
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the stabilization of monomeric stibinidene chalcogenides and stibine chalcogenides
John S.
Wenger
 and
Timothy C.
Johnstone
*
and
Timothy C.
Johnstone
*
Department of Chemistry and Biochemistry, University of California Santa Cruz, Santa Cruz, California 95064, USA. E-mail: johnstone@ucsc.edu
First published on 8th May 2024
Abstract
The elucidation of novel bonding situations at heavy p-block elements has greatly advanced recent efforts to access useful reactivity at earth-abundant main-group elements. Molecules with unsaturated bonds between heavier, electropositive elements and lighter, electronegative elements are often highly polarized and competent in small-molecule activations, but the reactivity of these molecules may be quenched by self-association of monomers to form oligomeric species where the polar, unsaturated groups are assembled in a head-to-tail fashion. In this Frontier, we discuss the synthetic strategies employed to isolate monomeric σ2,λ3-stibinidene chalcogenides (RSbCh) and monomeric σ4,λ5-stibine chalcogenides (R3SbCh). These classes of molecules each feature polarized antimony–chalcogenide bonds (Sb = Ch/Sb+–Ch−). We highlight how the synthesis and isolation of these molecules has led to the discovery of novel reactivity and has shed light on fundamental aspects of inorganic structure and bonding. Despite these advances, there are critical aspects of this chemistry that remain underdeveloped and we provide our perspective on yet-unrealized synthetic targets that may be achieved with the continued development of the strategies described herein.
Introduction
The chemistry of main-group elements is currently under intense investigation to discover novel molecular motifs that can be leveraged for small molecule activation and provide new insights into chemical bonding and reactivity.1,2 Different strategies have been developed to enable main-group elements to engage in useful reactivity. These strategies often involve the stabilization of molecules or molecular systems that possess unquenched reactivity. If a molecular motif or system exhibits ambiphilic reactivity, then deactivation can occur via self-association to form stable adducts, dimers, or oligomers. It should be noted, however, that self-association does not necessarily preclude interesting reactivity, especially if the adduct is in equilibrium with the dissociated form.3–6Unsaturated bonds between heavier electropositive elements and lighter electronegative elements are often highly polarized (Fig. 1a), and this polarization can be exploited for small molecule activation. The electronic structures of molecules with heavy main-group elements vary significantly from their first octal row congeners.7 As a group is descended, the valence orbitals of elements increase in size and diffuseness, resulting in diminished overlap with the relevant orbitals of bonding partners. Thus, π bonding tends to be less efficient with heavier main-group elements, causing a greater separation of charge and contributing ylidic character (Fig. 1a) to these polar, unsaturated bonds. These species can form head-to-tail dimers or oligomers to attenuate the separation of charge, exploiting the ability of the large heavy element to access an expanded coordination sphere (Fig. 1b).
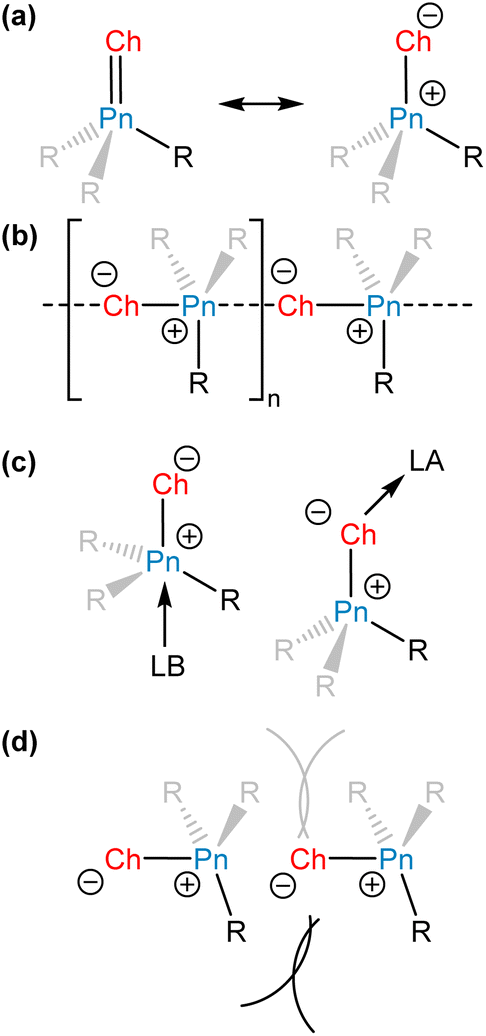 | ||
| Fig. 1 General strategies for the stabilization of pnictinidene chalcogenides and pnictine chalcogenides. (a) Resonance structures for unsaturated pnictogen–chalcogen bonding. (b) Self-association of pnictine chalcogenides or pnictinidene chalcogenides. (c) Self-association can be prevented via thermodynamic stabilization afforded by a Lewis acid or base. (d) Self-association can be prevented via kinetic stabilization using sterically demanding substituents. | ||
To isolate highly reactive molecules, chemists can employ an external Lewis acid or base to form an adduct with the reactive motif (Fig. 1c).8 By engaging with the loci of unquenched reactivity, this strategy thermodynamically stabilizes the reactive molecule, but also prevents direct investigation of its reactivity. The added acid or base can be a main-group compound but could also be a transition-metal complex and many reactive main-group bonding motifs have been captured for the first time in the primary coordination sphere of a metal atom.9
Chemists can alternatively employ a kinetic stabilization approach, in which reactive motifs are sterically protected using bulky substituents to prevent decomposition or self-association reactions (Fig. 1d).10 The strategy of kinetic stabilization is attractive because it enables access to unperturbed bonds. However, steric protection may limit the scope of substrates that can react with the fragment of interest and sterically encumbered species may be synthetically challenging to access. In addition to sterically hindering some self-association reactions, appropriately selected bulky substituents can also engage in significant attractive London dispersion interactions between one another and provide a driving force toward self-association.11 In this article we will focus on the ability of large substituents to prevent self-association, but even within that framework, the thermodynamic and kinetic stabilization strategies are not mutually exclusive.
The pnictogens have recently been identified as being particularly effective in mediating main-group redox catalysis.12 Among the pnictogens, antimony often exhibits characteristic reactivity. For example, investigations of comparable pnictogen-containing compounds found that Lewis acidity tends to increase with the pnictogen atomic number until antimony, and then decreases for bismuth.13–15 The Lewis acidity of antimony compounds has been exploited in applications ranging from ion sensing to catalysis.16–20 In light of recent developments in the chemistry of bismuth compounds, we anticipate that there are yet further exciting discoveries to be made in the chemistry of antimony.21 Our group has been particularly interested in the reactivity of unsaturated bonds involving group 16 elements and heavy group 15 elements. In this Frontier, we will discuss recent advances in the stabilization of σ2,λ3-stibinidene chalcogenides and σ4,λ5-stibine chalcogenides and highlight examples where the unquenched reactivity of polar, unsaturated antimony–chalcogen bonds has been leveraged for interesting substrate activations.
Monomeric stibinidene chalcogenides
Studies from the early-to-mid 20th century on the alkaline hydrolysis of RSbCl2 species afforded materials that analyzed as RSbO (e.g., PhSbO), but anomalous cryoscopic measurements and comparison to the analogous As species led to the conclusion that the species existed as (RSbO)n oligomers.22–24 This conclusion was supported by subsequent 121Sb Mössbauer isomer shifts, which were consistent with one aryl and two bridging oxide substituents,25 and single-crystal X-ray diffraction data.26,27 The use of larger substituents facilitated progress toward well-defined multimeric and, ultimately, monomeric σ2,λ3-stibinidene chalcogenides. The sterically encumbered stibinidene chalcogenides (RSbCh)n (R = CH(SiMe3)2, Ch = S, Se, Te) are predominantly trimeric in solution, but may engage in ring-ring equilibria with dimeric and tetrameric species.28 In the late 1990s, the dimeric species (L1Sb)2 with the bulky substituent L1 (Fig. 2) was accessed via the deselenation of (L1SbSe)3 by P(NMe2)3 (Fig. 3b).29,30 The (L1SbSe)3 starting material had been produced by reaction of L1SbCl2 with Li2Se (Fig. 3a); it exists as a trimeric species in which individual L1SbSe units associate in a head-to-tail fashion to form a 6-membered ring. (L1Sb)2 can serve as a starting material for new stibinidene chalcogenides. Exposure of (L1Sb)2 to oxygen quantitatively affords the corresponding dioxadistibetane (L1SbO)2 (Fig. 3c).30 Remarkably, the reaction of (L1Sb)2 with oxygen to form (L1SbO)2 can occur in a single-crystal-to-single-crystal transformation. (L1Sb)2 and L1SbH2 can also be oxidized with elemental sulfur and these reactions were proposed to proceed through an intermediate L1SbS species that could be trapped with MesCNO.31–33 Although not strictly stibinidene chalcogenides, we note that a series of other products were obtained from the oxidative chalcogenation of (L1Sb)2, including selenadistibiranes and telluradistibiranes.34–37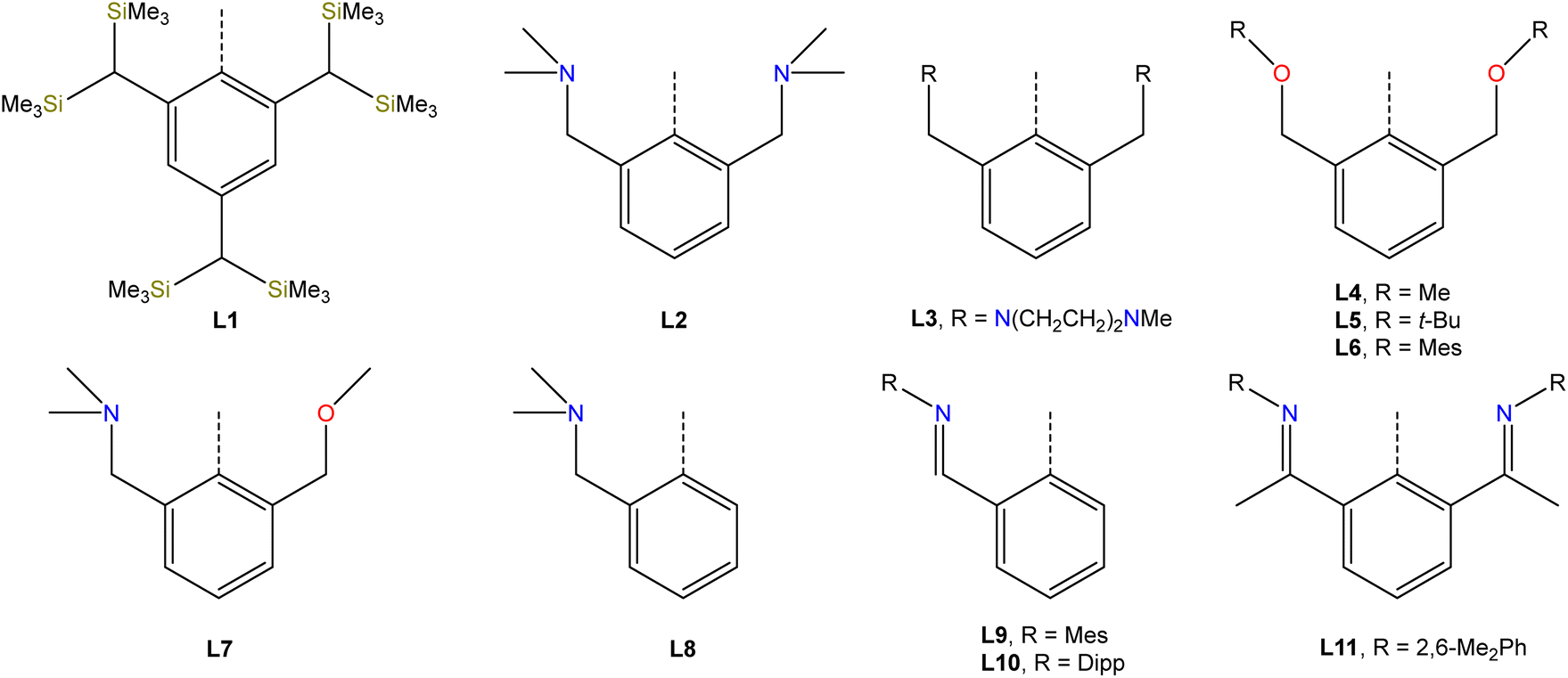 | ||
| Fig. 2 Ligands employed in the stabilization of stibinidene chalcogenides. | ||
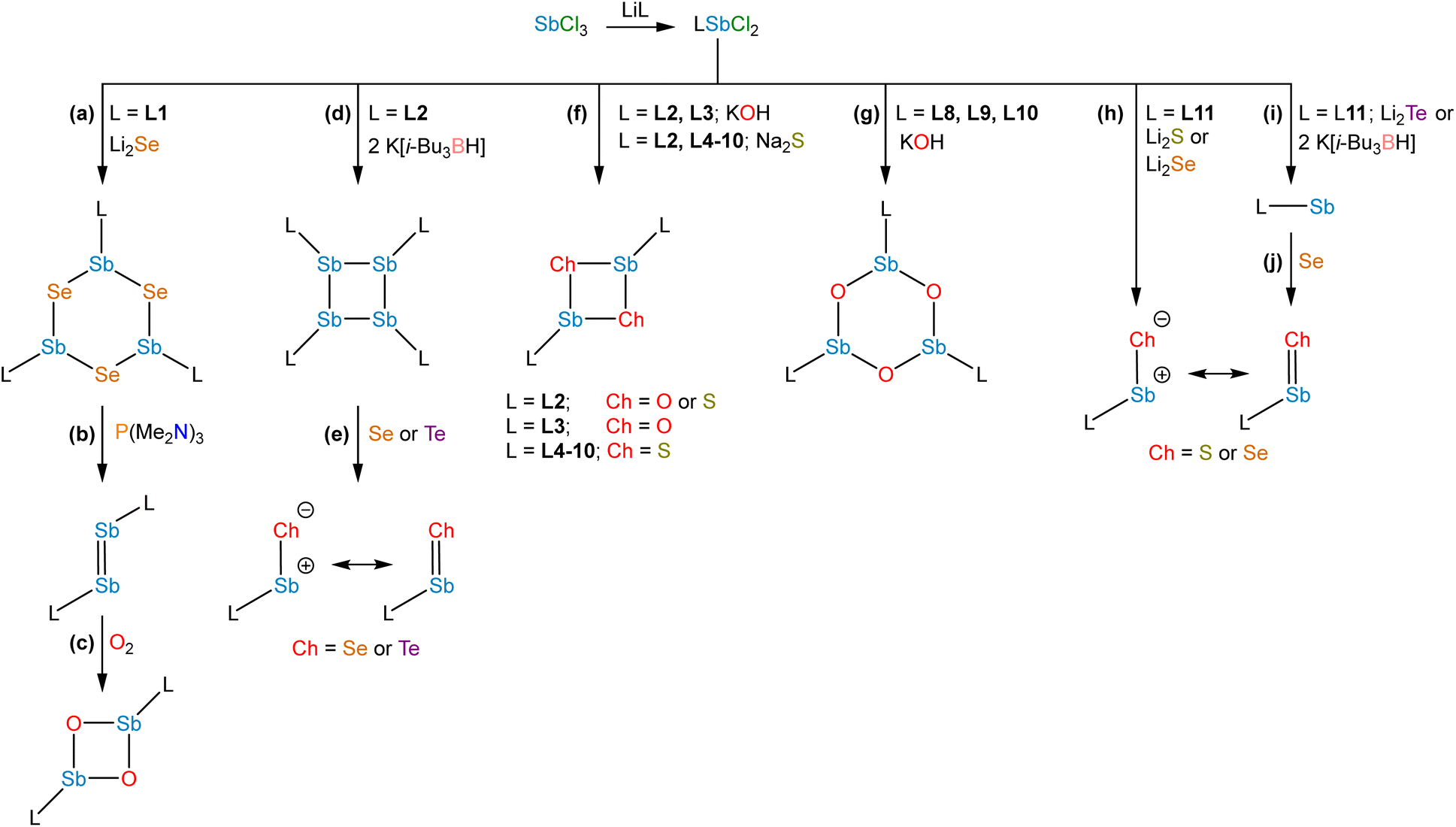 | ||
| Fig. 3 Strategies used to synthesize stibinidene chalcogenides. | ||
Beginning in the late 2000s, an extensive array of studies on the chemistry of pnictinidene chalcogenide compounds with aryl substituents bearing pendent donor groups were performed.38–40 Dehydrocoupling following the reaction between L2SbCl2 (L2 = C6H3-2,6-(CH2NMe2)2) and two equivalents of K[B(i-Bu)3H] afforded (L2Sb)4 (Fig. 3d),41 a tetrameric analog of (L1Sb)2 in which the pendent N-donors of the L2 substituent interact with the Sb(I) centers. These N-donor groups have a significant impact on the outcome of the oxidative chalcogenation of (L2Sb)4 as compared to (L1Sb)2. Oxidation of (L2Sb)4 with elemental selenium or tellurium resulted in clean formation of a product with the empirical formula L2SbCh (Ch = Se, Te) (Fig. 3e).42 Crystallographic analysis revealed that, unlike the oligomeric (L1SbSe)3 species described above, these L2SbCh products were monomeric and featured terminal Sb–Te and Sb–Se bonds (Fig. 4a and b). Comparison of the 77Se chemical shifts from NMR experiments in the solid state (δiso = −153 ppm) and in solution (δ = −197 ppm) confirmed that the compound remains monomeric in solution. If the compound oligomerized in solution, then the interaction between the Se atom and a Lewis acidic Sb center would be expected to result in a significant downfield shift. For example, the 77Se NMR signal of the trimeric compound (L1SbSe)3 appears at 179 ppm.43 Despite the much greater steric protection afforded by L1 relative to L2, L2SbCh is monomeric because of the N-donors that quench the Lewis acidity of the Sb center and preclude oligomerization. The natures of the terminal Sb–Se and Sb–Te bonds were probed with a suite of computational methods. The theoretical results suggested that the bonds are intermediate between polar-covalent single bonds and regular double bonds; substantial negative charge is localized on the chalcogen atom but there is significant back-donation from the Ch-centered lone pairs to vacant Sb-centered p orbitals. The positive charge localized on the Sb atom as a result of the bond polarization is also reflected by the presence of strong N → Sb dative interactions. It is suggested that this donation from the pendent amine groups attenuates the back-bonding from the Ch atom to the Lewis acidic Sb center and diminishes the double-bond character of the Sb–Ch bond. L2SbSe can also be synthesized by treating the organoantimony(III) bis(arylthiolate) species L2Sb(SC6H3-2,6-Me2)2 with elemental selenium.44 Interestingly, crystals of L2SbSe grown from a concentrated toluene solution at −20 °C contained (L2SbSe)2 as a dimeric species. There was notably Se/S substitutional disorder in the crystals of (L2SbSe)2 and, as described below, the L2-bearing stibinidene sulfide crystallizes as a dimer.
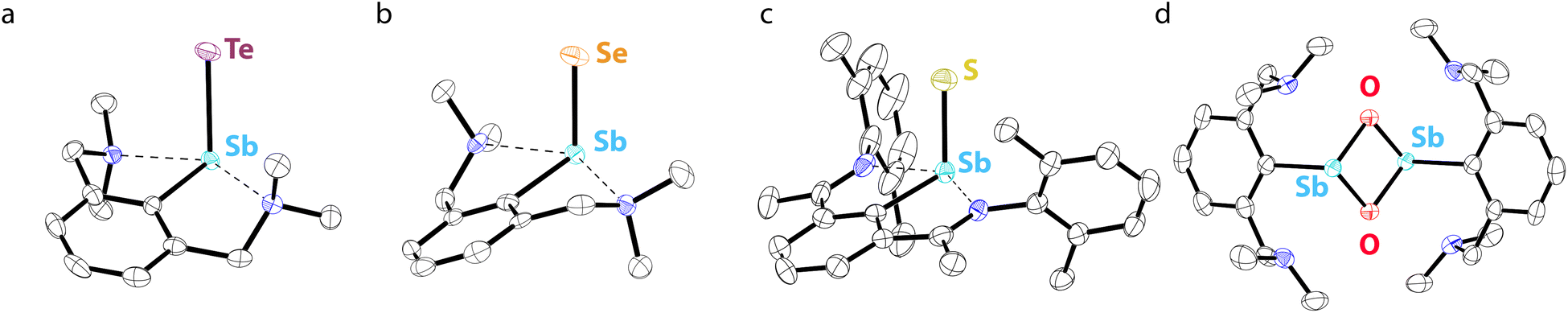 | ||
| Fig. 4 Thermal ellipsoid plots (50% probability) of (a) L2SbTe, (b) L2SbSe, (c) L11SbS, and (d) (L2SbO)2. Color code: Sb teal, Te purple, Se orange, S yellow, O red, N blue, and C black. Hydrogen atoms are omitted for clarity. | ||
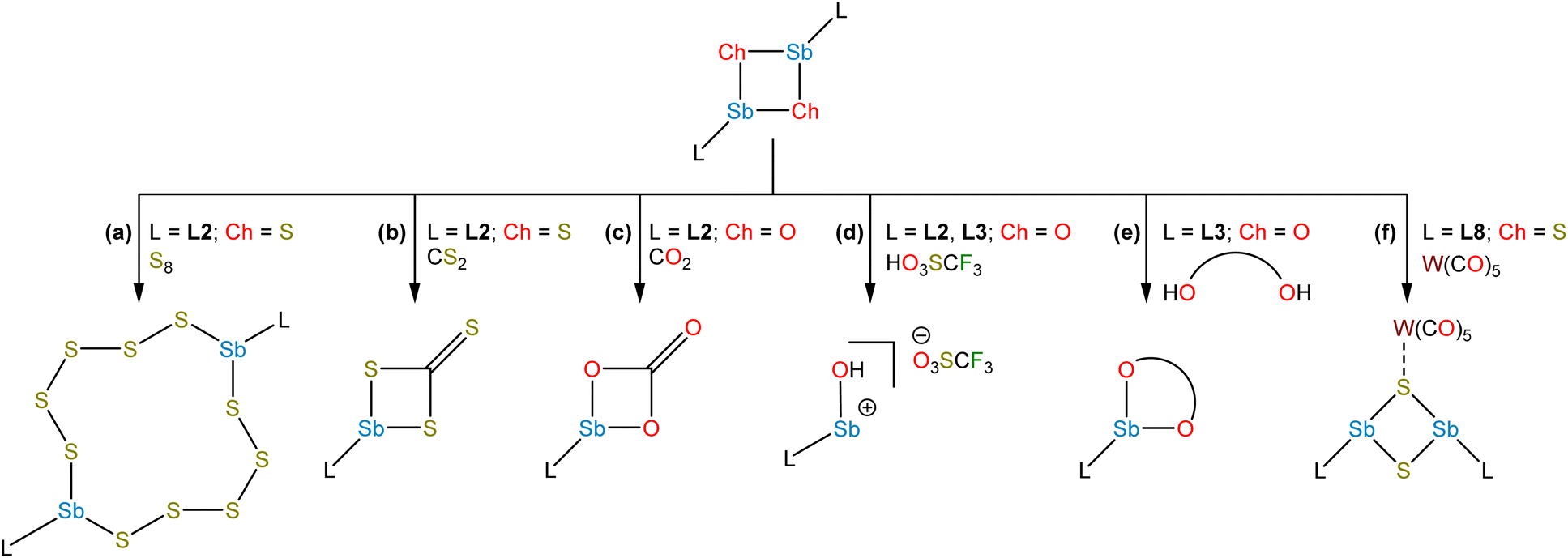
The lighter sulfide congener L2SbS was isolated, not by oxidation of the cyclo-Sb(I) species, but via the metathetical reaction of L2SbCl2 with Na2S (Fig. 3f).45 Crystallographic analysis of L2SbS unambiguously confirmed the species to exist as a centrosymmetric dimer in the solid state, in contrast to the analogous heavier stibinidene chalcogenides. Solution-phase NMR data collected on L2SbS were, however, reminiscent of the data obtained with the monomeric L2SbSe and L2SbTe species and were consistent with L2SbS existing as a monomer when dissolved in CHCl3 or CDCl3. Specifically, L2SbS did not produce the multiplicity of signals expected for the mixture of syn (C2-symmetric) and anti (Ci-symmetric) isomers that would be expected for a dimeric species. Furthermore, the most prominent ion in an electrospray ionization mass spectral measurement corresponded to the protonated monomer at m/z 345 and ebullioscopic experiments performed in CHCl3 determined the molecular weight to be 343 g mol−1. L2SbS reacts with elemental sulfur (S8) to yield a cyclic bis(pentasulfide), L2Sb(μ-S5)2SbL2, highlighting its ambiphilic reactivity (Fig. 5a). Moreover, L2SbS undergoes a [2 + 2] cycloaddition reaction with CS2 to form L2SbCS3 (Fig. 5b).46
 | ||
| Fig. 5 Reactivity of stibinidene chalcogenides. | ||
To complete the series, L2SbCl2 was treated with KOH to obtain (L2SbO)2 (Fig. 3f).47 Single-crystal X-ray diffraction revealed that (L2SbO)2, like (L2SbS)2, exists as a centrosymmetric dimer in the solid state (Fig. 4d). Unlike (L2SbS)2, however, (L2SbO)2 exhibited two distinct sets of signals in its solution-phase 1H and 13C NMR data, consistent with persistence of the dimeric form in solution, and demonstrated evidence of a dynamic equilibration between the syn and anti configurations. Bubbling CO2 into a toluene solution of (L2SbO)2 yielded L2SbCO3 (Fig. 5c). Similar to L2SbCS3, L2SbCO3 exists as a monomeric species with a 5-coordinate Sb(III) center. The addition could be reversed, and heating a solution of L2SbCO3 to 130 °C for 10 h afforded the parent stibinidene oxide dimer, (L2SbO)2. This reactivity nicely parallels the reactivity between the corresponding stibinidene sulfide and CS2, which raises the possibility that the predominantly dimeric (L2SbO)2 is in equilibrium with a sufficient amount of monomeric L2SbO to allow the reaction with CO2 to proceed readily. It was subsequently reported that treatment of (L2SbO)2 with trifluoromethanesulfonic or trifluoroacetic acid resulted in the disassociation of the dimer to form monomeric hydroxyorganoantimony(III) salts of the form [L2SbOH][X] (Fig. 5d).48 This reactivity was shared by the piperazinyl-substituted analogs (L3SbO)2. (L3SbO)2 can also react with pinacol and catechols to form pinacolato and catecholato species with the elimination of water (Fig. 5e).49 Similar ambiphilic reactivity of (L2SbO)2 with phosphonic acids,50 phosphoric acid,51 arsenic oxides,52 ChO2 (Ch = S, Se),53 silanols,54 boronic acids,55 and stannoxanes56 has also been explored.
These seminal studies of the stibinidene chalcogenides L2SbCh (Ch = Te, Se, S, O) provide insight into periodic trends in polar, unsaturated bonding interactions involving heavy pnictogens. To summarize some the key findings: L2SbTe and L2SbSe exist as monomers in both the solid state and in solution, L2SbS exists as a dimer in the solid state and a monomer in solution, and (L2SbO)2 exists as a dimer in both the solid state and solution, although it may exist in slight equilibrium with the monomer. As the chalcogen atom becomes lighter, its electronegativity increases relative to the Sb center resulting in a more polarized Sb+–Ch− bond and increasing the thermodynamic driving force to self-associate. The observation that the pendent N-donors afford a significant level of thermodynamic stabilization to prevent dimerization for L2SbTe and L2SbSe but not L2SbS and L2SbO, suggests that different ligands with more strongly interacting donors and/or enhanced steric shielding would allow monomeric stibinidene sulfides and oxides to be isolated and studied.
When the stibinidene dichlorides LSbCl2 with L = L4, L5, L6, which bear an O,C,O-pincer ligand of varying steric bulk, were treated with Na2S, dimeric species featuring the central (SbS)2 motif were observed in both the solid state and in solution (Fig. 3f).57 In this case, despite the relatively large size of the ethereal ligands, the pendent O atoms are too weakly donating to prevent dimerization. When the N,C,O-pincer ligand L7 was employed, dimeric sulfide and selenide species were similarly observed (Fig. 3f).58 When a chelator with a single NMe2-pendent donor, L8, was employed, a dimeric sulfide is also obtained.59 Stibinidene chalcogenides can act as ligands and (L8SbS)2 coordinates to W(CO)5, as a dimer, through a bridging sulfide (Fig. 5f). (RSbCh)2 (R = CH(SiMe3)2, Ch = S, Se) can also coordinate to W(CO)5 as dimers, but do so through the Sb(III) centers.60,61 The interaction of the donor arms of L8 with the Sb center likely favors interaction with W at the bridging sulfide. Treatment of L8SbCl2 with KOH afforded a cyclic trimeric stibinidene oxide (Fig. 3g). Similar results were obtained when chelators with a single pendent imine donor, L9 and L10, were employed.62
Treatment of L11SbCl2, featuring a substituent bearing two pendent imine groups, with Li2Se afforded a monomeric product (L11SbSe) analogous to that obtained with L2 (Fig. 3h).63 In contrast, however, treatment of L11SbCl2 with Li2S led to a product that not only analyzed as L11SbS, but was confirmed to be monomeric using single-crystal X-ray diffraction and solution-phase NMR spectroscopy (Fig. 3h and 4c).63 Interestingly, L11SbTe could not be similarly isolated, and the analogous reaction simply afforded L11Sb and elemental tellurium (Fig. 3i). Reaction of L11Sb with elemental selenium afforded L11SbSe (Fig. 3j). L11Sb could be independently prepared from L11SbCl2 and K[B(i-Bu)3H] (Fig. 3i), and crystallographic analysis unambiguously confirmed it to be a monomeric stibinidene.64 The difference in structure between the tetrameric (L2Sb)4 and the monomeric L11Sb, as well as between the dimeric (L2SbS)2 and the monomeric L11SbS, could arise in part from the change in the electronic nature of the donor (imine vs. amine), and in part from the enhanced steric shielding provided by the much bulkier substituents of L11. Examples of unsupported monomeric stibinidene and bismuthinidene compounds that lack pendent donors have now been accessed using extremely sterically protecting substituents and may provide access to a yet wider range of compounds.65–67
The natures of the terminal Sb+–S− and Sb+–Se− bonds in the L11SbCh species were investigated computationally and compared to those of the hypothetical monomeric molecules PhSbCh to assess the impact of the N-donor groups on stibinidene chalcogenide bonding.63L11SbCh feature more polarized, ylidic Sb+–Ch− bonds with lower double-bond character than those of PhSbCh. Donation from the imines to the Lewis acidic Sb center attenuates back-bonding from the Ch atom to the Sb atom, favoring a buildup of negative charge on the Ch atom. Analogous chemistry was realized for the imine-bearing compound with tert-butyl substituents as well.68 The isolation of the elusive monomeric stibinidene sulfide highlights the efficacy of bulky substituents in the kinetic stabilization of otherwise elusive polar unsaturated bonds involving heavy pnictogens; however, it remains unclear the extent to which the dative interactions between the N-donor groups and the Sb center cause the reactivity of these compounds to differ from as-yet unrealized species with unperturbed pnictinidene chalcogenide bonds.
A recent development centered on the observation that a stibinidene with an aryl substituent bearing one imine donor and one amine donor (L12Sb) could also support oxidation to form a monomeric oxide, sulfide, or selenide (Fig. 6).69 In the case of the oxide, however, a tautomerization occurred whereby the NH amine proton migrated to the O atom (Fig. 6). This tautomerization was not observed for the heavier chalcogenides and thermochemical calculations indicated that this experimental observation reflects the change in the relative stabilities of the tautomers as the group 16 element increases in atomic number.
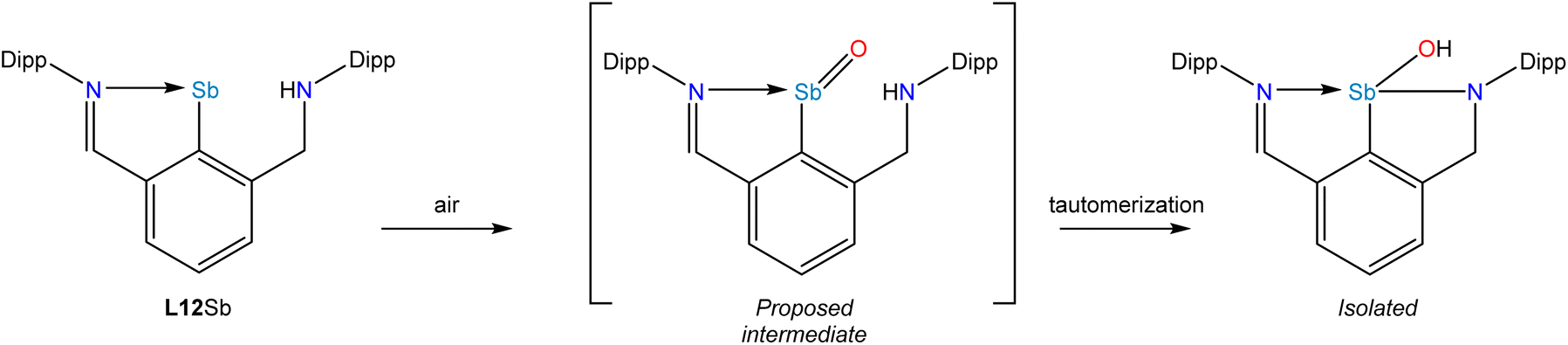 | ||
| Fig. 6 Oxidation of L12Sb to form a monomeric stibinidene oxide that undergoes a spontaneous NH to OH tautomerization. | ||
Monomeric stibine chalcogenides
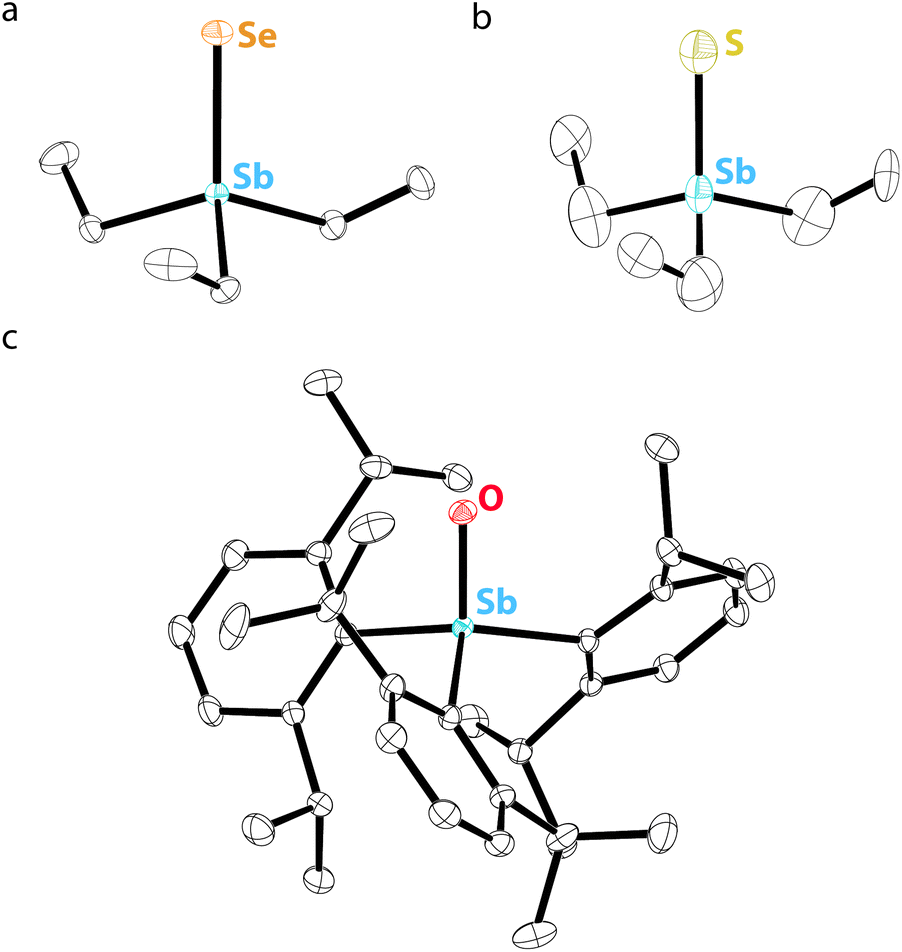
In contrast to σ2,λ3-stibinidene chalcogenides, some σ4,λ5-stibine chalcogenides of the form R3SbCh (Ch = S, Se) exist as stable monomers without the use of sterically demanding or thermodynamically stabilizing substituents. Compared to the exceedingly well-studied phosphine and arsine chalcogenides, however, there are only a few known examples of monomeric stibine chalcogenides. Ph3SbS can be accessed by reaction of Ph3SbBr2 with H2S (Fig. 7a).70 The tetrahedral, monomeric structure of Ph3SbS was confirmed by 121Sb Mössbauer spectroscopy and single-crystal X-ray diffraction.71 Me3SbS has been prepared by the reaction of Me3SbO with H2S, whereas Et3SbS and Cy3SbS have been prepared via reaction of the corresponding R3SbBr2 with Na2S (Fig. 7b).72 A wide range of trialkylstibine sulfides and selenides have been prepared by reaction of the corresponding stibine with the elemental chalcogen (Fig. 7c).73,74 Et3SbS and Et3SbSe were prepared in this way in the 1970s, but it was only within the last decade that their solid-state structures were reported (Fig. 8a and b).75 The direct oxidation of R3Sb with elemental sulfur to give R3SbS does not appear to be restricted to alkyl-substituted species; the extremely electron-deficient (C6F5)3Sb was reportedly oxidized to (C6F5)3SbS by refluxing with elemental sulfur in MeCN or C6H6.76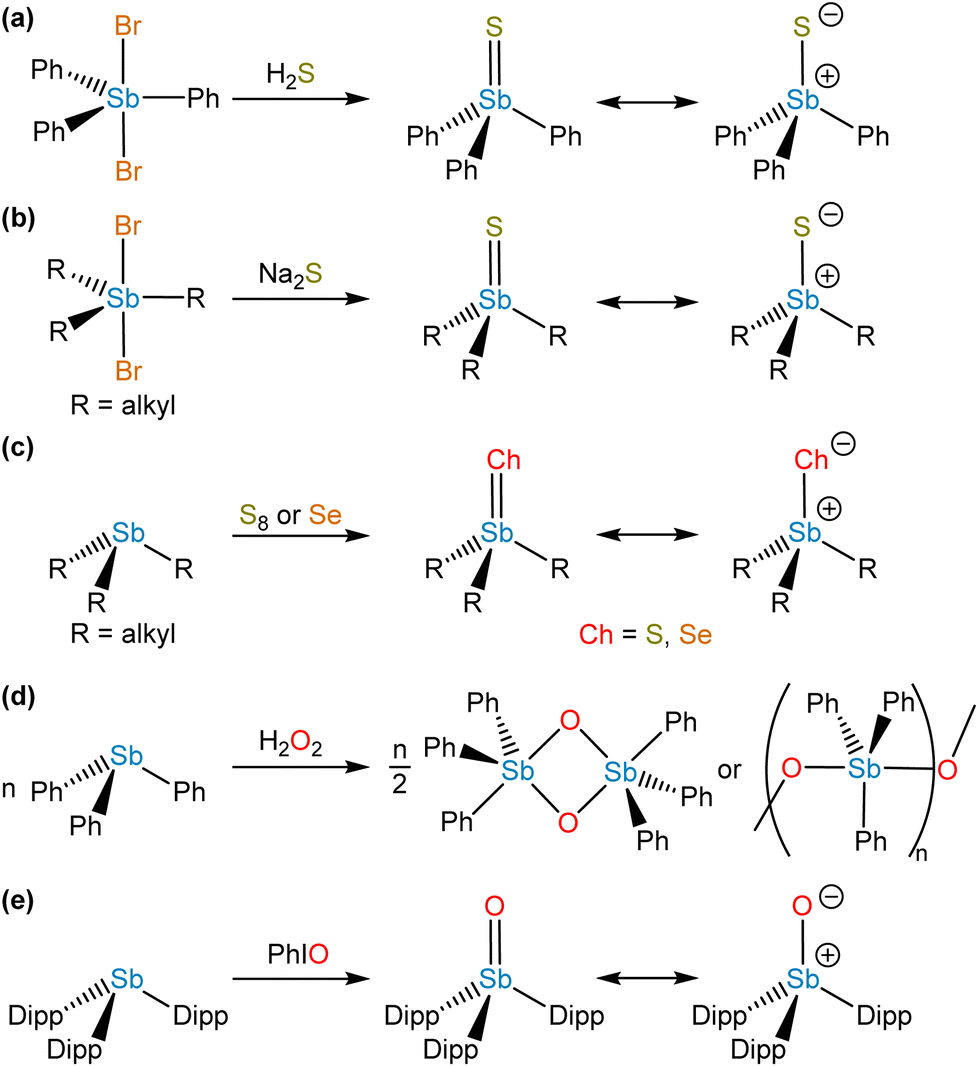 | ||
| Fig. 7 Synthesis of stibine chalcogenides. | ||
 | ||
| Fig. 8 Thermal ellipsoid plots (50% probability) of (a) Et3SbSe, (b) Et3SbS, and (c) Dipp3SbO. Color code: Sb teal, Se orange, S yellow, O red, and C black. Hydrogen atoms are omitted for clarity. | ||
Vibrational analyses of trialkylstibine chalcogenides found νSbS to range from 422–440 cm−1 and νSbSe to range from 272–300 cm−1.73,77 Values have also been reported for trialkylstibine oxides, but these data likely correspond to oligomeric species in light of contemporary knowledge (vide infra). NMR and UV-vis spectroscopic studies on Me3SbS provided early confirmation that the interaction between the Sb and S atoms is best described as a polar covalent bond.78 More recently, a set of thorough NPA, NBO, and ELF analyses of Et3SbCh (Ch = S, Se) provided results consistent with the presence of polar covalent single bonds between the Sb and Ch atoms.75 In each case, the bonding electrons are polarized significantly towards the chalcogen, with the Sb–S bond being more polarized than the Sb–Se bond. Although it is not explicitly described by the authors, the reported NBO analysis reveals that the two lone pairs of predominantly p character of the Ch atoms are significantly depopulated. Moreover, visual inspection of the HOMO and HOMO−1 suggests that they contain appreciable Sb–C σ* character, suggesting that there is a back-donation from the Ch-centered lone pairs to the Sb–C σ* antibonding orbitals that is analogous to the bonding in phosphine oxides (vide infra). To the best of our knowledge, there are no reported examples of well-characterized R3SbTe compounds.
In analogy to how stibine selenides and sulfides were prepared, early investigators conducted oxidative chalcogenation reactions to prepare species with the empirical formula R3SbO. Although these molecules were long formulated as monomeric stibine oxides in the literature, discrepancies arose in the data. For instance, whereas quadrupolar splitting measured by 121Sb Mössbauer spectroscopy revealed that stibine sulfides were tetrahedral (vide supra), analogous measurements on stibine oxides such as “Me3SbO” indicated that the Sb centers were trigonal bipyramidal.71 EXAFS and X-ray diffraction experiments ultimately revealed that these species had undergone self-association reactions of the type described above for stibinidene chalcogenides, forming either cyclic or linear head-to-tail oligomers (Fig. 7d).79–82 It is noteworthy that studies to date have indicated that “triphenylbismuthine oxide” similarly exists as dimers or oligomers.83,84 As with the stibinidene chalcogenides, a thermodynamic stabilization approach can be taken, but there are relatively few examples of Lewis acid-stabilized monomeric stibine oxides in the literature. Treatment of (Ph3SbO)2 with B(C6F5)3 results in the disassociation of the dimeric stibine oxide to form a stable Lewis adduct,85 and oxidation of 1,8-bis(diphenylstibino)biphenylene with o-chloranil in the presence of adventitious water resulted in the formation of a stibine oxide engaged in an intramolecular dative interaction with an adjacent stiborane unit.86 Finally, it has been reported that addition of oxygen to a mixture of Et3Sb and the tetrakis(3,5-difluorophenyl)stibonium tetrakis(pentafluorophenyl)borate resulted in the formation of a Lewis adduct between triethylstibine oxide and the Lewis acidic stibonium cation.87
A kinetic stabilization approach can also be taken. Treatment of Mes3Sb(OH)2 with sulfonic acids resulted in the elimination of a water molecule to form a crystalline product that was described as a hydrogen-bonded adduct of Mes3SbO and the sulfonic acid, Mes3SbO⋯HO3SR (R = Ph or CF3).88 Our group reevaluated these species using a variety of crystallographic, spectroscopic, and computational methods, and determined that the products were in fact hydroxystibonium salts, [Mes3SbOH][O3SR] (R = Ph or CF3).89 As a part of this effort, single-crystal neutron diffraction with [Mes3SbOH][O3SPh] was used to unambiguously confirm that the protic H atom is abutting the stiboryl O atom rather than the benzenesulfonate group.90 These results collectively suggested that monomeric stibine oxides bearing an unperturbed stiboryl group had remained unknown.
The absence of monomeric stibine oxides can be initially surprising given the prevalence of monomeric phosphine and arsine oxides. Indeed, the lighter congeners are exceedingly stable. Phosphine oxides exist as tetrahedral monomeric species bearing the unsaturated phosphoryl bond (P+–O−). The electronic structure of the phosphoryl bond has been rigorously investigated, and the currently accepted bonding model consists of a polar covalent single bond stabilized by cylindrically symmetrical back-donation from the O-centered lone pairs to vacant P–C σ* orbitals.91,92 Based on this description of the bonding, one can expect certain periodic trends for pnictoryl bonds.93 As the pnictogen atom becomes heavier, the size and diffuseness of the Pn-centered valence orbitals increases, resulting in diminished overlap between the filled O-centered p and the vacant Pn–C σ* orbitals. This disruption of back-bonding with the heavier pnictogens causes a decrease in the thermodynamic stability of the pnictoryl bond and an increase in the separation of charge across the bond. For Sb and Bi, the greater separation of charge in Sb+–O− and Bi+–O− relative to P+–O− and As+–O−, as well as the greater propensity for the larger pnictogens to expand their coordination spheres, can rationalize the favorability of self-associated species over monomeric species for stibine and bismuthine oxides.
We were recently successful in isolating the first unsupported monomeric stibine oxide. We employed a strategy in which sufficiently bulky substituents were installed on the Sb center to prevent both the self-association of monomers and the expansion of the coordination sphere.94 In this effort, we adopted a synthetic protocol reported by Sasaki and co-workers95 to isolate the sterically encumbered stibine Dipp3Sb (Dipp = 2,6-diisopropylphenyl). Treatment of Dipp3Sb with iodosobenzene afforded the monomeric stibine oxide Dipp3SbO as a bench-stable, crystalline solid (Fig. 7e). The solid-state structure of Dipp3SbO was probed by EXAFS and X-ray crystallography, revealing the species to exist as the expected tetrahedral monomer, with the extremely short Sb–O bond distance of 1.8372(5) Å (Fig. 8c). We prepared Dipp3AsO and Dipp3PO to assess trends in bonding and reactivity. Our topological analysis of Dipp3PnO (Pn = Sb, As, P) showed that the value of ρ at the bond critical point increases from Dipp3SbO < Dipp3AsO < Dipp3PO, suggesting that the pnictoryl bond is the weakest in the case of Dipp3SbO. The ellipticity of ρ along the Pn–O interatomic vector was negligible in all cases. Molecular orbital analysis of Dipp3PnO revealed that the HOMO energy was the highest and the LUMO energy was the lowest in the case of Dipp3SbO. The HOMO of Dipp3SbO features a large contribution from an O-centered lone pair with significant p character, and the LUMO features a large contribution from the Sb–O σ* antibonding orbital. NPA of Dipp3PnO found the greatest separation of charge in the case of Dipp3SbO. NBO analysis revealed that the stabilization afforded by delocalization of electron density from the O-centered lone pairs to the Sb–C σ* orbitals is lowest in the case of Dipp3SbO. Our theoretical analyses collectively confirm that, as the Pn atom becomes heavier and electronegativity decreases, back-donation from the O-centered lone pairs to the Pn–C σ* orbitals is disrupted leading to a greater separation of charge.
Dipp3SbO allowed us to investigate the reactivity of an unperturbed stiboryl (Sb+–O−) group for the first time. It engages in several classes of reactivity, including Brønsted base chemistry, coordination chemistry, addition chemistry, and oxo-transfer chemistry. Cooling a saturated solution of Dipp3SbO in neat 4-fluoroaniline afforded colorless crystals of the H-bonded adduct, Dipp3SbO⋯H2NPhF (Fig. 9a). Dipp3SbO could be protonated by benzenesulfonic acid to form the hydroxystibonium salt, [Dipp3SbOH][PhSO3] (Fig. 9b). Treatment of Dipp3SbO with acetic acid results in the formation of a cis-hydroxoacetatostiborane, Dipp3Sb(OH)(OAc), by addition of acetic acid across the unsaturated stiboryl group (Fig. 9c). The unexpected cis configuration of Dipp3Sb(OH)(OAc) is likely stabilized by an intramolecular H-bonding interaction between the hydroxo and acetato substituents. Dipp3SbO engaged in coordination chemistry with the first-, second-, and third-row coinage metals. (Dipp3SbO)CuCl, [(Dipp3SbO)2][AgCF3SO3], and [Dipp3SbOAuPPh3][CF3SO3] were each isolated as stable crystalline solids (Fig. 9d). Deoxygenation of Dipp3SbO with BF3·OEt2 formed the difluorostiborane trans-Dipp3SbF2 (Fig. 9e) and deoxygenation with phenylsilane formed the parent stibine, Dipp3Sb (Fig. 9f).
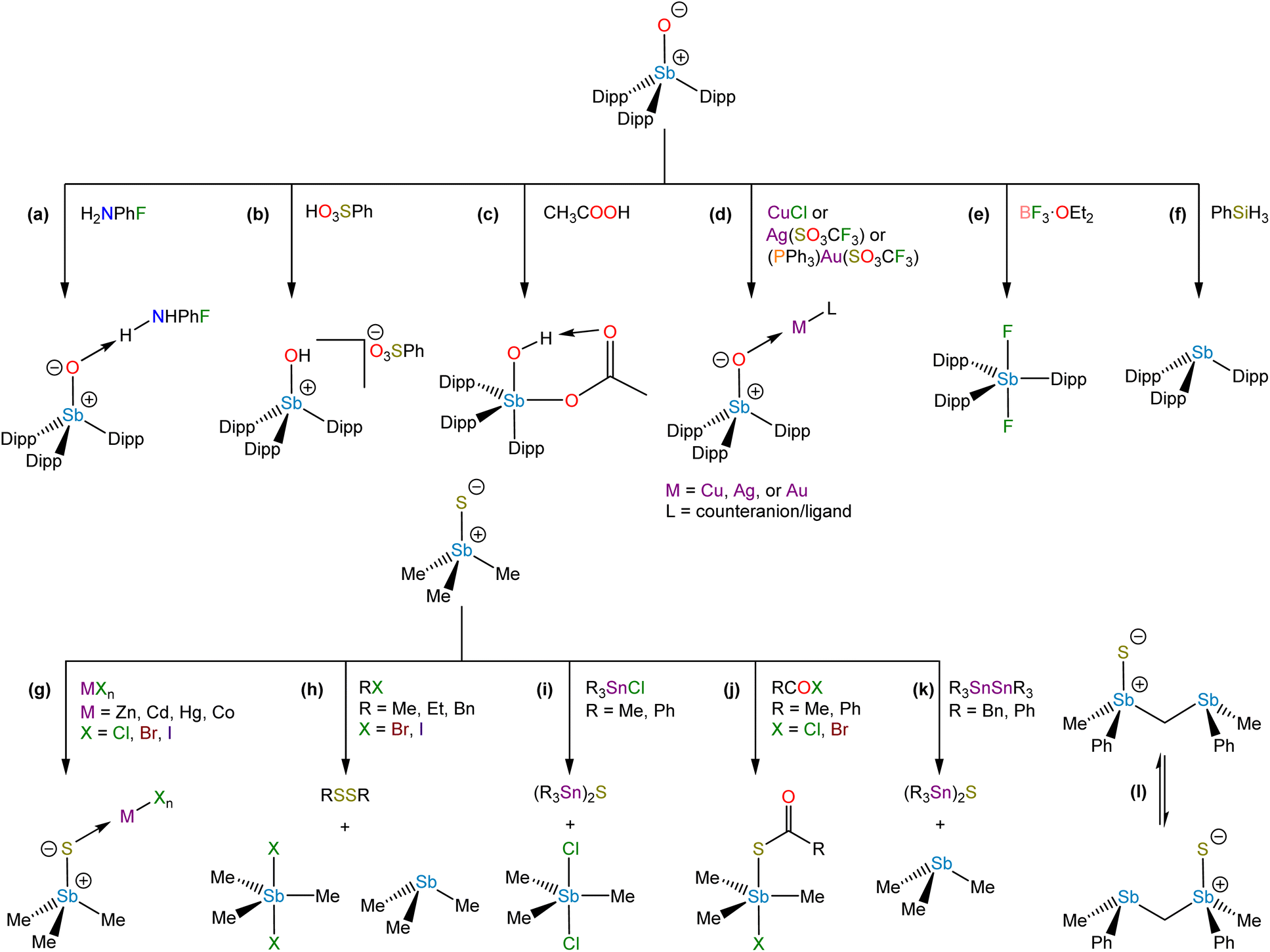 | ||
| Fig. 9 Reactivity of stibine chalcogenides. | ||
The relative Brønsted basicity of Dipp3PnO (Pn = P, As, Sb) was probed.96 Based on the theoretical analyses described above, we hypothesized that the Lewis basicity at the O atom would increase from Dipp3PO < Dipp3AsO < Dipp3SbO. Treatment of Dipp3PnO with triflic acid afforded the corresponding [Dipp3PnOH][CF3SO3] salt in all cases. Picric acid is a sufficiently protic acid to protonate Dipp3SbO and Dipp3AsO but not Dipp3PO. 2,4-Dinitrophenol only protonated Dipp3SbO. Treatment of Dipp3SbO with one equivalent of 4-nitrophenol afforded the H-bonded adduct, suggesting the pKaH of Dipp3SbO lies between that of 2,4-dinitrophenol and 4-nitrophenol. A 1H NMR spectrometric titration experiment determined the pKaH of Dipp3AsO to be 13.89(13) in acetonitrile. The pKaH of Dipp3SbO in acetonitrile is 19.81(5), reflecting the million-fold increase in basicity over that of the analogous arsine oxide.
Although monomeric stibine sulfides have been known for a longer time, there has not yet been a systematic investigation of their reactivity. They can function as Lewis bases, and early studies revealed that they could form a variety of complexes with p-block and d-block Lewis acids (Fig. 9g).72,97–99 Stibine selenides also demonstrated an ability to form complexes with transition metal centers.100,101 Sulfur-for-halide exchange was observed in some reactions of Me3SbS with metal halides or alkyl halides (Fig. 9h and i),98,102,103 and 1,2-addition across the Sb+–S− bond occurred on mixing with acyl halides (Fig. 9j).104 Stibine sulfides can also act as S-atom transfer reagents (Fig. 9k)105 and such reactivity may allow for intramolecular S transfer between Sb centers (Fig. 9l).106
Outlook
Efforts toward the isolation of monomeric σ2,λ3-stibinidene chalcogenides and σ4,λ5-stibine chalcogenides have yielded great insights into the bonding and reactivity of unsaturated Sb–Ch bonds. To date, all examples of monomeric stibinidene chalcogenides reported in the literature are thermodynamically stabilized by donation from a pendent Lewis base to the Lewis acidic Sb center, and no examples of monomeric stibinidene oxides have been reported. We expect that the isolation of unsupported stibinidene chalcogenides (i.e., without stabilization by a Lewis acid or base) could be achieved by using sufficiently sterically demanding substituents. Unsupported stibinidene chalcogenides have been predicted by theoretical calculations to have Sb–Ch bonds that are less polar and have more double-bond character than their Lewis-base-supported analogs because the greater electron deficiency at the Sb center favors increased back-donation from the Ch-centered lone pairs to the Sb-centered vacant p orbital. We expect that this variation in the electronic structure of the Sb–Ch bond could lead to interesting differences in reactivity relative to the currently known, Lewis-base-supported species. Furthermore, a yet still elusive monomeric stibinidene oxide is expected to have the most polarized Sb–Ch bond relative to all heavier chalcogens, which would invariably exhibit enhanced reactivity and unlock more challenging substrate activations.Monomeric stibine chalcogenides are now known for all Ch except Te. Whereas stibine sulfides and stibine selenides appear to readily exist as monomeric species, stibine oxides feature highly polarized Sb+–O− bonds that must be stabilized to prevent self-association. The kinetic stabilization of a monomeric stibine oxide, Dipp3SbO, allowed for the investigation of the bonding and reactivity of an unperturbed stiboryl group within an isolable molecule for the first time. The enhanced reactivity of Dipp3SbO relative to Dipp3PO and Dipp3AsO highlights the utility of the kinetic stabilization approach in unlocking unquenched reactivity at highly reactive main-group species. We expect that new monomeric stibine oxides featuring steric and electronic modulations (e.g., by using aryl groups with electron-donating or -withdrawing substituents) relative to Dipp3SbO will result in further insight into the bonding and reactivity of the stiboryl group. Despite the advances highlighted in this article, much of the chemistry of antimony still remains to be discovered.
The strategies developed thus far in the stabilization of stibinidene chalcogenides and stibine chalcogenides will invariably serve as a strong foundation of knowledge to researchers who continue to develop the rich topic of antimony chemistry. With no examples of a monomeric stibinidene oxide and only a single example of a monomeric stibine oxide reported to date, the utility of polar, unsaturated Sb–O bonds in synthetic chemistry has yet to be fully realized. The high polarity of these bonds, with a Lewis basic O atom and Lewis acidic Sb atom, coupled with the high propensity for the Sb atom to expand its coordination sphere, will invariably unlock new substrate activations that can be accessed through a biphilic mechanism. Specifically, we expect that these oxides will be potent in the breaking of polar covalent single bonds and the cycloaddition of unsaturated motifs to formally add substrates across the unsaturated Sb+–O− group. We expect that the realization of these transformations will ultimately lead to practical applications in sustainable catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF through CAREER award 2236365, the ACS through PRF grant 66098-ND3, and Arnold and Mabel Beckman Foundation through a Beckman Young Investigator Award to T. C. J.References
- R. L. Melen, Science, 2019, 363, 479–484 CrossRef CAS PubMed.
- D. You and F. P. Gabbaï, Trends Chem., 2019, 1, 485–496 CrossRef CAS.
- Y. Pang, M. Leutzsch, N. Nöthling and J. Cornella, Angew. Chem., Int. Ed., 2023, 62, e202302071 CrossRef CAS PubMed.
- S. Hino, M. Olmstead, A. D. Phillips, R. J. Wright and P. P. Power, Inorg. Chem., 2004, 43, 7346–7352 CrossRef CAS PubMed.
- C. A. Caputo and P. P. Power, Organometallics, 2013, 32, 2278–2286 CrossRef CAS.
- T. C. Johnstone, G. N. J. H. Wee and D. W. Stephan, Angew. Chem., Int. Ed., 2018, 57, 5881–5884 CrossRef CAS.
- L. Zhao, S. Pan, N. Holzmann, P. Schwerdtfeger and G. Frenking, Chem. Rev., 2019, 119, 8781–8845 CrossRef CAS PubMed.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- H. Hashimoto and K. Nagata, Chem. Lett., 2021, 50, 778–787 CrossRef CAS.
- E. Rivard and P. P. Power, Inorg. Chem., 2007, 46, 10047–10064 CrossRef CAS PubMed.
- D. J. Liptrot and P. P. Power, Nat. Rev. Chem., 2017, 1, 0004 CrossRef.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS.
- L. T. Maltz and F. P. Gabbaï, Inorg. Chem., 2023, 62, 13566–13572 CrossRef CAS.
- D. Sharma, S. Balasubramaniam, S. Kumar, E. D. Jemmis and A. Venugopal, Chem. Commun., 2021, 57, 8889–8892 RSC.
- L. S. Warring, J. E. Walley, D. A. Dickie, W. Tiznado, S. Pan and R. J. Gilliard, Inorg. Chem., 2022, 61, 18640–18652 CrossRef CAS PubMed.
- M. Yang, D. Tofan, C. H. Chen, K. M. Jack and F. P. Gabbaï, Angew. Chem., Int. Ed., 2018, 57, 13868–13872 CrossRef CAS PubMed.
- B. Pan and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 9564–9567 CrossRef CAS PubMed.
- M. Hirai and F. P. Gabbaï, Angew. Chem., Int. Ed., 2015, 54, 1205–1209 CrossRef CAS PubMed.
- B. L. Murphy and F. P. Gabbaï, J. Am. Chem. Soc., 2023, 145, 19458–19477 CrossRef CAS PubMed.
- M. Hirai, J. Cho and F. P. Gabbaï, Chem. – Eur. J., 2016, 22, 6537–6541 CrossRef CAS PubMed.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS.
- H. Jaffe and G. Doak, J. Am. Chem. Soc., 1949, 71, 602–606 CrossRef CAS PubMed.
- G. Doak and H. Jaffe, J. Am. Chem. Soc., 1950, 72, 3025–3027 CrossRef CAS.
- H. Jaffe and G. Doak, J. Am. Chem. Soc., 1950, 72, 3027–3029 CrossRef CAS.
- S. W. Hedges and L. H. Bowen, J. Chem. Phys., 1977, 67, 4706–4710 CrossRef CAS.
- H. J. Breunig, K. H. Ebert, M. A. Mohammed, J. Pawlik and J. Probst, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 93, 293–296 CrossRef.
- H. J. Breunig, M. A. Mohammed and K. H. Ebert, Z. Naturforsch., B: Chem. Sci., 1994, 49, 877–880 CrossRef CAS.
- M. A. Mohammed, K. H. Ebert and H. J. Breunig, Z. Naturforsch., B: Chem. Sci., 1996, 51, 149–152 CrossRef CAS.
- N. Tokitoh, Y. Arai, R. Okazaki and S. Nagase, Science, 1997, 277, 78–80 CrossRef CAS.
- N. Tokitoh, Y. Arai, T. Sasamori, R. Okazaki, S. Nagase, H. Uekusa and Y. Ohashi, J. Am. Chem. Soc., 1998, 120, 433–434 CrossRef CAS.
- N. Tokitoh, Y. Arai, J. Harada and R. Okazaki, Chem. Lett., 1995, 24, 959–960 CrossRef.
- N. Tokitoh, Y. Arai, T. Sasamori, N. Takeda and R. Okazaki, Heteroat. Chem., 2001, 12, 244–249 CrossRef CAS.
- T. Sasamori, E. Mieda, N. Takeda and N. Tokitoh, Chem. Lett., 2004, 33, 104–105 CrossRef CAS.
- T. Sasamori, E. Mieda, N. Takeda and N. Tokitoh, Angew. Chem., Int. Ed., 2005, 44, 3717–3720 CrossRef CAS PubMed.
- T. Sasamori, E. Mieda and N. Tokitoh, Bull. Chem. Soc. Jpn., 2007, 80, 2425–2435 CrossRef CAS.
- T. Sasamori, E. Mieda, A. Tsurusaki, N. Nagahora and N. Tokitoh, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 998–1002 CrossRef CAS.
- T. Sasamori and N. Tokitoh, Dalton Trans., 2008, 1395–1408 RSC.
- R. Jambor and L. Dostál, in Organometallic Pincer Chemistry, Springer Berlin Heidelberg, 2013, pp. 175–202 Search PubMed.
- C. I. Raţ, C. Silvestru and H. J. Breunig, Coord. Chem. Rev., 2013, 257, 818–879 CrossRef.
- H. J. Breunig, Rev. Roum. Chim., 2020, 65, 635–646 CrossRef.
- L. Dostál, R. Jambor, A. Růžička and J. Holeček, Organometallics, 2008, 27, 2169–2171 CrossRef.
- L. Dostál, R. Jambor, A. Růžička, A. Lyčka, J. Brus and F. de Proft, Organometallics, 2008, 27, 6059–6062 CrossRef.
- T. Sasamori, Y. Arai, N. Takeda, R. Okazaki, Y. Furukawa, M. Kimura, S. Nagase and N. Tokitoh, Bull. Chem. Soc. Jpn., 2002, 75, 661–675 CrossRef CAS.
- G. Duneş, A. Soran and C. Silvestru, Dalton Trans., 2022, 51, 10406–10419 RSC.
- L. Dostál, R. Jambor, A. Růžička, R. Jirásko, V. Lochař, L. Beneš and F. de Proft, Inorg. Chem., 2009, 48, 10495–10497 CrossRef PubMed.
- L. Dostál, R. Jambor, A. Růžička, R. Jirásko, E. Černošková, L. Beneš and F. de Proft, Organometallics, 2010, 29, 4486–4490 CrossRef.
- L. Dostál, R. Jambor, A. Růžička, M. Erben, R. Jirásko, E. Černošková and J. Holeček, Organometallics, 2009, 28, 2633–2636 CrossRef.
- A. l. Fridrichová, T. Svoboda, R. Jambor, Z. k. Padělková, A. Růžička, M. Erben, R. Jirásko and L. Dostál, Organometallics, 2009, 28, 5522–5528 CrossRef.
- G. Strîmb, A. Pöllnitz, C. I. Raţ and C. Silvestru, Dalton Trans., 2015, 44, 9927–9942 RSC.
- T. Svoboda, R. Jambor, A. Růžička, Z. Padělková, M. Erben, R. Jirásko and L. Dostál, Eur. J. Inorg. Chem., 2010, 2010, 1663–1669 CrossRef.
- T. Svoboda, L. Dostál, R. Jambor, A. Růžička, R. Jirásko and A. Lyčka, Inorg. Chem., 2011, 50, 6411–6413 CrossRef CAS PubMed.
- T. Svoboda, R. Jambor, A. Růžička, R. Jirásko, A. Lyčka, F. de Proft and L. Dostál, Organometallics, 2012, 31, 1725–1729 CrossRef CAS.
- B. Mairychová, T. Svoboda, M. Erben, A. Růžička, L. Dostál and R. Jambor, Organometallics, 2013, 32, 157–163 CrossRef.
- A. Fridrichová, B. Mairychová, Z. Padělková, A. Lyčka, K. Jurkschat, R. Jambor and L. Dostál, Dalton Trans., 2013, 42, 16403–16411 RSC.
- B. Mairychová, T. Svoboda, P. Štěpnička, A. Růžička, R. W. A. Havenith, M. Alonso, F. de Proft, R. Jambor and L. Dostál, Inorg. Chem., 2013, 52, 1424–1431 CrossRef PubMed.
- T. Svoboda, J. Warneke, A. Růžička, L. Dostál and J. Beckmann, J. Organomet. Chem., 2015, 797, 171–173 CrossRef CAS.
- M. Chovancová, R. Jambor, A. Růžička, R. Jirásko, I. Císařová and L. Dostál, Organometallics, 2009, 28, 1934–1941 CrossRef.
- J. Vrána, R. Jambor, A. Růžička, A. Lyčka and L. Dostál, J. Organomet. Chem., 2012, 718, 78–81 CrossRef.
- L. M. Opris, A. Silvestru, C. Silvestru, H. J. Breunig and E. Lork, Dalton Trans., 2004, 3575–3585 RSC.
- H. J. Breunig, I. Ghesner and E. Lork, Appl. Organomet. Chem., 2002, 16, 547–549 CrossRef CAS.
- H. J. Breunig, I. Ghesner and E. Lork, J. Organomet. Chem., 2002, 664, 130–135 CrossRef CAS.
- A. M. Preda, C. I. Raţ, C. Silvestru, H. J. Breunig, H. Lang, T. Rüffer and M. Mehring, Dalton Trans., 2013, 42, 1144–1158 RSC.
- P. Šimon, R. Jambor, A. Růžička, A. Lyčka, F. de Proft and L. Dostál, Dalton Trans., 2012, 41, 5140–5143 RSC.
- P. Šimon, F. de Proft, R. Jambor, A. Růžička and L. Dostál, Angew. Chem., Int. Ed., 2010, 49, 5468–5471 CrossRef PubMed.
- M. Wu, H. Li, W. Chen, D. Wang, Y. He, L. Xu, S. Ye and G. Tan, Chem, 2023, 9, 2573–2584 CAS.
- M. Wu, W. Chen, D. Wang, Y. Chen, S. Ye and G. Tan, Natl. Sci. Rev., 2023, nwad169 CrossRef CAS.
- Y. Pang, N. Nöthling, M. Leutzsch, L. Kang, E. Bill, M. van Gastel, E. Reijerse, R. Goddard, L. Wagner, D. SantaLucia, S. DeBeer, F. Neese and J. Cornella, Science, 2023, 380, 1043–1048 CrossRef CAS.
- C. Ganesamoorthy, C. Wölper, L. Dostál and S. Schulz, J. Organomet. Chem., 2017, 845, 38–43 CrossRef CAS.
- J. Zechovský, E. Kertész, V. Kremláček, M. Hejda, T. Mikysek, M. Erben, A. Růžička, R. Jambor, Z. Benkő and L. Dostál, Organometallics, 2022, 41, 2535–2550 CrossRef.
- L. Kaufmann, Ber. Dtsch. Chem. Ges., 1908, 41, 2762–2766 CrossRef CAS.
- J. Pebler, F. Weller and K. Dehnicke, Z. Anorg. Allg. Chem., 1982, 492, 139–147 CrossRef CAS.
- M. Shindo, Y. Matsumura and R. Okawara, J. Organomet. Chem., 1968, 11, 299–305 CrossRef CAS.
- G. N. Chremos and R. A. Zingaro, J. Organomet. Chem., 1970, 22, 637–646 CrossRef CAS.
- R. A. Zingaro and A. Merijanian, J. Organomet. Chem., 1964, 1, 369–372 CrossRef CAS.
- S. Heimann, D. Bläser, C. Wölper, R. Haack, G. Jansen and S. Schulz, Dalton Trans., 2014, 43, 14772–14777 RSC.
- P. Raj, A. K. Saxena, K. Singhal and A. Ranjan, Polyhedron, 1985, 4, 251–258 CrossRef CAS.
- G. N. Chremos and R. A. Zingaro, J. Organomet. Chem., 1970, 22, 647–651 CrossRef CAS.
- J. Otera and R. Okawara, Inorg. Nucl. Chem. Lett., 1970, 6, 855–857 CrossRef CAS.
- J. Bordner, G. O. Doak and T. S. Everett, J. Am. Chem. Soc., 1986, 108, 4206–4213 CrossRef CAS.
- D. L. Venezky, C. W. Sink, B. A. Nevett and W. F. Fortescue, J. Organomet. Chem., 1972, 35, 131–142 CrossRef CAS.
- G. Ferguson, C. Glidewell, B. Kaitner, D. Lloyd and S. Metcalfe, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 824–826 CrossRef.
- C. J. Carmalt, J. G. Crossley, N. C. Norman and A. G. Orpen, Chem. Commun., 1996, 1675–1676 RSC.
- H. Suzuki, T. Ikegami and Y. Matano, Tetrahedron Lett., 1994, 35, 8197–8200 CrossRef CAS.
- Y. Matano, H. Nomura, T. Hisanaga, H. Nakano, M. Shiro and H. Imahori, Organometallics, 2004, 23, 5471–5480 CrossRef CAS.
- R. Kather, T. Svoboda, M. Wehrhahn, E. Rychagova, E. Lork, L. Dostál, S. Ketkov and J. Beckmann, Chem. Commun., 2015, 51, 5932–5935 RSC.
- C.-H. Chen and F. P. Gabbaï, Dalton Trans., 2018, 47, 12075–12078 RSC.
- O. Coughlin, Doctor of Philosophy, Nottingham Trent University, 2021 Search PubMed.
- F. Huber, T. Westhoff and H. Preut, J. Organomet. Chem., 1987, 323, 173–180 CrossRef CAS.
- J. S. Wenger and T. C. Johnstone, Chem. Commun., 2021, 57, 3484–3487 RSC.
- J. S. Wenger, X. Wang and T. C. Johnstone, Inorg. Chem., 2021, 60, 16048–16052 CrossRef CAS PubMed.
- T. Yang, D. M. Andrada and G. Frenking, Phys. Chem. Chem. Phys., 2018, 20, 11856–11866 RSC.
- N. C. Norman and P. G. Pringle, Chemistry, 2022, 4, 1226–1249 CrossRef CAS.
- B. Lindquist-Kleissler, J. S. Wenger and T. C. Johnstone, Inorg. Chem., 2021, 60, 1846–1856 CrossRef CAS.
- J. S. Wenger, M. Weng, G. N. George and T. C. Johnstone, Nat. Chem., 2023, 15, 633–640 CrossRef CAS PubMed.
- S. Sasaki, K. Sutoh, F. Murakami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 14830–14831 CrossRef CAS PubMed.
- J. S. Wenger, A. Getahun and T. C. Johnstone, Dalton Trans., 2023, 52, 11325–11334 RSC.
- T. Maeda, G. Yoshida and R. Okawara, J. Organomet. Chem., 1972, 44, 237–241 CrossRef CAS.
- T. Saito, J. Otera and R. Okawara, Bull. Chem. Soc. Jpn., 1970, 43, 1733–1736 CrossRef CAS.
- R. Okawara, J. Otera and T. Osaki, Inorg. Chem., 1971, 10, 402–404 CrossRef CAS.
- N. Kuhn and H. Schumann, J. Organomet. Chem., 1985, 288, c51–c52 CrossRef CAS.
- N. Kuhn and H. Schumann, J. Organomet. Chem., 1986, 304, 181–193 CrossRef CAS.
- J. Otera and R. Okawara, J. Organomet. Chem., 1969, 16, 335–338 CrossRef CAS.
- M. Shindo, Y. Matsumura and R. Okawara, Bull. Chem. Soc. Jpn., 1969, 42, 265–266 CrossRef CAS.
- J. Otera and R. Okawara, J. Organomet. Chem., 1969, 17, 353–357 CrossRef CAS.
- J. P. Donahue, Chem. Rev., 2006, 106, 4747–4783 CrossRef CAS PubMed.
- S.-I. Sato and Y. Matsumura, J. Organomet. Chem., 1975, 96, 57–61 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |