
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBarium phosphidoboranes and related calcium complexes†
Gabriel
Duneş‡
 ,
Peter M.
Chapple‡
,
Samia
Kahlal
,
Thierry
Roisnel
,
Jean-François
Carpentier
,
Jean-Yves
Saillard
* and
Yann
Sarazin
*
,
Peter M.
Chapple‡
,
Samia
Kahlal
,
Thierry
Roisnel
,
Jean-François
Carpentier
,
Jean-Yves
Saillard
* and
Yann
Sarazin
*
Univ Rennes, CNRS, ISCR-UMR 6226, 35000 Rennes, France. E-mail: jean-yves.saillard@univ-rennes.fr; yann.sarazin@univ-rennes.fr
First published on 21st March 2024
Abstract
The attempted synthesis of [{Carb}BaPPh2] (1) showed this barium-phosphide and its thf adducts, 1·thf and 1·(thf)2, to be unstable in solution. Our strategy to circumvent the fragility of these compounds involved the use of phosphinoboranes HPPh2·BH3 and HPPh2·B(C6F5)3 instead of HPPh2. This allowed for the synthesis of [{Carb}Ae{PPh2·BH3}] (Ae = Ba, 2; Ca, 3), [{Carb}Ca{(H3B)2PPh2}·(thf)] (4), [{Carb}Ba{PPh2·B(C6F5)3}] (5), [{Carb}Ba{O(B(C6F5)3)CH2CH2CH2CH2PPh2}·thf] (6), [Ba{O(B(C6F5)3)CH2CH2CH2CH2PPh2}2·(thf)1.5] (7) and [Ba{PPh2·B(C6F5)3}2·(thp)2] (8) that were characterised by multinuclear NMR spectroscopy (thp = tetrahydropyran). The molecular structures of 4, 6 and 8 were validated by X-ray diffraction crystallography, which revealed the presence of Ba⋯F stabilizing interactions (ca. 9 kcal mol−1) in the fluorine-containing compounds. Compounds 6 and 7 were obtained upon ring-opening of thf by their respective precursors, 5 and the in situ prepared [Ba{PPh2·B(C6F5)3}2]n. By contrast, thp does not undergo ring-opening under the same conditions but affords clean formation of 8. DFT analysis did not highlight any specific weakness of the Ba–P bond in 1·(thf)2. The instability of this compound is instead thought to stem from the high energy of its HOMO, which contains the non-conjugated P lone pair and features significant nucleophilic reactivity.
Introduction
Soluble complexes of the large alkaline earth metals (= Ae) calcium, strontium and barium are increasingly useful in organic synthesis.1,2 They make excellent molecular catalysts for hydrophosphination reactions,3,4e.g. for the anti-Markovnikov addition of phosphines on activated alkenes5–11 where barium systems perform particularly well.12 The heteroleptic complex [{Carb}BaN(SiMe3)2] supported by the bulky bis(imino)carbazolato monoanionic ligand {Carb}− (Fig. 1) is a typical case of an efficient hydrophosphination precatalyst.11 | ||
| Fig. 1 Proligands used to produce heteroleptic Ba–phosphides. Among these, only {IAcrown}H has led to a structurally authenticated complex, [{IAcrown}Ba(PPh2)].5–11 | ||
In vinylarene hydrophosphination promoted by Ae precatalysts of the general composition [{Lig}AeX·(solv)n] (where X− is chosen among N(SiMe3)2−, CH(SiMe3)2−, H−etc., and {Lig}− is a monoanionic ancillary ligand), it is accepted that the active species responsible for catalytic turnovers is the heteroleptic phosphide [{Lig}Ae(PR2)·(D)n], where D is a molecule of the (donor) solvent or phosphine. This phosphido species undergoes insertion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unsaturated bond into the Ae–P bond, an elementary step followed by protonolysis of the resulting Ae–alkyl bond with an incoming phosphine to regenerate the active species.1 In hydrocarbons, the use of an ancillary ligand is required to suppress Schlenk redistribution to the insoluble homoleptic barium diphosphide [Ba(PR2)2]∞.13,14 A handful of soluble, homoleptic complexes [Ba(PAr2)2·(D)n] are known for D = thf, dme or 18-c-6.13–17 The rare examples of crystallographically characterised heteroleptic Ba-phosphides include the dimeric [Ba{P(H)SitBu3}{N(SiMe3)2}·(thf)2]2
C unsaturated bond into the Ae–P bond, an elementary step followed by protonolysis of the resulting Ae–alkyl bond with an incoming phosphine to regenerate the active species.1 In hydrocarbons, the use of an ancillary ligand is required to suppress Schlenk redistribution to the insoluble homoleptic barium diphosphide [Ba(PR2)2]∞.13,14 A handful of soluble, homoleptic complexes [Ba(PAr2)2·(D)n] are known for D = thf, dme or 18-c-6.13–17 The rare examples of crystallographically characterised heteroleptic Ba-phosphides include the dimeric [Ba{P(H)SitBu3}{N(SiMe3)2}·(thf)2]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and [{IAcrown}Ba(PPh2)], where {IAcrown}− is a multidentate, {amino-tetraether}-anilide (see Fig. 1 for the proligands used to produce heteroleptic Ba–phosphides). We found the synthesis of the latter to be erratic.10 In this context, it seemed desirable to assess whether the {Carb}− ligand framework could reliably stabilise the heteroleptic phosphido complex [{Carb}Ba(PPh2)]. As the following will show, it soon emerged that this ambition cannot be fulfilled.
15 and [{IAcrown}Ba(PPh2)], where {IAcrown}− is a multidentate, {amino-tetraether}-anilide (see Fig. 1 for the proligands used to produce heteroleptic Ba–phosphides). We found the synthesis of the latter to be erratic.10 In this context, it seemed desirable to assess whether the {Carb}− ligand framework could reliably stabilise the heteroleptic phosphido complex [{Carb}Ba(PPh2)]. As the following will show, it soon emerged that this ambition cannot be fulfilled.
A suite of heteroleptic phosphidoborane magnesium (D–F, Fig. 2) and calcium (G–I) complexes supported by the ubiquitous {BDIDipp}− β-diketiminato ligand have been described by the Hill group.18,19 The aim was to assess Ae catalysts for HPPh2·BH3 dehydrocoupling and to produce polyphosphinoboranes. In a related topic, the chemistry of the amidoborane derivatives of the Ae metals had provided a mechanistic understanding of Ae-catalysed amine–borane dehydrogenation.20–22 Of specific interest is that the reaction of [{BDIDipp}Ca{PPh2·BH3}] (H) with B(C6F5)3 gave [{BDIDipp}Ca{H3B·PPh2·B(C6F5)3}·(η6-toluene)] (I).19 Beyond Hill's complexes, known examples of phosphidoborane Ae compounds are limited to Izod's homoleptic Ca and Sr complexes and their Mg congeners, synthesised by the reaction of Li- and K-phosphides with AeI2, e.g. [{((Me3Si)2CH)(Ph)P(BH3)2}2Ae·(thf)4] (Ae = Ca, K; Sr, L).23,24 We are not aware of a previous barium-phosphidoborane complex.
 | ||
| Fig. 2 Known Mg-, Ca- and Sr-phosphidoboranes (A)–(L) characterised by single crystal X-ray diffraction.18,19,23,24 R = CH(SiMe3)2 and Dipp = 2,6-iPr2–C6H3. | ||
We report here our attempt at producing heteroleptic Ba-phosphides. Confronted by the instability of the sought compounds, we have used phosphinoboranes as starting materials in order to generate more stable Ba–P–B motifs, and the corresponding barium phosphidoboranes, along with related calcium complexes, are described. We also show that [{Carb}Ba{PPh2·B(C6F5)3}] ring-opens thf to provide [{Carb}Ba{O(B(C6F5)3)(CH2)4PPh2}·thf], an unusual Ba-alkoxide. By contrast, the six-member tetrahydropyran (= thp) is inert under similar conditions. The metal-assisted ring-opening of thf with phosphines or phosphides has been previously documented with oxophilic metals including Li,25 Mg,26 lanthanides/actinides27–29 or Zr,30 and for a number of phosphine-based frustrated Lewis pairs.31–34
Results and discussion
In an attempt to produce a stable heteroleptic barium–phosphido complex, HPPh2 was reacted with the solvent-free [{Carb}BaN(SiMe3)2] (Scheme 1). The bulky bis(imino)carbazolato ligand {Carb}− has been shown to meet the demanding coordination requirements of the large barium ion (rionic (Ba2+) = 1.35 Å).11,35 The addition of diphenylphosphine to the solution of the barium precursor in petroleum ether resulted in an instant colour change from yellow to dark orange, and the precipitation of a vibrant orange solid was observed within 5–10 minutes. Variable temperature NMR analysis of this orange powder revealed two distinct phosphido species, each with a carbazole fragment; no equilibrium between the two species was observed by NMR. One of these species was hypothesised to be the sought Ba–phosphide, [{Carb}Ba(PPh2)] (1). However, despite repeated attempts, we were unable to isolate or identify a clean material from this reaction, and neither product could be crystallised from the mixture.§ Instead of isolating the precipitate, direct addition of thf to the orange suspension in petroleum ether allowed for the isolation of small microcrystals. This material diffracted poorly, and the quality of the dataset is too low to warrant precise discussion of the metric parameters (Rint = 0.1219, R1 = 0.1235). Yet, the connectivity in the structure was confirmed to be the twice solvated, heteroleptic Ba–phosphide [{Carb}Ba(PPh2)·(thf)2] (1·(thf)2), a compound with a six-coordinate distorted octahedral geometry around Ba (Fig. 3). We were not able to record NMR data for this compound due to the insufficient amount of material. 1H and 31P NMR data collected in toluene-d8 for the in situ reaction of HPPh2 and the solvated [{Carb}BaN(SiMe3)2·(thf)] (see ESI, Fig. S1 and S2†) were consistent with the clean formation of the mono-thf adduct [{Carb}BaPPh2·(thf)] (1·thf). However, this product proved unstable and decomposed during collection of 13C NMR data to a number of other species, including Ph2P–PPh236 (the mechanism of formation of this compound, e.g. homolytic cleavage or dehydrocoupling of HPPh2 released in situ, could not be determined). Some of the unknown resonances were similar to those observed in the NMR spectra for the abovementioned compounds. The instability of the Ba–PPh2 bond is perhaps unsurprising. For instance, bond dissociation energies in a suite of terminal zirconium–phosphides have been calculated to be low (ca. 55.4–65.3 kcal mol−1),37 whereas the homolytic cleavage of Bi(III)–PPh2 in heteroleptic bismuth-phosphides has been reported to generate Bi(II) species.38 Despite repeated attempts, a sample of 1·thf could not be isolated to allow full characterisation. Overall, we concluded that the isolation of a stable Ba–phosphido species supported by the {Carb}− ligand could not be achieved. | ||
| Scheme 1 Attempted synthesis of a stable barium–phosphido complex. Dipp = 2,6-diisopropylphenyl. | ||
 | ||
| Fig. 3 Representation of the molecular solid-state structure of [{Carb}Ba(PPh2)·(thf)2] (1·(thf)2) showing connectivity. H atoms and two non-coordinating thf molecules are omitted for clarity. Rint = 0.1219, R1 = 0.1224. | ||
In order to better understand the stability and bonding within Ba–phosphido complexes, density functional theory (DFT) calculations were carried out on the isolated solvated complex 1·(thf)2 at the PBE0-ZORA-D3(BJ)/TZ2P level (see the ESI† for computational details). Selected computed data are provided in Table 1. The optimised geometry of 1·(thf)2 is consistent with its X-ray structure, in spite of the limitations mentioned above concerning the quality of the XRD data. Except perhaps for the somewhat large Ba–P interatomic distance (optimised value 3.206 Å vs. experimental one of ca. 3.296(3) Å, although it falls in the typical range for Ba–P bonds13–17), there is no indication that hints at a particularly weak Ba–P bond. To get a better insight, the hypothetical amido homologue [{Carb}Ba(NPh2)·(thf)2] was also calculated (selected computed data in Table 1). Its structural features are quite similar to those of 1·(thf)2, except that the NPh2 nitrogen atom is significantly less pyramidalised than its PPh2 counterpart in 1·(thf)2 (the sum of covalent angles 356° and 320° for N and P, respectively). This difference results from the substantial conjugation of the nitrogen lone pair with the phenyl rings. Although comparing the Wiberg bond indices (WBIs, see Table 1) between bonds involving different elements should be made with caution, the corresponding Ba–PPh2 and Ba–NPh2 values suggest that the covalent component of the Ba–P bond is weaker than its Ba–N homologue. The natural atomic charge distribution (Table 1) indicates also the weaker ionic bonding in the case of the Ba–P bond.
| [{Carb}Ba(PPh2)·(thf)2] (1·(thf)2) | [{Carb}Ba(NPh2)·(thf)2] (putative) | [{Carb}Ba{PPh2·B(C6F5)3}] (5) | ||
|---|---|---|---|---|
| a Wiberg bond indices. b Averaged values. | ||||
| HOMO–LUMO gap/eV | 2.16 | 3.70 | 3.76 | |
| Computed interatomic distances (Å) and [WBI]a (experimental values in italics for comparison) | Ba–P/N | 3.206 [0.103] | 2.683 [0.250] | 3.303 [0.062] |
| 3.296(3) | ||||
| Ba–Ncarb. | 2.647 [0.050] | 2.660 [0.047] | 2.590 [0.068] | |
| 2.629(9) | ||||
| Ba–Nimin.b | 2.861 [0.027] | 2.883 [0.025] | 2.740 [0.037] | |
| 2.824(9) | ||||
| Ba–O/Fb | 2.727 [0.021] | 2.772 [0.020] | 2.863 [0.010] | |
| 2.737(9) | ||||
| Natural atomic charges | Ba | +1.78 | +1.81 | +1.82 |
| P/N | −0.15 | −0.79 | +0.32 | |
| Ncarb. | −0.66 | −0.66 | −0.70 | |
| Nimin.b | −0.56 | −0.56 | −0.60 | |
| O/Fb | −0.60 | −0.60 | −0.37 | |
In addition to these qualitative indicators, a more quantitative comparative analysis of the Ba–EPh2 (E = P, N) bonds can be made by performing an energy decomposition analysis (EDA) of the interaction between two frozen molecular fragments, according to the Morokuma–Ziegler procedure.39–41 The decomposition of the total bonding energy (TBE) between the [{Carb}Ba·(thf)2]+ fragment and the [EPh2]− (E = P, N) ligand in the two complexes is provided in Table 2. TBE is expressed as the sum of four components: the Pauli repulsion (EPauli), the electrostatic interaction energy (Eelstat), the orbital interaction energy (Eorb) and the component associated with the dispersion forces (Edisp). It is of note that the TBE components are comparable between both complexes, those of the amido species indicating slightly stronger (positive or negative) interactions, except for the minor dispersion component. The dominating component is Eelstat, being about three times larger than Eorb. This is in line with a bonding possessing a largely ionic characteristic, but with non-negligible covalency. The TBE values confirm that the Ba–NPh2 bond is stronger than its Ba–PPh2 analogue, with both stronger covalent (Eorb) and electrostatic (Eelstat) interactions. The overall TBE difference of ca. 7 kcal mol−1 remains however modest, as in effect are also the differences across the two compounds for each of the four contributions.
| [{Carb}Ba(PPh2)·(thf)2] (1·(thf)2) | [{Carb}Ba(NPh2)·(thf)2] (putative complex) | [{Carb}Ba{PPh2·B(C6F5)3}] (5) | |
|---|---|---|---|
| a Values in eV, unless specified. b Total bonding energy (TBE) = EPauli + Eelstat + Eorb + Edisp. | |||
| Fragmentation | [{Carb}Ba·(thf)2]+ + [PPh2]− | [{Carb}Ba·(thf)2]+ + [NPh2]− | [{Carb}Ba]+ + [PPh2·B(C6F5)3]− |
| E Pauli | +2.43 | +2.57 | +1.84 |
| E elstat | −4.73 | −4.95 | −3.62 |
| E orb | −1.46 | −1.72 | −1.50 |
| E disp | −0.85 | −0.82 | −1.24 |
| TBEb | −4.61 (−106 kcal mol−1) | −4.92 (−113 kcal mol−1) | −4.52 (−104 kcal mol−1) |
The Quantum Theory of Atoms in Molecules (QTAIM) approach42,43 provides a different angle to look at these Ba–E (E = P, N) bonds. Selected QTAIM data are collated in Table 3. Surprisingly, the AIM charge on P in 1·(thf)2 is now significantly positive, whereas that of N in its amido analogue remains strongly negative. It is consistent with a more ionic (or very polar44) characteristic of the Ba–N bond. All Ba–EPh2 (E = P, N) bond critical point (bcp) indicators have small absolute values. The positive sign of the Laplacian density (larger in the case of N), the negative sign of the (very small) energy density and the |V|/G ratio somewhat larger for 1·(thf)2 are compatible with a weakly covalent, strongly ionic bonding.
| [{Carb}Ba(PPh2)·(thf)2] (1·(thf)2) | [{Carb}Ba(NPh2)·(thf)2] (putative complex) | [{Carb}Ba{PPh2·B(C6F5)3}] (5) | ||
|---|---|---|---|---|
| a Averaged values. b ρ, ∇2ρ, H, V and G are the electron density, Laplacian of ρ density, energy density, potential energy density and kinetic energy density values at the bcp, respectively. All values in a.u. | ||||
| Atom charge | Ba | +1.61 | +1.64 | +1.60 |
| P/N | +0.54 | −1.23 | +0.71 | |
| Ncarb. | −1.14 | −1.17 | −1.17 | |
| Nimin.a | −1.19 | −1.17 | −1.21 | |
| Oa | −1.09 | −1.09 | ||
| Delocalisation index | δ | 0.27 | 0.27 | 0.19 |
| bcp indicatorsa | ρ | 0.027 | 0.039 | 0.022 |
| ∇2ρ | 0.046 | 0.111 | 0.041 | |
| H | −0.003 | −0.004 | −0.001 | |
| V | −0.018 | −0.035 | −0.013 | |
| |V|/G | 1.22 | 1.12 | 1.13 | |
In any case, the above analyses do not underscore any particular weakness of the Ba–phosphide bond, even if it is not as strong as its Ba–amide homologue. We suggest that the relative instability of 1·thf or 1·(thf)2 is not of thermodynamic origin, but instead it is related to the reactivity of the lone pair on the P atom. In contrast to its Ba–NPh2 amido relative, where the nitrogen lone pair is stabilised through conjugation, the phosphido lone pair in 1·(thf)2 does not delocalise especially on the phenyl rings and remains rather unperturbed, hence constituting the HOMO in the complex. This HOMO sits isolated 1.60 eV above the HOMO−1, and confers to 1·(thf)2 a significant nucleophilic ability, especially as the lone pair is not protected. Very similar differences between the P and N derivatives were computed for the unsolvated species [{Carb}Ba(EPh2)] (E = P (1), N), as well as for the [{Carb}BaE(SiMe3)2] (E = P, N) pair.
Magnesium, calcium and even strontium phosphidoborane complexes that contain Ae–P bonds are known.18,19,23,24 We hence turned our attention to the use of phosphine–borane in order to achieve the stabilisation of a Ba–P bond in a heteroleptic complex. In an NMR-scale reaction, the data recorded in benzene-d6 after addition of one equivalent of HPPh2·BH3 to [{Carb}BaN(SiMe3)2] were consistent with formation of [{Carb}Ba{PPh2·BH3}] (2) (Scheme 2). The 1H NMR spectrum (Fig. S3†) showed the release of HN(SiMe3)2, while only one symmetrical {Carb}− ligand environment could be detected. As reported previously for [{BDIDipp}Ca{PPh2·BH3}] and [{BDIDipp}Mg{PPh2·BH3}]2 (H and D, respectively, in Fig. 2),18 the resonances for boron-bound H atoms in 2 could not be seen. The 31P NMR spectrum of the complex displayed a single broad singlet resonance at δP −32.3 ppm, shifted significantly upfield from the broad doublet of HPPh2·BH3 (1.6 ppm) and confirming the deprotonation of the phosphine. The 11B NMR spectrum also exhibited a broad singlet (δB −24.5 ppm), downfield shifted from HPPh2·BH3 (−40.3 ppm). Both chemical shifts are in line with those for [{BDIDipp}Ca{PPh2·BH3}] (toluene-d8, 48 °C: δP −45.0 ppm and δB −30.2 ppm) and [{BDIDipp}Mg{PPh2·BH3}]2 (toluene-d8, 76 °C: δP −57.1 and δB −34.0 ppm),18 though meaningful comparison is difficult due to the differences in metals, ligand environments, solvents and temperature. When the reaction was scaled up in petroleum ether, complex 2 could be isolated in near-quantitative yield as a bright yellow powder. However, in spite of multiple attempts, single-crystals suitable for analysis by XRD could not be obtained.§ A solution of 2 in thf rapidly lost its bright yellow colour, with concomitant formation of a yellow precipitate. The 1H NMR spectrum of the isolated powder revealed a mixture of unassignable {Carb}− environments, while no signal could be observed in the 31P and 11B NMR spectra. Again, despite several attempts, crystals could not be obtained from this solid to allow identification of any of its components by X-ray diffraction analysis.
 | ||
| Scheme 2 Synthesis of Ca and Ba phosphidoborane and phosphidodiborate complexes 2–4. | ||
In order to improve our understanding of the behaviour of barium complexes, the same experiments were carried out using calcium precursors, as Ca complexes, being less ionic in nature, are generally more robust in solution than their Sr and Ba congeners. Metalation of HPPh2·BH3 with [{Carb}CaN(SiMe3)2] in toluene-d8 resulted in the clean formation of [{Carb}Ca{PPh2·BH3}] (3). The 1H NMR spectrum displays a single {Carb}− ligand environment, though again no identifiable BH3 resonance could be detected. The 31P NMR spectrum displays a broad resonance at δP −41.3 ppm, while the 11B NMR spectrum features a broad signal at δB −26.9 ppm. Both match those already mentioned for [{BDIDipp}Ca{PPh2·BH3}].18 Again, despite repeated attempts, satisfactory crystals of 3 for XRD analysis could not be isolated.§ A small amount of thf was added to a sample of 3 in petroleum ether, in an attempt to produce a thf adduct that would perhaps allow for solid-state characterisation. Unexpectedly, the calcium–phosphidodiborate [{Carb}Ca{(H3B)2PPh2}·(thf)] (4) was isolated in 70% yield as pale yellow crystals. Its identity was established by X-ray diffraction (see Fig. 4) and NMR spectroscopy. In contrast, the addition of thf to a solution of [{BDIDipp}Ca{PPh2·BH3}] (H in Fig. 2) in benzene-d6 was reported to give the adduct [{BDIDipp}Ca{PPh2·BH3}·(thf)] (H·(thf)), a compound that was also obtained by reaction of [{BDIDipp}CaN(SiMe3)2·(thf)] with HPPh2·BH3.18
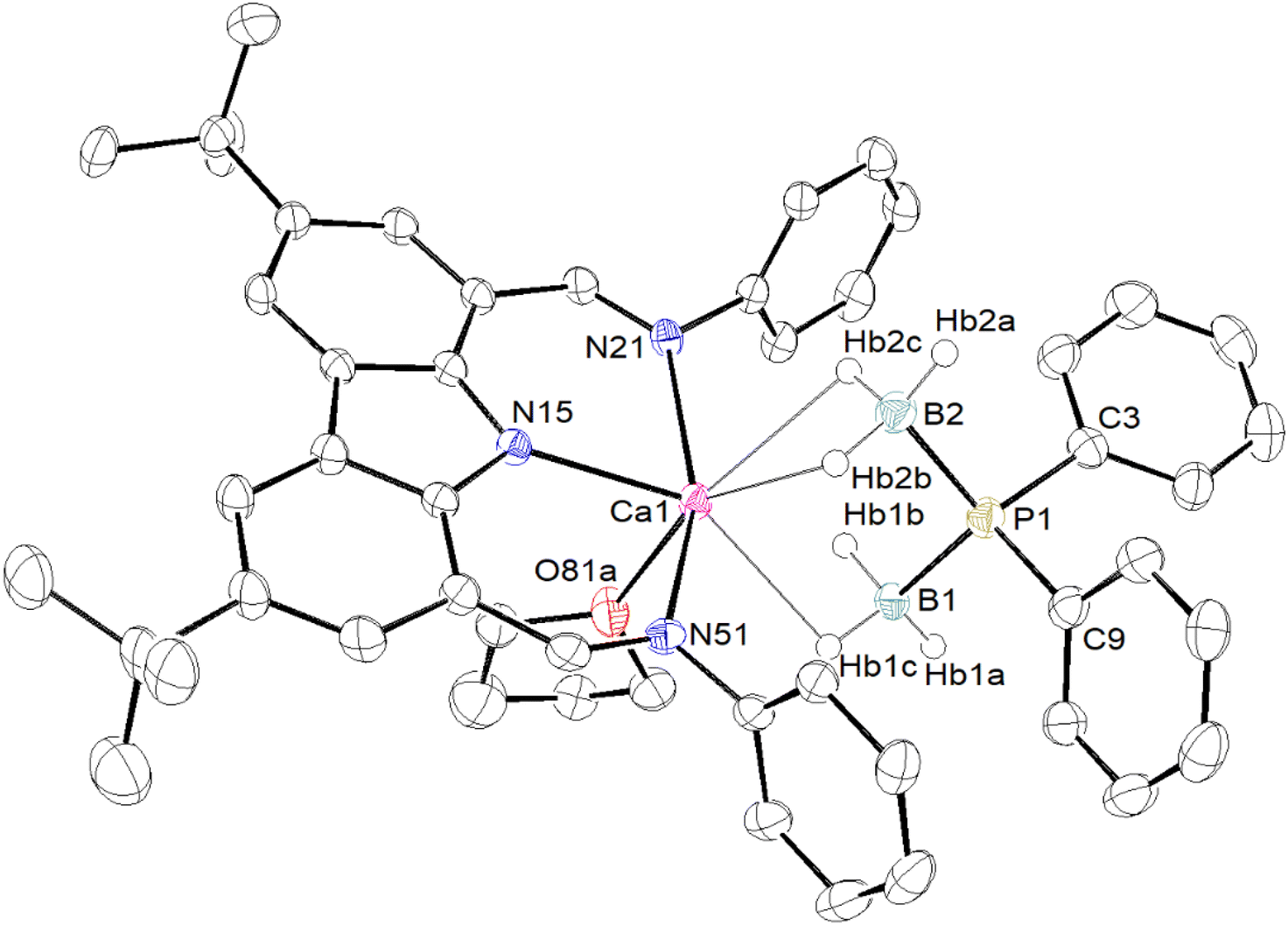 | ||
| Fig. 4 ORTEP representation of the molecular solid-state structure of [{Carb}Ca{(H3B)2PPh2}·(thf)] (4). Only the major components of disordered tBu group and thf molecule are shown. Hydrogen atoms except those bound to boron and iPr groups are omitted for clarity. Selected interatomic distances (Å) and angles (°): Ca1–HB1b = 2.55(3), Ca1–HB1c = 2.45(3), Ca1–HB2b = 2.43(3), Ca1–HB2c = 2.47(3), Ca1–N15 = 2.385(2), Ca1–N21 = 2.559(3), Ca1–N51 = 2.582(2), Ca1–O81a = 2.402(2), Ca1–P1 = 3.5483(10), P1–B1 = 1.949(3), P1–B2 = 1.937(3), B1–HB1a = 1.010(12), B1–HB1b = 1.066(12), B1–HB1c = 1.227(12), B2–HB2a = 1.133(9), B2–HB2b = 1.139(12), B2–HB2c = 1.10(3); N15–Ca1–N21 = 79.18(8), N15–Ca1–N51 = 79.58(8), N21–Ca1–N51 = 157.01(7), P1–Ca1–N15 = 155.93(6), P1–Ca1–N21 = 102.59(6), P1–Ca1–N51 = 93.31(6), P1–Ca1–O81a = 117.07(6), O81a–Ca1–N15 = 86.71(7), O81a–Ca1–N21 = 91.15(8), O81a–Ca1–N51 = 96.32(8), B1–P1–B2 = 109.22(16), B1–P1–C3 = 110.21(14), B1–P1–C9 = 111.82(15), B2–P1–C3 = 110.91(15), B2–P1–C9 = 110.93(14), C3–P1–C9 = 103.67(14). Distance from Ca1 to N15N21N51 plane: 0.243(2) Å. | ||
The formulation for complex 4 is reminiscent of that reported for [{BDIDipp}Mg{μ-(H3B·PPh2·BH3)}]2 (F in Fig. 2).18 However, the structures of these two complexes differ: whereas the magnesium compound is a dimer with a μ-bridging phosphidodiborate, complex 4 forms a monomeric, thf-solvated complex with a formally six-coordinated, pseudo-octahedral geometry around Ca (Fig. 4). The six boron-bound hydrogens were localised from their electron density in the Fourier difference map. The molecule has four clear Ca⋯H–B interactions (2.43(3)–2.55(3) Å) and one coordinated thf molecule. The Ca1–P1 interatomic distance is very long, 3.5483(10) Å, and clearly militates against the existence of a bond between the metal and the phosphorus atom. This is consistent with the near-perfect tetrahedral geometry about P1. By contrast, the Ca–P bond length is much shorter in the four-coordinated heteroleptic [{BDIDipp}CaPPh2·(thf)] (2.872(4) Å),5 in the six-coordinated [Ca(PPh2)2·(thf)4] (2.9865(6) Å),14 and in [{BDIDipp}Ca{PPh2·BH3}·(thf)] (2.8962(4) Å).18 At a mere 0.243(2) Å above the plane defined by N15, N15 and N51, the calcium centre in 4 fits well inside the sterically shielded cavity created by the bis(imino)carbazolato ligand.
The Mg–phosphidodiborate [{BDIDipp}Mg{μ-(H3B·PPh2·BH3)}]2 (F) was obtained by addition of two equivalents of HPPh2·BH3 to the dimeric [{BDIDipp}Mg{PPh2·BH3}]2 (D) via BH3-transfer from HPPh2·BH3.19 On the other hand, the formation of 4 most likely results from the transfer of a BH3 moiety from an already coordinated {PPh2·BH3}− ligand and concomitant formation of one equivalent of [{Carb}CaPPh2·(thf)]. Borane transfer of this type has also been documented for lithium compounds, e.g. in the formation of [{2-SMe–C6H4–P(BH3)2(CH(SiMe3)2)}Li].45 The 31P NMR spectrum of 4 displays a singlet at δP −22.6 ppm, a significant downfield chemical shift from complex 3 (δP −41.3 ppm). The 11B NMR resonance of δB −32.5 ppm is slightly upfield from that of 3 (δB −26.9 ppm), though similar to Hill's [{BDIDipp}Mg{μ-(H3B·PPh2·BH3)}]2 (F, δB −34.3 ppm).§
To circumvent the relative ease of BH3 migration, HPPh2·B(C6F5)3 was used due to its potential ability to set up stabilizing Ba⋯F secondary interactions. It is easily available by reaction of HPPh2 with B(C6F5)3·(Et2O)36 and provides a convenient 19F NMR spectroscopy handle. Addition of one equivalent of the colourless HPPh2·B(C6F5)3 to a stirred solution of [{Carb}BaN(SiMe3)2] in toluene at −78 °C followed by the removal of the volatiles afforded [{Carb}Ba{PPh2·B(C6F5)3}] (5) in 74% yield, as an extremely moisture-sensitive yellow powder (Scheme 3). The reaction can also be carried out in petroleum ether; however in this case it requires prolonged reaction times. The 1H NMR spectrum of 5 in toluene-d8 displays characteristic resonances for the {Carb}− ligand, a singlet at δH 8.23 (CHN), a heptet at 2.92 (CH(CH3)2), and two doublets at 1.02 and 0.94 ppm (3JH–H = 6.5 and 6.7 Hz, CH(CH3)2). The complex is characterised by a single broad resonance in the 31P NMR spectrum at δP −9.6 ppm, and another broad resonance in the 11B NMR spectrum (δB −12.6 ppm). Both values are substantially deshielded compared to those for 3 (δP −41.3 and δB −26.9 ppm), in agreement with the strongly electron-withdrawing nature of C6F5 groups. Three main resonances are visible in the 19F NMR spectrum (δF −128.0, −160.3 and −164.7 ppm for o-, p- and m-F atoms, respectively). The data for 5 are also different from those of the starting material, HPPh2·B(C6F5)3 (δP −0.4 ppm; δB −12.4 ppm; δF −128.1, −155.2 and −162.9 ppm). Taken collectively, these NMR data all point at the formation of a single product with a unique coordination sphere. Despite repeated recrystallisation attempts from a range of solvents, we could not obtain crystals of 5 suitable for XRD characterisation.§ Surprisingly, at the time of writing, there was no case of a metal complex containing the “[Met]–PPh2·B(C6F5)3” fragment in the CSD database,46 although the Ti(IV) phosphide (Cp)(η5-C5H4B(C6F5)2)(μ-PPh2)TiCl containing a Cp–B–P chelate was reported.47 The structure of 5 was then assessed by DFT calculations (Fig. 5). In the optimised geometry, the central metal is unsymmetrically five-coordinated by phosphorus, the three nitrogen atoms from the bis(imino)carbazolato ligand and one fluorine atom (Ba⋯F = 2.863 Å). The computed Ba–N interatomic distances are somewhat shorter than in 1·(thf)2 (see Table 1), whereas the Ba–P distance is larger (3.303 Å). Nonetheless, the EDA analysis (Table 2) does not suggest weaker bonding. This apparent contradiction finds its source in the fact that the considered metal–ligand interaction in 5 includes also one Ba⋯F contact. The Ba–P WBIs (Table 1) and bcp indicators (Table 3) are consistent with a somewhat weaker bond in 5 as compared to 1·(thf)2. The nature of Ba⋯F bonding is discussed in more detail below in the case of complexes 6 and 8.
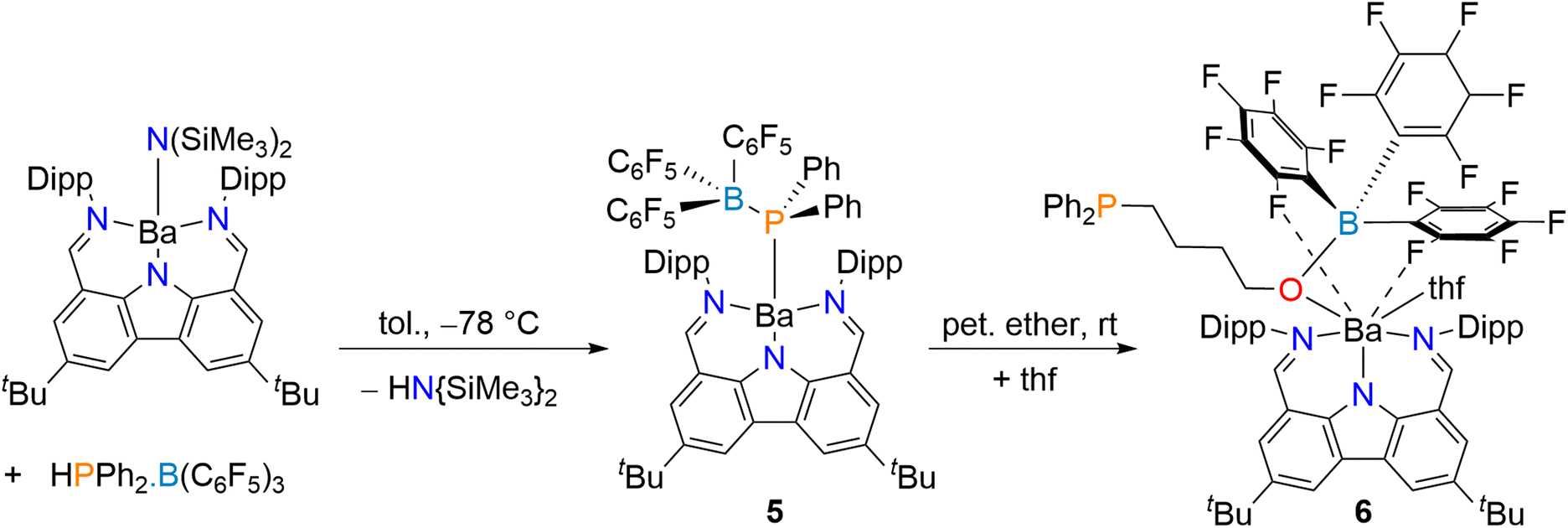 | ||
| Scheme 3 Synthesis of [{Carb}Ba{PPh2·B(C6F5)3}] (5) and [{Carb}Ba{O(B(C6F5)3)(CH2)4PPh2}·thf] (6). | ||
 | ||
| Fig. 5 The DFT-optimised geometry of [{Carb}Ba{PPh2·B(C6F5)3}] (5). See Table 1 for selected interatomic distances. Additional metrical data: P–B = 2.118 Å, B–C (av.) = 1.641 Å; P–Ba–F = 56°. | ||
The addition of a small amount of thf to a suspension of 5 in petroleum ether did not produce the expected [{Carb}Ba{PPh2·B(C6F5)3}·(thf)n], nor did it form a phosphidodiborate akin to 4, e.g. [{Carb}Ca{PPh2(B(C6F5)3)2}·(thf)n]. Instead, [{Carb}Ba{O(B(C6F5)3)CH2CH2CH2CH2PPh2}·thf] (6), a Ba–alkoxide resulting from the ring-opening of one thf molecule, was isolated in 61% yield as yellow crystals (Scheme 3).§ Complex 6 is a rare example of a heteroleptic, monometallic barium alkoxide. The ring-opening of thf has been previously reported for a number of systems built around oxophilic metals,25–30 frustrated Lewis pairs based on phosphines31–34 or NHCc,48 even middle-to-late transition metals.49,50 The formation of 6 presumably occurs via nucleophilic attack by the phosphorus atom on the thf α-carbon atom, resulting in ring opening, while the increased negative charge on the now Ba-coordinated oxygen atom also results in borane migration from the phosphine to generate a more stable Lewis adduct.
The 1H NMR spectrum of complex 6 in benzene-d6 displays characteristic ligand resonances at δH 8.47 (CHN), 3.00 (CH(CH3)2), and four isopropyl resonances at δH 1.06, 1.00 and 0.92 (two superimposed) ppm (CH(CH3)2), as well as four methylene resonances at δH 2.85 (OCH2), 1.22, 0.56 and −0.28 ppm. It also shows the presence of the protonated ligand {Carb}H which we could not remove. Yet, the 31P NMR spectrum displays a single resonance at δP −18.8 ppm, an upfield chemical shift from 5, while the 11B{1H} NMR spectrum has a single broad resonance at δB −2.6 ppm. This is consistent with the existence of single [Ba]–PPh2/B(C6F5)3 species. Both resonances are similar to those for the lithium alkoxide-bound borane [Li{O(B(C6F5)3)(CH2)4PMes2}] (Mes = mesityl; δB −3.1 and δP −21.4 ppm).25 Interestingly, the 19F NMR spectrum displays eight resonances in the δF range −132 to −165 ppm instead of the usually observed three signals. For instance, [Li{O(B(C6F5)3)(CH2)4PMes2}] features the common doublet at δF −137.6 ppm and triplets at −161.0 and −165.6 ppm. The unusual 19F NMR pattern for 6 suggests that at room temperature, one or more ortho-fluorine Ba⋯F–C interactions inhibit the free rotation of one or two of the C6F5 rings in the complex.
The crystal structure of 6 (Fig. 6) reveals the metal to be in a seven-coordinated environment, with coordination of the five N- and O-atoms, along with two clearly identifiable Ba⋯F–C interactions. At 7.696(2) Å, the phosphorus atom P1 is expulsed far away from the metal Ba1. The Ba1–F95 interatomic distance of 2.7497(17) Å testifies to a strong interaction with the fluorine atom, likely one that persists in solution, while the interaction with F73 is slightly weaker (Ba1–F73 = 2.9889(17) Å). The Ba⋯F95 distance is at the very low end of values for Ba⋯F secondary interactions, more commonly in the range 2.80–3.10 Å. It is for instance significantly shorter than in other previously reported complexes with strong Ba⋯F–C contacts, such as the bis(hydridoborate) [Ba{HB(C6F5)3}2·(thf)4] (2.8228(19)–2.8386(17) Å),51 the dimeric barium complex [Ba{μ-N(C6F5)2}{N(C6F5)2}·(toluene)]2 (2.825(2)–3.014(3) Å),52 or the polymeric [Ba{N(H)-2,6-F2C6H3}2·(thf)2]∞ (2.871(2)–2.901(3) Å).53 This short interatomic distance is consistent with the aforementioned 19F NMR data showing inequivalence at room temperature. As relative points of comparison, the sum of van der Waals radii for barium and fluorine (2.68 and 1.47 Å, respectively)54 is much greater than the Ba1–F73 and Ba1–F95 distances, whereas the added covalent radii for Ba and F amount to 2.72 Å. Note that it is now established that the nature of such Ba⋯F (and, more generally, Ae⋯F) interactions is essentially (albeit not exclusively) electrostatic.52,55 Yet, the C68–F73 (1.363(3) Å) and especially the C90–F95 (1.374(3) Å) bonds are stretched compared to the other C–F bonds with fluorine atoms that do not interact with the metal, typically between 1.338(4)–1.350(4) Å. Another notable feature in the structure of 6 is the long Ba1–O61 interatomic distance (2.788(2) Å). Remarkably, it is even longer than the shortest Ba⋯F contact in the complex (2.7497(17) Å). It is significantly greater than for other terminal alkoxides, such as the perfluorinated alkoxides [Ba{OC(CF3)3}2·(donor)n] (donor = thf, diglyme or 1,2-dimethoxyethane; Ba–O = 2.5335(9)–2.544(3) Å),56 [Ba2(OCPh3)(μ-OCPh3)3·(thf)3] (2.409(4) Å),57 and even the eight-coordinated [Ba{OC(CF3)2CH2N(CH2CH2OMe)2}2] (2.558(2)–2.562(2) Å).58 The Ba–Oalkoxide distance in 6 is also much longer than in the related O-ligated barium–boryloxide [Ba{OB(CH(SiMe3)2)2}2] (2.3832(16)–2.4098(16) Å)59 and barium–siloxide [Ba2{OSi(SiMe3)3}{μ-OSi(SiMe3)3}3] (2.414(7) Å)60 and [Ba2{OSitBu3}{μ-OSitBu3}3] (2.413(16) Å)57 complexes, despite the reduced donating ability of the O-atoms in these ligands.61 The Ba–Oalkoxide distance is still greater in 6 than in the eight-coordinated [Ba{OCH2CH2N(CH2CH2OH)2}2]·EtOH (2.73(1) Å),62 the first (and a very rare) structurally verified Ba–alkoxide featuring an unexpectedly long Ba–O length due to EtOH⋯OBa hydrogen bonding with lattice ethanol molecules. These comparisons are not ideal due to the structural differences between these compounds and 6. However, owing to the lack of structurally characterised monometallic barium alkoxides, there is no directly relevant comparison to be made at the current time.46 The long Ba1–O61 interatomic distance in 6 likely results from the transfer of electron density from the oxygen atom into the empty p-orbital of the borane. There is a distorted trigonal pyramidal geometry around the O61 atom (Σθ(O61) = 346.8(3)°). Such relative weakness of the Ba–O bond does raise the question of whether the alkoxide could be substituted for another ligand via protonolysis, and, if so, whether the ring-opening of thf could be turned advantageously into a catalytic manifold.
 | ||
| Fig. 6 ORTEP representation of the molecular structure of [{Carb}Ba{O(B(C6F5)3)CH2CH2CH2CH2PPh2}·thf] (6). H atoms, tBu an iPr groups are omitted for clarity. Only the major component of the disordered thf molecule is shown. Selected interatomic distances (Å) and angles (°): Ba1–F73 = 2.9889(17), Ba1–F95 = 2.7497(17), Ba1–N11 = 2.798(2), Ba1–N31 = 2.658(2), Ba1–N47 = 2.772(3), Ba1–O1a = 2.788(2), Ba1–O61 = 2.789(2), C64–F69 = 1.350(4), C65–F70 = 1.340(4), C68–F73 = 1.363(3), C86–F91 = 1.345(4), C88–F93 = 1.338(4), C90–F95 = 1.374(3); N11–Ba1–N31 = 70.07(7), N11–Ba1–N47 = 124.13(7), N11–Ba1–O1a = 89.19(8), N11–Ba1–O61 = 127.13(7), N31–Ba1–N47 = 70.12(7), N31–Ba1–O1a = 134.80(7), N31–Ba1–O61 = 98.75(7), N47–Ba1–O1a = 92.14(7), N47–Ba1–O61 = 96.33(7), O1a–Ba1–O61 = 124.97(7). Distance from Ba1 to N11N31N47 plane: 1.2349(4) Å. | ||
DFT calculations on complex 6 reproduce well its solid-state structure (ESI, Table S1†), with the shortest optimised Ba–F distance (2.813 Å) being slightly longer than its X-ray counterpart. In any case, both Ba⋯F contacts show similar characteristics, hence they are averaged in Table S1.† Their QTAIM descriptors are diagnostic of weak ionic bonds. Such Ae⋯F–C interactions have been investigated in low-coordinate Ae fluoroarylamides.52 Their very small covalent characteristic is also evidenced by the fact that the corresponding F-atoms are more negatively charged (−0.36 (natural), −0.69 (QTAIM), see Table S1†) than the other fluorines (range: −0.29 to −0.31 (natural), −0.65 to −0.67 (QTAIM)), indicating charge attraction from Ba onto the fluorine atoms, but very little electron transfer between them.
To further investigate the reactivity of the [Ba]–PPh2·B(C6F5)3 moiety and the nature of the Ba-to-alkoxide bonding in 6, the analogous homoleptic barium phosphidoborane starting material was synthesised. The protonolysis reaction between [Ba{N(SiMe3)2}2]2 and HPPh2·B(C6F5)3 (two equivalents vs. Ba) yields a pale yellow powder, presumed to be [Ba{PPh2·B(C6F5)3}2]n (Scheme 4). This solid is insoluble in aromatic or aliphatic hydrocarbons. Dissolution by addition of thf to a suspension in petroleum ether gives the product of composition [Ba{O(B(C6F5)3)CH2CH2CH2CH2PPh2}2·(thf)1.5] (7, probably an aggregate), where ring-opening of thf has occurred. This compound could not be crystallised to allow XRD analysis.§ Although it is a white solid, crystallisation attempts generated yellow clathrates that separated from the solvent, especially at low temperatures. By contrast, the six-member cyclic tetrahydropyran (thp) does not ring-open under the same conditions, and affords instead the thp-adduct [Ba{PPh2·B(C6F5)3}2·(thp)2] (8), which was characterised by NMR (Fig. S30–S35†) and XRD (Fig. 7) analyses.§
 | ||
| Scheme 4 Synthesis of [Ba{O(B(C6F5)3)CH2CH2CH2CH2PPh2}2·(thf)1.5] (7) and [Ba{PPh2·B(C6F5)3}2·(thp)2] (8). | ||
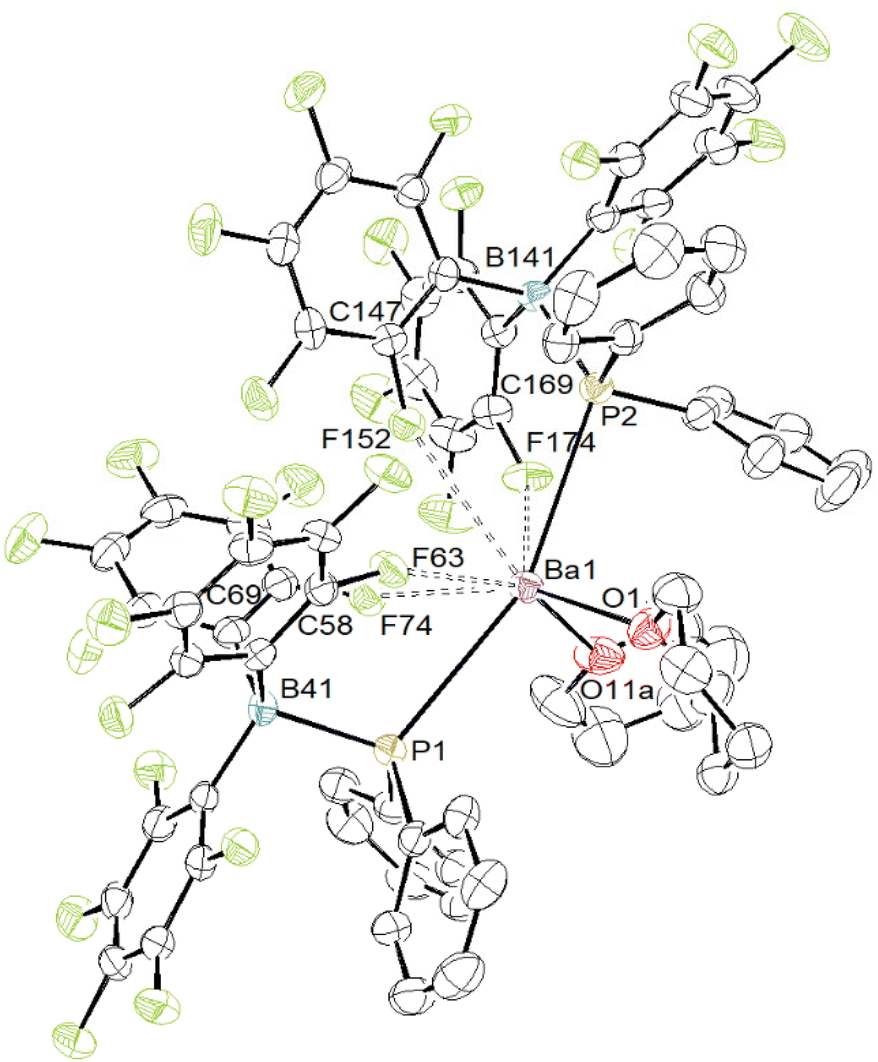 | ||
| Fig. 7 Representation of the molecular solid-state structure of [Ba{PPh2·B(C6F5)3}2·(thp)2] (8). Non-interaction lattice toluene molecule and H atoms are omitted for clarity. Only the main component of the disordered thp molecule is shown. Non-interacting F-atoms are shaded in green. Selected interatomic distances (Å) and angles (°): Ba1–F63 = 2.7557(17), Ba1–F74 = 2.7634(19), Ba1–F152 = 3.1120(18), Ba1–F174 = 2.7912(18), Ba1–O1 = 2.623(2), Ba1–O11a = 2.658(2), Ba1–P1 = 3.3080(8), Ba1–P2 = 3.2495(8), P1–B41 = 2.071(3), P2–B141 = 2.050(3), C58–F63 = 1.375(3), C69–F74 = 1.379(4), C147–F152 = 1.370(3), C169–F174 = 1.378(4); O1–Ba1–O11a = 97.31(8), O1–Ba1–P1 = 98.22(6), O1–Ba1–P2 = 87.00(6), O11a–Ba1–P1 = 96.21(6), O11a–Ba1–P2 = 106.93(6), P1–Ba1–P2 = 155.47(2). | ||
The 1H NMR spectrum of 7 in thf-d8 displays the resonances for four distinct methylene groups, along with the expected aromatic resonances for the phenyl moieties as two sets of multiplets at δH 7.34 (8H) and 7.23 (12H) ppm. The spectrum is consistent with the presence of 1.5 equivalents of non-deuterated thf per metal. The 31P NMR spectrum shows a broad singlet at δP −16.1 ppm, similar to the chemical shift in 6, while the 11B NMR spectrum presents a broad singlet at δB −2.9 ppm. It is probable that in the solid state, compound 7 creates a larger multimetallic species with bridging alkoxide ligands.12,57 The 1H NMR spectrum of 8 in thf-d8 displays resonances near-identical to those of 7 in this solvent, indicating that deuterated thf molecules displace the initially metal-bound thp ones in solution. Accordingly, three resonances for free thp are also observed. The NMR data confirm that complex 8 is the sole observable species. The 31P NMR spectrum displays a single resonance at δP −16.6 ppm, while the 11B NMR spectrum displays a sharp resonance at δB −2.9 ppm and the 19F NMR spectrum has three resonances, a doublet at δF −133.4 (3JF–F = 23.7 Hz) ppm, a sharp triplet at −165. 6 (3JF–F = 20.1 Hz) and a broad triplet at −168.4 (3JF–F = 19.8 Hz) ppm. All these data are almost identical to those of 7. Finally, when diethyl ether is added instead of a cyclic ether to a suspension of [Ba{PPh2·B(C6F5)3}2]n in petroleum ether, the metal species decomposes over the course of a few hours and the precipitation of a black solid is visible. The NMR analysis of the resulting mixture did not provide any insight into the decomposition process.
The solid-state structure of complex 8 depicted in Fig. 7 shows the metal to sit in an eight-coordinate geometry from coordination of the two P- and two O-atoms, along with four additional Ba⋯F contacts. The complex shows intriguing structural parameters, but there is no [Met]–PR2B(C6F5)3 metal fragment in the CSD database to allow for a direct comparison at the time of writing.46 The P1–Ba1–P2 angle is very obtuse (155.47(2) °). Approximately one half of the first coordination sphere around the metal is occupied by Ba⋯F interactions, whereas the other half is filled by the two coordinated thp molecules and phenyl rings. The Ba1–P1 and Ba1–P2 interatomic distances (3.2495(8)–3.3080(8) Å) are commensurate with those in [Ba(PPh2)2·(thf)5] (3.328(2) and 3.345(2) Å)14 and in the polymeric [Ba3(PPh2)6·(thf)4]∞ (3.2958(7)–3.3627(7) Å).13 This bonding situation suggests that coordination of the borane onto phosphorus does not affect significantly its electron-sharing properties with the metal. The geometry about the P- and B-atoms forms slightly distorted tetrahedra. The P1–B41 and P2–B141 bond lengths (2.071(3) and 2.050(3) Å) are greater than in the calcium complexes [({BDIDipp}Ca)3(μ2-H)(H3BPPh2)2] (G in Fig. 2, 1.945(3) and 1.946(3) Å) and [{BDIDipp}Ca{PPh2·BH3}·(thf)] (H·(thf), 1.9466(18) Å).18 They compare somewhat better with the P–B(C6F5)3 distance in [{BDIDipp}Ca{H3B·PPh2·B(C6F5)3}·(η6-toluene)] (I, 2.1368(18) Å), while the H3B–P distance to the κ3-bound BH3 group is shorter (1.9469(18) Å). However, the phosphorus atom is not metal-bound in this compound.19 The P–B interatomic distance is 2.054(4) Å in the chelate complex (Cp)(η5-C5H4B(C6F5)2)(μ-PPh2)TiCl where bonding involves a Ti–P σ-bond and a P → B donor interaction (Fig. 8),47 and where the closest Ti⋯F distances in this complexes are above 3.6 Å. The Ba1–O1 and Ba1–O11a bond lengths in 8 (2.623(2) and 2.658(2) Å, respectively) are substantially shorter than in the five-coordinated [Ba{N(SiMe3)2}2·(thp)3] (2.829(2)–2.869(2) Å)63 and in the seven-coordinated [BaI2·(thp)5] (2.723(6)–2.789(6) Å).64 Finally, three of the four Ba⋯F interactions in 8, in the range 2.7557(17)–2.7912(18) Å, compare well with those in complex 6 and can be considered as particularly strong. The last one, with a Ba1–F152 interatomic distance of 3.1120(18) Å, is weaker. As in 6, the C–F bond lengths for these four metal-interacting fluorine atoms, between 1.370(4) and 1.379(4) Å, are elongated compared to the other C–F bonds (1.342(4)–1.357(4) Å) in the compound. Besides, shortening of the Ba–F interatomic distance is accompanied by a lengthening of the corresponding C–F bond. It is not clear how heavily the multiple Ba⋯F contacts in 8 weigh on the other metric parameters and bonding properties in the complex, notably concerning the barium-to-phosphorus bonds.
 | ||
| Fig. 8 Structure of (Cp)(η5-C5H4B(C6F5)2)(μ-PPh2)TiCl.47 | ||
The X-ray structure of complex 8 is well reproduced by DFT calculations (ESI, Table S2†). In particular, the four short Ba⋯F contacts (2.854 Å in average) are also present in the gas phase-optimised geometry. The Ba–P and Ba⋯F bond indicators (Table S2†) are similar to those found in 1·(thf)2 and 6, respectively. In order to compare the strength of the Ba–P bonds in 8 with that in 1·(thf)2, an energy decomposition analysis was performed on 8, based on the [Ba·(thp)2]2+ + {[PPh2·B(C6F5)3]−}2 fragmentation (see Table 4). It should be pointed out that the reported bonding energies are not only associated with two Ba–P bonds, but also with four Ba⋯F contacts. In order to separate these two contributions, we also ran a single-point calculation on the frozen structure of a molecule of 8 in which the four F-atoms in contact with Ba were replaced by H-atoms (C–H = 1.09 Å). The corresponding EDA decomposition based on the [Ba·(thp)2]2+ + {PPh2·B[(C6H2F3)(C6F5)2]−}2 fragmentation is also given in Table 4. Its TBE values should approximate twice the Ba–P bonding energy and the difference with the TBE value in 8 should approximate four times the Ba⋯F bonding energy. From there, it ensues that the resulting Ba–P and Ba⋯F bond energies in 8 are ∼148 and ∼9 kcal mol−1, respectively. It thus appears that the Ba–P bond in 8 is substantially stronger than in 5 (104–9 = 95 kcal mol−1) and in 1·(thf)2 (106 kcal mol−1, see Table 2). It should be noted that replacing all F atoms in 8 by hydrogens results in an optimised geometry somehow different from that of 8, exhibiting a weak Ba-(η2-C6H5) interaction in addition to several Ba⋯H contacts. Finally, replacing the two thp ligands in 8 by thf molecules produces an optimised geometry with very similar metric data and electronic structure, indicating that the unsuccessful isolation of the 8(thf)2 relative is only the result of kinetic factors.
| [Ba{PPh2·B(C6F5)3}2·(thp)2] (8) | [Ba{PPh2·B[(C6H2F3)(C6F5)2]}2·(thp)2] (single point, see text) | |
|---|---|---|
| a Values in eV, unless specified. b Total bonding energy (TBE) = EPauli + Eelstat + Eorb + Edisp. | ||
| Fragmentation | [Ba·(thp)2]2+ + {PPh2·B[(C6F5)3]−}2 | [Ba·(thp)2]2+ + {PPh2·B[(C6H2F3)(C6F5)2]−}2 |
| E Pauli | 3.22 | 3.10 |
| E elstat | −11.74 | −10.48 |
| E orb | −4.64 | −4.24 |
| E disp | −1.21 | −1.19 |
| TBEb | −14.38 (332 kcal mol−1) | −12.82 (296 kcal mol−1) |
Conclusion
Our initial goal, the preparation of soluble and stable Ba–phosphides, in particular supported by the {Carb}− framework, has proved elusive. Compounds such as [{Carb}BaPPh2·(thf)] (1) and its thf adducts do not display sufficient stability in solution and can therefore not be used as catalysts for C–P bond forming reactions, such as the hydrophosphination of alkenes where other Ba precatalysts have, on the whole, exhibited very good performances.On the other hand, we have been able to prepare and, in some cases, crystallise, several barium and calcium phosphidoboranes using the “disguised” phosphines HPPh2·BH3 and HPP2·B(C6F5)3. All these complexes are very air-sensitive, and their stability in solution can be limited, which has in some cases hampered NMR analyses. Their structural features are unusual, in particular for complexes such as [{Carb}Ba{PPh2·B(C6F5)3}] (5), [{Carb}Ba{O(B(C6F5)3)CH2CH2CH2CH2PPh2}·thf] (6) and the remarkable [Ba{PPh2·B(C6F5)3}2·(thp)2] (8) that display strong intramolecular Ba⋯F secondary interactions. Although BH3 was involved in unwanted mobility eventually leading to the formation of [{Carb}Ca{(H3B)2PPh2}·(thf)] (4), we found that B(C6F5)3 produced stable Ba–phosphidoboranes where the transfer of the electron-depleted borane did not take place. Besides, in a way reminiscent of the behaviour exhibited by frustrated Lewis pairs, the latter compounds featured the ability to ring-open thf, and, in the presence of this solvent, to yield alkoxide derivatives such as 6 and [Ba{O(B(C6F5)3)CH2CH2CH2CH2PPh2}2·(thf)1.5] (7). No such reactivity was found when the more stable thp was used instead of thf, and we were hence able to isolate and characterise the bis(thp) adduct 8. We are now trying to exploit this reactivity and assess whether it is possible to turn it into a catalytic cycle for the production of P-functionalised alcohols.
DFT calculations performed to analyse the bonding and stability of the barium complexes do not reveal any particular weakness of the Ba–P bond in 1·(thf)2. The instability of this compound is better understood from the high energy of its HOMO, which contains the non-conjugated P lone pair, thus conferring it substantial nucleophilic reactivity. The structure of 5, assessed from DFT calculations, shows non-negligible Ba⋯F bonding. Calculations indicate that these ionic interactions, also present in 6 and 8, contribute significantly to the stabilisation of such architectures (Ba⋯F bond energy ∼9 kcal mol−1).
Author contributions
G. Duneş and P. M. Chapple performed the experimental and analytical work and participated in the planning of experiments, analysis of results and writing of the manuscript. S. Kahlal and J.-Y. Saillard performed the DFT calculations and participated in the preparation of the manuscript. T. Roisnel solved the crystallographic structures and prepared the XRD data. J.-F. Carpentier participated in the planning of experiments and analysis of the data. Yann Sarazin was the lead investigator, secured the funding, participated in the planning of experiments, data analysis and writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The French Agence Nationale de la Recherche is acknowledged for the provision of research grants to P. M. C. (ANR-17-CE07-0017-01) and to G. D. (ANR-21-CE07-0045-02).References
- For a general overview, see: Early Main Group Metal Catalysis, ed. S. Harder, Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, 2020 Search PubMed.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC and references therein.
- T. M. A. Al-Shboul, V. K. Pálfi, L. Yu, R. Kretschmer, K. Wimmer, R. Fischer, H. Görls, M. Reiher and M. Westerhausen, J. Organomet. Chem., 2011, 696, 216–227 CrossRef CAS.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2008, 27, 497–499 CrossRef CAS.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 2953–2956 CrossRef CAS.
- H. Hu and C. Cui, Organometallics, 2012, 31, 1208–1211 CrossRef CAS.
- M. He, M. T. Gamer and P. W. Roesky, Organometallics, 2016, 35, 2638–2644 CrossRef CAS.
- I. V. Lapshin, O. S. Yurova, I. V. Basalov, V. Yu Rad'kov, E. I. Musina, A. V. Cherkasov, G. K. Fukin, A. A. Karasik and A. A. Trifonov, Inorg. Chem., 2018, 57, 2942–2952 CrossRef CAS.
- I. V. Lapshin, I. V. Basalov, K. A. Lyssenko, A. V. Cherkasov and A. A. Trifonov, Chem. – Eur. J., 2019, 25, 459–463 CrossRef CAS PubMed.
- E. Le Coz, H. Roueindeji, T. Roisnel, V. Dorcet, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2019, 48, 9173–9180 RSC.
- P. M. Chapple, S. Kahlal, J. Cartron, T. Roisnel, V. Dorcet, M. Cordier, J.-Y. Saillard, J.-F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2020, 59, 9120–9126 CrossRef CAS PubMed.
- P. M. Chapple and Y. Sarazin, Eur. J. Inorg. Chem., 2020, 3321–3346 CrossRef CAS.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Inorg. Chem., 2007, 46, 10410–10415 CrossRef CAS PubMed.
- M. Gärtner, H. Görls and M. Westerhausen, Z. Anorg. Allg. Chem., 2007, 633, 2025–2031 CrossRef.
- M. Westerhausen, M. H. Digeser, M. Krofta, N. Wiberg, H. Nöth, J. Knizek, W. Ponikwar and T. Seifert, Eur. J. Inorg. Chem., 1999, 743–750 CrossRef CAS.
- M. Westerhausen and W. Schwarz, J. Organomet. Chem., 1993, 463, 51–63 CrossRef CAS.
- M. Westerhausen, M. Hartmann and W. Schwarz, Inorg. Chem., 1996, 35, 2421–2426 CrossRef CAS.
- L. J. Morris, M. S. Hill, M. F. Mahon, I. Manners and B. O. Patrick, Organometallics, 2020, 39, 4195–4207 CrossRef CAS.
- L. J. Morris, N. A. Rajabi, M. S. Hill, I. Manners, C. McMullin and M. F. Mahon, Dalton Trans., 2020, 49, 14584–14591 RSC.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and D. J. MacDougal, Chem. – Eur. J., 2010, 16, 8508–8515 CrossRef CAS.
- A. C. A. Ried, L. J. Taylor, A. M. Geer, H. E. L. Williams, W. Lewis, A. J. Blake and D. L. Kays, Chem. – Eur. J., 2019, 25, 6840–6846 CrossRef CAS.
- X. Zheng, J. Huang, Y. Yao and X. Xu, Chem. Commun., 2019, 55, 9152–9155 RSC.
- K. Izod, J. M. Watson, S. M. El-Hamruni, R. W. Harrington and P. G. Waddell, Organometallics, 2017, 36, 2218–2227 CrossRef CAS.
- K. Izod, J. M. Watson, R. W. Harrington and W. Clegg, Dalton Trans., 2021, 50, 1019–1024 RSC.
- G. C. Welch, J. D. Masuda and D. W. Stephan, Inorg. Chem., 2006, 45, 478–480 CrossRef CAS PubMed.
- J. Langer, I. Kosygin, R. Puchta, J. Pahl and S. Harder, Chem. – Eur. J., 2016, 22, 17425–17435 CrossRef CAS PubMed.
- H. Schumann, E. Palamidis and J. Loebel, J. Organomet. Chem., 1990, 384, C49–C52 CrossRef CAS.
- S. T. Liddle and P. L. Arnold, Dalton Trans., 2007, 3305–3313 RSC.
- F. Jaroschik, T. Shima, X. Li, K. Mori, L. Ricard, X.-F. Le Goff, F. Nief and Z. Hou, Organometallics, 2007, 26, 5654–5660 CrossRef CAS.
- T. L. Breen and D. W. Stephan, Inorg. Chem., 1992, 31, 4019–4022 CrossRef CAS.
- B. Birkmann, T. Voss, S. J. Geier, M. Ullrich, G. Kehr, G. Erker and D. W. Stephan, Organometallics, 2010, 29, 5310–5319 CrossRef CAS.
- A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 18463–18478 CrossRef CAS PubMed.
- S. L. Granville, G. C. Welch and D. W. Stephan, Inorg. Chem., 2012, 51, 4711–4721 CrossRef CAS PubMed.
- P. Federmann, C. Herwig, F. Beckmann, B. Cula and C. Limberg, Organometallics, 2021, 40, 4143–4149 CrossRef CAS.
- P. M. Chapple, M. Cordier, V. Dorcet, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2020, 49, 11878–11889 RSC.
- L. Wu, V. T. Annibale, H. Jiao, A. Brookfield, D. Collison and I. Manners, Nat. Commun., 2019, 10, 2786–2795 CrossRef PubMed.
- A. J. Roering, A. F. Maddox, L. T. Elrod, S. M. Chan, M. B. Ghebreab, K. L. Donovan, J. J. Davidson, R. P. Hughes, T. Shalumova, S. N. MacMillan, J. M. Tanski and R. Waterman, Organometallics, 2009, 28, 573–581 CrossRef CAS.
- R. J. Schwamm, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2016, 55, 14798–14801 CrossRef CAS PubMed.
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1558–1565 CrossRef CAS.
- F. M. Bickelhaupt and E. J. Baerends, Kohn-Sham Density Functional Theory: predicting and understanding chemistry, in Rev. Comput. Chem., ed. K. B. Lipkowitz and D. B. Boyd, Wiley, New York, 2000, vol. 15, pp. 1–86 Search PubMed.
- R. F. W. Bader, Atoms in molecules - A quantum theory, Oxford University Press, Oxford, England, 1990 Search PubMed.
- P. L. A. Popelier, in The chemical bond, ed. G. Frenking and S. Shaik, Wiley-VCH, 2014, vol. 1, pp. 271–308 Search PubMed.
- L. Zhao, S. Pan and G. Frenking, J. Chem. Phys., 2022, 157, 034105 CrossRef CAS PubMed.
- K. Izod, J. M. Watson, W. Clegg and R. W. Harrington, Dalton Trans., 2011, 40, 11712–11718 RSC.
- CSD Database 5.34, September 2023.
- S. J. Lancaster, A. J. Mountford, D. L. Hughes, M. Schormann and M. Bochmann, J. Organomet. Chem., 2003, 680, 193–205 CrossRef CAS.
- G. Kundu, S. Tothadi and S. S. Sen, Inorganics, 2022, 10, 97–109 CrossRef CAS.
- V. A. Stepanova, V. V. Dunina and I. P. Smoliakova, Organometallics, 2009, 28, 6546–6558 CrossRef CAS.
- M. Saldías, J. Manzur, R. E. Palacios, M. L. Gómez, J. De La Fuente, G. Günther, N. Pizarro and A. Vega, Dalton Trans., 2017, 46, 1567–1576 RSC.
- M. D. Anker, M. Arrowsmith, R. L. Arrowsmith, M. S. Hill and M. F. Mahon, Inorg. Chem., 2017, 56, 5976–5983 CrossRef CAS PubMed.
- H. Roueindeji, A. Ratsifitahina, T. Roisnel, V. Dorcet, S. Kahlal, J.-Y. Saillard, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2019, 25, 8854–8864 CrossRef CAS PubMed.
- M. Gärtner, H. Görls and M. Westerhausen, Dalton Trans., 2008, 1574–1582 RSC.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- C. A. Fischer, A. Rösch, H. Elsen, G. Ballmann, M. Wiesinger, J. Langer, C. Färber and S. Harder, Dalton Trans., 2019, 48, 6757–6766 RSC.
- W. D. Buchanan, M. A. Guino-o and K. Ruhlandt-Senge, Inorg. Chem., 2010, 49, 7144–7155 CrossRef CAS PubMed.
- S. R. Drake, W. E. Streib, K. Folting, M. H. Chisholm and K. G. Caulton, Inorg. Chem., 1992, 31, 3205–3210 CrossRef CAS.
- Y. Chi, S. Ranjan, T.-Y. Chou, C.-S. Liu, S.-M. Peng and G.-H. Lee, J. Chem. Soc., Dalton Trans., 2001, 2462–2466 RSC.
- E. Le Coz, V. Dorcet, T. Roisnel, S. Tobisch, J.-F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2018, 57, 11747–11751 CrossRef CAS PubMed.
- E. Le Coz, S. Kahlal, J.-Y. Saillard, T. Roisnel, V. Dorcet, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2019, 25, 13509–13513 CrossRef CAS PubMed.
- E. Le Coz, J. Hammoud, T. Roisnel, M. Cordier, V. Dorcet, S. Kahlal, J.-F. Carpentier, J.-Y. Saillard and Y. Sarazin, Chem. – Eur. J., 2021, 27, 11966–11982 CrossRef CAS PubMed.
- O. Poncelet, L. G. Hubert-Pfalzgraf, L. Toupet and J.-C. Daran, Polyhedron, 1991, 10, 2045–2050 CrossRef CAS.
- C. Müller, S. Krieck, H. Görls and M. Westerhausen, Eur. J. Inorg. Chem., 2016, 4637–4642 CrossRef.
- J. Langer and H. Görls, Dalton Trans., 2014, 43, 458–468 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic characterisation, and crystallographic and computational data. CCDC 2314377–2314380. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00487f |
| ‡ These two co-authors have contributed equally to the work and must both be considered as first co-authors. |
| § The barium and calcium complexes described herein were too air- and moisture sensitive to allow for elemental and mass spectrometry analyses. |
| This journal is © The Royal Society of Chemistry 2024 |