
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-induced spin-state switching in Fe(II) spin-crossover complexes with thiazole-based chelating ligands†
Minyoung
Jo
a,
Botagoz
Amanyazova
 ab,
Sandugash
Yergeshbayeva
a,
Miguel
Gakiya-Teruya
a,
Ökten
Üngör
a,
Paola
Lopez Rivera
ac,
Natalie
Jen
a,
Evgeny
Lukyanenko
d,
Alexander V.
Kurkin
d,
Rakhmetulla
Erkasov
b,
Mark W.
Meisel
e,
Andreas
Hauser
f,
Pradip
Chakraborty
*g and
Michael
Shatruk
*a
ab,
Sandugash
Yergeshbayeva
a,
Miguel
Gakiya-Teruya
a,
Ökten
Üngör
a,
Paola
Lopez Rivera
ac,
Natalie
Jen
a,
Evgeny
Lukyanenko
d,
Alexander V.
Kurkin
d,
Rakhmetulla
Erkasov
b,
Mark W.
Meisel
e,
Andreas
Hauser
f,
Pradip
Chakraborty
*g and
Michael
Shatruk
*a
aDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, Florida 32306, USA. E-mail: shatruk@chem.fsu.edu
bDepartment of Chemistry, L.N. Gumilyov Eurasian National University, Astana 010008, Kazakhstan
cDepartment of Chemistry, University of Puerto Rico at Humacao, Humacao, PR 00792, Puerto Rico
dDepartment of Chemistry, Lomonosov Moscow State University, Moscow, 119234, Russia
eDepartment of Physics and National High Magnetic Field Laboratory, University of Florida, Gainesville, Florida 32611, USA
fDepartment of Physical Chemistry, University of Geneva, CH-1211, Geneva 4, Switzerland
gDepartment of Chemistry, Indian Institute of Technology, Kharagpur 721 302, India. E-mail: pradipc@chem.iitkgp.ac.in
First published on 29th May 2024
Abstract
Homoleptic complexes [Fe(4bt)3](ClO4)2 (1), [Fe(2bt)3](ClO4)2 (2), and [Fe(3tpH)3](ClO4)2 (3) were obtained by a reaction between the Fe(II) precursor salt and the corresponding thiazole-based bidentate ligand (L = 4bt = 4,4′-bithiazole, 2bt = 2,2′-bithiazole, 3tpH = 3-(thiazol-2-yl)pyrazole). X-ray crystal structure determination revealed crystallization of solvent-free complex 1, a solvate 2·MeOH, and a co-crystal 3·2(3tpH). The crystal packing of all these complexes is dominated by one-dimensional interactions between the [Fe(L)3]2+ cations. These interactions are stronger in 2·MeOH and 3·2(3tpH), leading to cooperative and slightly hysteretic transitions between the high-spin and low-spin electronic configurations at ∼235 K and 159 K, respectively. In contrast, weaker intermolecular interactions in 1 result in a gradual spin crossover above 300 K, with the maximum fraction of the HS state ∼25% achieved at 400 K. Complexes 2 and 3·2(3tpH) exhibit light-induced excited spin state trapping (LIESST) under irradiation with white light or a 532 nm laser at 5 K. After the photoexcitation, the trapped metastable HS state relaxes to the ground LS state with the average relaxation temperature of 81 K and 68 K, respectively. Examination of the relaxation dynamics by optical absorption spectroscopy on a single crystal of 3·2(3tpH) revealed the sigmoidal shape of the relaxation curves at lower temperatures, attributed to cooperative effects, as well as a plateau at ∼10% of the HS fraction at intermediate temperatures, hinting at a more complex mechanism for the relaxation of the LIESST phase in this material.
Introduction
Recently, we have explored a correlation between the geometric parameters of chelating diimine-type ligands and the spin state of their homoleptic complexes with the Fe(II) ions.1 The outcome of that study was a simple yet effective empirical rule that predicted interconversion between the high-spin (HS) and low-spin (LS) electronic configurations for Fe(II) complexes formed with ligands in which the N⋯N separation between the N-donor sites ranges from 2.78 to 2.93 Å. Such spin crossover (SCO) is well known to be driven by changes in temperature, pressure, or light irradiation.2 Ligands with the N⋯N separation below or above this range typically yield Fe(II) complexes that exist only in the LS or HS state, respectively. The effectiveness of this simple rule for spin-state prediction is explained by the optimal alignment of the σ-donating orbitals on the N sites with the eg-type orbitals on the octahedrally coordinated metal site. The smaller/larger N⋯N separation in the free ligand is better suited for the geometry of the LS/HS state – as dictated by the typical Fe–N bond lengths in the corresponding electronic configuration,3 – thus resulting in the higher stability of that configuration.Interestingly, in the course of that study, we observed that the empirically found N⋯N separation boundaries between the LS and SCO (2.78 Å) or the HS and SCO (2.93 Å) regions are quite accurate, and even minor changes in the ligand geometry might be sufficient to cross from one type of behavior to the other. Thus, the 2,2′-biimidazole ligand, composed of two C–C bonded five-member rings, yields only HS complexes, in agreement with the larger separation between its N-donor sites (2.99 Å).4 Substituting the N-only heterocycle with a mixed N,S-heterocycle leads to an increase in the size of the 5-member ring, pushing the N-donor sites closer together and resulting in SCO behavior in homoleptic Fe(II) complexes with 4,4′-bithiazole (4bt), 2,2′-bithiazole (2bt), and 3-(thiazol-2-yl)pyrazole (3tpH) ligands (Scheme 1), for which the N⋯N separation equals 2.81 Å, 2.91 Å, and 2.85 Å, respectively.1
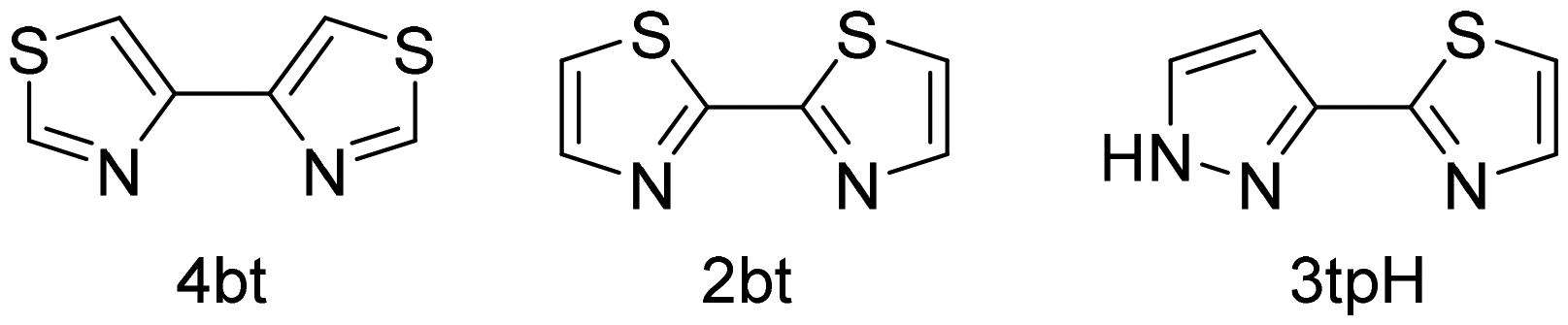 | ||
| Scheme 1 N,S-heterocyclic ligands used for the synthesis of homoleptic Fe(II) complexes in this work. | ||
Despite the simple structure of these thiazole-based ligands (Scheme 1), the magnetic properties of their Fe(II) complexes have remained relatively underexplored since the initial reports of SCO in [Fe(4bt)3](ClO4)2 (1),5 [Fe(2bt)3](ClO4)2 (2),6 and [Fe(3tpH)3](ClO4)2 (3).7 In particular, there is lack of any information regarding the possibility of light-induced excited spin state trapping (LIESST), which arguably represents one of the most appealing effects shown by SCO materials, both from the fundamental and applied viewpoints.8,9 Therefore, we decided to re-examine the SCO properties of complexes 1–3, especially as relevant to the possible LIESST behavior. In the present work, we report the study of structural and magnetic properties of these complexes and demonstrate a pronounced and quantitative LIESST effect in complexes 2 and 3. In addition, we observe an unusual two-step relaxation behavior for the metastable light-induced HS state generated in complex 3.
Results and discussion
Synthesis
Complexes 1–3 were prepared by direct reactions between [Fe(H2O)6](ClO4)2 and the corresponding ligand. Goodwin et al. reported the syntheses in refluxing ethanol,5–7 but we found that these complexes can be obtained by performing the reactions at room temperature (r.t.) in anhydrous methanol (MeOH, for 1 and 2) or acetonitrile (MeCN, for 3). While the earlier reports mentioned the products precipitating upon cooling of the ethanolic solutions, in our experiments the target complexes remained soluble in the corresponding solvents at r.t. Their crystallization was achieved by either slow evaporation of the solutions of 1 and 2 in MeOH or by slow diffusion of Et2O into the MeCN solution of 3. Once isolated by filtration and briefly dried by suction, the complexes appeared to be sufficiently air-stable, although the samples were kept in a desiccator for prolonged storage.Crystal structures
We note that only crystallization of 1 was reported in the earlier works.5 Attempts to crystallize 2 from EtOH were unsuccessful, leading to [Fe(2bt)2(H2O)2](ClO4)2·4(2bt),10 although analytically pure complex 2 was precipitated from a hot ethanolic solution.6 The cation of complex 3 was crystallized with a different anion, [Fe(3tpH)3](BF4)2·1.5H2O, while the perchlorate salt was obtained only with one of the ligands deprotonated, [Fe(3tpH)2(3tp)](ClO4).7 We did not encounter any substantial difficulties in crystallizing either of these homoleptic 2+ cations as perchlorate salts. Similar to the earlier findings,5 we observed crystallization of 1 as a solvent-free complex in the C2/c space group, with the r.t. monoclinic unit cell parameters (a = 17.2500(3) Å, b = 9.8245(2) Å, c = 16.8597(3) Å, β = 105.972(2)°) being in good agreement with the literature data (a = 17.250 Å, b = 9.817 Å, c = 16.867 Å, β = 105.94°). In contrast, complex 2 crystallized as solvate, 2·MeOH, and complex 3 co-crystallized with the free ligand as 3·2(3tpH). The crystal structures of these two complexes are reported here for the first time.In agreement with the crystal structure analysis discussed below, TGA experiments (Fig. S1†) revealed negligible mass losses for solvent-free powder samples of 1 and 3·2(3tpH) between r.t. and 500 K. On the other hand, a powder sample of 2·MeOH showed ∼5.1% decrease in mass when heated to 410 K, which corresponds to ∼1.3 interstitial MeOH molecule per formula unit of the complex, in reasonable agreement with the results of elemental analysis. The interstitial solvent, however, could not be removed from single crystals of 2·MeOH, even when they were exposed to vacuum (∼100 mbar) for 3 h at 380 K. This difference in the desolvation behavior of the single-crystal vs. powder samples can be attributed to the higher surface area of the latter, which facilitates the solvent removal.
Complex 1 crystallizes in the space group C2/c, as reported previously,5 and is isostructural with the analogous Ni(II) and Ru(II) complexes.5,11 The asymmetric unit (ASU) contains a half of the [Fe(4bt)3]2+ cation and one ClO4− anion (Fig. S2a†). The metal ion resides on a 2-fold rotation axis. The Fe–N bond lengths and the Σ90 angular distortion parameter1 observed at 300 K indicated the fully LS state for the Fe(II) ion (Table 1). Therefore, the crystal structure determination at lower temperatures was not pursued, since only increasing the temperature can drive the conversion to the HS state.
| Complex | 1 | 2·MeOH | 3·2(3tpH) | ||||
|---|---|---|---|---|---|---|---|
| temperature | 300 K | 300 K | 230 K | 100 K | 300 K | 230 K | 100 K![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
| a Due to the more complex crystal structure and poorer data quality, only the range of Fe–N distances is provided for each of the three Fe centers in the structure of 3·2(3tpH) at 100 K, and the dav(Fe–N) and Σ90 parameters have been averaged over the three Fe sites. | |||||||
| d(Fe–N), Å | 1.974(2) (×2) | 2.152(2) | 2.142(1) | 1.965(3) | 2.155(2) (×2) | 2.158(2) (×2) | 1.96(1)–1.99(1) for Fe(1) |
| 1.979(2) (×2) | 2.175(2) | 2.156(1) | 1.976(3) | 2.184(2) (×2) | 2.189(2) (×2) | 1.96(2)–2.02(2) for Fe(2) | |
| 1.974(2) (×2) | 2.186(2) | 2.169(1) | 1.976(3) | 2.195(2) (×2) | 2.194(2) (×2) | 1.97(1)–2.04(2) for Fe(3) | |
| 2.189(2) | 2.171(1) | 1.986(3) | |||||
| 2.210(2) | 2.194(1) | 1.986(3) | |||||
| 2.212(2) | 2.201(1) | 1.987(3) | |||||
| d av(Fe–N), Å | 1.976(1) | 2.187(1) | 2.172(1) | 1.979(1) | 2.178(1) | 2.180(1) | 1.982(2) |
| Σ 90 (°) | 49.8(3) | 97.1(2) | 94.8(2) | 57.5(4) | 85.9(3) | 85.1(3) | 61(1) |
The crystal packing of 1 is characterized by columns of the [Fe(4bt)3]2+ cations separated by the ClO4− anions (Fig. 1a). The columns run parallel to the c axis and feature π–π interactions between the 4bt ligands of neighboring cations (Fig. 1b), with the plane-to-plane distance of 3.53 Å at 300 K. The cohesion between the columns is provided by weak intermolecular O⋯H contacts between the ClO4− anions and the peripheral H atoms of the ligands (Fig. S3a†). These O⋯H distances vary from 2.477 Å to 2.681 Å, in agreement with the typical O⋯H van der Waals (vdW) interactions observed at 2.46–2.68 Å.12 Such crystal packing, with the weak intermolecular interactions, is not conducive to a cooperative spin transition, in agreement with the results of magnetic measurements discussed below.
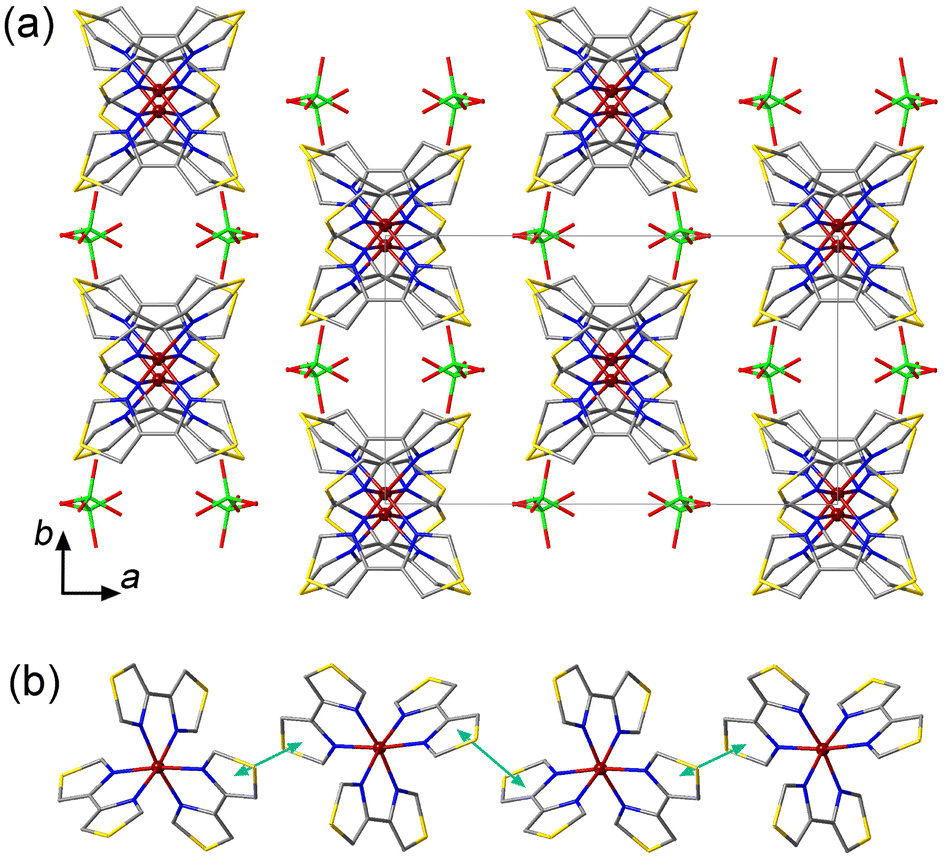 | ||
| Fig. 1 (a) The crystal packing of 1 viewed down the c axis to reveal the columns of [Fe(4bt)3]2+ cations. The unit cell is indicated with solid gray lines. The H atoms are omitted for clarity. (b) A side view of a single column of cations, highlighting the π–π interactions with light-blue arrows. Color scheme: Fe = garnet, S = gold, Cl = green, O = red, N = blue, C = gray. | ||
Complex 2 crystallized as a solvate 2·MeOH in the space group P21/n. The ASU contains one [Fe(2bt)3]2+ cation, two ClO4− anions, and one interstitial MeOH molecule (Fig. S2b†). In contrast to structure 1, the Fe–N bond lengths and the Σ90 angular distortion parameter at 300 K indicate the fully HS state for the Fe(II) ion (Table 1). At 230 K, the average Fe–N bond length and the Σ90 parameter decrease only slightly, but at 100 K, they become similar to the values observed for complex 1 at 300 K, thus indicating the conversion of complex 2 to the LS state.
Similar to the structure of 1, the crystal packing of 2·MeOH features columns of the [Fe(2bt)3]2+ cations extending along the c axis (Fig. 2a and b). The interactions between the cations along the columns, however, are more extensive as compared to those observed for 1. In the latter, the π–π contacts are observed only between single thiazole rings of the 4bt ligands, but in the structure of 2·MeOH the intermolecular π–π contacts involve both thiazole rings of the 2bt ligand (Fig. 2c). At 300 K, the average interplanar distance between the π–π-interacting 2bt ligands alternates between the values of 3.56 Å and 3.73 Å along the column. (At 100 K, these distances decrease only slightly, to 3.50 Å and 3.69 Å, respectively.) Due to the change in the character of the π–π interactions, the cationic columns in 2·MeOH also appear more undulated (Fig. 2b) as compared to those observed in 1 (Fig. 1b), which allows the ClO4− anions and MeOH molecules to occupy the space within the columns. As shown by magnetic studies described below, such change in the crystal packing has a pronounced effect on the SCO behavior.
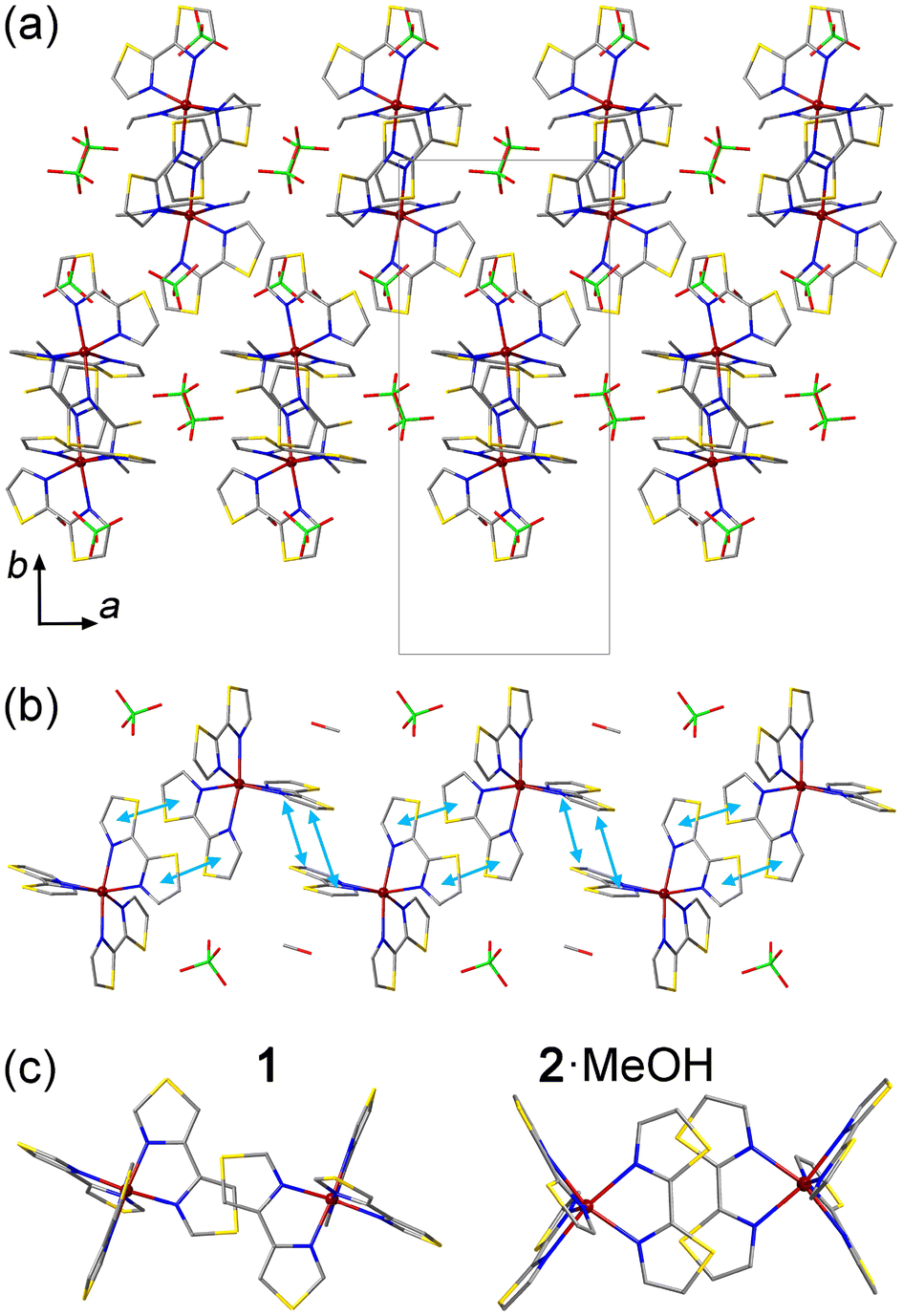 | ||
| Fig. 2 (a) The crystal packing of 2·MeOH viewed down the c axis to reveal the columns of [Fe(2bt)3]2+ cations. The unit cell is shown with solid gray lines. The H atoms are omitted for clarity. (b) A side view of a single column of cations, highlighting the π–π interactions with light-blue double arrows. (c) The top view of π–π overlaps between the 4bt and 2bt ligands in 1 and 2·MeOH, respectively. Color scheme: Fe = garnet, S = gold, Cl = green, O = red, N = blue, C = gray. | ||
Each ClO4− anion or MeOH molecule located within the columns of SCO cations exhibits vdW interactions with one of the surrounding 2-bt ligands (Fig. S3b†). The MeOH molecule forms a rather close H⋯S contact at 2.397 Å, but its vdW distances to other ligands or the nearest ClO4− anion exceed 3 Å, thus indicating negligible vdW forces. The ClO4− anion forms two notable O⋯H interactions with the peripheral H atoms of one 2-bt ligand, at 2.680 Å and 2.844 Å, but two other O⋯H distances substantially exceed 3 Å. Thus, the ClO4− anions and MeOH molecules located within the columns appear to be trapped by the vdW interactions, which are especially significant in the case of the interstitial solvent, thus justifying the difficulty in removing this solvent from the crystal structure.
The ClO4− anions located between the columns of cations form three vdW bonds to the pair of 2-bt ligands engaged in the π–π interaction, with the O⋯H distances of 2.497 Å, 2.551 Å, and 2.782 Å (Fig. S3c†), while the fourth O atom does not exhibit any noteworthy vdW interactions. Overall, we can conclude that the interstitial species do not provide any strong connections between the columns of cations, and the cooperativity of the spin transition, which will be discussed in the section on magnetic properties, arises primarily due to the interaction between the cations along the columns.
Complex 3 co-crystallized with a free ligand, affording crystals of 3·2(3tpH) in the space group Pbcn at 300 K. Although the only previously reported crystal structure of the [Fe(3tpH)3]2+ cation was obtained as a hydrate with the BF4− anion, [Fe(3tpH)3](BF4)2·1.5H2O,7 the Ni(II) salt was obtained as a co-crystal with the free ligand, [Ni(3tpH)3](BF4)2·2(3tpH),7 isostructural to 3·2(3tpH) obtained in our work. The ASU of 3·2(3tpH) contains a half of the [Fe(3tpH)3]2+ cation, one ClO4− anion, and one free 3tpH molecule (Fig. S2c†). The cation resides on a 2-fold rotation axis, which causes one of the coordinated 3tpH ligands to be disordered, due to its asymmetric nature. Crystallographic disorder was also observed for the non-coordinated 3tpH ligand and the ClO4− anion. The Fe–N bond lengths and the Σ90 parameter are characteristic of the HS Fe(II) ion at both 300 K and 230 K (Table 1).
The crystal data obtained at 100 K were of much lower quality. As the crystal of 3·2(3tpH) was cooled from 230 K to 100 K, its color changed from yellow to red and the diffraction spots became more diffuse, most likely, due to the strain introduced by the abrupt spin transition that takes place at ∼150 K (see below). Even relatively slow cooling through this temperature region (at 1 K min−1) did not alleviate the problem. Nevertheless, we were able to establish a reliable model of the structure (Table S1†). The spin transition causes lowering of the space group symmetry to Pna21 and tripling of the unit cell volume. The transformation of the unit cell parameters from the high-temperature HS phase to the low-temperature LS phase is described by aLS = cHS, bLS = bHS, cLS = 3aHS. The ASU of the low-temperature phase contains three unique [Fe(3tpH)3]2+ cations, six ClO4− anions, and six 3tpH molecules. The substantial decrease in both the Fe–N bond lengths and the Σ90 parameter (Table 1) clearly indicate the formation of the LS state at all Fe sites.
The crystal packing of 3·2(3tpH) bears features related to the crystal packings of both 1 and 2·MeOH. Similar to 1, the [Fe(3tpH)3]2+ cations form columns along the c axis of the HS structure, and the free 3tpH ligands and ClO4− anions are located only between the columns (Fig. 3a). Similar to 2·MeOH, both aromatic rings of the 3tpH ligands are involved in intermolecular π–π contacts (Fig. 3b), with the interplanar distance of 3.65 Å at 300 K. A comparison of the unit cell parameters (Table S1†), taking into account the relationships given above, indicates that the change in the Fe–N bond lengths caused by the spin transition mainly affects the parameters perpendicular to the direction of the columns of SCO cations, while the parameter cHS (aLS) along the columns remains essentially unchanged. Evidently, the stronger intermolecular interactions along the columns force the large SCO-induced structural changes to be accommodated in the direction perpendicular to these interactions.
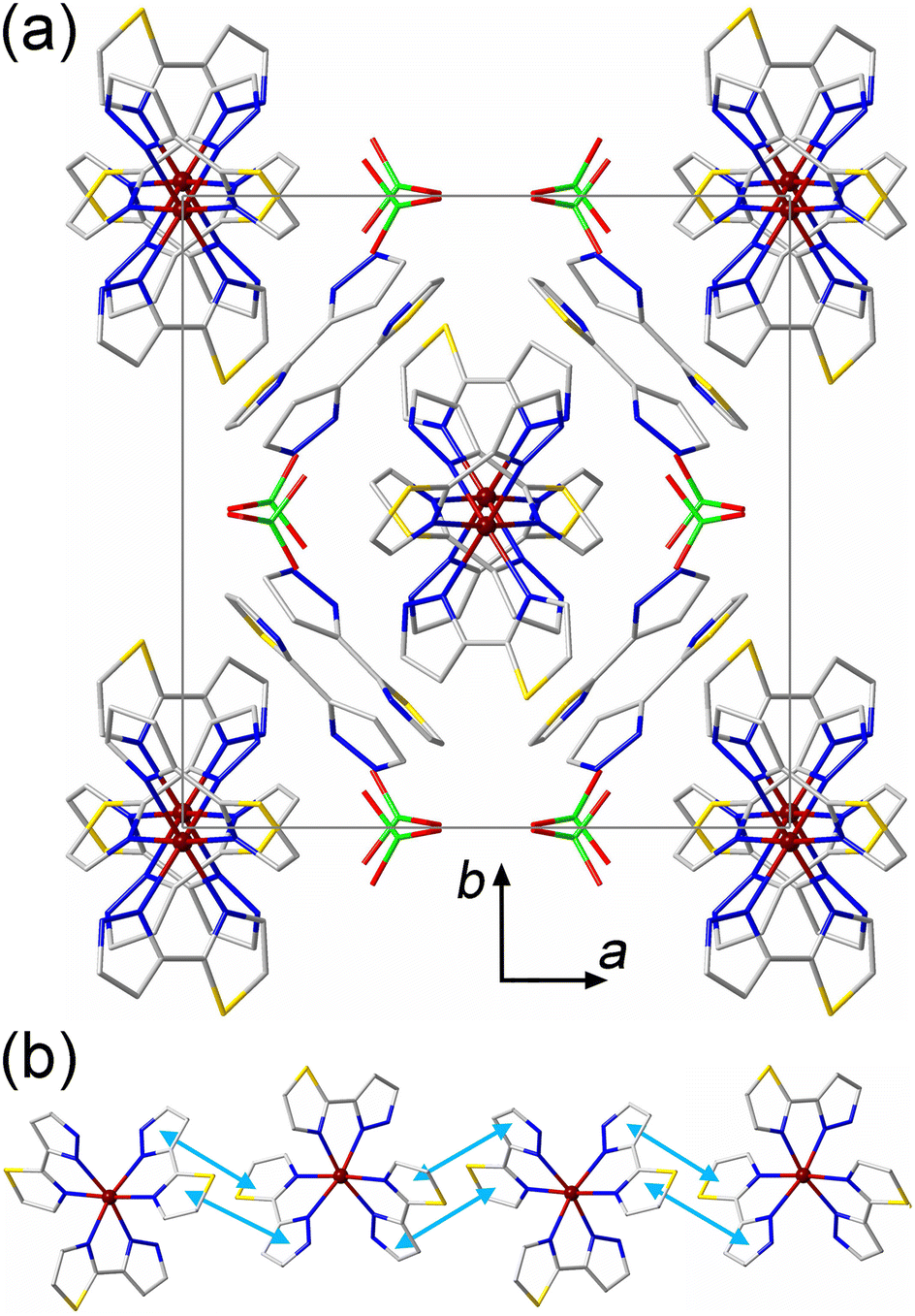 | ||
| Fig. 3 (a) The crystal packing of 3·2(3tpH) viewed down the c axis to reveal columns of [Fe(3tpH)3]2+ cations. The unit cell is indicated with solid gray lines. The H atoms are omitted for clarity. The disorders of the 3tpH ligands and ClO4− anions are not shown. (b) A side view of a single column of cations, showing π–π interactions with light-blue double arrows. Color scheme: Fe = garnet, S = gold, Cl = green, O = red, N = blue, C = gray. | ||
For each interstitial 3tpH ligand in the structure of 3·2(3tpH), the thiazole ring engages in a π–π interaction with a coordinated 3tpH ligand from one of the columns of cations, while the pyrazolyl ring exhibits a vdW contact with a 3tpH ligand from the other column at the N⋯H distance of 2.649 Å (Fig. S3d†). The ClO4− anions also show vdW interactions to the peripheral H atoms of coordinated 3tpH ligands with the O⋯H distances of 2.366 Å, 2.592 Å, and 2.686 Å. Thus, the crystal structure of 3·2(3tpH) exhibits more extensive vdW interactions between the interstitial species and the columns of cations as compared to the vdW forces observed in the crystal structure of 2·MeOH.
Magnetic properties
The powder samples of complexes 1–3 were studied by temperature-dependent measurements of magnetic susceptibility (χ). The magnetic behavior of complex 1 was previously reported only to 333 K.5 In agreement with the literature data and the LS state of the complex established by crystal structure determination at 300 K, the χT product was found to be close to zero (0.039 emu K mol−1) at this temperature. At higher temperatures, however, we observed a gradual SCO. While Baker and Goodwin reported the χT value of ∼0.27 emu K mol−1 at 333 K,5 we found a slightly lower value at this temperature (Fig. 4a). Nevertheless, we observed that χT reached 0.94 emu K mol−1 at 400 K. Based on the χT value observed for complex 2 in the HS state (see below), we estimate that complex 1 exhibits ∼25% conversion to the HS state at 400 K.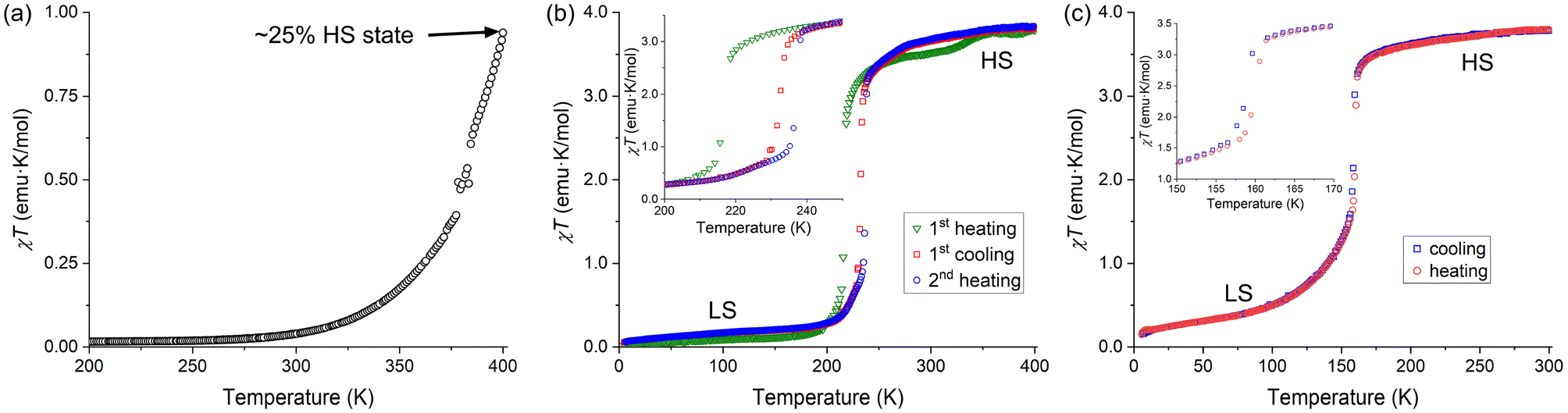 | ||
| Fig. 4 The temperature dependence of the χT product for complexes 1 (a), 2·MeOH (b), and 3·2(3tpH) (c), measured under the cooling or heating rate of 2 K min−1. The order of processes indicated in the legends corresponds to the actual sequence of measurements performed in the heating and cooling modes. The insets in panels (b) and (c) show an enlarged region of each plot around the spin-transition temperature (T1/2). Note that in the case of complex 2 the interstitial solvent has been lost by the end of the first heating cycle (see the discussion in the text for details). | ||
In contrast to 1, complexes 2 and 3 showed abrupt temperature-driven spin transitions. In the case of 2·MeOH, the initial heating of the sample from 5 K to 400 K revealed a transition from the LS to HS state with the midpoint at T1/2 = 217 K (the green triangles in Fig. 4b). The χT product reached 3.52 emu K mol−1 at 300 K. These findings agree with the LS and HS states of the complex established by crystal structure determination at 100 K and 300 K, respectively (Table 1). Heating the sample above 300 K revealed a small anomaly above 330 K, with the χT value increasing to ∼3.80 emu K mol−1 by 400 K. Next, the sample was cooled to 5 K and heated back to 400 K (the red squares and blue circles, respectively, in Fig. 4b). This cycle of measurements again revealed an abrupt spin transition with a small thermal hysteresis (T1/2,↓ = 232 K, T1/2,↑ = 237 K) and a disappearance of the high-temperature anomaly. Both the anomaly observed in the first heating branch and the minor shift in the spin-transition temperature in the following cooling–heating cycle can be signs of the solvent loss caused by the combination of heating and vacuum in the magnetometer chamber. Indeed, the TGA measurements on a powder sample of 2·MeOH showed that the material readily lost the interstitial solvent when heated to 400 K (Fig. S1b†). The sample obtained after magnetic measurements was subjected to TGA, which revealed no solvent loss to 475 K (Fig. S3†), thus corroborating our hypothesis.
It is interesting to compare our findings with those reported by Goodwin et al. for complex 2 obtained from ethanolic solutions.6 Although they were not able to determine the crystal structure of the complex, they did establish, by elemental analysis, the formation of analytically pure solvent-free samples precipitated from hot ethanol. They also found that 2, isolated in such a manner, existed as two different polymorphs that were quite unstable, difficult to prepare selectively, and readily hydrolyzed upon contact with water. One of these polymorphs showed a gradual SCO above 300 K, similar to our observation for complex 1. The other polymorph, however, showed an abrupt spin transition with T1/2,↓ = 230 K and T1/2,↑ = 235 K, which is very similar to our observations for 2·MeOH after it has been heated to 400 K. This comparison provides additional support for our hypothesis about the formation of solvent-free 2 under high temperature and vacuum of the magnetometer chamber. It is also possible that the presence of the interstitial MeOH molecules imparts additional stability to this complex under ambient conditions, as we did not observe any substantial deterioration of 2·MeOH, even when it was stored in air for several days.
An abrupt spin transition with a negligible thermal hysteresis (T1/2,↓ = 158 K, T1/2,↑ = 159 K) was also observed for 3·2(3tpH). In this case, the maximum χT value observed in the HS state was 3.80 emu K mol−1, very similar to the values observed in the HS state of complex 2. The occurrence of the complete spin transition as a function of temperature is also in agreement with the results of the crystal structure analysis presented above (Table 1). The very steep nature of the spin transition curve in cooling mode indicates that the cooperative effects are strong and the system is close to the point of hysteresis behavior. The small observed hysteresis of 1 K could be of kinetic origin, due to the scan rate of 2 K min−1. We also note that the SCO curve becomes rather gradual at temperatures below T1/2. Such behavior might be related to the lowering of crystallographic symmetry, which leads to appearance of three crystallographically unique Fe sites in the LS structure and emergence of extended defects, as evident from the decrease in the quality of the X-ray diffraction pattern.
Goodwin et al. obtained a related complex with the ClO4− anions but with deprotonation of one of the ligands.7 Both [Fe(3tpH)2(3tp)](ClO4) and [Fe(3tpH)2(3tp)](BF4) showed gradual SCO between 80 K and 300 K. Unfortunately, the crystal structures of these complexes were not established and the magnetic properties of [Fe(3tpH)3](BF4)2·1.5H2O, crystallized from an ethanolic solution, were not reported, which makes it impossible to make a reasonable comparison to the crystal structure and magnetic behavior of 3·2(3tpH) reported here.
Light-induced spin-state switching
The abrupt SCO observed for 2 and 3·2(3tpH) suggested that these materials could exhibit light-induced excited spin state trapping (LIESST). Consequently, they were subjected to photomagnetic studies by irradiating the samples in the magnetometer chamber. In each case, a thin powder sample was sandwiched between pieces of tape to ensure maximum light penetration. Upon cooling, these samples showed thermal spin transitions (Fig. S4†) whose temperatures agreed well with the values observed for the bulk samples. After being cooled to 5 K, the samples were irradiated with a broadband “white” light provided by a tungsten halogen lamp at ∼20 mW cm−2. Both 2 and 3·2(3tpH) showed rapid increase in the measured magnetization under irradiation, indicating photogeneration of a metastable HS state (Fig. S5†). When saturation was reached, the irradiation was stopped, and the sample was heated at 0.3 K min−1. As the temperature was increased from 5 K (Fig. 5), both complexes showed a slight increase in χT, which can be explained by zero-field splitting effects. The χT value reached a plateau of ∼3.7 emu K mol−1 for 2 and ∼3.2 emu K mol−1 for 3·2(3tpH), in agreement with nearly quantitative population of the HS state, i.e., an efficient LIESST effect in each case. Further heating caused rapid relaxation to the ground LS state. The characteristic relaxation temperature (TLIESST) was defined by the fastest rate of relaxation (the maximum −d(χT)/dT), with TLIESST = 81 K for 2 and 68 K for 3·2(3tpH).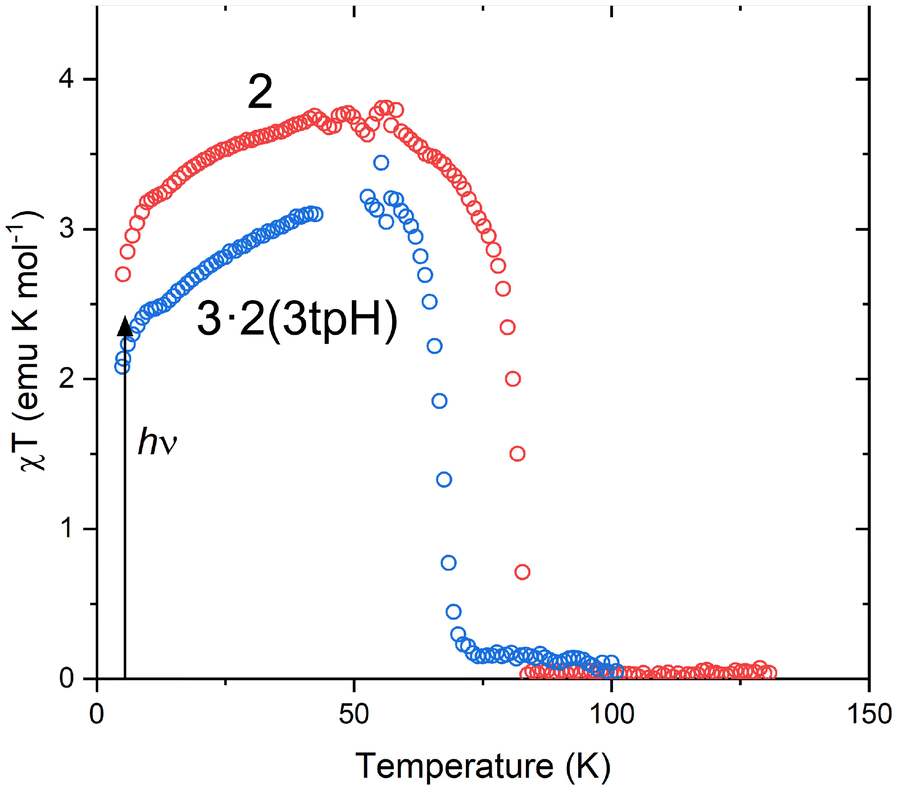 | ||
| Fig. 5 The LIESST effect in 2 and 3·2(3tpH). After irradiation at 5 K, the temperature was increased at 0.3 K min−1. (The noise in the curves at ∼50 K is due to leakage of oxygen through the fiberoptic sample rod. See Fig. S6 and explanation in the ESI.†) | ||
Given the observation of symmetry breaking due to strongly cooperative spin transition in 3·2(3tpH) (see the Crystal structures section), we decided to explore the LIESST effect by optical absorption spectroscopy on a single crystal of 3·2(3tpH), in particular, to assess relaxation dynamics for the light-induced HS state. The sample was cooled to 5 K and irradiated with a 532 nm laser of 2 mW power for 10 min. (The laser power was chosen based on the minimum power input that led to efficient photogeneration of the HS state – see Fig. S7.†) A comparison of the absorption spectra before and after irradiation (Fig. 6a) showed dramatic differences, with disappearance of the bands assigned to the 1A1 → 1T1 and 1A1 → 1T2 excitations of the LS state and appearance of a weaker low-energy band due to the 5T2 → 5E excitation of the HS state. These changes indicate quantitative light-induced LS → HS conversion and efficient trapping of the metastable HS state at 5 K, in agreement with the results of photomagnetic measurements described above.
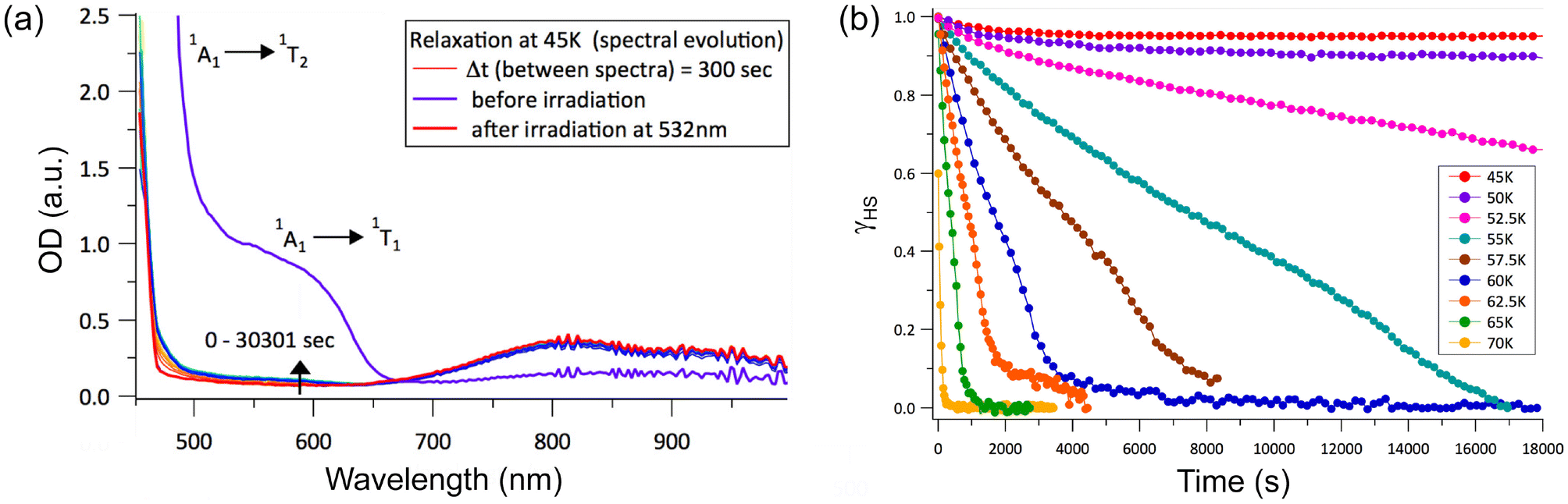 | ||
| Fig. 6 (a) A comparison of the optical absorption spectra measured on a single crystal of 3·2(3tpH) before and after irradiation at 5 K, as well as after rapid heating to 45 K and maintaining at this temperature for ∼500 min. (b) The time-dependent fraction of the light-induced HS state measured at constant temperature after turning off the laser. The HS fractions were extracted by analyzing the spectra shown in Fig. S8.† | ||
To investigate the stability of the LIESST phase, we carried out a series of time-dependent relaxation measurements, in which the sample was cooled to 5 K, irradiated for 10 min, then warmed rapidly to a desired temperature between 45 K and 70 K, and maintained at that temperature while optical absorption data were recorded as a function of time (Fig. S8†). After the completion of each relaxation measurement, the sample was warmed to 90 K to ensure the complete relaxation to the LS phase before cooling again to 5 K for subsequent irradiation and relaxation measurements at a different temperature. The HS → LS relaxation was quantified by monitoring evolution of the 1A1 → 1T1 ligand-field band of the LS state at λ = 590 nm. By referencing the time-dependent amplitude of the optical density, OD(t), of this band against its ODs in the spectra of the pure LS state (ODLS), recorded at 5 K before irradiation, and the pure HS state (ODHS), recorded at 5 K after irradiation, we can calculate the fraction of the HS state as a function of time:
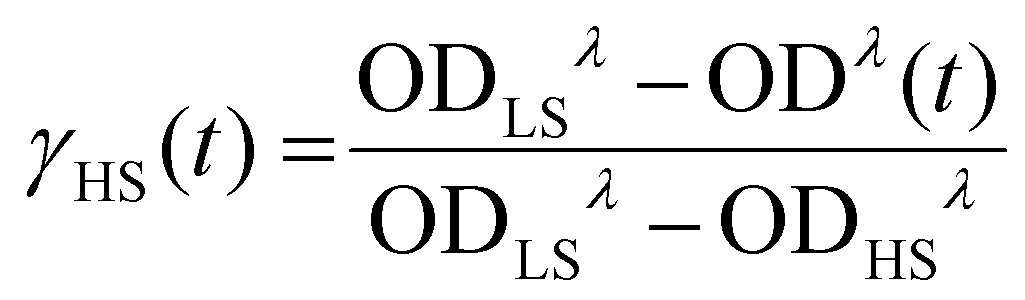 | (1) |
It is clear from the time evolution of the absorption spectra that heating to the individual relaxation temperature causes relaxation of the resultant photoexcited metastable HS state to the ground LS state at different rates (Fig. 6b), as illustrated by the intensification of the 1A1 → 1T1 and 1A1 → 1T2 ligand-field bands of the LS state (Fig. S8†). The relaxation observed at 45 K and 50 K is very slow and incomplete within the measured time interval of 5 h. Above 52.5 K, the relaxation visibly accelerates and proceeds to completion at a faster pace at higher temperatures. At 70 K, the relaxation is very fast and complete within a few minutes, in good agreement with TLIESST = 68 K determined from the magnetic data.
The shape of the relaxation curves, expressed as the time-dependent HS fraction, γHS, generally imitates the extent of cooperativity in SCO compounds. The single-exponential behavior indicates stochastic evolution of like-spin domains, while the sigmoidal relaxation points to the importance of cooperative effects that occur via probabilistic nucleation followed by self-accelerated deterministic growth of the like-spin domains. The step-sigmoidal character of the HS → LS relaxation curves shown in Fig. 6b is especially apparent at intermediate temperatures. Such relaxation behavior, undoubtedly, indicates the importance of cooperative effects of elastic origin with various incubation periods due to the nucleation of the LS domains followed by their growth, propagation, and mutual interactions.
In addition, a tail of sigmoidal relaxation is evidenced by plateaus observed around γHS ∼ 0.10 for measurements performed between 55 K and 62.5 K. The width and slope of the plateau are different for each temperature. At higher temperatures, the faster relaxation kinetics lead to the disappearance of the plateau. Overall, this interesting HS → LS relaxation behavior could be tentatively attributed to specific non-stochastic nearest-neighbor interactions, variable and finite non-negligible short-range interactions leading to the formation of domain structures, inhomogeneous zero-point energy distribution and self-organization of like-spin domains that cause various degrees of elastic frustration, and the influence of the like-spin lattice reorganization with possible crystallographic non-equivalency (which, indeed, was observed for the LS crystal structure of 3·2(3tpH)) on the kinetics of the spin relaxation.13–18 Understanding the true origin of the stepped sigmoidal relaxation behavior and the appearance of the plateau below 65 K will require a fully described elastic model, which we plan to apply to this system in future works.
Concluding remarks
In this work, we have demonstrated the SCO behavior of three homoleptic Fe(II) complexes with thiazole-based ligands. Importantly, we found that carrying out the reactions in methanol or acetonitrile facilitates crystallization of the desired [Fe(L)3](ClO4)2 complexes (L = 4bt, 2bt, 3tpH), in contrast to the problems encountered in the crystallization or even isolation of such complexes from reactions performed in hot ethanol.5–7 While chains of SCO cations interacting through π–π intermolecular contacts are a common feature in the structures of 1, 2·MeOH, and 3·2(3tpH), only the latter two compounds exhibit sufficient cooperativity of such interactions to lead to abrupt and weakly hysteretic spin transitions. We attribute this difference to the more efficient intermolecular overlaps between the thiazole-based ligands in structures 2·MeOH and 3·2(3tpH). As a result, these two complexes also exhibit quantitative light-induced LS → HS switching at low temperatures.The fact that the efficient one-dimensional packing of the cationic complexes is sufficient to achieve the abrupt spin transition suggests that the [Fe(2bt)3]2+ and [Fe(3tpH)3]2+ cations can be used to assemble more complex structures with potentially abrupt SCO behavior. For example, inclusion of organic radical anions can promote conductivity in the anionic part of the structure,19–25 thus leading to multifunctional conductors with magnetic and optical bistability. Efforts in this direction are currently underway in our laboratories, and their results will be reported in due course.
Materials and methods
Synthesis
All reactions were performed under inert atmosphere using standard Schlenk techniques. The starting materials, [Fe(H2O)6](ClO4)2, 2-bromothiazole, n-tetrabutylammonium bromide, diisopropylethylamine, and Pd(OAc)2, were purchased from Millipore Sigma and used as received. Anhydrous commercial solvents were additionally purified by passing through a double-stage drying/purification system (Pure Process Technology). Elemental analyses were carried out by Atlantic Microlab, Inc. (Norcross, GA, USA).Caution! The complexes between metal ions and organic ligands with perchlorate counter ions are potentially explosive. The compounds should be prepared in small amounts and handled with great care!
:hexanes = 9:1 v/v, Rf = 0.2). The solvent was removed by rotary evaporation to yield 2bt as yellow powder (665 mg, 90%). 1H NMR (CDCl3), δ, ppm: 7.92 (d, 2H), 7.46 (d, 2H).
Author contributions
Conceptualization, M. J. and M. S.; methodology, M. J., B. A., S. Y., M. G.-T., Ö. Ü., P. L. R., N. J., E. L., A. V. K., M. W. M., A. H., and P. C.; validation, M. J., S. Y., M. G.-T., A. V. K., M. W. M., P. C., and M. S.; formal analysis, M. J., S. Y., M. G.-T., M. W. M., P. C., and M. S.; investigation, M. J., B. A., S. Y., M. G.-T., Ö. Ü., P. L. R., N. J., E. L., and P. C.; resources, A. V. K., M. W. M., A. H., P. C., and M. S.; writing – original draft preparation, M. J., P. C., and M. S.; writing – review and editing, M. G.-T., A. V. K., M. W. M., and M. S.; visualization, S. Y., M. G.-T., M. W. M., P. C., and M. S.; supervision, A. V. K., R. E., M. W. M., and M. S.; funding acquisition, A. V. K., R. E., M. W. M., P. C., and M. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. National Science Foundation (awards CHE-2300779 and CHE-1955754 to M.S.). P. L. R. and M. S. also acknowledge support from the NSF Research Experiences for Undergraduates program (award CHE-1659661). P. C. sincerely acknowledges the financial support from the Science and Engineering Research Board (award ECR/2018/000923) and the Indian Institute of Technology, Kharagpur (award IIT/SRIC/CY/ENE/2018-19/194). The Rigaku Synergy-S single-crystal X-ray diffractometer used for crystallographic work and the Quantum Design MPMS-3 system used for magnetic measurements were acquired with support of the NSF MRI program (CHE-1828362 and DMR-2216125, respectively). A portion of this work was supported by NSF DMR-1708410 and used MagLab facilities supported by NSF DMR-2128556. The project also used resources provided by the X-ray Crystallography Center (FSU075000XRAY) and the Materials Characterization Laboratory (FSU075000MAC) at the Department of Chemistry and Biochemistry, Florida State University. We also acknowledge Dr Mikhail Lebedev for helpful discussions of some aspects of the project.References
- H. Phan, J. J. Hrudka, D. Igimbayeva, L. M. Lawson Daku and M. Shatruk, A simple approach for predicting the spin state of homoleptic Fe(II) tris-diimine complexes, J. Am. Chem. Soc., 2017, 139, 6437–6447 CrossRef CAS PubMed.
- P. Gütlich, A. B. Gaspar and Y. Garcia, Spin state switching in iron coordination compounds, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef PubMed.
- P. Guionneau, M. Marchivie, G. Bravic, J. F. Létard and D. Chasseau, Structural aspects of spin crossover. Example of the [FeIILn(NCS)2] complexes, Top. Curr. Chem., 2004, 234, 97–128 CrossRef CAS.
- A. S. Abushamleh and H. A. Goodwin, Coordination of 2,2′-biimidazole with iron, cobalt, nickel and copper, Aust. J. Chem., 1979, 32, 513–518 CrossRef CAS.
- A. T. Baker and H. A. Goodwin, Iron(II) and nickel(II) complexes of 4,4′-bithiazole: spectral, magnetic and structural studies, Aust. J. Chem., 1985, 38, 851–863 CrossRef CAS.
- D. C. Craig, H. A. Goodwin, D. Onggo and A. D. Rae, Coordination of 2,2′-bithiazole. Spectral, magnetic and structural studies of the iron(II) and nickel(II) complexes, Aust. J. Chem., 1988, 41, 1625–1644 CrossRef CAS.
- L. S. Harimanow, K. H. Sugiyarto, D. C. Craig, M. L. Scudder and H. A. Goodwin, Magnetic, spectral and structural aspects of spin transitions in iron(II) complexes of 2-(pyrazol-3-yl)pyridine and 3-(thiazol-2-yl)pyrazole, Aust. J. Chem., 1999, 52, 109–122 CrossRef CAS.
- A. Hauser, Light-induced spin crossover and the high-spin → low-spin relaxation, Top. Curr. Chem., 2004, 234, 155–198 CrossRef CAS.
- G. Chastanet, M. Lorenc, R. Bertoni and C. Desplanches, Light-induced spin crossover - solution and solid-state processes, C. R. Chim., 2018, 21, 1075–1094 CrossRef CAS.
- D. Onggo, M. L. Scudder, D. C. Craig and H. A. Goodwin, Structural and Mössbauer spectral studies of iron(II) complexes of thiazole-containing bidentates, Aust. J. Chem., 2000, 53, 153–158 CrossRef CAS.
- H. Yu, S. Huang, J. Kou, L. Li, H. Jia, H. Chao and L. Ji, Synthesis, DNA-binding and photocleavage studies of ruthenium(II) complexes [Ru(btz)3]2+ and [Ru(btz)(dppz)2]2+, Sci. China, Ser. B: Chem., 2009, 52, 1504–1511 CrossRef CAS.
- R. S. Rowland and R. Taylor, Intermolecular nonbonded contact distances in organic crystal structures: comparison with distances expected from van der Waals radii, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- I. Krivokapic, C. Enachescu, R. Bronisz and A. Hauser, Spin transition and relaxation dynamics coupled to a crystallographic phase transition in a polymeric iron(II) spin-crossover system, Chem. Phys. Lett., 2008, 455, 192–196 CrossRef CAS.
- P. Chakraborty, C. Enachescu, C. Walder, R. Bronisz and A. Hauser, Thermal and light-induced spin switching dynamics in the 2D coordination network of {[Zn1−xFex(bbtr)3](ClO4)2}∞: the role of cooperative effects, Inorg. Chem., 2012, 51, 9714–9722 CrossRef CAS PubMed.
- K. W. Törnroos, M. Hostettler, D. Chernyshov, B. Vangdal and H.-B. Bürgi, Interplay of spin conversion and structural phase transformations: re-entrant phase transitions in the 2-propanol solvate of tris(2-picolylamine)iron(II) dichloride, Chem. – Eur. J., 2006, 12, 6207–6215 CrossRef PubMed.
- J. Kusz, H. Spiering and P. Gütlich, X-ray structure study of the light-induced metastable states of the spin-crossover compound [Fe(mtz)6](BF4)2, J. Appl. Crystallogr., 2001, 34, 229–238 CrossRef CAS.
- C. Chong, A. Slimani, F. Varret, K. Boukheddaden, E. Collet, J.-C. Ameline, R. Bronisz and A. Hauser, The kinetics features of a thermal spin transition characterized by optical microscopy on the example of [Fe(bbtr)3](ClO4)2 single crystals: size effect and mechanical instability, Chem. Phys. Lett., 2011, 504, 29–33 CrossRef CAS.
- C. Enachescu, M. Nishino, S. Miyashita, A. Hauser, A. Stancu and L. Stoleriu, Cluster evolution in spin crossover systems observed in the frame of a mechano-elastic model, Europhys. Lett., 2010, 91, 27003 CrossRef.
- Ö. Üngör and M. Shatruk, Transition metal complexes with fractionally charged TCNQ radical anions as structural templates for multifunctional molecular conductors, Polyhedron, 2020, 177, 114254 CrossRef.
- H. Phan, S. M. Benjamin, E. Steven, J. S. Brooks and M. Shatruk, Photomagnetic response in highly conductive iron(II) spin-crossover complexes with TCNQ radicals, Angew. Chem., Int. Ed., 2015, 54, 823–827 CrossRef CAS PubMed.
- X. Zhang, Z.-X. Wang, H. Xie, M.-X. Li, T. J. Woods and K. R. Dunbar, A cobalt(II) spin-crossover compound with partially charged TCNQ radicals and an anomalous conducting behavior, Chem. Sci., 2016, 7, 1569–1574 RSC.
- Y. N. Shvachko, D. V. Starichenko, A. V. Korolyov, E. B. Yagubskii, A. I. Kotov, L. I. Buravov, K. A. Lyssenko, V. N. Zverev, S. V. Simonov, L. V. Zorina, O. G. Shakirova and L. G. Lavrenova, The conducting spin-crossover compound combining Fe(II) cation complex with TCNQ in a fractional reduction state, Inorg. Chem., 2016, 55, 9121–9130 CrossRef CAS PubMed.
- Y. N. Shvachko, N. G. Spitsyna, D. V. Starichenko, V. N. Zverev, L. V. Zorina, S. V. Simonov, M. A. Blagov and E. B. Yagubskii, Magnetism, conductivity and spin-spin interactions in layered hybrid structure of anionic radicals [Ni(dmit)2] alternated by iron(III) spin-crossover complex [Fe(III)(3-OMe-Sal2trien)] and ferric moiety precursors, Molecules, 2020, 25, 4922 CrossRef CAS PubMed.
- Ö. Üngör, E. S. Choi and M. Shatruk, Optimization of crystal packing in semiconducting spin-crossover materials with fractionally charged TCNQδ− anions (0 < δ < 1), Chem. Sci., 2021, 12, 10765–10779 RSC.
- Ö. Üngör, E. S. Choi and M. Shatruk, Solvent-dependent spin-crossover behavior in semiconducting co-crystals of [Fe(1-bpp)2]2+ cations and TCNQδ− anions (0<δ<1), Eur. J. Inorg. Chem., 2021, 2021, 4812–4820 CrossRef.
- H. Erlenmeyer and M. Erne, On the knowledge of polythiazole compounds, Helv. Chim. Acta, 1946, 29, 275–279 CrossRef CAS.
- J. Hassan, C. Gozzi and M. Lemaire, Palladium-catalysed symmetrical and unsymmetrical coupling of aryl halides, C. R. Acad. Sci., Ser. Gen. Vie Sci., 2000, 3, 517–521 CAS.
- CrysAlis, Oxford Diffraction Ltd., Abingdon, England, 2006 Search PubMed.
- SCALE3 ABSPACK - An Oxford Diffraction program (1.0.4,gui:1.0.3), Oxford Diffraction Ltd., Abingdon, England, 2005 Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. A. Bain and J. F. Berry, Diamagnetic corrections and Pascal's constants, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- E. S. Knowles, Ph.D. Thesis, University of Florida, 2013.
Footnote |
| † Electronic supplementary information (ESI) available: Single-crystal X-ray data collection and crystal structure refinements, (CCDC 2325289-2325295), thermogravimetric curves, ORTEP-style images of molecular structures, and time-dependent optical absorption spectra used to monitor the HS → LS relaxation of the light-induced HS state in 3·2(3tpH). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00308j |
| This journal is © The Royal Society of Chemistry 2024 |