
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An immunogenic anti-cancer stem cell bi-nuclear copper(II)-flufenamic acid complex†
Yue
Li‡
,
Jiaxin
Fang‡
,
Kuldip
Singh
,
Fabrizio
Ortu
 * and
Kogularamanan
Suntharalingam
*
* and
Kogularamanan
Suntharalingam
*
School of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: k.suntharalingam@leicester.ac.uk; fabrizio.ortu@leicester.ac.uk
First published on 7th March 2024
Abstract
An asymmetric bi-nuclear copper(II) complex with both cytotoxic and immunogenic activity towards breast cancer stem cells (CSCs) is reported. The bi-nuclear copper(II) complex comprises of two copper(II) centres bound to flufenamic acid and 3,4,7,8-tetramethyl-1,10-phenanthroline. The bi-nuclear copper(II) complex exhibits sub-micromolar potency towards breast CSCs grown in monolayers and three-dimensional cultures. Remarkably, the bi-nuclear copper(II) complex is up to 25-fold more potent toward breast CSC mammospheres than salinomycin (a gold standard anti-breast CSC agent) and cisplatin (a clinically administered metallodrug). Mechanistic studies showed that the bi-nuclear copper(II) complex readily enters breast CSCs, elevates intracellular reactive oxygen species levels, induces apoptosis, and promotes damage-associated molecular pattern release. The latter triggers phagocytosis of breast CSCs by macrophages. As far as we are aware, this is the first report of a bi-nuclear copper(II) complex to induce engulfment of breast CSCs by immune cells.
Introduction
Metastasis and relapse, the major causes of cancer-related deaths, are heavily linked to the existence of a sub-set of tumour cells called cancer stem cells (CSCs).1 CSCs display both cancer and stem cell-like properties, the latter enables them to differentiate, self-renew, and reform tumours.2 CSCs are able to survive conventional oncology treatments.3 Their slow cell cycle profile allows them to overcome therapies that target fast growing cells such as chemotherapy and radiation.4 The fact that CSCs tend to reside in hard-to-reach pockets means that they are often missed by surgical interventions.5 Despite the time and resources invested into the development of clinically viable anti-CSC agents over the last few decades, none have been approved for human use.6 Most small molecule anti-CSC agents undergoing (pre-)clinical investigation are fully organic in nature.6 We and others have developed a number of metal complexes with promising anti-CSC activities, both in in vitro and in vivo models.7The redox status in CSCs is finely controlled, more so than in bulk cancer cells and non-proliferating cells. Therefore any downstream effects of reactive oxygen species (ROS) elevation is exacerbated in CSCs.8 This provides an opportunity to elicit selective CSC death by ROS modulation. Redox active metal complexes have been used to successfully perturb the ROS status in CSCs and thereby induce selective CSC death (through oxidative stress).9 A number of copper(II)-phenanthroline and -Schiff base complexes containing auxiliary nonsteroidal anti-inflammatory drugs (NSAIDs) have been reported to kill CSCs in the sub-micromolar to micromolar range.10,11 Detailed mechanistic studies suggest that the copper(II)-NSAID complexes induce CSC cytotoxicity by elevating ROS levels through the copper(II)-phenanthroline or -Schiff base motifs (by Fenton-like redox chemistry) and inhibiting the inducible cyclooxygenase isoenzyme, COX-2 through the NSAID moieties.10,11 NSAIDs are established, clinically used inhibitors of COX-2.12 COX-2 is an enzyme that catalyses the production of the inflammation mediator prostaglandin. COX-2 is overexpressed in certain CSCs and thought to be vital to their regulation and maintenance.13
An emerging approach to durable therapeutic outcomes is activation of the immune system to recognise and remove bulk cancer cells and CSCs.14 Immune cells can be stimulated by bulk cancer cells and CSCs that have undergone immunogenic cell death (ICD) through the release of damage-associated molecular patterns (DAMPs).15 ICD can be blocked by inhibitory DAMPs (iDAMPs) such as prostaglandin.16 Therefore, the reduction of prostaglandin levels through the inhibition of COX-2 is envisaged to enhance DAMP release. The reduction of iDAMPs by ROS-generating copper(II)-NSAID complexes to prompt ICD in CSCs has not been attempted before. Here we have sought to use copper(II)-phenanthroline complexes containing flufenamic acid to trigger a cytotoxic and immunogenic response against breast CSCs. The flufenamic acid moiety is expected to enhance immunogenicity by facilitating DAMP release (through COX-2 downregulation).
Results and discussion
Synthesis and characterisation of copper(II)-flufenamic acid complexes
The mono- and bi-nuclear copper(II) complexes 1 and 2 investigated in this study are depicted in Fig. 1A. Reaction of 1,10-phenanthroline or 3,4,7,8-tetramethyl-1,10-phenanthroline with CuCl2·2H2O and two equivalents of flufenamic acid in methanol, under basic conditions, yielded the mono-nuclear complex 1 and the bi-nuclear complex 2, respectively. Both copper(II) complexes were isolated as green solids in good yields (57–59%) and were characterised by infra-red spectroscopy, high-resolution mass spectrometry, and elemental analysis (Fig. S1–S3†). According to the ATR-FTIR spectra of 1 and 2, the difference between the νasym(CO2) and νsym(CO2) stretching bands for 1 and 2 were 200 cm−1 and 258 cm−1 (Fig. S1†), indicative of a monodentate and a mixed monodentate-bridging binding mode, respectively, for the carboxylate group on flufenamic acid to the respective copper(II) centres. Distinctive molecular ion peaks corresponding to 1 and 2 with the appropriate isotopic pattern were observed in the positive mode of the corresponding HR-ESI mass spectra (Fig. S2 and S3†). The purity and composition of 1 and 2 were confirmed by elemental analysis.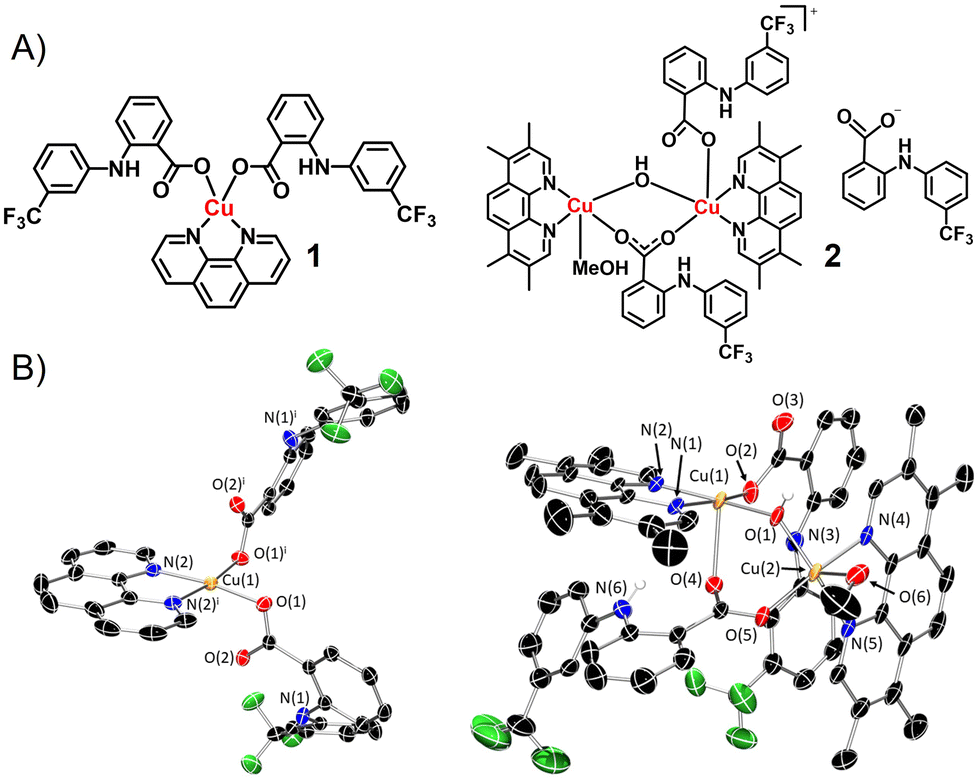 | ||
| Fig. 1 (A) Chemical structures of copper(II)-flufenamic acid complexes 1 and 2. (B) X-ray structures of 1 and 2. Ellipsoids are shown at 50% probability, H atoms and the counter anion for 2 have been omitted for clarity. Symmetry operation to generate equivalent atoms in 1: i = 1 − x, +y, 3/2 − z. | ||
Single crystals of 1 and 2 suitable for X-ray diffraction studies were obtained by slow evaporation of a methanolic solution of 1 and 2 (CCDC 2324436–2324437, Fig. 1B and Table S1†). Selected bond distances and bond angles are presented in Tables S2 and S3.† The mono-nuclear copper(II) complex 1 exhibits a distorted square planar geometry with the copper centre coordinated to 1,10-phenanthroline (forming a five-membered chelated ring) and two flufenamato moieties (via one of the carboxylato oxygen atoms). The Cu atom resides on the N2O2 plane and the average angle between atoms cis to one another around the Cu atom is 90.12°, consistent with a distorted square planar geometry. The structure of 1 is in line with a previously reported structure of the same complex.17 The cationic component of the bi-nuclear copper(II) complex 2 contains two copper(II) centres with different coordinating environments. Cu(1) exhibits a five coordinate, distorted square-based pyramidal structure (τ5 = 0.04) with bonds to 3,4,7,8-tetramethyl-1,10-phenanthroline, a bridging hydroxyl ligand, and a terminal and bridging flufenamato moiety. Cu(2) displays a five coordinate, distorted square-based pyramidal structure (τ5 = 0.02) with bonds to 3,4,7,8-tetramethyl-1,10-phenanthroline, methanol, a bridging hydroxyl ligand, and a bridging flufenamato moiety. The average Cu–N and Cu–O bond lengths found in 1 and 2 are consistent with bond parameters for related copper(II) complexes.17–20
Stability of the copper(II) complexes in solution
The experimentally determined log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value for 1 and 2 was 0.29 ± 0.01 and 0.70 ± 0.04, respectively. This suggests that both 1 and 2 are amphiphilic and thus should be readily soluble in aqueous solutions. The stability of 1 and 2 in solutions relevant for cell-based studies was probed using time-course UV-vis spectroscopy and mass spectrometry studies. In DMSO and mammary epithelial cell growth medium (MEGM):DMSO (95:5) the bands associated to 1 and 2 (25 μM) remained largely unchanged over the course of 24 h, indicative of stability (Fig. S4–S7†). In PBS:DMSO (95:5) with the added presence of ascorbic acid or glutathione (10 equivalence), the absorbance associated to 1 and 2 remained largely unaltered over 24 h, suggesting that 1 and 2 are stable under biologically reducing conditions (Fig. S8–S11†). In the presence of bathocuproine disulfonate (2 equivalents), a strong copper(I) chelator, and ascorbic acid or glutathione (10 equivalence), in PBS:DMSO (95:5), the UV-vis traces for 1 and 2 (25 μM) both displayed a distinctive absorbance band at 480 nm corresponding to [CuI(BCS)2]3− (Fig. S12–S15†). This suggests that the copper centres in 1 and 2 undergo reduction to the copper(I) form in the presence of cellular reductants. The presence of BCS allows the Cu(I) form of 1 and 2 to be trapped as [CuI(BCS)2]3− and detected by UV-vis spectroscopy. Therefore, the data presented in Fig. S12–S15† does not imply that 1 and 2 are unstable in the presence of ascorbic acid or glutathione but merely that they undergo reduction. The ESI (positive) mass spectrum of 1 (500 μM) in the presence of ascorbic acid or glutathione (10 equivalents), in H2O:DMSO (10:1) after 24 h incubation, was dominated by a distinctive molecular ion peak corresponding to [CuI(1,10-phenanthroline)2]+ (423 m/z), with the appropriate isotopic pattern (Fig. S16 and 17†). This suggests that the copper(II) centre in 1 undergoes reduction to copper(I), by ascorbic acid or glutathione, and subsequent ligand exchange. The ESI (positive) mass spectrum of 2 (500 μM) under identical conditions displayed a molecular ion peak corresponding to [CuII(3,4,7,8-tetramethyl-1,10-phenanthroline)(flufenamato)]+ (580 m/z) as well as [CuI(3,4,7,8-tetramethyl-1,10-phenanthroline)2]+ (535 m/z) (Fig. S16 and 17†). This suggests that 2 is less prone to liberating the flufenamato moiety upon reduction compared to 1. Next, the ability of 1 and 2 to undergo reduction by glutathione and subsequent oxidation by air was determined. UV-vis spectroscopy studies showed that upon incubation of 1 or 2 (1 mM) with glutathione (1 mM) in DMSO at 37 °C, the d–d transition band associated to the copper(II) centre disappeared rapidly within minutes implicative of reduction to copper(I) (Fig. S18 and S19†). The disappearance of the d–d band does not indicate structural instability, only that 1 and 2 have been reduced to the Cu(I) form. Exposure of the same sample to air for 24 h led to the reappearance of the d–d transition band suggesting oxidation to the copper(II) form (Fig. S18 and S19†). Further addition of glutathione (1 mM) followed by exposure to air for 24 h led to the disappearance and subsequent re-appearance of the d–d transition band (Fig. S18 and S19†). Collectively, this shows that 1 and 2 have the potential to redox cycle between the copper(II) and copper(I) states by undergoing reduction by glutathione and oxidation by air (multiple times) while maintaining a suitable coordination structure.
P value for 1 and 2 was 0.29 ± 0.01 and 0.70 ± 0.04, respectively. This suggests that both 1 and 2 are amphiphilic and thus should be readily soluble in aqueous solutions. The stability of 1 and 2 in solutions relevant for cell-based studies was probed using time-course UV-vis spectroscopy and mass spectrometry studies. In DMSO and mammary epithelial cell growth medium (MEGM):DMSO (95:5) the bands associated to 1 and 2 (25 μM) remained largely unchanged over the course of 24 h, indicative of stability (Fig. S4–S7†). In PBS:DMSO (95:5) with the added presence of ascorbic acid or glutathione (10 equivalence), the absorbance associated to 1 and 2 remained largely unaltered over 24 h, suggesting that 1 and 2 are stable under biologically reducing conditions (Fig. S8–S11†). In the presence of bathocuproine disulfonate (2 equivalents), a strong copper(I) chelator, and ascorbic acid or glutathione (10 equivalence), in PBS:DMSO (95:5), the UV-vis traces for 1 and 2 (25 μM) both displayed a distinctive absorbance band at 480 nm corresponding to [CuI(BCS)2]3− (Fig. S12–S15†). This suggests that the copper centres in 1 and 2 undergo reduction to the copper(I) form in the presence of cellular reductants. The presence of BCS allows the Cu(I) form of 1 and 2 to be trapped as [CuI(BCS)2]3− and detected by UV-vis spectroscopy. Therefore, the data presented in Fig. S12–S15† does not imply that 1 and 2 are unstable in the presence of ascorbic acid or glutathione but merely that they undergo reduction. The ESI (positive) mass spectrum of 1 (500 μM) in the presence of ascorbic acid or glutathione (10 equivalents), in H2O:DMSO (10:1) after 24 h incubation, was dominated by a distinctive molecular ion peak corresponding to [CuI(1,10-phenanthroline)2]+ (423 m/z), with the appropriate isotopic pattern (Fig. S16 and 17†). This suggests that the copper(II) centre in 1 undergoes reduction to copper(I), by ascorbic acid or glutathione, and subsequent ligand exchange. The ESI (positive) mass spectrum of 2 (500 μM) under identical conditions displayed a molecular ion peak corresponding to [CuII(3,4,7,8-tetramethyl-1,10-phenanthroline)(flufenamato)]+ (580 m/z) as well as [CuI(3,4,7,8-tetramethyl-1,10-phenanthroline)2]+ (535 m/z) (Fig. S16 and 17†). This suggests that 2 is less prone to liberating the flufenamato moiety upon reduction compared to 1. Next, the ability of 1 and 2 to undergo reduction by glutathione and subsequent oxidation by air was determined. UV-vis spectroscopy studies showed that upon incubation of 1 or 2 (1 mM) with glutathione (1 mM) in DMSO at 37 °C, the d–d transition band associated to the copper(II) centre disappeared rapidly within minutes implicative of reduction to copper(I) (Fig. S18 and S19†). The disappearance of the d–d band does not indicate structural instability, only that 1 and 2 have been reduced to the Cu(I) form. Exposure of the same sample to air for 24 h led to the reappearance of the d–d transition band suggesting oxidation to the copper(II) form (Fig. S18 and S19†). Further addition of glutathione (1 mM) followed by exposure to air for 24 h led to the disappearance and subsequent re-appearance of the d–d transition band (Fig. S18 and S19†). Collectively, this shows that 1 and 2 have the potential to redox cycle between the copper(II) and copper(I) states by undergoing reduction by glutathione and oxidation by air (multiple times) while maintaining a suitable coordination structure.
Potency of the copper(II) complexes towards breast cancer stem cells
The antiproliferative potential of 1 and 2 against bulk breast cancer cells (HMLER) and breast CSC-enriched cells (HMLER-shEcad) grown in monolayer systems was determined using the MTT assay. The IC50 values were determined from dose–response curves (Fig. S20 and S21†) and are summarised in Table 1. The mono-nuclear complex 1 was equipotent towards HMLER and HMLER-shEcad cells within the micromolar range. The bi-nuclear complex 2 was also equipotent towards HMLER and HMLER-shEcad cells but within the sub-micromolar range. Notably, 2 was 11-fold and 15-fold more potent towards breast CSCs than salinomycin (the most advanced anti-breast CSC agent to date) and cisplatin (a clinically used metallodrug), respectively (Table 1).10,21 Flufenamic acid was up to 24-fold and 81-fold less toxicity towards HMLER and HMLER-shEcad cells than 1 and 2 (Fig. S22† and Table 1), implying that the copper(II)-phenathroline motif in 1 and 2 is responsible for its potency. To gauge therapeutic potential, the cytotoxicity of 1 and 2 towards non-cancerous bronchial epithelial BEAS-2B cells was determined. The complexes 1 and 2 displayed similar toxicity to BEAS-2B cells compared to HMLER and HMLER-shEcad cells (IC50 = 1.68 ± 0.07 μM for 1 and 0.18 ± 0.01 μM for 2) (Fig. S23†). Therefore, according to the cytotoxicity studies in monolayer systems, 1 and 2 kill breast CSCs, bulk breast cancer cells, and non-cancerous bronchial epithelial cells to a comparable extent.| Compound | HMLER IC50/μM | HMLER-shEcad IC50/μM | Breast CSC spheroid IC50/μM |
|---|---|---|---|
| a Reported in ref. 10, 11, 21 and 22. | |||
| 1 | 3.57 ± 0.03 | 3.04 ± 0.09 | 9.17 ± 2.47 |
| 2 | 0.31 ± 0.01 | 0.38 ± 0.17 | 0.74 ± 0.11 |
| Flufenamic acid | 7.32 ± 1.04 | 30.86 ± 2.65 | >133 |
| Cisplatina | 2.57 ± 0.02 | 5.65 ± 0.30 | 13.50 ± 2.34 |
| Salinomycina | 11.43 ± 0.42 | 4.23 ± 0.35 | 18.50 ± 1.50 |
The ability of 1 and 2 to inhibit breast CSC spheroid (mammosphere) formation and viability was also determined. The addition of 1 and 2 (IC20 value for 5 days) to single cell suspensions of HMLER-shEcad cells markedly reduced the number and size of mammospheres formed (Fig. 2A and B), to a similar or better extent than salinomycin and cisplatin (Fig. 2A and S24†). Dosage with flufenamic acid (IC20 value for 5 days) did not significantly change the number or size of mammospheres formed (Fig. 2A and B). The colorimetric resazurin-based reagent, TOX8 was used to probe mammosphere viability. The IC50 value for 1 and 2 was in the micromolar to sub-micromolar range. Strikingly, the IC50 value of 2 was 25-fold and 18-fold lower than salinomycin and cisplatin, respectively (Fig. S25† and Table 1).11,22 Flufenamic acid did not display any observable mammosphere potency (IC50 > 133 μM).23 Taken together the breast CSC spheroid studies show that 2 is effectively able to reduce mammosphere formation and viability, and that its capacity to do so is greater or comparable to salinomycin and cisplatin.
 | ||
| Fig. 2 (A) Quantification of mammosphere formation with HMLER-shEcad cells untreated and treated with 1, 2, flufenamic acid, cisplatin, or salinomycin (at their IC20 values, 5 days) (B) representative bright-field images (×10) of HMLER-shEcad mammospheres in the absence and presence of 1, 2 or flufenamic acid (at their IC20 values, 5 days). | ||
Modulation of reactive oxygen species and cyclooxygenase-2 levels in breast cancer stem cells
Further cell-based studies were conducted to shed light on the mechanism of action of the most effective copper(II) complex 2. Cellular uptake studies showed that 2 (1 μM) was readily taken up by HMLER-shEcad cells upon incubation for 24 h (88.6 ± 1.1 ng of Cu per million cells). The majority of internalised 2 was detected in the cytoplasm (75%) with little present in the nucleus and membrane (Fig. S26†). The copper(II) complex 2 was, in part, envisaged to kill breast CSCs by increasing intracellular ROS levels. To determine the ability of 2 to produce ROS in HMLER-shEcad cells, 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate was used. HMLER-shEcad cells treated with 2 (2 × IC50 value) displayed a significant increase in ROS levels compared to untreated control cells after short (0.5 h) and long exposure times (6–24 h) (Fig. S27†). Cytotoxicity studies in the presence of N-acetylcysteine (2 mM, 72 h) showed that the potency of 2 towards breast CSCs decreased significantly (IC50 value = 3.18 ± 0.01 μM, 8-fold, p < 0.05) (Fig. S28†). Taken together, this suggests that 2-induced breast CSC death is related to intracellular ROS generation. Intracellular ROS production can lead to activation of p38 MAP kinase (MAPK) or Jun-amino-terminal kinase (JNK) pathways.24 HMLER-shEcad cells treated with 2 (0.5–2 μM for 48 h) exhibited enhanced phosphorylation of p38 and its respective downstream effector MAP kinase-activated protein kinase 2 (MAPKAPK-2) (Fig. S29†). Although 2 induced phosphorylation of JNK, the downstream effector c-Jun was not phosphorylated (Fig. S29†). Activation of the p38 and JNK pathways can lead to apoptosis.25 Apoptosis can lead to changes in cell morphology, where the cell membrane becomes compromised and phosphatidylserine residues are translocated from the membrane interior to the exterior. According to the FITC Annexin V-propidium iodide flow cytometry assay, treatment of HMLER-shEcad cells with 2 (2 × IC50 value and 4 × IC50 value for 72 h) induced a sizeable population of cells to expose phosphatidylserine on the cell membrane exterior and take up propidium iodide, indicative of early- and late-stage apoptosis (Fig. S30†). This was comparable to treatment with cisplatin (25 μM for 72 h), an established apoptosis inducer (Fig. S30†). Cytotoxicity studies in the presence of z-VAD-FMK (5 μM), a peptide-based caspase-dependent apoptosis inhibitor showed that the potency of 2 towards HMLER-shEcad cells decreased significantly (IC50 value = 0.76 ± 0.02 μM, 2-fold, p < 0.05) (Fig. S31†). This confirms that 2 induces apoptotic breast CSC death.Given that the copper(II) complex 2 contains three flufenamato moieties, flow cytometric studies were conducted to determine if 2 could downregulate COX-2. HMLER-shEcad cells pre-treated with lipopolysaccharide (LPS) (2.5 μM for 24 h), to increase basal COX-2 levels, were treated with 2 (2 × IC50 value for 48 h) and flufenamic acid (20 μM for 48 h), and the COX-2 expression was determined. COX-2 expression decreased significantly upon treatment with 2 and flufenamic acid (Fig. S32†). To determine if 2 induces COX-2-dependent breast CSC death, cytotoxicity studies were performed with HMLER-shEcad cells co-treated with prostaglandin E2 (20 μM). The potency of 2 towards HMLER-shEcad cells decreased significantly (IC50 value = 0.75 ± 0.01 μM, 2-fold, p < 0.05) under these conditions (Fig. S33†). Collectively, the flow cytometric and cytotoxicity studies suggest that the mechanism of action of 2 could be related to COX-2 downregulation.
Copper(II)-flufenamic acid complex induced immunogenic cell death and phagocytosis
COX-2 inhibition and subsequent prostaglandin reduction has been shown to effect ICD in bulk cancer cells and CSCs.16,23 As 2 is able to downregulate COX-2 in breast CSCs, we investigated whether it can induce DAMP release from breast CSCs. The three major DAMPs linked to ICD, calreticulin (CRT), adenosine triphosphate (ATP), and nuclear high mobility group box 1 (HMGB-1)15 were monitored in breast CSCs upon treatment with 2. HMLER-shEcad cells dosed with 2 (3 μM or 6 μM for 24 h) displayed noticeably higher levels of CRT on their cell membrane than untreated control cells (Fig. 3A). As expected, HMLER-shEcad cells dosed with cisplatin (150 μM for 24 h) and thapsigargin (7 μM for 24 h) also displayed an increase in CRT on their cell membrane (Fig. 3B). A luciferase-based assay was used to determine the release of ATP from HMLER-shEcad cells treated with 2. HMLER-shEcad cells treated with 2 (IC50 value, 2 × IC50 value or 4 × IC50 value for 24 h) released ATP in a dose-dependent manner (up to 1.9-fold more than untreated cells) (Fig. 3C). As expected, cisplatin (50 μM for 24 h) also induced significant ATP release (2.3-fold more than untreated cells) (Fig. 3C). Immunoblotting studies showed that the relative amount of HMGB-1 in HMLER-shEcad cells markedly decreased upon treatment with 2 (0.5–2 μM for 48 h) (Fig. S34†). Overall, the DAMP detection studies suggest that the mode of cell death induced by 2 is consistent with ICD. | ||
| Fig. 3 Representative histograms displaying the green fluorescence emitted by anti-CRT Alexa Fluor 488 nm antibody-stained HMLER-shEcad cells (A) untreated (red), and treated with 2 (3 μM) (green) or 2 (6 μM) (orange) for 24 h and (B) HMLER-shEcad cells untreated (red) and treated with cisplatin (150 μM) and thapsigargin (7 μM) (blue) for 24 h. (C) Normalised extracellular ATP released from HMLER-shEcad cells untreated and treated with 2 (IC50 value, 2 × IC50 value, and 4 × IC50 value) or cisplatin (50 μM) for 24 h. | ||
Next, we determined the ability of breast CSCs treated with 2 to be engulfed by macrophages using an in vitro phagocytosis assay. HMLER-shEcad cells pre-stained with CellTracker Green were treated with 2 (5 μM for 24 h) and then incubated with macrophages pre-stained with CellTracker Orange for 2 h. The population of double-stained macrophages and engulfed HMLER-shEcad cells is indicated in the two-dimensional scatter plots shown in Fig. 4. This analysis showed that 2 (5 μM for 24 h) was able to significantly enhance phagocytosis of HMLER-shEcad cells by macrophages (15-fold increase compared to untreated cells, Fig. 4). HMLER-shEcad cells treated with flufenamic acid (20 μM for 24 h) did not prompt phagocytosis by macrophages (Fig. 4). As expected, cisplatin (150 μM for 24 h) and thapsigargin (7 μM for 24 h) hugely enhanced phagocytosis of HMLER-shEcad cells by macrophages (Fig. 4).
 | ||
| Fig. 4 Representative two-dimensional scatter plots of CellTracker Green-stained HMLER-shEcad cells (A) untreated and treated with (B) 2 (5 μM) or (C) flufenamic acid (20 μM) or (D) cisplatin (150 μM) and thapsigargin (7 μM) for 24 h and then co-cultured with CellTracker Orange-stained THP-1 macrophages for 2 h. | ||
Conclusions
In summary, we report a bi-nuclear copper(II) complex 2 that kills breast CSCs in a manner that instigates engulfment by immune cells. The bi-nuclear copper(II) complex 2 displayed sub-micromolar potency towards breast CSCs grown in monolayers, 11-fold and 15-fold greater than salinomycin and cisplatin, respectively. The bi-nuclear copper(II) complex 2 also displayed up to 25-fold greater potency towards breast CSC mammospheres than salinomycin and cisplatin. Cell-based mechanistic studies indicated that 2 is able enter breast CSCs, localise in the cytoplasm, increase intracellular ROS levels, and prompt caspase-dependent apoptosis. The bi-nuclear copper(II) complex 2 was also shown to downregulate COX-2 expression in breast CSCs using flow cytometric studies. Moreover, cytotoxicity studies in the presence of prostaglandin E2 indicated that the mechanism of cytotoxicity of 2 could be related to COX-2 downregulation. COX-2 downregulation (or reduction in prostaglandin E2 levels) can promote ICD, therefore the ability of 2 to promote DAMP release from breast CSCs was probed. Breast CSCs treated with the bi-nuclear copper(II) complex 2 displayed typical DAMP signals, indicative of ICD. Phagocytosis studies showed that breast CSCs treated with 2 were effectively engulfed by macrophages, highlighting the promising immunogenic potential of 2. To the best of our knowledge this is first report of a bi-nuclear copper(II)-NSAID complex to induce ICD of breast CSCs and promote their engulfment by macrophages. This study reinforces the therapeutic potential of copper complexes and opens the door for the development of other bi-nuclear metal complexes with NSAIDs as immunotherapeutic agents.Author contributions
Conceptualisation, Y. L., J. F., and K. S.; methodology, Y. L., J. F., K. S., F. O., and K. S.; formal analysis, Y. L., J. F., K. S., F. O., and K. S.; investigation, Y. L., J. F., K. S., F. O., and K. S.; writing—original draft preparation, Y. L., J. F., F. O., and K. S.; writing—review and editing, Y. L., J. F., K. S., F. O., and K. S.; supervision, K. S. and F. O.; project administration, K. S. and F. O.; funding acquisition, K. S. and F. O.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. S. and F. O. are supported by EPSRC New Investigator Awards (EP/S005544/1 and EP/W00691X/1). XRD crystallography at the University of Leicester is supported by an EPSRC Core Equipment Award (EP/V034766/1). The Advanced Imaging Facility (RRID:SCR_020967) at the University of Leicester is also thanked.References
- J. C. Chang, Medicine, 2016, 95, S20–S25 CrossRef CAS PubMed.
- L. V. Nguyen, R. Vanner, P. Dirks and C. J. Eaves, Nat. Rev. Cancer, 2012, 12, 133–143 CrossRef CAS PubMed.
- M. Dean, T. Fojo and S. Bates, Nat. Rev. Cancer, 2005, 5, 275–284 CrossRef CAS PubMed.
- W. Chen, J. Dong, J. Haiech, M. C. Kilhoffer and M. Zeniou, Stem Cells Int., 2016, 2016, 1740936 Search PubMed.
- V. D'Andrea, S. Guarino, F. M. Di Matteo, M. Maugeri Sacca and R. De Maria, G. Chir., 2014, 35, 257–259 Search PubMed.
- F. Y. Du, Q. F. Zhou, W. J. Sun and G. L. Chen, World J. Stem Cells, 2019, 11, 398–420 CrossRef PubMed.
- J. Northcote-Smith and K. Suntharalingam, Curr. Opin. Chem. Biol., 2023, 72, 102237 CrossRef CAS PubMed.
- X. Shi, Y. Zhang, J. Zheng and J. Pan, Antioxid. Redox Signal., 2012, 16, 1215–1228 CrossRef CAS PubMed.
- K. Laws and K. Suntharalingam, ChemBioChem, 2018, 19, 2246–2253 CrossRef CAS PubMed.
- J. N. Boodram, I. J. McGregor, P. M. Bruno, P. B. Cressey, M. T. Hemann and K. Suntharalingam, Angew. Chem., Int. Ed., 2016, 55, 2845–2850 CrossRef CAS PubMed.
- C. Lu, K. Laws, A. Eskandari and K. Suntharalingam, Dalton Trans., 2017, 46, 12785–12789 RSC.
- S. B. Abramson and G. Weissmann, Arthritis Rheum., 1989, 32, 1–9 CrossRef CAS PubMed.
- L. Y. Pang, E. A. Hurst and D. J. Argyle, Stem Cells Int., 2016, 2016, 11 Search PubMed.
- Q. Pan, Q. Li, S. Liu, N. Ning, X. Zhang, Y. Xu, A. E. Chang and M. S. Wicha, Stem Cells, 2015, 33, 2085–2092 CrossRef CAS PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef CAS PubMed.
- K. Hayashi, F. Nikolos, Y. C. Lee, A. Jain, E. Tsouko, H. Gao, A. Kasabyan, H. E. Leung, A. Osipov, S. Y. Jung, A. V. Kurtova and K. S. Chan, Nat. Commun., 2020, 11, 6299 CrossRef CAS PubMed.
- L. Hudecova, K. Jomova, P. Lauro, M. Simunkova, S. H. Alwasel, I. M. Alhazza, J. Moncol and M. Valko, Open Chem., 2020, 18, 1444–1451 CAS.
- P. Zheng, A. Eskandari, C. Lu, K. Laws, L. Aldous and K. Suntharalingam, Dalton Trans., 2019, 48, 5892–5896 RSC.
- C. Tolia, A. N. Papadopoulos, C. P. Raptopoulou, V. Psycharis, C. Garino, L. Salassa and G. Psomas, J. Inorg. Biochem., 2013, 123, 53–65 CrossRef CAS PubMed.
- P. B. Osei, J. Northcote-Smith, J. Fang, K. Singh, F. Ortu and K. Suntharalingam, Chem. – Eur. J., 2023, 29, e202301188 CrossRef CAS PubMed.
- A. Eskandari and K. Suntharalingam, Chem. Sci., 2019, 10, 7792–7800 RSC.
- A. Eskandari, A. Kundu, S. Ghosh and K. Suntharalingam, Angew. Chem., Int. Ed., 2019, 58, 12059–12064 CrossRef CAS PubMed.
- J. Fang, O. Orobator, C. Olelewe, G. Passeri, K. Singh, S. Awuah and K. Suntharalingam, Angew. Chem., Int. Ed., 2024, 63, e202317940 CrossRef CAS PubMed.
- J. A. McCubrey, M. M. Lahair and R. A. Franklin, Antioxid. Redox Signal., 2006, 8, 1775–1789 CrossRef CAS PubMed.
- M. R. Junttila, S. P. Li and J. Westermarck, FASEB J., 2008, 22, 954–965 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2324436 and 2324437. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00384e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |