1995, 39, 375–395 CrossRef CAS PubMed.
- R. D. Bourke, R. N. Simpson, R. J. Cooling and J. R. Sparrow, Arch. Ophthalmol., 1996, 114, 537–544 CrossRef CAS PubMed.
- J. Pastor, R. M. Coco, I. Fernandez-Bueno, M. L. Alonso-Alonso, J. Medina, A. Sanz-Arranz, F. Rull, M. J. Gayoso, A. Dueñas, M. T. Garcia-Gutierrez, L. Gonzalez-Buendia, S. Delgado-Tirado, E. Abecia, M. Ruiz-Miguel, M. A. Serrano, J. M. Ruiz-Moreno and G. K. Srivastava, Retina, 2017, 37, 1140–1151 CrossRef CAS PubMed.
- I. Georgalas, I. Ladas, I. Tservakis, S. Taliantzis, E. Gotzaridis, D. Papaconstantinou and C. Koutsandrea, Cutaneous Ocul. Toxicol., 2011, 30, 251–262 CrossRef CAS PubMed.
- C. Gatto, P. Ruzza, L. Giurgola, C. Honisch, O. Rossi, M. R. Romano, E. Ragazzi and J. D'Amato Tóthová, ACS Omega, 2023, 8, 365–372 CrossRef CAS PubMed.
- T. Stahl, D. Mattern and H. Brunn, Environ. Sci. Eur., 2011, 23, 38 CrossRef.
- P. Hermann, J. Blahut, J. Kotek and V. Herynek, Met. Ions Life Sci., 2021, 22, 239–270 Search PubMed.
- P. K. Senanayake, A. M. Kenwright, D. Parker and S. K. van der Hoorn, Chem. Commun., 2007, 2923–2925 RSC.
- A. M. Kenwright, I. Kuprov, E. De Luca, D. Parker, S. U. Pandya, P. K. Senanayake and D. G. Smith, Chem. Commun., 2008, 2514–2516 RSC.
- K. H. Chalmers, E. De Luca, N. H. M. Hogg, A. M. Kenwright, I. Kuprov, D. Parker, M. Botta, J. I. Wilson and A. M. Blamire, Chem. – Eur. J., 2010, 16, 134–148 CrossRef CAS PubMed.
- Z.-X. Jiang, Y. Feng and Y. B. Yu, Chem. Commun., 2011, 47, 7233–7235 RSC.
- K. H. Chalmers, M. Botta and D. Parker, Dalton Trans., 2011, 904–913 RSC.
- P. Harvey, K. H. Chalmers, E. De Luca, A. Mishra and D. Parker, Chem. – Eur. J., 2012, 18, 8748–8757 CrossRef CAS PubMed.
- P. Harvey, I. Kuprov and D. Parker, Eur. J. Inorg. Chem., 2012, 2015–2022 CrossRef CAS.
- M. P. Placidi, M. Botta, F. K. Kálmán, G. E. Hagberg, Z. Baranyai, A. Krenzer, A. K. Rogerson, I. Tóth, N. K. Logothetis and G. Angelowski, Chem. – Eur. J., 2013, 19, 11644–11660 CrossRef CAS PubMed.
- N. Cakić, T. Savić, J. Stricker-Shaver, V. Truffault, C. Platas-Iglesias, C. Mirkes, R. Pohmann, K. Scheffler and G. Angelovski, Chem. Commun., 2016, 52, 9224–9227 RSC.
- K. Srivastava, E. A. Weitz, K. L. Peterson, M. Marjańska and V. C. Pierre, Inorg. Chem., 2017, 56, 1546–1557 CrossRef CAS PubMed.
- R. Pujales-Paradela, T. Savić, D. Esteban-Gómez, G. Angelovski, F. Carniato, M. Botta and C. Platas-Iglesias, Chem. – Eur. J., 2019, 25, 4782–4792 CrossRef CAS PubMed.
- R. Pujales-Paradela, T. Savić, P. Pérez-Lourido, D. Esteban-Gómez, G. Angelovski, M. Botta and C. Platas-Iglesias, Inorg. Chem., 2019, 58, 7571–7583 CrossRef CAS PubMed.
- V. Herynek, M. Martinisková, Y. Bobrova, A. Gálisová, J. Kotek, P. Hermann, F. Koucký, D. Jirák and M. Hájek, Magn. Reson. Mater. Phys., Biol. Med., 2019, 32, 115–122 CrossRef CAS PubMed.
- R. B. Lauffer, Chem. Rev., 1987, 87, 901–927 CrossRef CAS.
- D. Xie, M. Yu, R. T. Kadakia and E. L. Que, Acc. Chem. Res., 2020, 53, 2–10 CrossRef CAS PubMed.
- K. Srivastava, G. Ferrauto, V. G. Young Jr., S. Aime and V. C. Pierre, Inorg. Chem., 2017, 56, 12206–12213 CrossRef CAS PubMed.
- M. Yu, B. S. Bouley, D. Xie and E. L. Que, Dalton Trans., 2019, 48, 9337–9341 RSC.
- NIST Standard Reference Database 46 (Critically Selected Stability Constants of Metal Complexes), Version 7.0, 2003, distributed by NIST standard Reference Data, Gaithersburg, MD 20899, USA.
- M. Yu, D. Xie, K. P. Phan, J. S. Enriquez, J. J. Luci and E. L. Que, Chem. Commun., 2016, 52, 13885–13888 RSC.
- A. E. Thorarinsdottir, A. I. Gaudette and T. D. Harris, Chem. Sci., 2017, 8, 2448–2456 RSC.
- A. I. Gaudette, A. E. Thorarinsdottir and T. D. Harris, Chem. Commun., 2017, 53, 12962–12965 RSC.
- M. Yu, B. S. Bouley, D. Xie, J. S. Enriquez and E. L. Que, J. Am. Chem. Soc., 2018, 140, 10546–10552 CrossRef CAS PubMed.
- J. Blahut, P. Hermann, A. Gálisová, V. Herynek, I. Císařová, Z. Tošner and J. Kotek, Dalton Trans., 2016, 45, 474–478 RSC.
- J. Blahut, K. Bernášek, A. Gálisová, V. Herynek, I. Císařová, J. Kotek, J. Lang, S. Matějková and P. Hermann, Inorg. Chem., 2017, 56, 13337–13348 CrossRef CAS PubMed.
- R. Pujales-Paradela, T. Savić, I. Brandariz, P. Pérez-Lourido, G. Angelovski, D. Esteban-Gómez and C. Platas-Iglesias, Chem. Commun., 2019, 55, 4115–4118 RSC.
- D. Xie, L. E. Ohman and E. L. Que, Magn. Reson. Mater. Phys., Biol. Med., 2019, 32, 89–96 CrossRef CAS PubMed.
- J. Blahut, L. Benda, J. Kotek, G. Pintacuda and P. Hermann, Inorg. Chem., 2020, 59, 10071–10082 CrossRef CAS PubMed.
- Z. Kotková, F. Koucký, J. Kotek, I. Císařová, D. Parker and P. Hermann, Dalton Trans., 2023, 52, 1861–1875 RSC.
- F. Koucký, J. Kotek, I. Císařová, J. Havlíčková, V. Kubíček and P. Hermann, Dalton Trans., 2023, 52, 12208–12223 RSC.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth Heinemann, An imprint of Elsevier Science, 5th edn, 2003 Search PubMed.
- T. M. Gøgsig, L. S. Søbjerg, A. T. Lindhardt, K. L. Jensen and T. Skrydstrup, J. Org. Chem., 2008, 73, 3404–3410 CrossRef PubMed.
- E. T. McBee, D. H. Campbell and C. W. Roberts, J. Am. Chem. Soc., 1955, 77, 3149–3151 CrossRef CAS.
- S. Xia, Y.-B. Shi, F.-F. He and H.-B. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o2779 CrossRef CAS PubMed.
- K. Wieghardt, U. Bossek, P. Chaudhuri, W. Herrmann, B. C. Menke and J. Weiss, Inorg. Chem., 1982, 21, 4308–4314 CrossRef CAS.
- M. Krejčik, M. Daněk and J. Hartl, J. Electroanal. Chem., 1991, 317, 179–187 CrossRef.
- M. Försterová, I. Svobodová, P. Lubal, P. Táborský, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2007, 535–549 RSC.
- C. F. Baes Jr. and R. E. Mesmer, The Hydrolysis of Cations, Wiley, New York, 1976 Search PubMed.
- M. Kývala and I. Lukeš, International conference, chemometrics × 95, abstract book p. 63, Pardubice (Czech Republic), 1995; full version of ™OPIUM.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, SHELXT2014/5. Program for Crystal Structure Solution from Diffraction Data, University of Göttingen, Göttingen, 2014 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- (a) C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt graphical user interface for SHELXL, University of Göttingen, Göttingen, 2014 Search PubMed; (b) C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed; (d) G. M. Sheldrick, SHELXL-2017/1. Program for Crystal Structure Refinement from Diffraction Data, University of Göttingen, Göttingen, 2017 Search PubMed.
- K. Bazakas and I. Lukeš, Dalton Trans., 1995, 1133–1137 RSC.
- J. Šimeček, M. Schulz, J. Notni, J. Plutnar, V. Kubíček, J. Havlíčková and P. Hermann, Inorg. Chem., 2012, 51, 577–590 CrossRef PubMed.
- J. Huskens and A. D. Sherry, J. Am. Chem. Soc., 1996, 118, 4396–4404 CrossRef CAS.
- I. Lukeš, J. Kotek, P. Vojtíšek and P. Hermann, Coord. Chem. Rev., 2001, 216–217, 287–312 CrossRef.
- B. Drahoš, V. Kubíček, C. S. Bonnet, P. Hermann, I. Lukeš and E. Tóth, Dalton Trans., 2011, 40, 1945–1951 RSC.
- V. Kubíček, Z. Böhmová, R. Ševčíková, J. Vaněk, P. Lubal, Z. Poláková, R. Michalicová, J. Kotek and P. Hermann, Inorg. Chem., 2018, 57, 3061–3072 CrossRef PubMed.
- C. F. G. C. Geraldes, A. D. Sherry and W. P. Cacheris, Inorg. Chem., 1989, 28, 3336–3341 CrossRef CAS.
- J. Gao, H. He, W. Yang, J.-G. Hou and J. Kang, Lab. Rob. Autom., 1998, 10, 229–233 CrossRef CAS.
- M. I. Kabachnik, T. Ya. Medved, Yu. M. Polikarpov, B. K. Tscherbakov, F. I. Belskij, E. I. Mamrosov and M. P. Pasechnik, Izv. Akad. Nauk SSSR, Ser. Khim., 1984, 835–843 CAS.
- H. F. Bauer and W. C. Drinkard, J. Am. Chem. Soc., 1960, 82, 5031–5032 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- D. M. Corsi, C. Platas-Iglesias, H. van Bekkum and J. A. Peters, Magn. Reson. Chem., 2001, 39, 723–726 CrossRef CAS.
- T. L. Hatfield, R. J. Staples and D. T. Pierce, Inorg. Chem., 2010, 49, 9312–9320 CrossRef CAS PubMed.
- S. Aime, A. S. Batsanov, M. Botta, J. A. K. Howard, D. Parker, K. Senanayake and G. Williams, Inorg. Chem., 1994, 33, 4696–4706 CrossRef CAS.
- S. Aime, M. Botta, M. Fasano, M. P. M. Marques, C. F. G. C. Geraldes, D. Pubanz and A. E. Merbach, Inorg. Chem., 1997, 36, 2059–2068 CrossRef CAS PubMed.
- S. Aime, A. Barge, F. Benetollo, G. Bombieri, M. Botta and F. Uggeri, Inorg. Chem., 1997, 36, 4287–4289 CrossRef CAS.
- F. Benetollo, G. Bombieri, L. Calabi, S. Aime and M. Botta, Inorg. Chem., 2003, 42, 148–157 CrossRef CAS PubMed.
- M. Woods, M. Botta, S. Avedano, J. Wang and A. D. Sherry, Dalton Trans., 2005, 3829–3837 RSC.
- P. Vojtíšek, P. Cígler, J. Kotek, J. Rudovský, P. Hermann and I. Lukeš, Inorg. Chem., 2005, 44, 5591–5599 CrossRef PubMed.
- J. Kotek, J. Rudovský, P. Hermann and I. Lukeš, Inorg. Chem., 2006, 45, 3097–3102 CrossRef CAS PubMed.
- P. Urbanovský, J. Kotek, I. Císařová and P. Hermann, Dalton Trans., 2020, 49, 1555–1569 RSC.
- W. D. Kim, G. E. Kiefer, J. Huskens and A. D. Sherry, Inorg. Chem., 1997, 36, 4128–4134 CrossRef CAS.
- J. Rohovec, I. Lukeš and P. Hermann, New J. Chem., 1999, 23, 1129–1132 RSC.
- Z. Kotková, G. A. Pereira, K. Djanashvili, J. Kotek, J. Rudovský, P. Hermann, L. Vander Elst, R. N. Muller, C. F. G. C. Geraldes, I. Lukeš and J. A. Peters, Eur. J. Inorg. Chem., 2009, 119–136 CrossRef.
- Y.-H. Su, S.-S. Bao and L.-M. Zheng, Inorg. Chem., 2014, 53, 6042–6047 CrossRef CAS PubMed.
- M. I. Kabachnik, M. Y. Antipin, B. K. Tscherbakov, A. P. Baranov, Yu. T. Struchkov, T. Y. Medved and Yu. M. Polikarpov, Koord. Khim., 1988, 14, 536–542 CAS.
- E. Cole, D. Parker, G. Ferguson, J. F. Gallagher and B. Kaitner, J. Chem. Soc., Chem. Commun., 1991, 1473–1475 RSC.
- E. Cole, R. C. B. Copley, J. A. K. Howard, D. Parker, G. Ferguson, J. F. Gallagher, B. Kaitner, A. Harrison and L. Royle, J. Chem. Soc., Dalton Trans., 1994, 1619–1629 RSC.
- S.-S. Bao, G.-S. Chen, Y. Wang, Y.-Z. Li, L.-M. Zheng and Q.-H. Luo, Inorg. Chem., 2006, 45, 1124–1129 CrossRef CAS PubMed.
- M. Zalewski, D. Janasik, A. Wierzbicka and T. Krawczyk, Inorg. Chem., 2022, 61, 19524–19542 CrossRef CAS PubMed.
- K. H. Chalmers, A. M. Kenwright, D. Parker and A. M. Blamire, Magn. Reson. Med., 2011, 66, 931–936 CrossRef CAS PubMed.
- F. Schmid, C. Höltke, D. Parker and C. Faber, Magn. Reson. Med., 2013, 69, 1056–1062 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC data, details on refinement of the crystal structures, discussion of the crystal structures of organic intermediates, molecular structures found in the crystal structures, selected geometric parameters, NMR spectra of the prepared compounds, UV-Vis spectra of selected complexes, distribution diagrams of the studied systems, and visualization of spectro-electrochemical experiments. CCDC 2327147–2327158. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00507d |
| This journal is © The Royal Society of Chemistry 2024 |