
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of reducibility vis-à-vis oxygen vacancies of doped Co3O4/CeO2 in the oxygen evolution reaction†
Saraswati
Roy
a,
Preeti
Dahiya
b,
Tapas Kumar
Mandal
bc and
Sounak
Roy
 *ad
*ad
aDepartment of Chemistry, Birla Institute of Technology and Science Pilani, Hyderabad Campus, Hyderabad-500078, India. E-mail: sounak.roy@hyderabad.bits-pilani.ac.in
bDepartment of Chemistry, Indian Institute of Technology Roorkee, Roorkee – 247 667, India
cCentre for Nanotechnology, Indian Institute of Technology Roorkee, Roorkee – 247 667, India
dMaterials Center for Sustainable Energy & Environment, Birla Institute of Technology and Science Pilani, Hyderabad Campus, Hyderabad – 500078, India
First published on 17th February 2024
Abstract
Electrochemical water splitting, which is a highly promising and environmentally friendly technology for H2 fuel production, faces significant hurdles due to the sluggish kinetics of the oxygen evolution reaction. Co -based oxides have garnered significant attention as alternative catalysts for the oxygen evolution reaction owing to the Co2+/Co3+ redox couple. Enhancing the challenging Co2+ → Co3+ oxidation process can further improve the catalytic oxygen evolution reaction. The aim of our work was to design a Co3O4-based catalyst to enhance reactivity by increasing the number of Co3+ active sites, serving as an excellent platform for facilitating the oxygen evolution reaction. To drive the effectiveness of the catalyst, in this study, we synthesized Co3O4 anchored on CeO2 (Co3O4/CeO2). The kinetics and efficacy of the oxygen evolution reaction catalysed by Co3O4/CeO2 was significantly improved by aliovalent doping of Sr into Ce sites and Cu into Co sites. The reducible nature of Ce stimulates the formation of Co3+ ions, resulting in an increased production of intermediate –OOH species, thus expediting the reaction. The transformation of Co2+ to Co3+ consequently leads to an increase in anion vacancies, which, in turn, promotes the adsorption of more intermediate species at the active site. The Sr- and Cu-doped Co3O4/CeO2 catalyst exhibited a high current density of 200 mA cm−2 at 580 mV and a low overpotential of 297 mV at 10 mA cm−2. The study functions as a key indicator to establish a connection between oxygen vacancies and metal oxidation states in order to investigate the mechanistic aspects of the oxygen evolution reaction on mixed metal oxides. Moreover, this study is expected to pave the way for the development of innovative oxygen evolution reaction catalysts with reducible supports, thus offering a new pathway for their design.
1. Introduction
The transient nature of the primary feedstock obtained from the spontaneous combustion of non-renewable fossil fuels has spurred the advancement of the sustainable H2 energy economy. Water electrolyzers enable the conversion of water into the energy carrier H2 through an electrochemical process. However, the bottleneck of this technology is the sluggish anodic oxygen evolution reaction (OER) characterized by a four-electron transport process, the breaking of the O–H bond, and the subsequent formation of the O–O bond. These complex processes result in high kinetic energy barriers and, consequently, significant overpotentials.1,2 In addition to water splitting, the OER is also a crucial half-reaction in processes such as CO2 and N2 electroreduction as well as in metal–air batteries. The practical implementation of common OER catalysts, such as Pt, Ru, and Ir, in large-scale applications is constrained by their high cost and limited availability. Therefore, extensive research is being conducted for developing affordable, stable, and efficient OER catalysts. In recent times, significant attention has been devoted to exploring non-noble metals as potential electrocatalysts for the OER.3–5Among the transition metals, Co-based materials have garnered significant attention as alternative catalysts for the OER owing to their crucial redox couple, which promotes OER kinetics through surface reconstruction.6–8 For instance, the bimetallic hetero-structured hybrid nanorod NiSe2–CoSe2 has been explored as an excellent OER catalyst.9 Transitional metal boride-based materials are well-known catalysts for the water-splitting reaction.10 Based on the literature, it is widely believed that during the OER, the first redox feature of the Co2+/Co3+ couple facilitates the formation of essential Co3+–OOH species under basic conditions, while the second redox feature of the Co3+/Co4+ couple promotes the formation of Co4+O2.11,12 In our earlier report, we have exclusively shown that Co2+/Co3+ oxidation in Co3O4 aids the facile occurrence of the OER.13 However, the oxidation of Co to its highest possible oxidation state i.e. Co2+/Co3+ and Co3+/Co4+ must occur alongside parallel reduction reactions, which require a support material with facile reducibility.
To create an efficient Co2+/Co3+ and Co3+/Co4+ redox system in Co3O4, in this work, we have developed cerium-dioxide-supported cobalt oxide (Co3O4/CeO2) by a solution combustion method. CeO2 can improve the electrical conductivity and optical activity of Co3O4 as the oxidation of cobalt is facilitated by the reduction of cerium and/or lattice oxygen evolution (O2− → ½O2 + 2e−).14 The ability of CeO2 to transit between the Ce3+ and Ce4+ oxidation states allows it to facilitate reversible surface oxygen ion exchange. This property serves as an oxygen buffer for effective oxygen supply and also creates the potential for robust electron interactions with other materials.15 Moreover, density functional theory calculations suggest that CeOx can influence the electronic properties of cobalt species.16,17 We further substituted Ce with Sr (Co3O4/Ce1−ySryO2−δ) to enhance the oxidizing power of CeO2 as Sr with a lower oxidation potential is a judicious choice for ameliorating the redox behaviour of cerium and cobalt.18–23 To further enhance the impact of oxygen vacancies, we partially substituted the cobalt sites with another transition metal, namely Cu (Co3−xCuxO4/Ce1−ySryO2−δ). The substitution of cobalt with copper will elevate the oxidation state of cobalt. In addition, it can also accelerate the formation of oxygen vacancies to maintain the charge balance.24,25 The study shows the crucial roles of the reducible CeO2 support and Cu2+ doping in Co3O4 in facilitating the oxidation of Co2+/Co3+ and Co3+/Co4+ and their impact on the catalytic oxygen evolution reaction.
2. Experimental methods
The Co3O4/CeO2 catalysts were synthesized using a novel solution combustion method, which is known for its energy efficiency and low-temperature initiation.13 During synthesis, water-soluble Ce(NO3)3·6H2O (SRL chemicals, 99%), Co(NO3)2·6H2O (S.D. Fine Chem Limited, 99%), Sr(NO3)2 (Sigma Aldrich, 99%), and Cu(NO3)2·6H2O (S.D. Fine Chem Limited, 99%) were utilized as oxidizers, while glycine served as the fuel. The synthesis of 40% Co3O4/CeO2 was carried out by dissolving 0.58 g of Co(NO3)2·6H2O, 0.87 g of Ce(NO3)3·6H2O, and 0.2 g of glycine in 50 mL of water. The resulting homogeneous aqueous solution was then placed in a preheated furnace at 450 °C for 15 minutes. Initially, the solution exhibited boiling and foaming; the exothermic redox reaction generated sufficient heat of combustion, compensating for the energy required for oxide formation and resulting in a burning flame at the ignition point. Subsequently, the complete dehydration of the solution yielded the solid powder product of pristine Co3O4/CeO2, accompanied by the evolution of N2, and CO2. Henceforth, Co3O4/CeO2 is denoted as CoCe in the manuscript. About 0.58 g of Co(NO3)2·6H2O, 0.34 g of Ce(NO3)3·6H2O, 0.04 g of Sr(NO3)2 and 0.29 g of glycine were used for the synthesis of 40% Co3O4/Ce0.9Sr0.1O2−δ (designated as CoSrCe) and 0.78 g of Co(NO3)2, 0.08 g of Cu(NO3)2·6H2O, 0.34 g of Ce(NO3)3·6H2O, 0.04 g of Sr(NO3)2 and 0.37 g of glycine were used to synthesise 40% Co3−xCuxO4/Ce0.9Sr0.1O2−δ (designated as CoCuSrCe) in a similar manner.The structure and crystallinity of the prepared mixed metal oxides were characterized using X-ray diffraction (XRD) data obtained using a Rigaku Ultima IV instrument with Cu Kα radiation. The XRD scans were performed at a scan rate of 0.02° min−1 with a step size of 0.01°. The nanocrystalline size of the synthesized metal was determined using Debye–Scherrer's formula (D = 0.9λ/B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ), where λ is the wavelength of the radiation, B is the full-width at half-maximum, and θ is the corresponding angle. The surface morphology and composition analyses were conducted using a Field Emission Scanning Electron Microscope (FE-SEM, FEI-Apreo S) equipped with an Energy Dispersive X-ray Spectroscopy (EDX) unit. The instrument was operated at an acceleration voltage of 20 kV. The Brunauer–Emmett–Teller (BET) method was employed to determine the surface area and pore size of the synthesized catalysts. The X-ray photoelectron spectra (XPS) of the synthesized catalysts were recorded using a Thermo Scientific K-Alpha surface analysis spectrometer with Al Kα radiation (1486.6 eV). The binding energies were reported with respect to C (1s) at 284.8 eV. The EPR analysis was conducted using a Bruker ESR 5000. A UniRam Raman spectroscopy system was used for Raman spectroscopy.
cosθ), where λ is the wavelength of the radiation, B is the full-width at half-maximum, and θ is the corresponding angle. The surface morphology and composition analyses were conducted using a Field Emission Scanning Electron Microscope (FE-SEM, FEI-Apreo S) equipped with an Energy Dispersive X-ray Spectroscopy (EDX) unit. The instrument was operated at an acceleration voltage of 20 kV. The Brunauer–Emmett–Teller (BET) method was employed to determine the surface area and pore size of the synthesized catalysts. The X-ray photoelectron spectra (XPS) of the synthesized catalysts were recorded using a Thermo Scientific K-Alpha surface analysis spectrometer with Al Kα radiation (1486.6 eV). The binding energies were reported with respect to C (1s) at 284.8 eV. The EPR analysis was conducted using a Bruker ESR 5000. A UniRam Raman spectroscopy system was used for Raman spectroscopy.
The electrochemical tests of the synthesized materials were conducted using a conventional three-electrode system. The working electrode was prepared by coating the synthesized catalyst onto Ni foam (NF). The reference electrode used was Hg/HgO saturated with an aqueous KOH solution, and a Pt wire served as the counter electrode. For the preparation of the working electrode, 3 mg of the synthesized catalyst and 0.5 mg of carbon black powder were dispersed in 1 mL of methanol. Additionally, 10 μL of 5% aqueous Nafion (Sigma-Aldrich) was added to the solution. The mixture was sonicated for 60 minutes to achieve a homogenized dispersion of the synthesized metal oxides and carbon. The active mass ratio of the catalyst to carbon black was maintained at 6:1.5. The NF electrode was cut into pieces of 1 × 0.3 cm2 and soaked in 0.3 M H2SO4 for 15 minutes. It was then ultrasonically washed with acetone followed by distilled water. An ink prepared with 50 μL of the catalyst was drop-casted onto the clean NF electrode, with an active mass loading of 0.1 mg cm−2. The electrode was left to dry at room temperature. Cyclic voltammetry (CV), linear sweep voltammetry (LSV), and electrochemical impedance spectroscopy (EIS) measurements were performed in a basic electrolyte medium of 1 M KOH using a scan rate of 10 mV s−1. The CV cycles and OER polarization curves from LSV traces were recorded between 0.9 and 1.9 V (vs. RHE). The obtained potentials were converted to values corresponding to the reversible hydrogen electrode (RHE) using the equation:  . Here, ERHE represents the final converted potential with respect to RHE, EHg/HgO is the experimental potential with respect to the Hg/HgO electrode, and
. Here, ERHE represents the final converted potential with respect to RHE, EHg/HgO is the experimental potential with respect to the Hg/HgO electrode, and  is the standard reduction potential of the Hg/HgO electrode. To evaluate material stability, chronoamperometric (CA) measurements were performed in a three-electrode system for 12 hours in 1 M KOH. EIS was carried out in the frequency range of 100 kHz to 10 mHz, by applying the overpotential versus Hg/HgO. The gaseous product O2 was measured using a portable Gas Chromatograph (GC) from Mayura Analytical Pvt. Limited, India.
is the standard reduction potential of the Hg/HgO electrode. To evaluate material stability, chronoamperometric (CA) measurements were performed in a three-electrode system for 12 hours in 1 M KOH. EIS was carried out in the frequency range of 100 kHz to 10 mHz, by applying the overpotential versus Hg/HgO. The gaseous product O2 was measured using a portable Gas Chromatograph (GC) from Mayura Analytical Pvt. Limited, India.
3. Results and discussion
3.1 Structure and surface of the catalysts
The XRD patterns and the refinements of the synthesized composite metal oxides are shown in Fig. 1(a–c). The Rietveld refinements were carried out by using the Fullprof program suite. The diffraction patterns of all the synthesized catalysts exhibited sharp peaks, indicating high crystallinity, and based on the Debye–Scherrer formula, the crystallite sizes were found to be ∼30 nm for all the materials. From Fig. 1a, it is evident that crystals of both Co3O4 with space group Fd3m (S.G. no. 227, JCPDS #43-1003) and CeO2 with space group Fm3m (S.G. no. 225, JCPDS #34-0394) were clearly present in CoCe. Similarly, the Rietveld refinement of the diffraction data of CoSrCe confirmed the coexistence of Co3O4 and Sr-doped CeO2 (Fig. 1b). On the other hand, the CoCuSrCe data in Fig. 1c exhibit the presence of four phases, namely, Sr-doped CeO2 with space group Fm3m (S.G. no. 225, JCPDS #34-0394), Cu-doped Co3O4 with space group Fd3m (S.G. no. 227, JCPDS #43-1003), tetragonal SrCoO2.8 with space group I4/mmm (S.G. no. 139, JCPDS #39-1084) and monoclinic CuO with space group C2/c (S.G. no. 15, JCPDS #05-0661). The Rietveld refinement of the compound, incorporating mixed phases, has successfully achieved a well-fitting profile for the data. The reliability factors along with the refined lattice parameters and constraints of all the phases from the three synthesized composite catalysts are provided in Tables S1–S3.† Elemental doping was corroborated by the XRF studies, as shown in Table S4.† | ||
| Fig. 1 Rietveld refinement profiles of (a) CoCe, (b) CoSrCe and (c) CoCuSrCe. The blue vertical lines represent the Bragg positions of CeO2 and Sr-doped CeO2 fluorite phases, and the red lines indicate those of the Co3O4 and Cu-doped Co3O4 spinel phases. The green vertical lines represent the Bragg positions of SrCoO2.8, and the pink lines represent those of CuO. The corresponding FESEM images are provided as insets. The EDX mappings and elemental analysis of (d) CoCe, (e) CoSrCe and (f) CoCuSrCe. | ||
As electrocatalysis is a surface phenomenon, next, we studied the surface properties of the combustion-synthesized materials. The FESEM micrographs in the insets of Fig. 1(a–c) show the surface morphologies of pristine CoCe, doped CoSrCe and CoCuSrCe. All three materials showed a similar spherical particle morphology with an average diameter of ∼0.17 μm. EDX elemental mapping indicated the surface enrichment of dopants Cu and Sr in the matrix (Fig. 1(d–f) and Table S4†). This was further verified by the other spot EDX elemental mappings shown in Fig. S1.† The surface area was also studied, and all the materials exhibited type II N2 adsorption–desorption isotherms with negligible hysteresis, specifying the nanoporous nature of the surface (Fig. S2†). The average surface areas of the materials estimated using the BET equation were found to be in the 1–3 m2 g−1 range.
The electrochemical active surface area (ECSA) plays an important role in the electrocatalytic activity of a material. The ECSA was calculated from the electrochemical double layer capacitance (Cdl) and specific capacitance (CS) according to the equation: ECSA = Cdl/Cs.26,27 The Cdl values were obtained from the CV curves obtained in the non-faradaic region (0.72 to 1.32 V vs. RHE) in 1 M KOH at various scan rates (10–50 mV s−1), as shown in Fig. S3.† The current is linearly proportional to the active surface area owing to the charging of the double layer, and the slope obtained from the plot of half of the capacitive current (ΔJ/2 = (Janodic − Jcathodic)/2) against the scan rate is defined as Cdl.28 The representative Cdl values of the synthesized materials were obtained from the curves presented in Fig. 2a. The CS was calculated using the following equation:  , where
, where  (mA mV) is the area under the CV curve cycled at the scan rate of 10 mV s−1, m (0.1 mg) is the active mass of the catalyst loaded on the electrode (area = 0.6 cm2), VS is the scan rate (mV s−1) and ΔV is the potential window (mV). Thus, the obtained CS values with respect to unit mass loading (mF mg−1) were converted to mF cm−2. The representative ECSAs were found to be 2.36, 5.7 and 6.9 cm2 for CoCe, CoSrCe and CoCuSrCe, respectively (Fig. 2b). An apparent 3-fold increase in electrochemical surface area was observed with the concomitant doping of Sr and Cu in CoCe.
(mA mV) is the area under the CV curve cycled at the scan rate of 10 mV s−1, m (0.1 mg) is the active mass of the catalyst loaded on the electrode (area = 0.6 cm2), VS is the scan rate (mV s−1) and ΔV is the potential window (mV). Thus, the obtained CS values with respect to unit mass loading (mF mg−1) were converted to mF cm−2. The representative ECSAs were found to be 2.36, 5.7 and 6.9 cm2 for CoCe, CoSrCe and CoCuSrCe, respectively (Fig. 2b). An apparent 3-fold increase in electrochemical surface area was observed with the concomitant doping of Sr and Cu in CoCe.
 | ||
| Fig. 2 (a) Linear regression between current density differences vs. scan rates based on CV in a potential window of 1.16 V, and (b) ECSAs of the three composite catalysts. | ||
XPS was used to probe the surface elemental composition and oxidation states of the synthesized catalysts. The survey spectra of all the synthesized catalysts displayed the expected elements (Fig. S4a†). The areas under the peaks of the corresponding elements were derived using the peak fitting software Avantage, and the values were normalized against the X-ray energy used. The surface atomic percentages were calculated by dividing the normalized peak area by the respective photoionization cross-section of the corresponding elements.29 Table S4† indicates surface enrichment of Sr and Cu than the bulk. The deconvoluted Ce 3d core level spectra (3d5/2 is labelled as v, and 3d3/2 is labelled as u) of CoCe, CoSrCe, and CoCuSrCe are presented in Fig. 3a. The spectra exhibited a total of 5 spin–orbit coupled peaks, of which v–u, v′′–u′′, and v′′′–u′′′ belong to Ce4+, and vo–uo and v′–u′ belong to Ce3+. The Ce4+ doublets designated as v–u (882.2–901.0 eV), v′′–u′′ (889.3–907.3 eV) and v′′′–u′′′ (897.0–916.5 eV) are associated with the Ce 3d94f2O 2p4, Ce 3d94f1O 2p5 and Ce 3d94f0O 2p6 final states, respectively. The other two doublets of Ce3+ positioned at vo–uo (879.6–898.5 eV) and v′–u′ (885.3–903.0) correspond with the Ce 3d94f2O 2p5 and Ce 3d94f1O 2p6 final states.30–32 The relative surface concentrations (C) of Ce3+ and Ce4+ were estimated from the total area under the deconvoluted peak (A) by using the following equations:
| ACe3+ = Avo + Auo + Av′ + Au′ |
| ACe4+ = Av + Au + Av′′ + Au′′ + Av′′′ + Au′′′ |
| CCe4+ = ACe4+/ACe4+ + ACe3+ × 100% |
 | ||
| Fig. 3 (a) The Ce 3d core level spectra, (b) Co 2p core level spectra, (c) Cu 2p core level spectra, and (d) O 1s core level spectra of the synthesized pristine and doped oxides. | ||
According to the calculations, the percentage of Ce4+ present in CoCe, CoSrCe, and CoCuSrCe were 65.6, 68.0 and 74.1%, respectively. Apparently, the occurrence of Ce4+ increased with Sr and Cu doping. Fig. 3b exhibits the high-resolution Co 2p core level spin–orbit doublet peaks of the three catalysts, which demonstrate the 2p3/2 and 2p1/2 peaks associated with the Co2+ and Co3+ ions. The Co2+ peak appeared at a higher binding energy (2p3/2–2p1/2 at 782.5–796.2 eV) than the Co3+ peak (2p3/2–2p1/2 at 780.0–795 eV).13,33 The percentage of Co3+ present in CoCe, CoSrCe, and CoCuSrCe were 61.02, 67.9, and 71.1%, respectively. It is interesting to observe that with the incorporation of lower valent Sr and Cu at the Ce and Co sites, respectively, the Ce and Co ions underwent oxidation for charge neutralization. We also collected the core-level Cu 2p spectrum of CoCuSrCe (Fig. 3c), which showed the presence of bivalent Cu2+ with the peaks of Cu 2p3/2 and Cu 2p1/2 associated with the Cu2+ and Cu1+ ions at 933.5 and 953.3 eV, respectively.32,34–38 The Cu2+ peak appeared at a higher binding energy (2p3/2–2p1/2 at 934.9–954.7 eV) than the Cu1+ peak (2p3/2–2p1/2 at 933–952.8 eV).39 The Cu L3M45M45 Auger spectrum in Fig. S4b† distinctively shows the Cu1+ peaks at 916 eV and Cu2+ at 917 eV. The percentage of Cu2+ present in CoCuSrCe was found to be 50.29%. We also checked the Cu 2p core level spectrum of CoCuSrCe after the OER study (Fig. S4c†). The XPS data revealed that lattice doping with lower-valent elements, such as Sr2+ and Cu2+, induced further oxidation of Ce and Co. This phenomenon suggests the potential creation of oxygen vacancies as well. Therefore, a detailed XPS study of O 1s was performed for all three catalysts, and the deconvoluted spectra are plotted in Fig. 3d. The spectra showed three distinct peaks: one at 529 eV due to lattice oxygen (OL), one at 531 eV due to the adsorbed oxygen species at the surface defect sites i.e. oxygen vacancies (OV), and one at 533 eV due to the hydroxyl species of the surface-adsorbed water molecules (OW). The percentage of surface oxygen vacancies in each catalyst was quantified from the area under the OV peak with respect to the total area under the O 1s core level spectrum (OT)40–43 with the help of the following equation:

The percentage of oxygen vacancies was found to be 27% in pristine CoCe. Interestingly, the OV content (%) increased to 65 and 74% in CoSrCe and CoCuSrCe, respectively. The OL content (%) in the materials was also calculated in a similar manner, and the values were found to be 69.3, 36.2 and 21.8% for CoCe, CoSrCe, and CoCuSrCe, respectively. Apparently, lattice doping of bivalent Sr2+ and Cu2+ not only made Ce and Co undergo further oxidation but also created sufficient surface oxygen vacancies. Surface oxygen vacancies are known to facilitate the formation of the −OOH species, a key OER intermediate.
A correlation between the occurrence of oxygen vacancies and doping was also established using the fluorescence spectra. Fig. S5a† illustrates a gradual decrease in fluorescence intensity with Sr and Cu doping, indicating that the absorbed light is trapped by oxygen vacancies (OV). Among the three oxides, CoCuSrCe exhibited the lowest intensity, suggesting the formation of oxygen vacancies.42 To further confirm the presence of OV, we conducted Raman spectroscopy. Fig. S5b† shows that the Raman peak intensity at 548 cm−1 corresponding to OV (A1g mode)41 increased in the order of CoCe < CoSrCe < CoCuSrCe, indicating an increase in the formation of oxygen vacancies with doping. The yellow-highlighted region in the range of 440–460 cm−1 in the Raman spectra indicates the presence of Co–O and Ce–O bonds.44,45
3.2 OER
The catalytic OER activity of the synthesized materials deposited on Ni foam electrodes was measured in 1 M KOH using LSV polarization curves, as shown in Fig. 4a. For clarity, the activity of the bare Ni foam electrode is also plotted in the graph. The three catalysts exhibited a prominent peak at ∼1.4 V vs. RHE before the OER onset potential. This peak can be attributed to the oxidation of Co2+ to Co3+.46 Interestingly, from the peak current density, it was observed that the extent of Co oxidation increased with Sr doping at the Ce sites and consequently Cu doping at the Co sites. Notably, the highly oxidized Co3+ facilitates the formation of the key OER intermediate species –OOH. Fig. 4a represents that compared to pristine CoCe, CoSrCe exhibited a lower onset potential, as well as a lower OER overpotential. Among the three catalysts, CoCuSrCe displayed the lowest overpotential. The higher extent of Co2+ → Co3+ oxidation, which leads to the formation of the intermediate –OOH species with the help of oxygen vacancies (as observed from the XPS studies), is apparently proportional to the OER catalytic activity of pristine and doped oxides. For benchmarking, a commercial RuO2 sample was measured under similar conditions, and the oxides clearly outperformed the commercial RuO2 catalyst. The overpotential required to achieve a current density of 10 mA cm−2 is a widely accepted metric for solar fuel production. Hence, the overpotentials at 10, 50 and 100 mA cm−2 were plotted for the synthesized materials, as shown in Fig. 4b. Fig. 4b demonstrates that the best OER activities of CoCuSrCe with the lowest overpotential were 297, 410 and 470 mV at 10, 50 and 100 mA cm−2, respectively. We further compared the OER activity data with other reported Co-based catalysts, and Table 1 demonstrates that CoCuSrCe exhibits comparable or even superior performance to the other Co-based catalysts. For a global comparison and practical applicability, good mass activity, as well as intrinsic activity, of a catalyst is highly desirable. The mass activity of electrochemical OER catalysts is determined by normalizing the current density to the loaded amount of catalyst, whereas, the intrinsic activity is the current density per unit electrochemical active surface area. Fig. 4c shows the mass activity and intrinsic activity of the three catalysts at the OER onset potential. Evidently, CoCuSrCe exhibited a far superior OER catalytic activity to the pristine and only Sr-doped oxides. To corroborate the superiority of CoCuSrCe, we further measured the oxygen evolved during OER with the help of gas chromatography, and thereby calculated the faradaic efficiency using the following equation:where n is the total number of electrons transferred, F is the Faraday constant, i is the amount of current produced and t denotes time. The calculated FEO2% values are plotted in Fig. 4d. Apparently, the FEO2% of CoCuSrCe (84%) turned out to be better than those of the other two catalysts.

 | ||
| Fig. 4 (a) OER polarisation curves of the three catalysts synthesized with bare Ni foam and the commercial RuO2 catalyst; (b) overpotentials, (c) mass activity and intrinsic activity, and (d) FE (%) of oxygen evolution of the synthesized materials. | ||
| Catalysts | Measurement conditions | OER activity | Ref. | |
|---|---|---|---|---|
| Overpotential at 10 mA cm−2 | Tafel slope | |||
| a At 100 mA cm−2. | ||||
| Co3O4 nanoarrays on carbon | 0.1 M KOH | 0.29 V | 70 mV dec−1 | 47 |
| Co3O4 nanoparticles | 1 M KOH with 5 mV s−1 scan rate | 0.38 V | 153 mV dec−1 | 48 |
| Co3O4-ZIF 8 | 1 M KOH with 10 mV s−1 | 0.40 V | 82 mV dec−1 | 49 |
| Co(OH)2 on GC | 1 M NaOH with 10 mV s−1 scan rate | 0.4 V | 90 mV dec−1 | 50 |
| Co3O4 nanocubes | 1 M KOH with 1 mV s−1 scan rate | 0.5 V | 59 mV dec−1 | 51 |
| Co3O4@CoO | 0.5 M KOH | 0.43 V | 89 mV dec−1 | 52 |
| CoOx/CeOx | 1 M NaOH with 10 mV s−1 scan rate | 0.31 V | 66 mV dec−1 | 17 |
| Co3O4/CeO2 nanohybrids (NHs) | 1 M NaOH with 10 mV s−1 scan rate | 0.27 V | 60 mV dec−1 | 14 |
| Co3O4/CeO2 heterojunction | 1 M KOH | 0.25 Va | 34.4 mV dec−1 | 53 |
| CoCuSrCe | 1 M KOH | 0.29 V | 42 mV dec−1 | This work |
To understand the kinetics of the OER over the three synthesized catalysts, the scan rate dependency of their OER activity and the activation energy of the OER were evaluated. Scan rate dependency studies were carried out by varying the scan rate from 10 to 50 mV s−1 in the presence of 1 M KOH as the electrolyte. Fig. 5a unveils that the OER current density linearly increased with the square root of the scan rate, indicating a diffusion-controlled electrocatalytic reaction over these catalysts.54 The Arrhenius plots of CoCe, CoSrCe, and CoCuSrCe showing current density (lnj) vs. temperature (T−1) at the OER potential were charted to determine the activation energy. The temperature was varied from 25 to 50 °C with a gap of 5 °C. The activation energy (Ea) was obtained from the slope values in Fig. 5b. The activation energy of pristine CoCe was 14.8 kJ mol−1, whereas CoSrCe and CoCuSrCe exhibited activation energies of 12.3 and 11.7 kJ mol−1, respectively. The lower activation energy indicates faster OER kinetics over CoCuSrCe.
 | ||
| Fig. 5 (a) Scan rate dependency study and (b) activation energy plots of the oxygen evolution reactions over the synthesized materials. | ||
A good electrocatalyst with considerable charge transfer ability should have a low Tafel slope. Fig. 6a shows the lowest Tafel slope of 42 mV dec−1 for CoCuSrCe compared with 78 mV dec−1 over pristine CoCe. The low Tafel slope indicates an improved charge transfer between the doped electrode material and the electrolyte, rationalizing its efficient OER activity. The Nyquist impedance plots (Fig. 6b) also supported the OER efficacy trend of the materials. EIS measurements were performed under OER conditions (1 M KOH), and the data were fitted to the equivalent circuit shown in the inset of Fig. 6b. The equivalent circuit revealed one polarisation resistance (Rct) with a parallel constant-phase resistance. CoCuSrCe showed a smaller semicircle than those of CoSrCe and CoCe, suggesting faster shuttling of charges due to higher conductivity in the double-doped composite catalyst.55,56
 | ||
| Fig. 6 (a) Tafel plots and (b) Nyquist plots of the three composite catalysts. | ||
The electrochemical stability of the best catalyst was evaluated by chronoamperometry studies in a 1 M KOH solution for 22 h at the corresponding OER overpotential. A remarkably stable OER current density was observed with CoCuSrCe (Fig. 7). To corroborate this further, we also collected the LSV traces of CoCuSrCe before and after the 22 h chronoamperometry study. The inset in Fig. 7 demonstrates the almost identical current densities of the fresh and exhausted catalysts, signifying the outstanding stability of the catalyst even at a considerably strong alkaline condition.
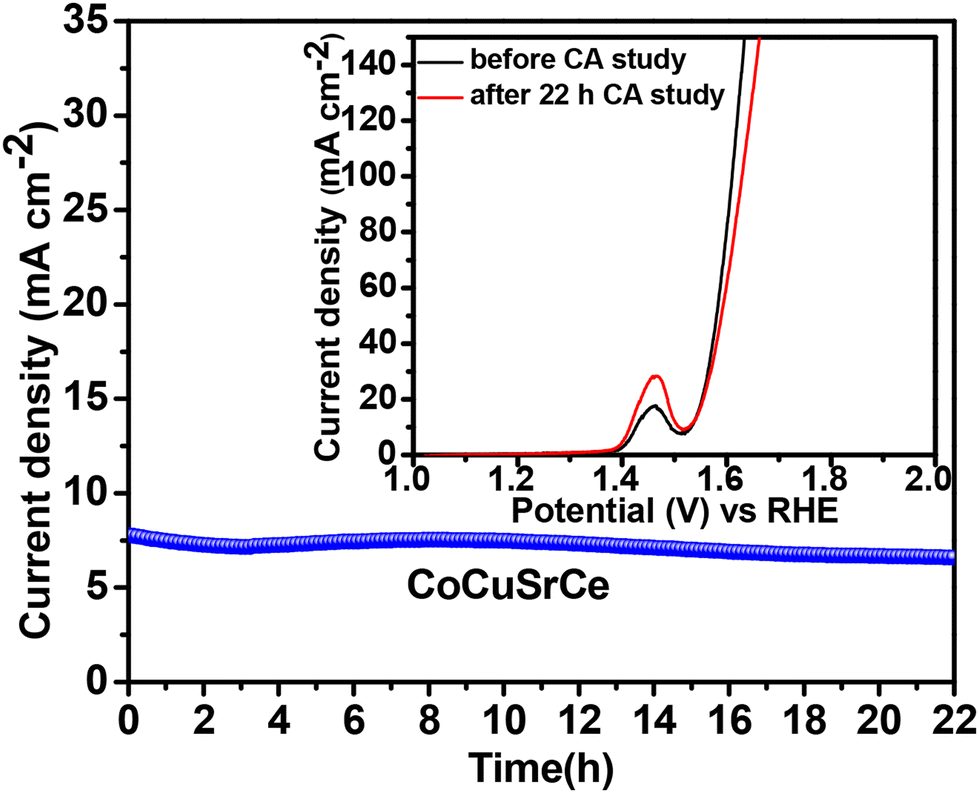 | ||
| Fig. 7 Chronoamperometry plots of the three synthesized catalysts. The inset shows the polarisation curves of CoCuSrCe before and after the 22 h chronoamperometry study. | ||
3.3 Mechanism
The superiority of the OER catalytic activity of double-doped CoCuSrCe intrigued us to probe the mechanism of the reaction in the presence of this material. In order to understand the role of the doped oxides, we thoroughly characterized the exhausted CoCuSrCe catalyst after 12 h of OER chronoamperometry. Fig. 8a shows the XRD pattern of the exhausted material along with the FESEM image in the inset. While the bulk crystalline structure remained unchanged, some minor additional peaks at 2θ = 35.4°, 39.06° and 40.9° were observed. These peaks correspond to the crystalline structures of Co(OH)2, Cu(OH)2 and Co–OOH formed during the OER.57–59 The crystalline peaks of M(OH)2 confirm the active involvement of Co and Cu in the OER reaction mechanism. The formation of M(OH)2 and their apparent oxidation to form M–OOH species evidently modifies the surface morphology of the exhausted CoCuSrCe, as observed in the FESEM image. | ||
| Fig. 8 (a) XRD pattern of CoCuSrCe and its FESEM image in the inset; the core level (b) Ce 3d (c) Co 2p spectra of all three exhausted composite oxides after 12 h of chronoamperometry OER; (d) O 1s of exhausted CoCuSrCe after the 12 h chronoamperometry study. | ||
The core-level deconvoluted Ce 3d XPS spectra of the exhausted catalysts are shown in Fig. 8b. The spectra reveal the same 5 spin–orbit coupled peaks corresponding to the Ce4+ and Ce3+ oxidation states as the fresh catalyst (Fig. 4a). However, after OER, the concentration of Ce4+ in the exhausted catalysts reduced to 61.3, 58.6 and 51% for CoCe, CoSrCe and CoCuSrCe, respectively. Notably, 65.6, 68.0 and 74.1% of Ce4+ were present in the as-prepared CoCe, CoSrCe and CoCuSrCe catalysts, respectively. This signifies the highly reducible nature of Ce4+ in these catalysts and that the extent of reduction of Ce4+ increased from CoCe to CoSrCe and CoCuSrCe. The reduction of Ce4+ → Ce3+ during the positive potential sweep of OER must occur alongside parallel oxidation reactions. These parallel oxidation reactions can be attributed to the oxidation of Co2+ → Co3+ and/or the evolution of lattice oxygen (O2− → ½O2 + 2e−) to create oxygen vacancies. Therefore, either the Co3+ ions or the adjacent oxygen vacancies are responsible for the formation of the –OOH species, which was also observed evidently in the XRD pattern of the exhausted catalyst. To corroborate this hypothesis, we collected the core-level Co 2p spectra of the exhausted catalysts (Fig. 8c). Interestingly, the relative ratios of Co3+/Co2+ increased to 66.5, 75.2, and 79.1% in CoCe, CoSrCe and CoCuSrCe, respectively. We further probed the O 1s spectra, and the deconvoluted spectrum is plotted in Fig. 8d. The extra peak at 535 eV is attributed to the intermediate –OOH formed during the OER. The corresponding peak, however, could be attributed to the generation of Co–OOH and/or Cu–OOH. The surface OL content (%) was found to be significantly reduced to 7.9% after the OER reaction. The comprehensive structural and surface study revealed that the facile reduction of Ce4+ → Ce3+ during the positive potential of OER helped the parallel occurrence of both oxidation of Co2+ → Co3+ and the evolution of lattice oxygen to create oxygen vacancies primarily on the surface of the oxides, and the extent of reduction of Ce4+ along with the oxidation of Co3+ was found to be the maximum in CoCuSrCe (Fig. 9). The formation of Co3+ along with oxygen vacancies due to the electronic interaction of the catalyst with the support helped the formation of the key intermediate –OOH, which in turn reduced the activation energy barrier and thereby enhanced the kinetics of oxygen evolution over the CoCuSrCe catalyst. However, in addition to Co3+, the presence of Cu2+ on the surface of CoCuSrCe also accelerated the OER efficacy, as observed from the XRD pattern and the core level O 1s XPS.
 | ||
| Fig. 9 Change in the content (%) of Ce4+, Co3+ and Cu2+ during the OER over the three catalysts. | ||
4. Conclusion
The three composite catalysts were synthesized by a single-step solution combustion route. The Rietveld refinement of the powder XRD pattern of CoCe revealed the co-existence of Co3O4/CeO2. Similarly, CoSrCe showed the presence of Co3O4/Ce0.9Sr0.1O2−δ and CoCuSrCe was composed of Co3−xCuxO4, Ce0.9Sr0.1O2−δ, SrCoO2.8 and CuO. The electrochemical surface area of doped CoCuSrCe was found to be 3 times higher than that of CoCe. In the electrocatalytic OER studies, the doped material CoCuSrCe outperformed the other two composites, showing the highest current density and highest faradaic efficiency at the lowest overpotential. CoCuSrCe also exhibited the lowest Tafel slope of 42 mV dec−1 and the lowest OER activation energy of 11.7 kJ mol−1. The outstanding stability of the catalyst CoCuSrCe was observed in the 12 h OER study at a considerably strong alkaline condition. The detailed mechanistic studies indicate that the favourable reducibility of cerium introduces a higher concentration of Co3+ ions at the catalytic sites, leading to enhanced formation of the Co–OOH species. Further, the lower-valence Sr and Cu ions doped at the Ce and Co sites, respectively, accelerate the formation of the active Co3+ species, as well as oxygen vacancies, aiding the efficient and facile progression of the oxygen evolution reaction. In addition to Co3+, the presence of Cu2+ on the surface of CoCuSrCe might also accelerate the OER kinetics, as evidenced by the XRD pattern and XPS spectrum of the exhausted catalyst. Overall, these findings underscore the significance of the electronic interactions between the active catalytic sites and the support, as well as the involvement of oxygen vacancies, in realizing sustainable advancement of the H2 economy in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank BITS Pilani Hyderabad Campus for the financial support.References
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Eng. Mater., 2017, 7, 1601275 Search PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- P. C. Meenu, S. Roy, C. Chakraborty and S. Roy, Adv Powder Technol, 2021, 32, 2663–2689 CrossRef CAS.
- A. Adruzzaman, A. Yuda, A. Ashok and A. Kumar, Inorg. Chim. Acta, 2020, 511, 119854 CrossRef.
- Y. Li, F. M. Li, X. Y. Meng, S. N. Li, J. H. Zeng and Y. Chen, ACS Catal., 2018, 8, 1913–1920 CrossRef CAS.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- M. Li and L. Feng, Chin. J. Struct. Chem., 2022, 41, 2201019–2201024 CAS.
- F. G. Wang, X. Liu, Q. X. Lv, B. Liu, Y. M. Chai and B. Dong, Chin. J. Struct. Chem., 2022, 41, 2209008–2209044 CAS.
- J. Qi, W. Zhang and R. Cao, Chem. Commun., 2017, 53, 9277–9280 RSC.
- A. Moysiadou, S. Lee, C. S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- S. Roy, N. Devaraj, K. Tarafder, C. Chakraborty and S. Roy, New J. Chem., 2022, 46, 6539–6548 RSC.
- Y. Liu, C. Ma, Q. Zhang, W. Wang, P. Pan, L. Gu, D. Xu, J. Bao and Z. Dai, Adv. Mater., 2019, 31, 1900062 CrossRef PubMed.
- J. X. Feng, S. H. Ye, H. Xu, Y. X. Tong and G. R. Li, Adv. Mater., 2016, 28, 4698–4703 CrossRef CAS PubMed.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS PubMed.
- J. H. Kim, K. Shin, K. Kawashima, D. H. Youn, J. Lin, T. E. Hong, Y. Liu, B. R. Wygant, J. Wang, G. Henkelman and C. B. Mullins, ACS Catal., 2018, 8, 4257–4265 CrossRef CAS.
- P. C. Meenu and S. Roy, ACS Appl. Energy Mater., 2023, 6, 11212–11225 CrossRef CAS.
- P. C. Meenu and S. Roy, ACS Appl. Mater. Interfaces, 2023, 15, 36154–36166 CrossRef.
- J. Kim, P. C. Shih, K. C. Tsao, Y. T. Pan, X. Yin and C. J. Sun, J. Am. Chem. Soc., 2017, 139, 12076–12083 CrossRef CAS PubMed.
- K. Sardar, S. C. Ball, J. D. B. Sharman, D. Thompsett, J. M. Fisher, R. A. P. Smith, P. K. Biswas, M. R. Lees, R. J. Kashtiban, J. Sloan and R. I. Walton, Chem. Mater., 2012, 24, 4192–4200 CrossRef CAS.
- J. Park, M. Park, G. Nam, M. G. Kim and J. Cho, Nano Lett., 2017, 17, 3974–3981 CrossRef CAS PubMed.
- J. B. Goodenough, R. Manoharan and M. Paranthaman, J. Am. Chem. Soc., 1990, 112, 2076–2082 CrossRef CAS.
- P. Bothra and S. K. Pati, ACS Energy Lett., 2016, 1, 858–862 CrossRef CAS.
- Q. Wang, X. Xue, Y. Lei, Y. Wang, Y. Feng, X. Xiong, D. Wang and Y. Li, Small, 2020, 16, 2001571 CrossRef CAS.
- Y. Zheng, S. Chen, K. A. I. Zhang, J. Zhu, J. Xu, C. Zhang and T. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 13328–13337 CrossRef CAS.
- X. Jiang, Y. Dong, Z. Zhang, J. Li, J. Qian and D. Gao, J. Alloys Compd., 2021, 878, 160433 CrossRef CAS.
- A. Valipour, N. Hamnabard, S. M. H. Meshkati, M. Pakan and Y. H. Ahn, Dalton Trans., 2019, 48, 5429–5443 RSC.
- J. H. Scofield and J. Electron, Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- R. Rameshan, P. Pentyala, S. A. Singh, P. A. Deshpande and S. Roy, J. Environ. Chem. Eng., 2022, 10, 108966 CrossRef CAS.
- S. Payra and S. Roy, J. Phys. Chem. C, 2021, 125, 8497–8507 CrossRef CAS.
- T. Baidya, T. Mazumder, K. Y. Koltunov, P. R. Likhar, A. H. Clark, K. Tiwari, V. I. Sobolev, S. Payra, T. Murayama, M. Lin, P. Bera, S. Roy, K. Biswas, O. Safonova, B. Srinivasa Rao and M. Haruta, J. Phys. Chem. C, 2020, 124, 14131–14146 CrossRef CAS.
- C. Wang, L. Zeng, W. Guo, C. Gong and J. Yang, RSC Adv., 2019, 9, 35646–35654 RSC.
- M. A. Khan, N. Nayan, Shadiullah, M. K. Ahmad and C. F. Soon, Nanomaterials, 2020, 10, 1298 CrossRef CAS PubMed.
- S. Payra, S. Kanungo and S. Roy, Nanoscale, 2022, 14, 13352–13361 RSC.
- S. Payra, N. Devaraj, K. Tarafder and S. Roy, ACS Appl. Energy Mater., 2022, 5, 4945–4955 CrossRef CAS.
- V. S. Kirankumar and S. Sumathi, J. Mater. Sci.: Mater. Electron., 2018, 29, 8738–8746 CrossRef CAS.
- S. K. Shinde, D. P. Dubal, G. S. Ghodake and V. J. Fulari, RSC Adv., 2015, 5, 4443–4447 RSC.
- F. C. de Godoi, E. Rodriguez-Castellon, E. Guibal and M. M. Beppu, Chem. Eng. J., 2013, 234, 423–429 CrossRef CAS.
- A. Hezam, K. Namratha, Q. A. Drmosh, D. Ponnamma, J. Wang, S. Prasad, M. Ahamed, C. Cheng and K. Byrappa, ACS Appl. Nano Mater., 2020, 3, 138–148 CrossRef CAS.
- T. Ye, W. Huang, L. Zeng, M. Li and J. Shi, Appl. Catal., B, 2017, 210, 141–148 CrossRef CAS.
- D. Jiang, W. Wang, L. Zhang, Y. Zheng and Z. Wang, ACS Catal., 2015, 5, 4851–4858 CrossRef CAS.
- S. Payra, S. K. Ganeshan, S. Challagulla and S. Roy, Adv. Powder Technol., 2020, 31, 510–520 CrossRef CAS.
- A. K. Venugopal, A. T. Venugopalan, P. Kaliyappan and T. Raja, Green Chem., 2013, 15, 3259–3267 RSC.
- C. W. Tang, C. B. Wang and S. H. Chien, Thermochim. Acta, 2008, 473, 68–73 CrossRef CAS.
- A. Mehboob, S. R. Gilani, A. Anwar, A. Sadiqa, S. Akbar and J. Patujo, J. Appl. Electrochem., 2021, 51, 691–702 CrossRef CAS.
- S. Sun, H. Li and Z. J. Xu, Joule, 2018, 2, 1024–1027 CrossRef.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- H. Y. Wang, S. F. Hung, H. Y. Chen, T. S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
- X. Xiao, C. T. He, S. Zhao, J. Li, W. Lin, Z. Yuan, Q. Zhang, S. Wang, L. Dai and D. Yu, Energy Environ. Sci., 2017, 10, 893–899 RSC.
- C. Alex, S. C. Sarma, S. C. Peter and N. S. John, ACS Appl. Energy Mater., 2020, 3, 5439–5447 CrossRef CAS.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CrossRef CAS PubMed.
- Z. Zhao, M. Yu, Y. Liu, T. Zeng, R. Ye, Y. Liu, J. Hu and A. Li, Adv. Energy Sustainability Res., 2023, 4, 2300123 CrossRef CAS.
- P. C. Meenu, P. K. Samanta, T. Yoshida, N. J. English, S. P. Datta, S. A. Singh, S. Dinda, C. Chakraborty and S. Roy, ACS Appl. Energy Mater., 2022, 5, 503–515 CrossRef.
- Q. Xu, H. Jiang, H. Zhang, H. Jiang and C. Li, Electrochim. Acta, 2018, 259, 962–967 CrossRef CAS.
- Y. Xiao, Y. Wang, M. Xiao, C. Liu, S. Hou, J. Ge and W. Xing, NPG Asia Mater., 2020, 12, 73 CrossRef CAS.
- Y. Si, C. Guo, C. Xie and Z. Xiong, Materials, 2018, 11, 1912 CrossRef PubMed.
- Z. Zhai, Y. You, L. Ma, D. Jiang, F. Li, H. Yuan, M. Zheng and W. Shen, Nanoscale Res. Lett., 2019, 14, 167 CrossRef PubMed.
- S. Song, H. Bao, X. Lin, X. L. Du, J. Zhou, L. Zhang, N. Chen, J. Hu and J. Q. Wang, J. Energy Chem., 2020, 42, 5–10 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The reliability factors along with refined lattice parameters of the catalysts, N2 adsorption isotherm, CV curves in the non-faradaic region at different scan rates, XPS, fluorescence and Raman spectra of the catalysts. See DOI: https://doi.org/10.1039/d4dt00315b |
| This journal is © The Royal Society of Chemistry 2024 |