
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilica-based monoliths functionalized with DTPA for the removal of transition and lanthanide ions from aqueous solutions†
Gioele
Ancora
 a,
Stefano
Marchesi
a,
Mauro
Botta
a,
Leonardo
Marchese
a,
Fabio
Carniato
*a and
Chiara
Bisio
*ab
a,
Stefano
Marchesi
a,
Mauro
Botta
a,
Leonardo
Marchese
a,
Fabio
Carniato
*a and
Chiara
Bisio
*ab
aDipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale, Viale Teresa Michel 11, 15121 Alessandria, AL, Italy. E-mail: gioele.ancora@uniupo.it; stefano.marchesi@uniupo.it; fabio.carniato@uniupo.it; mauro.botta@uniupo.it; leonardo.marchese@uniupo.it; chiara.bisio@uniupo.it
bCNR-SCITEC Istituto di Scienze e Tecnologie Chimiche, “Giulio Natta”, Via C. Golgi 19, 20133 Milano, MI, Italy
First published on 8th April 2024
Abstract
Transition and rare earth metals serve as indispensable raw materials across a broad spectrum of technological applications. However, their utilization is frequently linked to substantial waste production. Consequently, the recycling and recovery of these metals from end-of-life products or metal-contaminated aqueous environments hold significant importance within the framework of a circular economy. In our investigation, we employed synthetic mesoporous silica monoliths, synthesized via the sol–gel method and functionalized with chelating groups, for the efficient recovery of metal ions from aqueous matrices. The monoliths were characterized using a multi-technique approach and were tested in the recovery of paramagnetic Gd3+, Cu2+ and Co2+ ions from aqueous solutions, using 1H-NMR relaxometry to evaluate their uptake performance in real time and under simple conditions. Detailed information on the kinetics of the capture process was also highlighted. Finally, the possibility to regenerate the solid sorbents was evaluated.
Introduction
Rare earth metals (REEs), transition metals and their complexes exhibit distinctive magnetic, optical, and catalytic properties, rendering them indispensable to the technology industry and, consequently, integral to everyday human life. Over the last three decades, REEs and their compounds have found widespread applications in various technological domains, including rechargeable batteries, superconductors, computer memory devices, magnets, mobile phones, catalysts, illumination systems (i.e. light-emitting diodes, LEDs), and for the preparation of magnetic resonance imaging (MRI) probes based on Gd3+-doped complexes.1–3Presently, the world reserves of REEs by major countries (China, Brazil, Vietnam, Russia and India) stand at about 120 Mt,1 and these raw materials are being used and consumed at an unprecedented rate.4 REEs are, however, not homogeneously distributed all over our planet, and are generally found at low concentrations in mineral ores.5 In particular, China owns about one-third of the world's REE reserves, and is the largest producer in the current market.1 Transition metals have similar importance to REEs in scientific technology and are therefore employed in a wide range of applications.6–8 Globally, Chile and Congo are the largest producers of copper and cobalt transition metals,1 while China is the world's leading consumer of cobalt and plays a prominent role by producing about 43% of worldwide refined copper.1
Among these metals, REEs have no clear biological role,4 but both their organic and environmental accumulation could represent a future problem. It is crucial to consider that contaminated wastewaters from electronic waste or healthcare facilities can introduce REEs and transition metals into the environment.9,10 This renders them emerging contaminants, and their overall environmental impact is not yet fully understood.4 As an example, anomalously high quantities of Gd3+-based MRI probes have been recently observed in San Francisco Bay11 and in the underground sewers of Berlin.12
Currently, most transition metals and REEs are obtained by extraction procedures from their mineral ores, while recycling techniques are still poorly explored.5 Nowadays, the most used recycling procedures are the liquid–liquid extraction (LLE) or solid–liquid extraction (SLE) techniques, which allow recovery of most of these metals in the form of ions from different solution-based media.
SLE, which involves the use of solid materials that employ different mechanisms for the sequestration of metal ions from aqueous matrices, represents a suitable alternative to LLE methods, i.e. ion exchange, adsorption and complexation. These methodologies, as opposed to those of LLE, present some advantages: (i) they necessitate fewer procedural steps and circumvent the need for different solvents or specific molecules, which pose challenges for disposal and treatment;13–18 and (ii) solid supports can be regenerated and used again across numerous capture cycles, thereby enhancing the sustainability of the process.
A variety of solid sorbents have been studied with respect to this aim, such as porous silica,19–21 clays,22–24 metal–organic frameworks (MOFs),25–27 nanoparticles,28,29 metal oxides30–32 and composite 2D materials.33,34
Among porous materials, mesoporous silica monoliths have been recently proposed for the removal of several contaminants in water and gaseous media (i.e. organic pollutants or volatile organic compounds, VOCs),35 due to their chemical stability and easy-to-functionalize structure. It is important to note that silica monoliths also have the advantage of being easy to handle and recover. Hence, their development represents a promising strategy to enhance the adsorption, recovery, and reusability for subsequent treatment cycles of water and soil containing heavy or precious metals.
Nevertheless, the silica surface of monoliths can be easily modified by introducing specific organic functionalities to mitigate the conventional issues related to adsorption-driven metal sequestration processes, such as poor selectivity and reversibility.36 To this aim, a possible strategy is to chemically modify the monoliths’ surface with chelating agents, which are able to coordinate lanthanide and transition metal ions.37 These organo-functional groups generally possess specific binding sites that generate a stronger interaction with metal ions or the contaminant under examination, increasing the selectivity of the overall uptake process.36,38 This strategy has also been adapted for other materials on a few occasions, e.g., chitosan–silica hybrid materials functionalized with chelating agents, such as DTPA (diethylenetriamine pentaacetic acid) and EDTA (ethylenediaminetetraacetic acid),39 and magnetic iron-nanoparticles coated with a silica shell and loaded with DTPA (dMNP–DTPA).40
Recently, Gossuin et al. studied the adsorption of paramagnetic Cu2+ ions from aqueous matrices on alumina substrates in real time using low-resolution 1H-NMR relaxometry as an alternative to traditional elemental techniques.41 This technique is based on the fact that the coupling between water 1H magnetic moments and paramagnetic ions in solution leads to changes in the longitudinal (R1) and transverse (R2) relaxation rates of the water protons,41 which are proportional to the concentration of free paramagnetic ions through the relaxivity parameter.41 Therefore, by measuring R1 or R2 relaxation rates of the 1H nuclei of water molecules in the presence of paramagnetic species,42,43 it is possible to make real time evaluations of the paramagnetic ion concentrations in water media,44–46 based on which relevant information on the capture performance of several solid sorbents can be gathered. Low-resolution NMR relaxometry can thus serve as an ideal analytical tool for monitoring the recovery processes of paramagnetic species via SLE processes. This is particularly advantageous considering that this procedure generally does not require expensive instruments, and measurements can be easily performed in real time using a small amount of sample without pretreatment procedures,47 as is required for conventional analytical techniques generally used for the determination of metal-ion concentrations in solution (e.g. ICP-OES).
Based on these assumptions, in this study, for the first time, cylindrical-shaped mesoporous silica monoliths functionalized with the diethylenetriamine pentaacetic acid (DTPA) ligand have been synthesized and tested for the recovery of paramagnetic Gd3+, Cu2+ and Co2+ ions from aqueous solutions, considering the high affinity DTPA has for these metals (logK of 19.3 and 21.5 for Co2+-DTPA and Cu2+-DTPA, respectively, at pH 7.4 and 295 K; log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K of 22.5 for Gd3+-DPTA, at pH 7.4 and 298 K).48 The capture process was primarily followed over time using the 1H-NMR relaxometry technique, obtaining important information about the uptake mechanism and related kinetic parameters. In addition, the regeneration of the silica-based materials under acid conditions was also evaluated.
K of 22.5 for Gd3+-DPTA, at pH 7.4 and 298 K).48 The capture process was primarily followed over time using the 1H-NMR relaxometry technique, obtaining important information about the uptake mechanism and related kinetic parameters. In addition, the regeneration of the silica-based materials under acid conditions was also evaluated.
Results and discussion
Synthesis and characterization of silica monoliths
Synthetic silica-based monoliths were prepared using the sol–gel method optimized in our laboratories,35,49 followed by calcination at high temperature to remove the structure templating agent of polyethylene oxide (PEO). The calcined solids (MONO) were then functionalized with 3-aminopropyltriethoxysilane (APTS) to introduce amino functionalities on their surface (MONO-APTS).Thus, the NH2 groups were exploited to anchor the DTPA molecules on the surface, through the formation of amide bonds with the carboxylic arms of DTPA (MONO-DTPA).
The structural features of the siliceous monoliths were studied by X-ray powder diffraction analysis. As shown in Fig. S1,† no reflections assigned to well-defined crystalline plane families are discernible. As expected, the presence of a broad signal between 2.5° and 6.5° 2θ denotes a non-ordered arrangement of the siliceous framework, which is typical of amorphous materials.50
The morphological features of the materials were studied by scanning electron microscopy (SEM). In Fig. 1, typical micrographs of calcined (frame A) and functionalized monoliths (frames B and C) are reported. The materials exhibit a general sponge-like morphology engendered by a continuous siliceous structure, in agreement with already reported literature evidence.49,51,52
 | ||
| Fig. 1 SEM micrographs of MONO (A and A′), MONO-APTS (B and B′) and MONO-DTPA (C and C′) collected at different magnifications (3000× and 20000×, respectively). On the right is a photograph of the monoliths along with the average dimensions of a single pellet. | ||
CHN elemental analyses were performed before and after the functionalization steps to collect more information about the chemical composition of the prepared materials (Table S1†). The data show that no organic compounds are present in the MONO sample, thereby confirming the complete removal of the PEO template by thermal treatment. The amount of C% and N% increased after each grafting step, further confirming the successful functionalization steps.
The amounts of APTS and DTPA functionalities, estimated by CHN analyses, were found to be 1.35 ± 0.02 and 0.31 ± 0.01 mmol g−1 in MONO-APTS and MONO-DTPA, respectively. The amount of DTPA chemically anchored on the silica surface suggests that not all –NH2 groups of APTS were effectively used during the functionalization with the ligand molecule, as clearly monitored by FTIR spectroscopy (Fig. 2).
 | ||
| Fig. 2 FT-IR spectra of MONO (a), MONO-APTS (b) and MONO-DTPA (c) collected in a vacuum (residual pressure < 10−3 mbar) at rt. | ||
The FT-IR spectrum of the MONO sample (Fig. 2, curve a) is characterized by the presence of an intense band at 3745 cm−1, which is associated with the O–H stretching (ν) modes of the isolated silanol (Si–OH) groups on the silica surface, and a broad band between 3600 and 3000 cm−1 generated by νO–H of the surface hydroxyl groups interacting with each other via hydrogen bonding.53 In the low frequency region, two bands at 1986 and 1866 cm−1 due to overtones and combination modes of the silica framework vibrations53 are present. In the spectrum of the MONO-APTS sample (Fig. 2, curve b) new bands, which are associated with the introduced amino functionalities, appear: at 3371 and 3316 cm−1 due to asymmetric and symmetric νN–H of amino groups;54 at 2939 and 2864 cm−1 related to asymmetric and symmetric νC–H bond vibrations of the alkyl chain of APTS;54,55 and at 1650 and 1592 cm−1 assigned to the bending (δ) modes of N–H oscillators of NH3+ and NH2 groups of the silane, respectively.54 In the spectrum of the MONO-DTPA sample, three new bands appear: at 1735 and 1400 cm−1, which are associated with the stretching modes of the COOH and COO− species of DTPA, respectively, and at 1630 cm−1 due to the amide groups introduced during the functionalization steps (Fig. 2, curve c).56 In both spectra of functionalized materials (Fig. 2, curves b and c), a strong decrease of the signal for silanols (both free and H-bonded) is also observed. This is a clear indication that the grafting procedure was successful. The textural features of the MONO samples, before and after the functionalization steps, were investigated by N2 physisorption analysis at 77 K (Fig. S2†). The samples showed type IV isotherms (IUPAC classification), typical of meso-porosity, with H2 hysteresis loops, commonly observed for mesostructured siliceous materials.35 The specific surface area (SSA) was found to be 430 and 254 m2 g−1 for the MONO and MONO-DTPA samples, respectively, as estimated by the multi-point Brunauer–Emmett–Teller (BET) model (Table S2†). The total pore volume decreased from 0.999 cm3 g−1 for the MONO sample to 0.739 cm3 g−1 for the MONO-DTPA sample, as well as the average pore diameter (see Table S2†). The decrease of these parameters is attributable to the presence of DTPA molecules inside the pores of the material, which partially occlude them, as a result of the functionalization steps.
Uptake tests of lanthanide and transition metal ions from aqueous solutions
Gadolinium (Gd3+), copper (Cu2+) and cobalt (Co2+) ions were selected from among lanthanides and transition metals for the evaluation of the uptake performance of DTPA-functionalized silica monoliths (MONO-DTPA) in metal-polluted aqueous solutions. It has already been shown in our recent study that low-resolution 1H-NMR relaxometry is an excellent substitute for conventional ICP-OES and ICP-MS techniques normally used for the determination of the concentration of metal ions in solution. Furthermore, the results obtained by ICP-OES and NMR relaxometry shows that the concentrations determined by the two techniques are strictly comparable.47 Thus, the low-resolution 1H-NMR relaxometry technique was employed to study the uptake processes in real time and under simple experimental conditions (10 MHz, 298 K, pH = 5), without any pre-treatment of the samples. The metal ions possess different ionic radii and coordination numbers (Gd3+ = 1.05 Å with CN = VIII/IX, Cu2+ = 0.73 Å with CN = VI and Co2+ = 0.65 Å with CN = VI) and exhibit paramagnetic properties with a magnetic moment of 7.93 μB, 1.73 μB and 3.88 μB, respectively.57 The uptake mechanism is formally based on the formation of a complex between the DTPA chelating molecule and the metal ions.57The uptake tests were performed at pH = 5 to avoid precipitation of the metal ions as hydroxide species (Fig. S3†).58–60 Acidic conditions are typically used in industrial applications for the extraction of heavy metals from polluted wastewater and waste devices.61
The experimental setup was conceived as follows: 20 mg of the MONO-DTPA solid material was introduced into 10 mm NMR tubes and 1 mL of a 10 mM aqueous solution of Gd3+, Cu2+ or Co2+ ions at pH = 5 was carefully added to each tube. Once the sorbent was put into contact with the metal solution, the tubes were inserted into the relaxometer probe, operating at a fixed magnetic field of 0.235 T and temperature of 298 K. Without performing any pre-treatment or further manipulation of the samples, the metal capture process was studied in static contact conditions by measuring the longitudinal relaxation rate (R1) of each metallic aqueous solution over time. The concentration of free metal ions in solution and, consequently, the concentration captured by functionalized silica monoliths were then calculated using eqn (2) reported in the Experimental section.62
Quantitative uptake data are shown in Fig. 3. An exponential decrease of the R1 values is observed in a time span of about 8 h in all cases, which corresponds to an increase in the amount of captured metal ions from aqueous solution by the MONO-DTPA sorbent (Table 1).
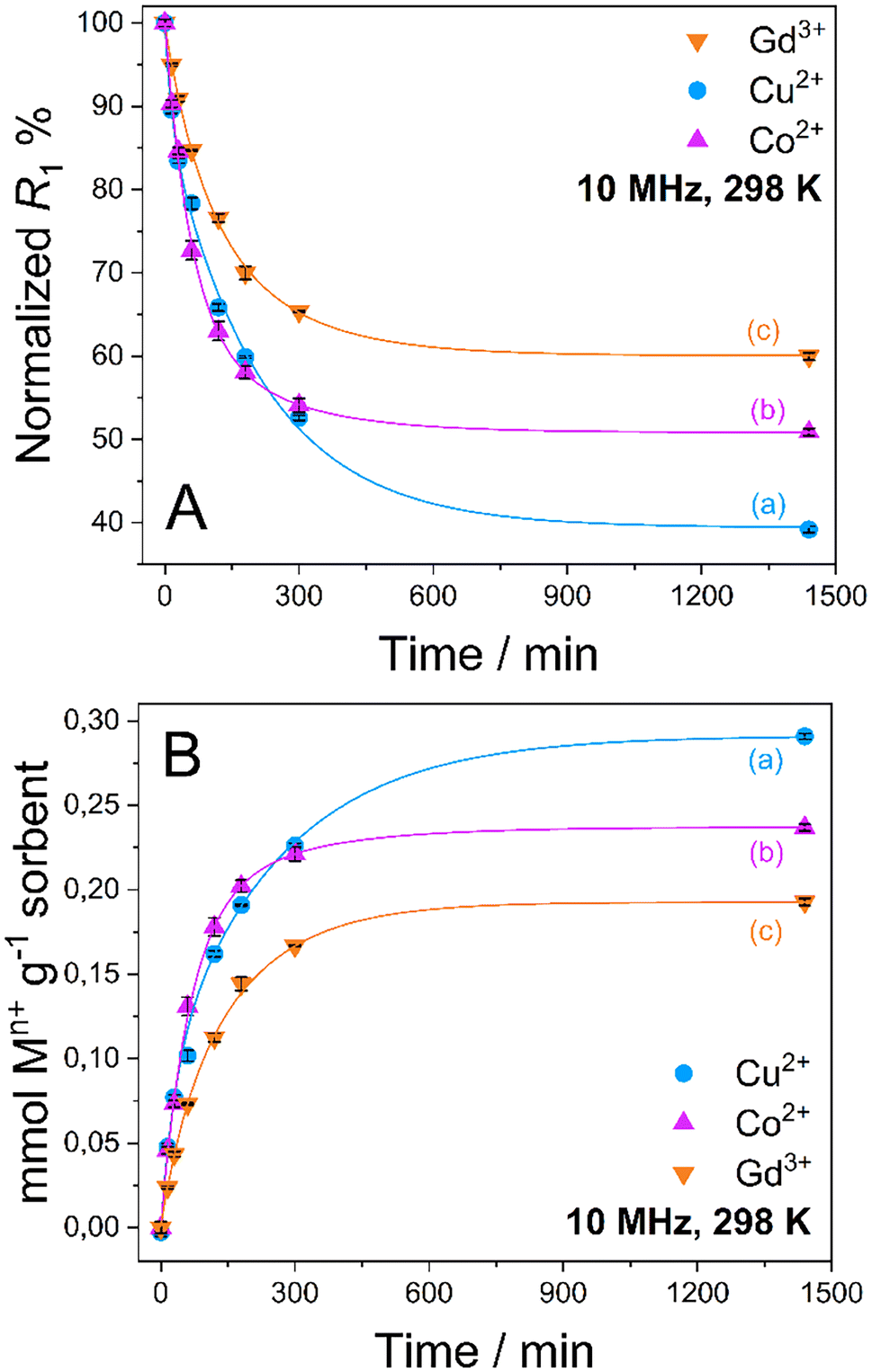 | ||
| Fig. 3 (A) R1 percentage decrease over time and (B) mmol amounts of Cu2+ (a), Co2+ (b) and Gd3+ (c) captured by 1 g of MONO-DTPA over time (the points represent the mean value and the error bars indicate the standard deviation; n = 3). | ||
| Sample | mmol Mn+/ g sorbent | Captured Mn+a % |
mg Mn+/g sorbent | [DTPA] (mmol g−1) |
|---|---|---|---|---|
| a In reference to the initial concentration value of each metal solution used (10 mM). | ||||
| MONO-DTPA + Cu2+ | 0.29 ± 0.01 | 62.7 ± 0.3 | 18.48 ± 0.11 | 0.31 ± 0.01 |
| MONO-DTPA + Co2+ | 0.24 ± 0.01 | 49.2 ± 0.4 | 13.96 ± 0.12 | 0.31 ± 0.01 |
| MONO-DTPA + Gd3+ | 0.19 ± 0.01 | 40.0 ± 0.4 | 30.31 ± 0.34 | 0.31 ± 0.01 |
As reported in Table 1, MONO-DTPA was able to remove from the aqueous solutions (and after 24 h of contact) 62.7%, 49.2% and 40.0% of Cu2+, Co2+ and Gd3+ ions, respectively. By comparing the millimoles of the captured metal ions per gram of sorbent with the concentration of available ligand on the surface of the MONO-DTPA material (also expressed in mmol g−1, as shown in Table 1), it can be inferred that there is a 1:1 interaction between the metal ions and the ligand molecules during the complexation process in all the studied cases. In addition, by comparing these two quantities it is possible to observe that not all DTPA ligand molecules “reacted” with the metal ions in solution.
Uptake tests were also carried out on as-synthetized monoliths (MONO) and APTS-functionalized monoliths (MONO-APTS) to assess the actual role and efficiency of the DTPA ligand in metal-ion capture processes. For this purpose, relaxometry measurements were performed with Gd3+, Cu2+ and Co2+ aqueous solutions. As reported in Fig. S4 and Table S3,† the as-synthetized monoliths (Fig. S4,† frames A and A′) are unable to capture metal ions, while the addition of the APTS ligand (MONO-APTS) led to an increase in its capture performance (Fig. S4,† frames B and B′), although not comparable to that of MONO-DTPA solids (Fig. 3). This suggests that the MONO-APTS samples can establish an interaction with the metal ions, but complexation phenomena with DTPA are fundamental to the increase in metal-ion recovery.
Assessments were made on the kinetics of the metal uptake processes in the presence of MONO-DTPA and MONO-APTS sorbents. The data obtained from 1H-NMR relaxometry were quantitatively analyzed by using different mathematical models (zero-order model, first-order model, Bhaskara equation, parabolic diffusion model, and a modified Freundlich model)63 to obtain a deeper insight into the metal sequestration mechanism. Among all the models used, the best fit of the experimental data was obtained by applying the parabolic diffusion model63 (Fig. 4 and Fig. S5†). This model, applied to our cases, describes a capture mechanism governed by diffusive phenomena47 associated with the disordered structure of the silica monolith, and eqn (3) (see Experimental section) mathematically defines the model. It is important to highlight that diffusive phenomena are limiting factors particularly in the early stages of the capture process. For this reason, the parabolic model correctly describes the experimental data collected in the first phase of the uptake process.
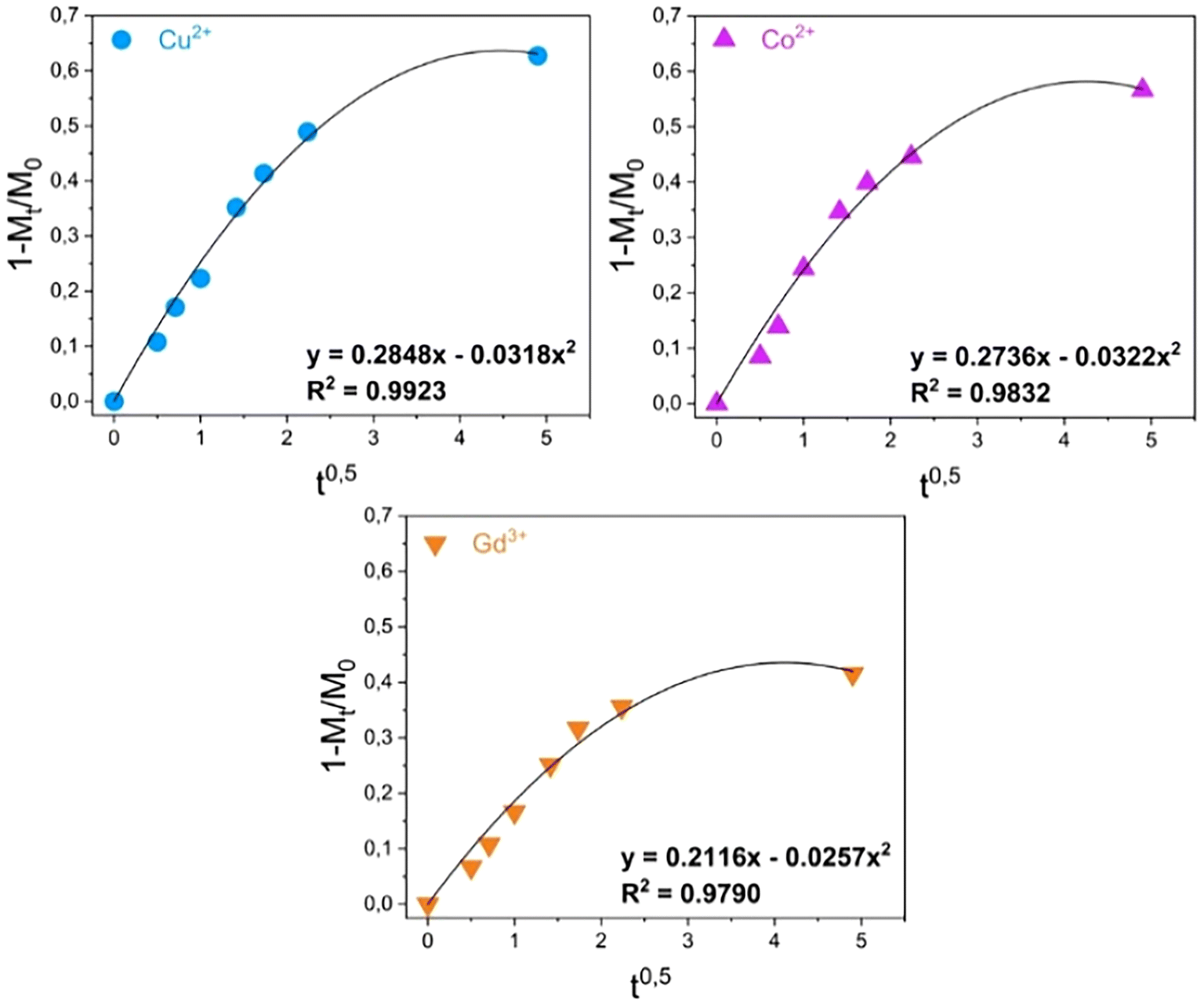 | ||
| Fig. 4 Kinetic analysis of the uptake data for the MONO-DTPA monolith.63 | ||
Kinetic constants were quantitatively calculated from the application of the model and are reported in Table 2 for MONO-DTPA and in Table S4† for the reference MONO-APTS. From the analyses of these values, the capture process appears to be faster for the Cu2+ ions (k = 0.2848 s−1), followed by Co2+ (0.2736 s−1) and Gd3+ (0.2116 s−1). The same behavior is observed for the MONO-APTS samples (Table S4†).
| Sample | Cu2+ | Co2+ | Gd3+ |
|---|---|---|---|
| MONO-DTPA | k = 0.2848 s−1 | k = 0.2736 s−1 | k = 0.2116 s−1 |
| R 2 = 0.9923 | R 2 = 0.9832 | R 2 = 0.97903 |
Extraction performances
An evaluation of the MONO-DTPA extraction efficiency in relation to the various treated metal ions was carried out using the mass-weighted distribution coefficient (Kd, mL g−1) (Fig. 5), calculated according to eqn (1):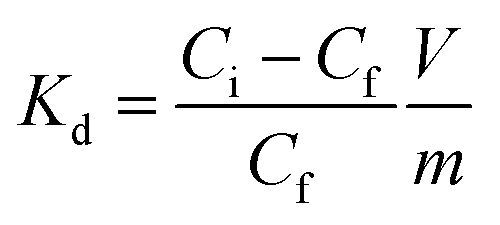 | (1) |
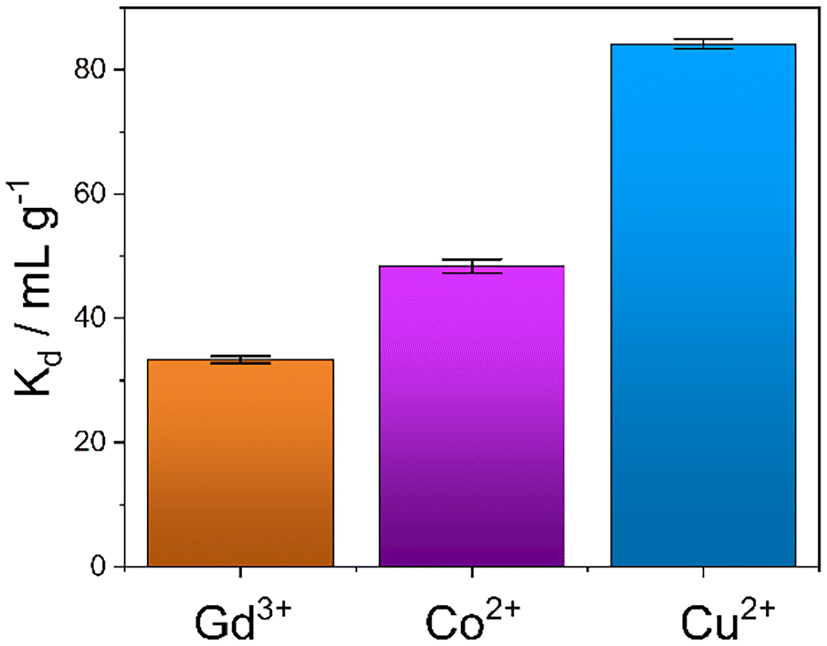 | ||
| Fig. 5 Distribution coefficient (Kd) values of the MONO-DTPA samples used in the capture of the Gd3+, Cu2+ and Co2+ ions from 10 mM aqueous solutions (the bars represent the mean value and the error bars indicate the standard deviation; n = 3). | ||
MONO-DTPA has higher affinity toward Cu2+ compared to the other two tested metals (Fig. 5), with a Kd value of 84.13 ± 1.12 mL g−1, thus supporting our hypothesis of a stronger interaction between the DTPA ligand (grafted on the monolith) and the Cu2+ ions. Considering the complexity of our system, in which a chelating agent is anchored onto the inner silica surface of the material, i.e. within its pores, our hypothesis is that the observed behavior could result from diffusional phenomena. In this context, the Cu2+ ion seems to diffuse better inside the channels of the solid with respect to the other metal ions. A similar trend was also observed in a previous work carried out by our research group in which Na-SAP, a clay having cation-exchange properties, was used for the uptake of the same three metal ions (Gd3+, Cu2+ and Co2+).47 Even in that case, although the uptake mechanism is different, the least sequestered metal ion was the Gd3+ ion, probably due to the size and charge of the ion in an environment where diffusion phenomena control the capture process.47 In addition, we observed a correlation between the kinetics and uptake data as having the same trend: the fastest capture process is in the case of Cu2+ ions, followed by Co2+ ions and then Gd3+ ions.
Regeneration tests
The possible regeneration of silica monoliths was investigated in order to use them in subsequent metal capture cycles. For this purpose, the MONO-DTPA samples used in the uptake tests were treated in acid solution to induce the release of metal ions from the metal complexes formed during the capture processes, since generally DTPA-based metal complexes are subjected to decomplexation at very acidic pH values.64 The regeneration procedure was monitored by 1H-NMR relaxometry, in a similar way to that described in the previous subsection. During material regeneration, the release of metal ions into the acid solution leads to an exponential increase in R1 values over time, which is associated with the gradual formation of metal aquaions.41 In detail, MONO-DPTA monoliths were placed in contact with 1 mL of 0.01 M HCl solution at pH = 2, and the amount of released metal ions was determined after 24 h (Fig. 6). Under these conditions, the material is stable; indeed, CHN elemental analysis of a sample of MONO-DTPA placed at pH = 2 for 24 h showed no percentage decrease in the organic fraction of the material.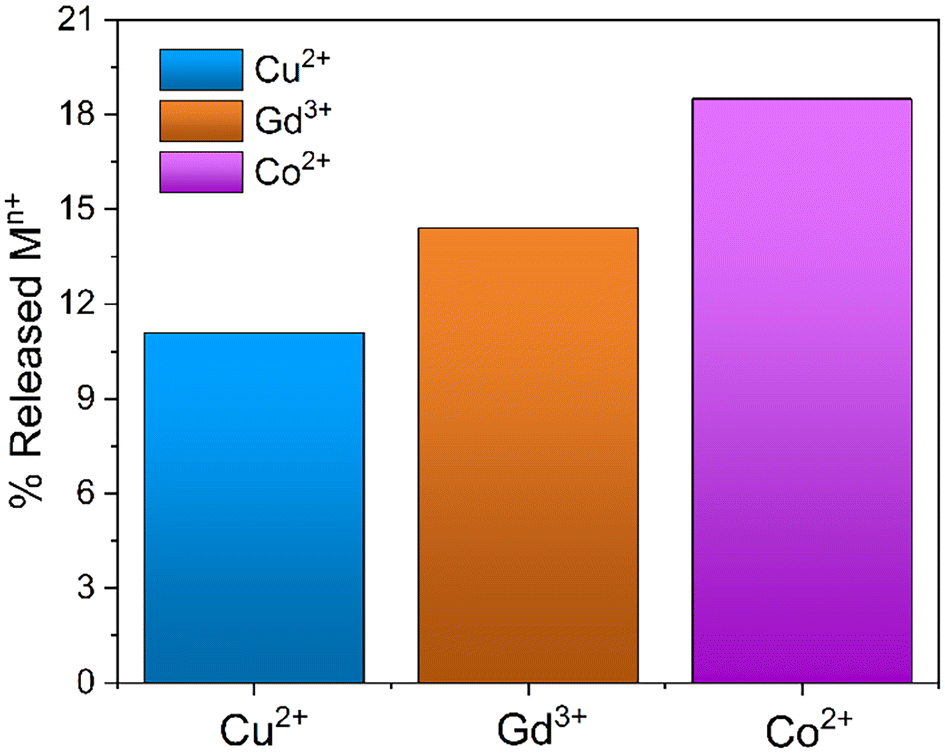 | ||
| Fig. 6 Percentage of metal ions released from the MONO-DTPA samples following the regeneration procedure at pH = 2 after 24 h. | ||
The MONO-DTPA samples released an amount of 11.1%, 14.4% and 18.5% of Cu2+, Gd3+ and Co2+ ions, respectively, in aqueous solution. The percentages were calculated starting from the previously captured quantities of each metal ion (Table 3). Based on the measurements of metal-ion uptake and kinetics, it appears that the Cu2+ ions exhibit a greater affinity for the DTPA ligand when compared to the other two metal ions. The strong affinity observed is likely to impact the regeneration. As expected for the DTPA and EDTA-based sorbents,39 MONO-DTPA regeneration is generally mild due to the high stability of the complexes formed by the chelating agent used (the DTPA molecule) and the metal ions tested.
| Sample | % Released [Mn+] pH = 2 | % Released [Mn+] pH = 1 |
|---|---|---|
| MONO-DTPA + Cu2+ | 11.1 ± 0.1 | 28.5 ± 0.2 |
| MONO-DTPA + Gd3+ | 14.4 ± 0.4 | 63.8 ± 0.3 |
| MONO-DTPA + Co2+ | 18.5 ± 0.2 | 61.9 ± 0.1 |
More encouraging results have been obtained by treating MONO-DTPA at pH = 1 (Table 3). Unfortunately, these strong acidic conditions led to the removal of the chelating agent linked to the monolith surface. However, the siliceous monolith remained intact after the treatment at lower pH and can be eventually used for a novel functionalization with the chelating agents.
Comparison with other literature materials
A comprehensive comparison was conducted between the maximum uptake capacities for Cu2+ ions of MONO-DTPA synthesized in this study and other natural and synthetic materials frequently employed for the removal of metal ions, given their characteristics as cation exchangers (Fig. S7 and Table S5†).47,65–67 The capture performances of MONO-DTPA are better than those of clay materials, and it is important to consider that the latter occur as powders. On the other hand, the functionalized silica monoliths developed in this work are massive solids, and this feature makes them easier to use: it is only required to set the material in contact with the metal-ion solution and remove it after 24 h. Therefore, fewer operational steps are required in polluted water cleanup treatment processes. In addition, since this material occurs as a massive solid, real time monitoring of metal-ion sequestration is possible through relaxometric techniques using inexpensive instruments and through nondestructive analyses, which would not be possible using powder materials that are suspended in solution, making relaxometric measurement difficult to be carried out.Conclusions
In this study, DTPA-functionalized silica monoliths, prepared by the sol–gel technique followed by two functionalization steps, were used as solid sorbents for the recovery of different paramagnetic metal ions (Gd3+, Cu2+, and Co2+) from aqueous solutions. The metal sequestration process was monitored in real time by 1H-NMR relaxometry under simple experimental conditions (10 MHz, room temperature and pH = 5), without any pretreatment of the samples. The modified silica monoliths were able to recover an appreciable amount of both di- and trivalent metal ions. The best results were obtained in the case of Cu2+ after 24 h of contact, with a recovered amount of 0.29 mmol g−1 corresponding to 18.48 mg g−1.The regeneration of silica sorbents was then evaluated in an HCl solution at pH = 2 to facilitate the release of the selected captured metal ion and thus the regeneration of the complex itself. The gentle regeneration of the material is attributed to the robust stability of the DTPA complexes. Consequently, the effective capture of metal ions in solution is achievable; however, regenerating the metal-binding complex necessitates highly acidic conditions. Experiments conducted at pH = 1 resulted in the complete removal of the DTPA functionalizing molecule from the material surface. Nevertheless, the material itself remained intact even under severe acidic conditions, allowing for its reuse in subsequent functionalization processes. The development of this study will necessarily encompass the efficient capture and release of metal ions under controlled conditions. This approach requires an accurate design of the ligands to be able to: (i) generate metal chelates that are less thermodynamically stable but more selective and (ii) release the ion under mild and controlled pH conditions.
Silica monoliths have shown higher capture performance than that of clay materials commonly used for the same purpose. Furthermore, they possess the advantage of occurring as a massive solid rather than as a powder, making it possible to employ the 1H-NMR relaxometry technique. These features make MONO-DTPA an excellent material for metal-ion sequestration from aqueous media, which is easier to use and handle compared to conventional powder systems. Future perspectives encompass several crucial aspects: (i) refining the steps involved in material functionalization to enhance the quantity of introduced ligands; (ii) exploring ligands capable of efficiently coordinating metal ions while permitting decomplexation under non-destructive acidic conditions for the inorganic support; and (iii) assessing the metal-uptake performance across multiple consecutive cycles.
Experimental section
Materials
After 3 days, the formed silica monoliths were removed from the tubes and placed in a water bath at room temperature (rt), which was regenerated every 30 min until neutral pH of the monoliths was reached. The silica monoliths were then added to a 0.1 M ammonia solution (NH3, 28–30%; Merck KGaA) inside a Teflon® bottle and placed in an oven at 45 °C for 24 h to catalyze the Ostwald ripening mechanism of the weakly condensed silica units.35 After 24 h, the monoliths were washed again until neutral pH with ultrapure water and slowly dried at rt for 3 days. After 3 days, they were cut into pellets of 1 cm in length and calcined in an air flow at 650 °C for 6 h at 3 °C min−1 to remove the organic template (PEO). The obtained silica monoliths are hereafter named MONO (Fig. S6†).
The mixture was then placed under slow stirring at 50 °C for 24 h. After 24 h, the monoliths were filtered on a Büchner filter and then washed with toluene and diethyl ether to remove the unreacted APTS. Finally, the monoliths were dried at rt in air for 24 h. The obtained functionalized silica monoliths were named MONO-APTS.
Uptake tests of Gd3+, Cu2+ and Co2+ ions from aqueous metal solutions
In detail, 20 mg of MONO-DTPA was placed in 10 mm NMR tubes with 1 mL of 10 mM aqueous solutions of Gd3+, Cu2+ or Co2+ at pH = 5. Each sample tube was placed in the probe of a fast-field cycling (FFC) Stelar SmarTracer relaxometer. Measurement of R1 values over time of the aqueous metal solution above the solid sorbent was then performed for each tube at fixed magnetic field of 10 MHz, temperature of 298 K and under static conditions.In the presence of paramagnetic ions, an increase in the R1 (and R2) values of water protons (1H) can be observed due to the coupling of their magnetic moments with paramagnetic ions: this change is proportional to the concentration of free paramagnetic ions in aqueous solution through the parameter named relaxivity, as shown in eqn (2):62
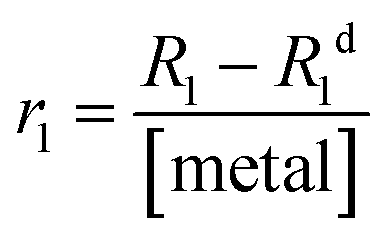 | (2) |
Mathematical evaluation of the uptake kinetics
Assessments regarding the kinetics of uptake processes were made. The experimental NMR data were analyzed quantitatively by using different mathematical models to understand the kinetics of the metal sequestration process for all three metals studied in more detail: (1) zero-order model, (2) first-order model, (3) Bhaskar equation, (4) parabolic diffusion model and (5) a modified Freundlich model. A complete description and physical meaning of each model are detailed in the work of Zhang et al.63The best fitting of relaxometric uptake data in all the cases explored experimentally was obtained using the parabolic diffusion model, which describes a diffusion-controlled event/metal uptake process, and is mathematically defined by eqn (3):63
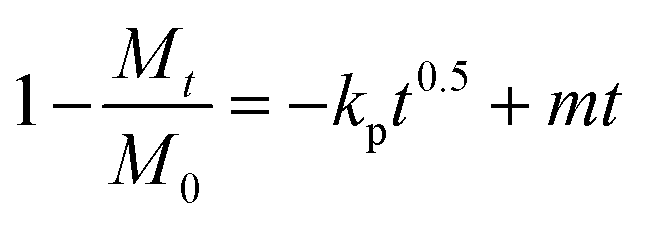 | (3) |
Regeneration of silica monoliths after uptake tests
The MONO-DTPA solids used for the metal uptake tests (after 24 h) were first dried in air for 3 days and then dispersed in 1 mL of a HCl solution at pH = 2 to dissociate the formed metal complexes (with DTPA) from the silica monoliths. The material regeneration process was followed by 1H-NMR relaxometry at 10 MHz and 298 K, using eqn (2) to calculate the metal-ion concentration released in solution following the decomplexation of the DTPA–metal complexes on the silica monoliths. The R1 values were collected after 24 h.Characterization methods
− CHN elemental analyses were performed using an EA3000 CHN elemental analyzer (EuroVector, Milano, Italy). Acetanilide, purchased from EuroVector (Milan, Italy), was used as the calibration standard (C % = 71.089, H % = 6.711, N % = 10.363).− X-ray powder diffractograms (XRPD) were collected on unoriented ground powders on a Bruker D8 Advance powder diffractometer (Karlsruhe, Germany), operating in Bragg–Brentano geometry, with a Cu anode target equipped with a Ni filter (used as an X-ray source) and with a Linxeye XE-T high-resolution position-sensitive detector. Trio and twin/twin optics are mounted on the DaVinci design modular XRD system. The X-ray tube of the instrument operates with Cu-Kα1 monochromatic radiation (λ = 1.54062 Å), with current intensity and operative electric potential difference set to 40 mA and 40 kV, respectively, and with automatic variable primary divergent slits and primary Soller slits of 2.5°. The X-ray profiles were recorded at room temperature in the 0.7°–10° 2θ range with a coupled 2θ − θ method, continuous PSD fast scan mode, time per step (rate or scan speed) of 0.100 s per step, and 2θ step size (or increment) of 0.02°, with automatic synchronization of the air scatter (or anti-scatter) knife and slits and with a fixed illumination sample set at 15 mm.
− The water proton longitudinal relaxation rates (R1) were measured by using a fast-field cycling (FFC) Stelar SmarTracer relaxometer (Mede, Italy) operating at fixed magnetic field strength of 10 MHz proton Larmor frequency range (0.235 T) and at 298 K. The measurements were carried out using standard non-polarized NP/S sequences with a typical 90° pulse width of 3.5 μs and reproducibility of data within ±0.5%. The temperature was controlled with a Stelar VTC-91 heater airflow equipped with a copper-constantan thermocouple (uncertainty of ±0.1 °C).
− Infrared spectra were collected using a Thermo Electron Corporation FT Nicolet 5700 spectrometer (Waltham, MA, USA) operating in the 4000–400 cm−1 range with a 4 cm−1 resolution. The measurements were made using self-supporting pellets of the MONO and MONO-DTPA samples obtained by grinding a piece of silica monolith and compressing the obtained powder with a mechanical press at 6 tons cm−2. The pellets were then introduced into a suitable IR cell equipped with KBr windows, which was permanently attached to a vacuum line (residual pressure ≤ 1 × 10−3 mbar) to perform the analyses. All IR spectra were collected at rt, and the samples were pre-treated at rt for 1 h in a vacuum to remove any traces of physisorbed water. The absorbance values in the spectra shown in Fig. 2 are normalized by the density of each sample's pellet to ensure accurate comparison among them.
− Nitrogen N2 physisorption measurements were conducted at the N2 cryogenic temperature (77 K) under relative pressure from 1 × 10−6 to 1.0 P/P0 by using a Quantachrome Autosorb 1MP/TCD instrument (Florida, USA). Prior to the analysis, the samples were outgassed under the following conditions: 343 K for 90 min, 373 K for 8 h (residual pressure lower than 10−6 Torr). Specific surface areas (SSAs) were determined using the Brunauer–Emmett–Teller (BET) equation within the relative pressure range of 0.01 to 0.1 P/P0. Pore-size distributions were obtained by applying the non-localized density functional theory (NLDFT) method (N2 silica kernel based on a cylindrical pore model applied to the desorption branch).
− Scanning electron micrographs were recorded with a Quanta 200 FEI (Hillsboro, OR, USA) scanning electron microscope (SEM) operating at 30 kV with an EDAX (Mahwah, NJ, USA) 60 mm2 Octane Super EDS detector attachment. Before SEM analysis, the samples were sputtered with a 20 nm layer of platinum to increase the conductivity of their surface.
Author contributions
Conceptualization: G. A., S. M., F. C. and C. B.; methodology: G. A., S. M., F. C. and C. B.; formal analysis: G. A. and S. M.; investigation: G. A. and S. M.; data curation: G. A. and S. M.; writing – original draft preparation: G. A. and S. M.; writing – review and editing: G. A., S. M., F. C., M. B., L. M., and C. B. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are fully grateful to Dr Elena Perin (DiSIT, Università del Piemonte Orientale, Alessandria, Italy) for the CHN analyses and to Dr Federico Begni for the N2 physisorption analyses. The authors thank the NODES project (Nord Ovest Digitale e Sostenibile; Spoke 2 “Green Technologies and Sustainable Industries”) for its financial support.References
- U. S. G. Survey, Mineral commodity summaries 2022, U.S. Geological Survey, 2022 Search PubMed.
- C. S. K. Raju, A. Cossmer, H. Scharf, U. Panne and D. Lück, J. Anal. At. Spectrom., 2009, 25, 55–61 RSC.
- F. Carniato, L. Tei and M. Botta, Eur. J. Inorg. Chem., 2018, 2018, 4936–4954 CrossRef CAS.
- V. Balaram, Geosci. Front., 2019, 10, 1285–1303 CrossRef CAS.
- A. Tsamis , M. C. (Centre for Strategy and E. S. Llp).
- M. Halka and B. Nordstrom, Transition Metals, Infobase Holdings, Inc, 2nd edn, 2019 Search PubMed.
- E. P. Beaumier, A. J. Pearce, X. Y. See and I. A. Tonks, Nat. Rev. Chem., 2019, 3, 15–34 CrossRef PubMed.
- E. Boros and A. B. Packard, Chem. Rev., 2019, 119, 870–901 CrossRef CAS PubMed.
- H. Herrmann, J. Nolde, S. Berger and S. Heise, Ecotoxicol. Environ. Saf., 2016, 124, 213–238 CrossRef CAS PubMed.
- G. Klaver, M. Verheul, I. Bakker, E. Petelet-Giraud and P. Négrel, Appl. Geochem., 2014, 47, 186–197 CrossRef CAS.
- V. Hatje, K. W. Bruland and A. R. Flegal, Environ. Sci. Technol., 2016, 50, 4159–4168 CrossRef CAS PubMed.
- A. Knappe, P. Möller, P. Dulski and A. Pekdeger, Geochemistry, 2005, 65, 167–189 CrossRef CAS.
- A. Roca-Sabio, M. Mato-Iglesias, D. Esteban-Gómez, É. Tóth, A. de Blas, C. Platas-Iglesias and T. Rodríguez-Blas, J. Am. Chem. Soc., 2009, 131, 3331–3341 CrossRef CAS PubMed.
- L. Tei, Z. Baranyai, E. Brücher, C. Cassino, F. Demicheli, N. Masciocchi, G. B. Giovenzana and M. Botta, Inorg. Chem., 2010, 49, 616–625 CrossRef CAS PubMed.
- J. Florek, A. Mushtaq, D. Larivière, G. Cantin, F.-G. Fontaine and F. Kleitz, RSC Adv., 2015, 5, 103782–103789 RSC.
- A. Leoncini, P. K. Mohapatra, A. Bhattacharyya, D. R. Raut, A. Sengupta, P. K. Verma, N. Tiwari, D. Bhattacharyya, S. Jha, A. M. Wouda, J. Huskens and W. Verboom, Dalton Trans., 2016, 45, 2476–2484 RSC.
- P. Kishor, A. Sengupta, N. K. Gupta and S. Biswas, Sep. Sci. Technol., 2018, 53, 286–294 CrossRef CAS.
- Y. Hu, E. Drouin, D. Larivière, F. Kleitz and F.-G. Fontaine, ACS Appl. Mater. Interfaces, 2017, 9, 38584–38593 CrossRef CAS PubMed.
- E. Bou-Maroun, G. J. Goetz-Grandmont and A. Boos, Sep. Sci. Technol., 2007, 42, 1913–1930 CrossRef CAS.
- P. D. Hopkins, T. Mastren, J. Florek, R. Copping, M. Brugh, K. D. John, M. F. Nortier, E. R. Birnbaum, F. Kleitz and M. E. Fassbender, Dalton Trans., 2018, 47, 5189–5195 RSC.
- D. L. Ramasamy, V. Puhakka, S. Iftekhar, A. Wojtuś, E. Repo, S. B. Hammouda, E. Iakovleva and M. Sillanpää, J. Hazard. Mater., 2018, 348, 84–91 CrossRef CAS PubMed.
- S. Marchesi, F. Carniato, M. Guidotti, M. Botta, L. Marchese and C. Bisio, New J. Chem., 2020, 44, 10033–10041 RSC.
- M.-S. Song, K. Vijayarangamuthu, E. Han and K.-J. Jeon, J. Nanosci. Nanotechnol., 2016, 16, 4469–4473 CrossRef CAS PubMed.
- V. B. Yadav, R. Gadi and S. Kalra, J. Environ. Manage., 2019, 232, 803–817 CrossRef CAS PubMed.
- X. Zhao, M. Wong, C. Mao, T. X. Trieu, J. Zhang, P. Feng and X. Bu, J. Am. Chem. Soc., 2014, 136, 12572–12575 CrossRef CAS PubMed.
- P. A. Kobielska, A. J. Howarth, O. K. Farha and S. Nayak, Coord. Chem. Rev., 2018, 358, 92–107 CrossRef CAS.
- M. Feng, P. Zhang, H.-C. Zhou and V. K. Sharma, Chemosphere, 2018, 209, 783–800 CrossRef CAS PubMed.
- C. Basualto, J. Gaete, L. Molina, F. Valenzuela, C. Yañez and J. F. Marco, Sci. Technol. Adv. Mater., 2015, 16, 035010 CrossRef PubMed.
- M. A. Tahoon, S. M. Siddeeg, N. S. Alsaiari, W. Mnif and F. B. Rebah, Processes, 2020, 8, 645 CrossRef CAS.
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 211–212, 317–331 CrossRef CAS PubMed.
- K. Gupta, P. Joshi, R. Gusain and O. P. Khatri, Coord. Chem. Rev., 2021, 445, 214100 CrossRef CAS.
- R. Kumar and J. Chawla, Water Qual., Exposure Health, 2014, 5, 215–226 CrossRef CAS.
- V. D. Gohel, A. Rajput, S. Gahlot and V. Kulshrestha, Macromol. Symp., 2017, 376, 1700050 CrossRef.
- C.-Z. Zhang, Y. Yuan and T. Li, Environ. Eng. Sci., 2018, 35, 978–987 CrossRef CAS.
- V. Miglio, C. Zaccone, C. Vittoni, I. Braschi, E. Buscaroli, G. Golemme, L. Marchese and C. Bisio, Molecules, 2021, 26, 1316 CrossRef CAS PubMed.
- I. Sierra and D. Pérez-Quintanilla, Chem. Soc. Rev., 2013, 42, 3792–3807 RSC.
- J. R. Kumar, J.-S. Kim, J.-Y. Lee and H.-S. Yoon, Sep. Purif. Rev., 2011, 40, 77–125 CrossRef CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- J. Roosen, J. Spooren and K. Binnemans, J. Mater. Chem. A, 2014, 2, 19415–19426 RSC.
- H. Zhang, R. G. McDowell, L. R. Martin and Y. Qiang, ACS Appl. Mater. Interfaces, 2016, 8, 9523–9531 CrossRef CAS PubMed.
- Y. Gossuin and Q. L. Vuong, Sep. Purif. Technol., 2018, 202, 138–143 CrossRef CAS.
- I. Foley, S. A. Farooqui and R. L. Kleinberg, J. Magn. Reson., Ser. A, 1996, 123, 95–104 CrossRef CAS PubMed.
- S. H. Koenig and R. D. Brown III, Magn. Reson. Med., 1984, 1, 478–495 CrossRef CAS PubMed.
- R. Kimmich, Field-cycling NMR Relaxometry: Instrumentation, Model Theories and Applications, Royal Society of Chemistry, 2018 Search PubMed.
- R. M. Steele, J.-P. Korb, G. Ferrante and S. Bubici, Magn. Reson. Chem., 2016, 54, 502–509 CrossRef CAS PubMed.
- S. M. Nagel, C. Strangfeld and S. Kruschwitz, J. Magn. Reson. Open, 2021, 6–7, 100012 CrossRef.
- S. Marchesi, S. Nascimbene, M. Guidotti, C. Bisio and F. Carniato, Dalton Trans., 2022, 51, 4502–4509 RSC.
- G. Anderegg, F. Arnaud-Neu, R. Delgado, J. Felcman and K. Popov, Pure Appl. Chem., 2005, 77, 1445–1495 CrossRef CAS.
- A. Galarneau, A. Sachse, B. Said, C.-H. Pelisson, P. Boscaro, N. Brun, L. Courtheoux, N. Olivi-Tran, B. Coasne and F. Fajula, C. R. Chim., 2016, 19, 231–247 CrossRef CAS.
- A. Chauhan and P. Chauhan, J. Anal. Bioanal. Tech., 2014, 5, 1–5 Search PubMed.
- B. Fotoohi, M. Kazemzad and L. Mercier, Ceram. Int., 2018, 44, 20199–20210 CrossRef CAS.
- T. Amatani, K. Nakanishi, K. Hirao and T. Kodaira, Chem. Mater., 2005, 17, 2114–2119 CrossRef CAS.
- G. Gatti, C. Vittoni, D. Costenaro, G. Paul, E. Mangano, S. Brandani, L. Marchese and C. Bisio, Phys. Chem. Chem. Phys., 2017, 19, 29449–29460 RSC.
- L. Etgar, G. Schuchardt, D. Costenaro, F. Carniato, C. Bisio, S. M. Zakeeruddin, M. K. Nazeeruddin, L. Marchese and M. Graetzel, J. Mater. Chem. A, 2013, 1, 10142–10147 RSC.
- G. Gatti, D. Costenaro, C. Vittoni, G. Paul, V. Crocellà, E. Mangano, S. Brandani, S. Bordiga, M. Cossi, L. Marchese and C. Bisio, Phys. Chem. Chem. Phys., 2017, 19, 14114–14128 RSC.
- M. Laprise-Pelletier, M. Bouchoucha, J. Lagueux, P. Chevallier, R. Lecomte, Y. Gossuin, F. Kleitz and M.-A. Fortin, J. Mater. Chem. B, 2015, 3, 748–758 RSC.
- P. Atkins, Shriver and Atkins’ Inorganic Chemistry, OUP, Oxford, 2010 Search PubMed.
- P. Djurdjevic, R. Jelic, L. Joksović, I. Lazarevic and M. Jelikić-Stankov, Acta Chim. Slov., 2010, 57, 386–397 CAS.
- B. Yang, X. Tong, Z. Deng and X. Lv, J. Chem., 2016, 2016, e4627929 Search PubMed.
- K. A. Krishnan and T. S. Anirudhan, Chem. Eng. J., 2008, 137, 257–264 CrossRef CAS.
- R. W. Gaikwad, Appl. Ecol. Environ. Res., 2008, 6, 81–98 CrossRef.
- S. Aime, M. Botta, M. Fasano and E. Terreno, Chem. Soc. Rev., 1998, 27, 19–29 RSC.
- H. Zhang, D. Pan and X. Duan, J. Phys. Chem. C, 2009, 113, 12140–12148 CrossRef CAS.
- E. Repo, T. A. Kurniawan, J. K. Warchol and M. E. T. Sillanpää, J. Hazard. Mater., 2009, 171, 1071–1080 CrossRef CAS PubMed.
- Ö. Yavuz, Y. Altunkaynak and F. Güzel, Water Res., 2003, 37, 948–952 CrossRef PubMed.
- M. Šljivić, I. Smičiklas, S. Pejanović and I. Plećaš, Appl. Clay Sci., 2009, 43, 33–40 CrossRef.
- C. O. Ijagbemi, M.-H. Baek and D.-S. Kim, J. Hazard. Mater., 2009, 166, 538–546 CrossRef CAS PubMed.
- C. Luchinat, Magn. Reson. Chem., 1993, 31, S145–S153 CrossRef CAS.
- K. Micskei, D. H. Powell, L. Helm, E. Brücher and A. E. Merbach, Magn. Reson. Chem., 1993, 31, 1011–1020 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt00388h |
| This journal is © The Royal Society of Chemistry 2024 |