
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel, copper, and zinc dinuclear helicates: how do bulky groups influence their architecture?†
Sandra
Fernández-Fariña
 b,
Marcelino
Maneiro
b,
Guillermo
Zaragoza
c,
José M.
Seco
d,
Rosa
Pedrido
*a and
Ana M.
González-Noya
*a
b,
Marcelino
Maneiro
b,
Guillermo
Zaragoza
c,
José M.
Seco
d,
Rosa
Pedrido
*a and
Ana M.
González-Noya
*a
aDepartamento de Química Inorgánica, Facultade de Química, Campus Vida, Universidade de Santiago de Compostela, Santiago de Compostela, Galicia E-15782, Spain. E-mail: rosa.pedrido@usc.es; ana.gonzalez.noya@usc.es
bDepartamento de Química Inorgánica, Facultade de Ciencias, Campus Terra, Universidade de Santiago de Compostela, E-27002, Lugo, Spain
cUnidade de Difracción de Raios X, Edificio CACTUS, Universidade de Santiago de Compostela, Campus Sur, Santiago de Compostela, Galicia E-15782, Spain
dDepartamento de Química Orgánica Facultade de Química, Campus Vida, Universidade de Santiago de Compostela, Santiago de Compostela, Galicia E-15782, Spain
First published on 6th March 2024
Abstract
The ligand design factors that may influence the isolation of metallosupramolecular helicates or mesocates still deserve to be investigated. In this sense, dinuclear nickel(II), copper(II) and zinc(II) compounds were obtained by electrochemical synthesis using a family of five Schiff base ligands, H2Ln (n = 1–5), derived from bisphenylmethane and functionalized with bulky tert-butyl groups in the periphery and ethyl groups in the spacer. Six of the new complexes were characterized by X-ray crystallography, thus demonstrating that the helicate structure is predominant in the solid state. 1H NMR studies were performed for the zinc complexes to analyze if the helical architecture of the metal complexes is retained in solution. These studies reveal that the presence of a tert-butyl group in the ortho position with respect to the OH group is an essential factor identified for the existence of a helicate conformation in solution.
Introduction
The knowledge and understanding of how different elements and molecules combine through diverse self-assembly processes has led to the emergence of a new field called supramolecular chemistry.1 Supramolecular chemistry is focused on the design and study of those systems formed by the spontaneous union of two or more components through the interaction of non-covalent bonds, for instance, hydrogen bonds, van der Waals forces or π–π interactions.2–4 These interactions are often found in nature, such as in proteins or DNA, and are essential for the development of their main functions.5Metal–ligand interactions are also important tools within the supramolecular chemistry field. Depending on the orientation of the parent ligand, a wide variety of metallosupramolecular architectures showing interesting properties and applications5–7 can be obtained. Among them, helicates and mesocates have emerged as two of the most promising functional metallosupramolecular architectures. Helicates are formed by one or more organic ligands helically wrapped around a series of metal ions defining the axis of the helix.1,8–10 In contrast, mesocates are formed by two or more ligands coordinated to metal ions without crossing each other.11 Currently, in contrast to mesocates, helicates have been extensively studied. In this sense, a few reviews on helicates can be found in the literature.10,12–15 Although the preparation of helicate/mesocate architectures is relatively simple, a single controlled route to achieving helicate versus mesocate has not yet been established. In recent years, some of the factors that govern the self-assembly processes of helicates and/or mesocates have been identified: the coordinative preferences of the metal ion, its size16 and hard or soft characteristic,17 the inner design of the organic ligand,3,16,18–23 the temperature and the presence of anions.24 It should be highlighted that understanding and controlling these factors are crucial because helicate/mesocate isomers may exhibit different biological behaviours.25 Additionally, it has been found that the isolation of helicoidal or meso-helicoidal architectures can be controlled by intra- and intermolecular interactions that occur between the ligands.26,27 In the literature there is a large variety of examples of helicoidal architectures derived from ligands whose formation is favoured by the existence of weak non-covalent π–π or CH⋯π interactions.28–31 Along this line, it was found that the existence of non-covalent CH⋯π interactions favoured the helicate-type structure, contrary to expectations, since increasing the distance between the linking domains of the ligand should favour the mesocate conformation.32
With this in mind, we have decided to delve deeper into the factors that influence both the metal ions and the ligand design for obtaining helical or meso-helical compounds. To achieve that, the coordination chemistry of a family of bisphenylmethane-derived Schiff base ligands (H2Ln, Fig. 1) has been explored towards nickel(II), copper(II) and zinc(II) metal ions. Schiff base ligands33 constitute one of the most used approaches for synthesizing helicates and mesocates, as they require cheap or easy-to-prepare starting materials and short preparation times.15,19,34
 | ||
| Fig. 1 Synthesized Schiff base ligands H2Ln. | ||
The long and semi-rigid bisphenylmethane-derived spacers were successfully employed by Hannon and co-workers in the parent ligands to prepare helicates and/or mesocates.28,35–37 In fact, these pioneering works established that functionalization with ethyl groups in the aromatic rings of the spacer could foreseeably favour the formation of helicoidal species instead of mesocates.37 In addition, different bisphenylmethane-derived ligands leading to helical-type species are found in the literature.31,38–40
Besides, the Schiff base branches in our series H2Ln (Fig. 1) have been functionalized with tert-butyl groups in the salicyloyl groups (Fig. 1). We will modify the position of the tert-butyl group with respect to the hydroxy group (para for H2L1 and H2L4, and ortho for H2L2 and H2L5) and introduce a second tert-butyl group (H2L3) to determine whether the position and number of bulky groups prevent and/or determine the formation of helicates or mesocates. The presence of the alkyl groups in the skeleton of these ligands could also improve the solubility of the species formed in apolar solvents and thus favour their crystallization.
Experimental section
Materials and methods
All solvents, p-toluenesulfonic acid, 4,4′-methylenedianiline, 4,4′-methylenebis(2,6-diethylaniline), 3-tert-butyl-2-hydroxybenzaldehyde, 5-tert-butyl-2-hydroxybenzaldehyde, 3,5-di-tert-butyl-2-hydroxybenzaldehyde and nickel, copper and zinc plates were purchased from commercial sources and were used without purification. Melting points were determined using a Buchi 560 instrument. Elemental analysis of compounds (C, N and H) was carried out on a Fisons EA model 1108 analyser. Infrared spectra were recorded from 4000 to 500 cm−1 on a Bruker FT-MIR spectrophotometer model VERTEX 70V in the solid state using KBr pellets. Mass spectra were obtained using Bruker Microtof spectrometers for the ESI+ technique (electrospray ionization in positive mode) and Bruker Autoflex for the MALDI technique (matrix assisted laser desorption/ionization), both coupled to a time-of-flight (TOF) analyser. Room-temperature magnetic susceptibilities were measured using a digital measurement system MSB-MKI vibrating magnetometer. Tetrakis(isothiocyanato)cobaltate(II) was employed as a susceptibility standard. A Varian Inova 400 spectrometer was employed to record the 1H NMR spectra operating at room temperature using acetone-d6 as a deuterated solvent. Chemical shifts are reported as δ (in ppm).Synthesis and characterization of the Schiff base ligands H2Ln
The Schiff base ligands H2Ln (n = 1–5) have been prepared by a condensation reaction between two equivalents of the corresponding hydroxyl-benzaldehyde functionalized with tert-butyl groups and one equivalent of the amine compound (Fig. S13†), following the same procedure previously reported for H2L1 1 and H2L2 2 ligands.41 All the ligands have been fully characterized by melting point determination, elemental analysis, infrared spectroscopy, mass spectrometry and 1H NMR spectroscopy techniques, as well as by X-ray diffraction in the cases where it was possible to obtain quality crystals.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1250 m (C–O), 820 w (CH2); MALDI-TOF (m/z) 631.1 [H2L3 + H]+; 1H-NMR (400 MHz, acetone-d6, δ (m, nH, Hx, J)): 13.88 (s, 2H, H1), 8.90 (s, 2H, H2), 7.50 (bs, J = 2.3 Hz, 2H, H3), 7.46 (bs, J = 2.3 Hz, 2H, H4), 7.31 (bs, 8H, H5 + H6), 4.08 (s, 2H, H7), 1.47 (s, 18H, H8), 1.33 (s, 18H, H9).
N), 1265 s (C–O), 824 m (CH2); ESI+ (m/z) 631.4 [H2L4 + H]+; 1H-NMR (400 MHz, acetone-d6, δ (m, nH, Hx, J)): 12.79 (s, 2H, H1), 8.56 (s, 2H, H2), 7.61 (d, J = 2.2 Hz, 2H, H3), 7.52 (dd, J = 8.7, 2.4 Hz, 2H, H4), 7.09 (s, 4H, H5), 6.94 (d, J = 8.7 Hz, 2H, H6), 3.94 (s, 2H, H7), 2.53 (q, J = 7.5 Hz, 8H, H8), 1.47 (s, 18H, H9); 1.12 (t, J = 7.5 Hz, 12H, H10). By slow evaporation of the mother liquor during the synthesis of the ligand in ethanol, colourless prisms suitable for X-ray diffraction studies were obtained H2L4 4*.
N), 1190 s (C–O), 750 m (CH2); ESI+ (m/z) 631.4 [H2L5 + H]+; 1H-NMR (400 MHz, acetone-d6, δ (m, nH, Hx, J)): 13.82 (s, 2H, H1), 8.54 (s, 2H, H2), 7.45 (dd, J = 7.8, 1.2 Hz, 2H, H3), 7.41 (dd, J = 7.8, 1.5 Hz, 2H, H4), 7.11 (s, 4H, H5), 6.93 (t, J = 7.8 Hz, 2H, H6), 3.95 (s, 2H, H7), 2.54 (c, J = 7.5 Hz, 8H, H8), 1.47 (s, 18H, H9); 1.13 (t, J = 7.5 Hz, 12H, H10). By slow evaporation of the mother liquor during the synthesis of the ligand in ethanol, colourless prisms suitable for X-ray diffraction studies were obtained H2L5 5*.
N), 1250 m (C–O), 820 w (CH2); MALDI-TOF (m/z) 631.1 [H2L3 + H]+; 1H-NMR (400 MHz, acetone-d6, δ (m, nH, Hx, J)): 13.88 (s, 2H, H1), 8.90 (s, 2H, H2), 7.50 (bs, J = 2.3 Hz, 2H, H3), 7.46 (bs, J = 2.3 Hz, 2H, H4), 7.31 (bs, 8H, H5 + H6), 4.08 (s, 2H, H7), 1.47 (s, 18H, H8), 1.33 (s, 18H, H9).
N), 1265 s (C–O), 824 m (CH2); ESI+ (m/z) 631.4 [H2L4 + H]+; 1H-NMR (400 MHz, acetone-d6, δ (m, nH, Hx, J)): 12.79 (s, 2H, H1), 8.56 (s, 2H, H2), 7.61 (d, J = 2.2 Hz, 2H, H3), 7.52 (dd, J = 8.7, 2.4 Hz, 2H, H4), 7.09 (s, 4H, H5), 6.94 (d, J = 8.7 Hz, 2H, H6), 3.94 (s, 2H, H7), 2.53 (q, J = 7.5 Hz, 8H, H8), 1.47 (s, 18H, H9); 1.12 (t, J = 7.5 Hz, 12H, H10). By slow evaporation of the mother liquor during the synthesis of the ligand in ethanol, colourless prisms suitable for X-ray diffraction studies were obtained H2L4 4*.
N), 1190 s (C–O), 750 m (CH2); ESI+ (m/z) 631.4 [H2L5 + H]+; 1H-NMR (400 MHz, acetone-d6, δ (m, nH, Hx, J)): 13.82 (s, 2H, H1), 8.54 (s, 2H, H2), 7.45 (dd, J = 7.8, 1.2 Hz, 2H, H3), 7.41 (dd, J = 7.8, 1.5 Hz, 2H, H4), 7.11 (s, 4H, H5), 6.93 (t, J = 7.8 Hz, 2H, H6), 3.95 (s, 2H, H7), 2.54 (c, J = 7.5 Hz, 8H, H8), 1.47 (s, 18H, H9); 1.13 (t, J = 7.5 Hz, 12H, H10). By slow evaporation of the mother liquor during the synthesis of the ligand in ethanol, colourless prisms suitable for X-ray diffraction studies were obtained H2L5 5*.
Synthesis and characterization of helicates
The electrochemical synthesis of nickel, copper, and zinc neutral helicates was performed using an electrochemical cell and a power supply to regulate the intensity (10 mA) and the potential (10–15 V) of the reaction.41 The cell contains a solution of the Schiff base ligand in acetonitrile, along with a small quantity of tetraethylammonium perchlorate (10 mg) to act as a conductive electrolyte. The electrochemical reaction involves reduction of the ligand at the platinum cathode and oxidation of the metal anode. The electrochemical cell is depicted as follows:| Pt(−)|H2Ln + acetonitrile|M(+) |
The proposed mechanism for the formation of the neutral helicates [M2(L)2] involves two electrons per ligand, as shown below.
| Cathode: 2H2Ln + 4e− → 2(Ln)2− + 2H2(g) |
| Anode: 2M → 2M2+ + 4e− |
| Global: 2(Ln)2− + 2M2+ → [M2(Ln)2] |
The main analytical and characterization data of the complexes are given below.
N); 1261 m (C–O); 833 m (CH2); MALDI-TOF (m/z): 1151.3 [Ni2(L1)2 + H]+, 1726.5 [Ni3(L1)3 + H]+; μeff = 3.1 B.M.
N); 1271 w (C–O); 752 m (CH2); MALDI-TOF (m/z): 1151.5 [Ni2(L2)2 + H]+; μeff = 3.0 B.M. By slow evaporation of the mother liquor during the synthesis, brown crystals suitable for X-ray diffraction studies were obtained [Ni2(L2)2]·CH3CN 7*.
N); 1255 m (C–O); 837 w (CH2); MALDI-TOF (m/z): 688.7 [Ni(L3) + H]+; μeff = 3.4 B. Recrystallization in dichloromethane yielded yellow prisms suitable for X-ray diffraction studies [Ni2(L3)2]·2CH2Cl28*.
N); 1267 m (C–O); 831 w (CH2); MALDI-TOF (m/z): 1375.8 [Ni2(L4)2 + H]+; μeff = 3.2 B.M.
N); 1269 w (C–O); 853 w (CH2); MALDI-TOF (m/z): 1375.7 [Ni2(L5)2 + H]+; μeff = 3.3 B.M.
N) 1614 (f), (C–O) 1255 (m), (CH2) 833 (d); MALDI-TOF (m/z) 694.5 [Cu(L3) + H]+, 1386.9 [Cu2(L3)2 + H]+; μeff = 1.7 B.M. By slow evaporation of the mother liquor during the synthesis, brown crystals suitable for X-ray diffraction studies were obtained [Cu2(L3)2]·2CH3CN 11*.
N) 1618 (m), (C–O) 1267 (d), (CH2) 835 (d); MALDI-TOF (m/z) 691.5 [Cu(L4) + H]+, 1386.9 [Cu2(L4)2 + H]+; μeff = 1.9 B.M.
N) 1603 (mf), (C–O) 1184 (m), (CH2) 750 (d); MALDI-TOF (m/z) 691.3 [Cu(L5) + H]+, 1386.6 [Cu2(L5)2 + H]+. μeff = 1.8 B.M. By slow evaporation of the mother liquor during the synthesis, brown crystals suitable for X-ray diffraction studies were obtained [Cu2(L5)2]·2CH3CN 13*.
N); 1263 m (C–O); 833 m (CH2); MALDI-TOF (m/z): 581.9 [Zn(L1) − H]+, 1165.3 [Zn2(L1)2 + H]+, 1745.4 [Zn3(L1)3 + H]+; 1H-NMR (400 MHz, acetone-d6) δ/ppm: 8.63 (sb, 2H, H2), 7.50 (sb, 2H, HAr), 7.39 (sb, 2H, HAr), 6.94 (sb, 8H, HAr), 6.84 (sb, 2H, HAr), 3.87 (s, H8), 1.32 (sb, 18H, H9). Recrystallization in a mixture of dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (1:1) yielded yellow prisms suitable for X-ray diffraction studies [Zn2(L1)2]·2.8CH3OH 14*.
N); 1292 vw (C–O); 752 w (CH2); MALDI-TOF (m/z): 581.9 [Zn(L2) − H]+, 1165.3 [Zn2(L2)2 + H]+, 1745.4 [Zn3(L2)3 + H]+; 1H-NMR (400 MHz, acetone-d6) δ/ppm: 8.59 (s, 2H, H2), 7.42 (m, HAr), 7.28 (d, 2H, HAr), 6.94 (s, 8H, H5 + H6), 6.63 (t, 2H, H7), 3.87 (s, H8), 1.48 (s, 18H, H9).
N); 1253 m (C–O); 789 w (CH2); MALDI-TOF (m/z): 1389.0 [Zn2(L3)2 + H]+; 1H-NMR (400 MHz, acetone-d6) δ/ppm: 8.60 (s, 2H, H2), 7.56 (sa, 2H, H3), 7.26 (sa, 2H, H4), 6.94 (sa, 8H, HAr), 3.87 (s, H5), 1.50 (s, 18H, H6), 1.33 (s, 18H, H7).
N); 1260 m (C–O); 833 w (CH2); MALDI-TOF (m/z): 1389.6 [Zn2(L4)2 + H]+; 1H-NMR (400 MHz, acetone-d6) δ/ppm: 8.25 (s, 2H, H2), 7.61–6.66 (m, 10H, HAr), 3.99 (s, H7), 2.53 (c, 8H, H8), 1.27 (m, 18H, H9), 0.77 (t, 12H, H10).
N); 1180 m (C–O); 750 m (CH2); MALDI-TOF (m/z): 1388.6 [Zn2(L5)2 + H]+; 1H-NMR (400 MHz, acetone-d6) δ/ppm: 8.22 (s, 2H, H2), 7.46–7.36 (m, HAr), 7.20 (d, HAr), 7.15–7.13 (m, HAr), 6.69 (sa, HAr), 6.55 (t, 2H, H6), 4.01 (s, H7), 2.54 (c, H8), 1.44 (s, H9), 0.84 (t, H10). By slow evaporation of the mother liquor during the synthesis, brown crystals suitable for X-ray diffraction studies were obtained [Zn2(L5)2]·2CH3CN 18*.
:methanol (1:1) yielded yellow prisms suitable for X-ray diffraction studies [Zn2(L1)2]·2.8CH3OH 14*.
N); 1292 vw (C–O); 752 w (CH2); MALDI-TOF (m/z): 581.9 [Zn(L2) − H]+, 1165.3 [Zn2(L2)2 + H]+, 1745.4 [Zn3(L2)3 + H]+; 1H-NMR (400 MHz, acetone-d6) δ/ppm: 8.59 (s, 2H, H2), 7.42 (m, HAr), 7.28 (d, 2H, HAr), 6.94 (s, 8H, H5 + H6), 6.63 (t, 2H, H7), 3.87 (s, H8), 1.48 (s, 18H, H9).
N); 1253 m (C–O); 789 w (CH2); MALDI-TOF (m/z): 1389.0 [Zn2(L3)2 + H]+; 1H-NMR (400 MHz, acetone-d6) δ/ppm: 8.60 (s, 2H, H2), 7.56 (sa, 2H, H3), 7.26 (sa, 2H, H4), 6.94 (sa, 8H, HAr), 3.87 (s, H5), 1.50 (s, 18H, H6), 1.33 (s, 18H, H7).
N); 1260 m (C–O); 833 w (CH2); MALDI-TOF (m/z): 1389.6 [Zn2(L4)2 + H]+; 1H-NMR (400 MHz, acetone-d6) δ/ppm: 8.25 (s, 2H, H2), 7.61–6.66 (m, 10H, HAr), 3.99 (s, H7), 2.53 (c, 8H, H8), 1.27 (m, 18H, H9), 0.77 (t, 12H, H10).
N); 1180 m (C–O); 750 m (CH2); MALDI-TOF (m/z): 1388.6 [Zn2(L5)2 + H]+; 1H-NMR (400 MHz, acetone-d6) δ/ppm: 8.22 (s, 2H, H2), 7.46–7.36 (m, HAr), 7.20 (d, HAr), 7.15–7.13 (m, HAr), 6.69 (sa, HAr), 6.55 (t, 2H, H6), 4.01 (s, H7), 2.54 (c, H8), 1.44 (s, H9), 0.84 (t, H10). By slow evaporation of the mother liquor during the synthesis, brown crystals suitable for X-ray diffraction studies were obtained [Zn2(L5)2]·2CH3CN 18*.
Crystallographic data collection
Crystallographic data for ligands H2L4 4* and H2L5 5* and compounds 7*, 8*, 11*, 13*, 14* and 18* were collected at 100 K on a Bruker D8 VENTURE diffractometer equipped with a CCD detector, using a MoK(α) graphite monochromator (λ = 0.71073 Å). The data were treated using APPEX3v2018.7-2 software for all compounds, except for complex 8* and 11*, which were treated using APPEX2 (Bruker AXS). In all cases, an absorption correction (SADABS)42 was applied to the measured reflections. The H2L5 structure was solved using SHELXT 2014/5,43 while the remaining structures were solved with SHELXT2018/2.2. All structures were refined using SHELXL2018/3,44 with the exception of the 11* structure, which was refined using SHELXL2016/6.43 Hydrogen atoms were included in the model at geometrically calculated and refined positions. The images included in this chapter were prepared using Mercury.45 CCDC 2321087–2321094† contain the supplementary crystallographic data for the compounds.Results and discussion
Schiff base ligands H2Ln
The family of Schiff base ligands named H2Ln (n = 1–5, Fig. 1) is potentially dianionic with two bidentate [NO] domains separated by a semi-rigid aromatic spacer. The ligands were fully characterized using a wide variety of techniques, as detailed in the Experimental section and in Fig. S1–S3 and Tables S1 and S2, (ESI).†The study of the ligand crystal structures is relevant since it allows us to explore the conformational changes that should be undergone to coordinate to the metal ions. Previous studies showed that the presence of a large spacer determined that each of the [NO] linker domains of the ligand coordinated to different metal ions, giving rise to dinuclear helicoidal or meso-helicoidal structures.46
Quality crystals valid for X-ray diffraction studies of ligands H2L4 4* (Fig. 2) and H2L5 5* (Fig. 3) were obtained by slow evaporation of the ethanol mother liquors during the synthesis. Due to the similarity of their structures, both will be discussed together below.
 | ||
| Fig. 2 Stick representation of the ligand H2L4 4* showing the syn configuration of its branches and the intramolecular hydrogen bonds. | ||
 | ||
| Fig. 3 Stick representation of the ligand H2L5 5* showing the syn configuration of its branches and the intramolecular hydrogen bonds. | ||
The structures show discrete molecules crystallizing in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) system. In both ligands the two branches exhibit an E configuration with respect to the imine bonds and a syn-type conformation, with respect to the spacer, with the two ligand branches oriented toward the same side. This conformation is achieved by the establishment of strong intramolecular hydrogen bonds47 involving the imine nitrogen of each ligand branch and the phenol group (H2L4 4*: O2–H20⋯N2 2.574 Å, O1–H10⋯N1 2.615 Å and H2L5 5*: O2–H20⋯N1 2.586 Å, O1–H10⋯N2 2.565 Å). These interactions are different in the two ligands due to the opposite orientation adopted by the phenol ring as a consequence of the position of the tert-butyl group (ortho or para). In the case of an ortho substitution a rotation around the CN bond is required to avoid unfavourable steric hindrance. The imine and phenol distances are in the usual range found in Schiff base ligands.48,49
system. In both ligands the two branches exhibit an E configuration with respect to the imine bonds and a syn-type conformation, with respect to the spacer, with the two ligand branches oriented toward the same side. This conformation is achieved by the establishment of strong intramolecular hydrogen bonds47 involving the imine nitrogen of each ligand branch and the phenol group (H2L4 4*: O2–H20⋯N2 2.574 Å, O1–H10⋯N1 2.615 Å and H2L5 5*: O2–H20⋯N1 2.586 Å, O1–H10⋯N2 2.565 Å). These interactions are different in the two ligands due to the opposite orientation adopted by the phenol ring as a consequence of the position of the tert-butyl group (ortho or para). In the case of an ortho substitution a rotation around the CN bond is required to avoid unfavourable steric hindrance. The imine and phenol distances are in the usual range found in Schiff base ligands.48,49
Assembly of nickel(II), copper(II) and zinc(II) complexes
The next step was the preparation of the nickel, copper and zinc derived complexes with the ligands of the series H2Ln. It is well known that metal ions would have their own coordinative preferences and different affinities to donor atoms, so the use of metal ions with different electronic configurations could lead to distinct metallosupramolecular structures.The Cu2+ metal ion is a d9 system that could exhibit different coordination geometries: square-planar or tetrahedral (four-coordinate), square pyramidal or trigonal bipyramidal (five-coordinate) or octahedral (six-coordinate). It is important to mention that the Cu(II) complexes derived from the ligands H2L1 and H2L2 were previously reported by us.41 Both complexes were found to be similar tetracoordinated helicates [Cu2(L1|2)2]·xCH3CN showing a distorted tetrahedral environment. In that work we observed that the position of the bulky t-butyl groups influenced the intermolecular Cu–Cu distance in the extended structures and their magnetic behaviour.
Our study was completed with Ni2+, whose electronic configuration is d8. The preferential coordination spheres of Ni2+ are six-coordinate (octahedral geometry)19,50 or four-coordinate complexes with planar-square if the ligand-field is strong.51 However, there are also some examples of tetrahedral geometries.52 We have also chosen the softer acid Zn2+ ion, a d10 system whose geometrical preferences are more variable: octahedral (six-coordinate),53 pentacoordinate with a square-based pyramidal geometry,54,55 or the most common type, the tetrahedral geometry (four-coordinate).16,54
The neutral nickel, copper and zinc complexes derived from the Schiff base ligand series H2Ln were prepared using an electrochemical methodology.41 The isolated complexes are powdery solids stable to light and air. Both analytical and spectroscopic data allow us to propose dinuclear stoichiometry [M2(Ln)2] for the complexes, with the ligands being bound to the metal ions in their dianionic [Ln]2− form. The IR spectra of the complexes exhibit some shifting in the ν(CN) and ν(C–O) bands, thus indicating that the ligand coordinates to the metal ion via the imine nitrogen and phenolic oxygen atoms. The dinuclear nature of these complexes was additionally confirmed by MALDI-TOF (+) mass spectrometry experiments as peaks corresponding to the dinuclear fragments [M2(Ln)2 + H]+ were observed (Fig. S4–S8†).
X-ray diffraction studies
The X-ray diffraction technique allowed us to analyse whether the number and position of the bulky groups of the ligands and the nature and coordinative preferences of the metal ion influence the final architecture of the complexes. Slow evaporation of the mother liquors during the synthesis of 7, 11, 13 and 18, and recrystallization in dichloromethane (solid 8) or dichloromethane:methanol (solid 14) allowed us to obtain valid crystals for X-ray diffraction studies.
All the structures show four-coordinate dinuclear neutral helicate-type architecture (Fig. 4) formed by two strands of the dianionic ligand [Ln]2−, which cross-coordinate around the two M(II) ions [M = Ni, Cu, and Zn].
 | ||
| Fig. 4 Spacefill representation of the [Ni2(L2)2]·CH3CN 7* complex, showing the helicoidal structure common to all the complexes studied by X-ray diffraction. | ||
Specifically, the structures revealed the formation of dinuclear neutral helicates [Ni2(L2)2]·CH3CN 7* (Fig. 5), [Ni2(L3)2]·2CH2Cl28* (Fig. 6), [Cu2(L3)2]·2CH3CN 11* (Fig. 7), [Cu2(L5)2]·3CH3CN 13* (Fig. 8), [Zn2(L1)2]·2.8CH3OH 14* (Fig. 9) and [Zn2(L5)2]·2CH3CN 18* (Fig. 10). Tables S3–S8† summarize the most relevant distances and angles.
 | ||
| Fig. 5 Crystal structure of the nickel(II) helicate [Ni2(L2)2]·CH3CN 7*. Solvent molecules and hydrogen atoms have been omitted for clarity. | ||
 | ||
| Fig. 6 Crystal structure of the nickel(II) helicate [Ni2(L3)2]·2CH2Cl28*. Solvent molecules and hydrogen atoms have been omitted for clarity. | ||
 | ||
| Fig. 7 Crystal structure of the copper(II) helicate [Cu2(L3)2]·2CH3CN 11*. Solvent molecules and hydrogen atoms have been omitted for clarity. | ||
 | ||
| Fig. 8 Crystal structure of the copper(II) helicate [Cu2(L5)2]·3CH3CN 13*. Solvent molecules and hydrogen atoms have been omitted for clarity. | ||
 | ||
| Fig. 9 Crystal structure of the zinc(II) helicate [Zn2(L1)2]·2.8CH3OH 14*. Solvent molecules and hydrogen atoms have been omitted for clarity. | ||
 | ||
| Fig. 10 Crystal structure of the zinc(II) helicate [Zn2(L5)2]·2CH3CN 18*. Solvent molecules and hydrogen atoms have been omitted for clarity. | ||
The six crystal structures are similar so, in order to avoid repetitive descriptions, we will only describe in detail the zinc complex [Zn2(L1)2]·2.8CH3OH 14* (Fig. 9), highlighting only some structural facts that could be attributed to the substitution in the remaining complexes.
The molecular structure of 14* (Fig. 9) shows a non-centrosymmetric dinuclear neutral Zn2+ complex. The two H2L1 ligands are helically arranged around the two Zn2+ ions, so these metal centres have the same absolute configuration. The two enantiomers are present in the crystal cell as a racemate. The Schiff base ligands act in such a way that each of their bidentate [NO] branches coordinate to a different metal ion giving rise to a distorted tetrahedral geometry [≠109.50]. The O–M–N bond angles clearly show the distortion of the tetrahedral geometry (Table S7†). The main bond distances Zn–O and Zn–N are in the expected ranges for complexes derived from Schiff base ligands with phenol groups56 with the Zn–O distance being slightly shorter than Zn–N distance (Table S7†). The intermetallic distance Zn⋯·Zn (11.639 Å) is similar to the distance between the zinc ions in dinuclear helicoidal compounds and does not deserve further comments.56
On the other hand, hydrogen bonds are established between the O3 and O4 phenolic oxygens of the helicate ligands and two methanol molecules. In line with this, one of the solvent molecules forms a hydrogen bond with an adjacent methanol molecule [O6–H6⋯O3 2.74 Å, O5–H5⋯O4 2.76 Å and O6–H6⋯O7 2.74 Å] (Fig. S9†). Additionally, the existence of eight aromatic rings in each helicate makes it necessary to explore aromatic π–π or CH⋯·π stacking interactions. Thus, there are weak π–π interactions between the aromatic rings of the two ligands that contribute to the stabilization of the helicoidal structure [the distance between centroids: 4.48 Å and 4.63 Å] (Fig. S9†).
In the case of the crystal lattice of [Zn2(L1)2]·2.8CH3OH 14* (Fig. 11), intermolecular π–π interactions are observed between the aromatic rings of the spacer of adjacent helicates (a centroid–centroid distance of 3.95 Å), with these interactions being stronger than the intramolecular ones (centroid to centroid distances: 4.2 and 4.6 Å).57 Additionally, these interactions are observed between one of the phenyl rings of the spacer and the aromatic ring of a linker domain (centroid–centroid distances of 3.59 and 3.44 Å).
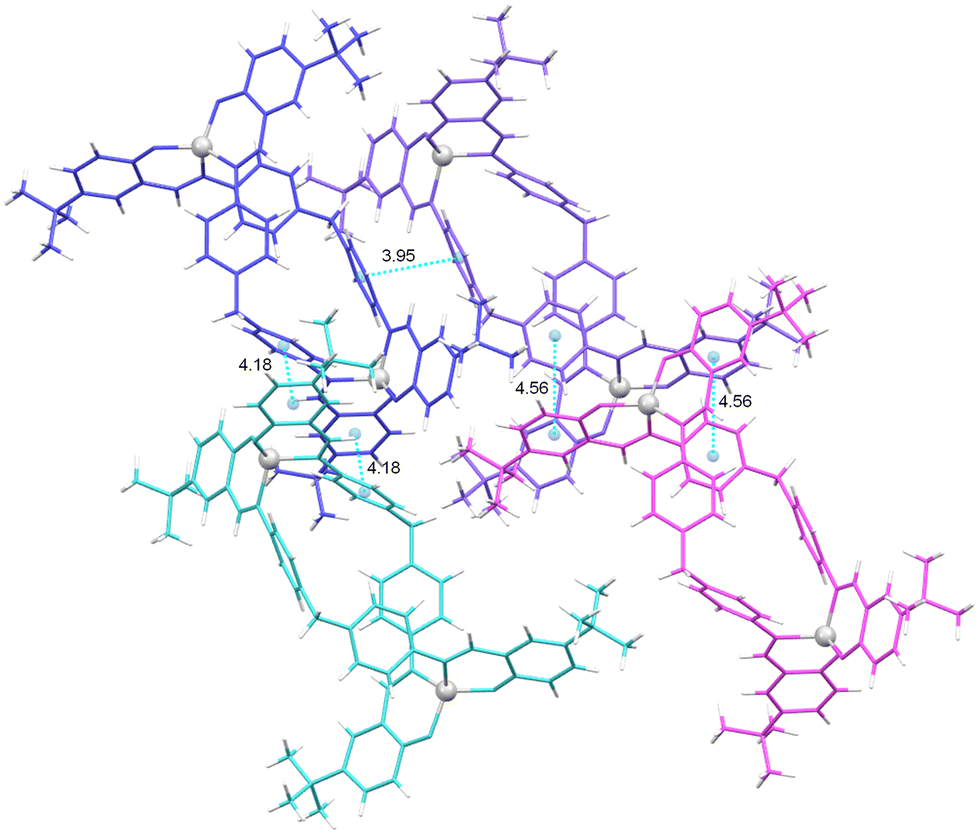 | ||
| Fig. 11 Intermolecular π-stacking interactions in the crystal lattice of the complex [Zn2(L1)2]·2.8CH3OH 14*. | ||
In addition, CH⋯π interactions between the aromatic ring of one of the ligand branches and one of the tert-butyl substituents of the adjacent helicate can be observed in the crystalline packing of [Zn2(L1)2]·2.8CH3OH 14*, (Fig. S10†).
It should be noted that the six helicoidal structures studied exhibit similar packing patterns to the 14* helicate, except in the case of the [Zn2(L5)2]·2CH3CN 18* helicate (Fig. 10), where the stacking distances are much longer.
Additionally, as can be seen in Fig. S11–S14†, the complexes with tert-butyl groups in the ortho position to the phenol groups (7*, 8*, 11*, 13*, and 18*) exhibit hydrogen bonds between the CH3 of the tert-butyl groups and the phenolic oxygens. Additionally, the crystal structure derived from the H2L5 ligand functionalized with ethyl groups on the spacer exhibits CH⋯π interactions between the ethyl groups and the aromatic rings of the adjacent ligand spacer (Fig. S13 and S14†).
The obtainment of dinuclear helicates in all these cases shows that the position and/or the number of bulky groups in the ligands does not affect the type of structure isolated, but it does affect the microstructure of the helicates.
Our results demonstrated that the functionalization of the ligands with ethyl substituents in the spacer (H2L4 and H2L5) generates new intramolecular non-covalent CH⋯π interactions between the CH2 of the ethyl groups and the aromatic rings of the spacer of the adjacent ligand, which could additionally favour the helicoidal conformation in the compounds.
Furthermore, the results demonstrate that the use of metal ions with different natures (Cu2+, Ni2+ and Zn2+) does not affect the arrangement of the ligands around the metal centres. In all cases, helicate architectures were achieved, indicating that the variation from Cu2+ to Ni2+ to a similar-sized metal ion Zn2+, with no ligand-field stabilization energy, does not affect the macrostructure of the compounds.
If we check the coordination kernels in the two pairs of Ni(II), Cu(II) and Zn(II) helicates we can see that the Zn(II) helicates show a distorted tetrahedral geometry in both cases (14*: τZn1 = 0.85, τZn2 = 0.76, Table S7;†18*: τZn1 = 0.82, Table S8†), and the metal ions in the Cu(II) helicates exhibit intermediate τ4 values (11*: τCu1 = 0.43; 13*: τCu1 = 0.58, τCu2 = 0.42).58 However, in the case of the Ni(II) helicate [Ni2(L2)2]·CH3CN 7*, this exhibits a distorted tetrahedral geometry around one of the nickel ions (Ni1) and a distorted square-planar geometry around the second metal ion (Table S3,†τNi1 = 0.76; τNi2 = 0.30) whereas a distorted square-planar environment around both Ni2+ metal ions was found in the case of [Ni2(L3)2]·2CH2Cl28* (Table S4,†τNi1 = 0.28). In this case, the presence of one (H2L2) or two tert-butyl groups (H2L3) in the periphery of the ligands does affect the microstructure of the nickel complexes obtained.
1H NMR studies
Hannon and co-workers37 have a wide experience in the preparation of helicate/mesocate complexes with bisphenylmethane derived ligands. In these studies, they stated that a careful analysis of the 1H NMR spectrum in the aliphatic region may be indicative of the exclusive presence of the mesocate and helicate conformations (Fig. 12) in solution, or the coexistence of both forms. | ||
| Fig. 12 Helicate (left) and mesocate (right) conformations. | ||
In this sense, they concluded that if the ligand spacer is not functionalised with alkyl substituents in the phenyl rings, a mixture of helicate–mesocate is shown, whereas when adding bulky groups (ethyl and methyl) to the ligand spacer, the mesocate architecture is precluded. This could be explained by the ligand twisting induced by the alkyl groups and also by the establishment of new CH⋯π intermolecular interactions which favour the helicate-type architecture (dinuclear and trinuclear with Me, and dinuclear with Et).
What happens if we also introduce bulky groups in the ligand branches? Trying to go a step further we studied the aliphatic methylene region for the series of the zinc complexes 14, 15, 16, 17 and 18 in order to explore whether the helical structure observed in the solid state is maintained in solution.
The zinc complex 14 [Zn2(L1)2] derived from the ligand with the para tert-butyl group position shows a central singlet signal corresponding to the helicate-type conformation and four additional satellite signals that could correspond to mesocate species (Fig. 13), thus confirming that both species co-exist in solution. Therefore, the presence of an external tert-butyl group together with the non-substituted spacer does not tip the balance towards the mesocate or helicate species that co-exist in solution.
 | ||
| Fig. 13 1H NMR spectrum of the CH2 group aliphatic region of [Zn2(L1)2] 14 (400 MHz, r.t., acetone-d6). | ||
In contrast, the zinc complexes 15, 16, 17 and 18 show one singlet, thus indicating the exclusive presence of the helicate species in solution only (Fig. 14). These experimental observations indicate that in the absence of bulky groups in the spacer, the equilibrium of helicate/mesocate is displaced to helicate after functionalization of the external rings with a tert butyl group in the ortho position to the phenol.
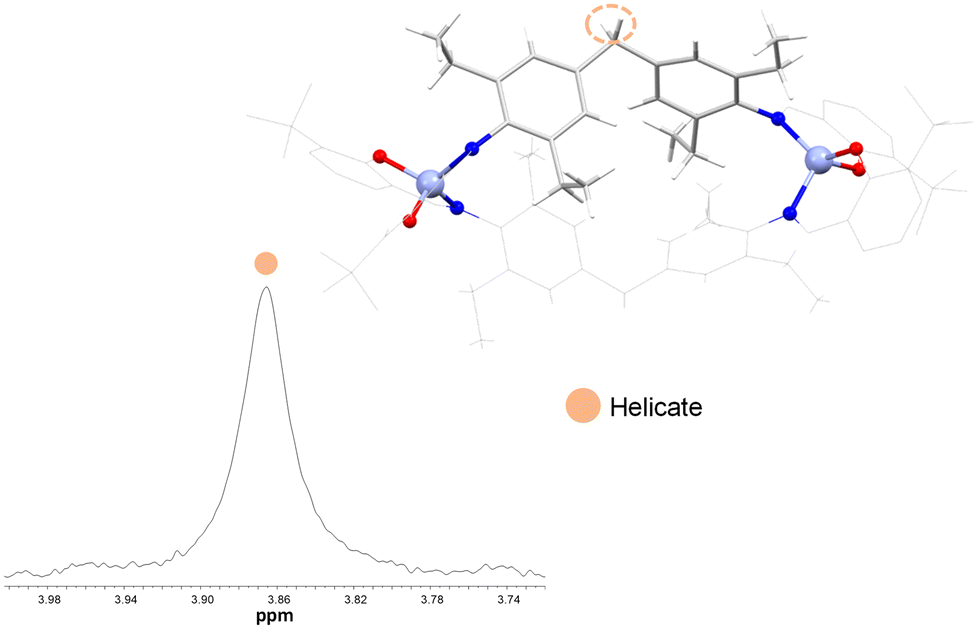 | ||
| Fig. 14 1H NMR spectrum of the CH2 group aliphatic region of [Zn2(L5)2] 18 (400 MHz, r.t., acetone-d6). | ||
Likewise, in the case of the zinc complexes derived from the ligands functionalised with the ethyl groups in the spacer rings, [Zn2(L4)2] 17 and [Zn2(L5)2] 18, the helicate-type structure is also confirmed by the appearance of four signals corresponding to diastereotopic ethyl CH2 protons (Fig. 15).
 | ||
| Fig. 15 1H NMR spectrum of the aliphatic region of [Zn2(L5)2] 18 (400 MHz, r.t., acetone-d6). | ||
Thus, two of the signals appear at low field with respect to the free ligand and the remaining two signals appear at high field. In addition, two signals corresponding to the CH3 of the ethyl groups are observed, one of which appears unshielded with respect to the free ligand due to the existence of π-stacking interactions along the crystal lattice.37
Therefore, the results of these NMR studies allow us to deduce that the driving factor determining the existence in solution of helicate or a helicate–mesocate mixture does not reside exclusively in the spacer, as the introduction of an ortho bulky group on the external surface of the helicate displaces the equilibrium of helicate–mesocate towards the helicate (Fig. 14 and Table 1). However, the introduction of ethyl groups in the ligand spacer still sterically favours the formation of helicates, independently of the presence of bulky groups in the periphery of the complex (Fig. 15 and Table 1).
| Complex | Species | Spacer rings | Salicyloyl rings |
|---|---|---|---|
| Hel = Helicate; Mes = Mesocate. | |||
| 14 | Hel/Mes |

|

|
| 15 | Hel |
|

|
| 16 | Hel |

|

|
| 17 | Hel |

|

|
| 18 | Hel |

|

|
Conclusions
Dinuclear nickel(II), copper(II) and zinc(II) complexes were isolated using an electrochemical methodology from a family of five Schiff base ligands derived from bisphenylmethane and functionalized with bulky tert-butyl groups. The introduction of bulky groups on both the spacer and ligand branches favours the formation of dinuclear helicates. From the six crystal structures reported in this work, we can conclude that small modifications of the ligands do not affect the macrostructure of the compounds, all of them being helicates. However, the microstructure, e.g., the metal ion environment, could be altered in some cases. Besides, the introduction of ethyl substituents in the spacer provides new intramolecular interactions that stabilize the helicate-type architecture of the compounds. Furthermore, the 1H NMR study reveals that the presence of the tert-butyl group in the ortho position with respect to the OH group is essential for the maintenance of the helicate conformation in solution.Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the following FEDER co-funded grants: Consellería de Cultura, Educación e Ordenación Universitaria, Xunta de Galicia GRC GI-1584 (ED431C 2023/02), MetalBIONetwork (ED431D2017/01), and Ministerio de Ciencia e Innovación, Project PID2021-127531NB-I00 (AEI/10.13039/501100011033/FEDER/UE).References
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- M. Albrecht, M. Fiege and O. Osetska, Coord. Chem. Rev., 2008, 252, 812–824 CrossRef CAS.
- J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- G. F. Swiegers and T. J. Malefetse, Chem. Rev., 2000, 100, 3483–3537 CrossRef CAS PubMed.
- H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS PubMed.
- E. Chinnaraja, R. Arunachalam, E. Suresh, S. K. Sen, R. Natarajan and P. S. Subramanian, Inorg. Chem., 2019, 58, 4465–4479 CrossRef CAS PubMed.
- V. Martínez-Agramunt and E. Peris, Inorg. Chem., 2019, 58, 11836–11842 CrossRef PubMed.
- J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565–2569 CrossRef CAS PubMed.
- C. T. McTernan, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2021, 143, 664–670 CrossRef CAS PubMed.
- C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed.
- S. Fernández-Fariña, M. Martínez-Calvo, M. Maneiro, J. M. Seco, G. Zaragoza, A. M. González-Noya and R. Pedrido, Inorg. Chem., 2022, 61, 14121–14130 CrossRef PubMed.
- H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977–6991 RSC.
- M. Albrecht, X. Chen and D. Van Craen, Chem. – Eur. J., 2019, 25, 4265–4273 CrossRef CAS PubMed.
- J. L. Greenfield and J. R. Nitschke, Acc. Chem. Res., 2022, 55, 391–401 CrossRef CAS PubMed.
- M. Albrecht, Chem. Rev., 2001, 101, 3457–3497 CrossRef CAS PubMed.
- M. J. Romero, M. Martínez-Calvo, M. Maneiro, G. Zaragoza, R. Pedrido and A. M. González-Noya, Inorg. Chem., 2019, 58, 881–889 CrossRef CAS PubMed.
- T. L. Ho, Chem. Rev., 1975, 75, 1–20 CrossRef CAS.
- J. M. Lehn, Eur. Rev., 2009, 17, 263–280 CrossRef.
- A. A. Escuer, J. Mayans, L. Di Bari, M. Font-, L. Arrico and F. Zinna, Chem. – Eur. J., 2018, 24, 7653–7663 CrossRef PubMed.
- N. Wu, C. F. C. Melan, K. A. Stevenson, O. Fleischel, H. Guo, F. Habib, R. J. Holmberg, M. Murugesu, N. J. Mosey, H. Nierengarten and A. Petitjean, Dalton Trans., 2015, 44, 14991–15005 RSC.
- M. Bhol, R. L. Borkar, B. Shankar, S. K. Panda, M. Wolff and M. Sathiyendiran, Inorg. Chem., 2023, 62, 11554–11569 CrossRef CAS PubMed.
- J. E. Niklas, E. A. Hiti, G. R. Wilkinson, J. T. Mayhugh, J. D. Gorden and A. E. V. Gorden, Inorg. Chim. Acta, 2022, 529, 120653 CrossRef CAS.
- (a) K. L. Flint, D. M. Huang, O. M. Linder-Patton, C. J. Sumby and F. R. Keene, Eur. J. Inorg. Chem., 2022, e202200225 CrossRef CAS; (b) U. Phukon, B. Shankar and M. Sathiyendiran, Dalton Trans., 2022, 51, 16307–16315 RSC.
- F. Cui, S. Li, C. Jia, J. S. Mathieson, L. Cronin, X. Yang and B. Wu, Inorg. Chem., 2012, 51, 179–187 CrossRef CAS.
- S. J. Allison, D. Cooke, F. S. Davidson, P. I. P. Elliott, R. A. Faulkner, H. B. S. Griffiths, O. J. Harper, O. Hussain, P. J. Owen-Lynch, R. M. Phillips, C. R. Rice, S. L. Shepherd and R. T. Wheelhouse, Angew. Chem., Int. Ed., 2018, 57, 9799–9804 CrossRef CAS PubMed.
- D. J. Cooke, J. M. Cross, R. V. Fennessy, L. P. Harding, C. R. Rice and C. Slater, Chem. Commun., 2013, 49, 7785–7787 RSC.
- T. K. Ronson, H. Adams, T. Riis-Johannessen, J. C. Jeffery and M. D. Ward, New J. Chem., 2006, 30, 26–28 RSC.
- J. Malina, M. J. Hannon and V. Brabec, Chem. – Eur. J., 2015, 21, 11189–11195 CrossRef CAS PubMed.
- M. J. Hannon and L. J. Childs, Supramol. Chem., 2004, 16, 7–22 CrossRef CAS.
- J. Mayans, M. Font-Bardia, L. Di Bari, L. Arrico, F. Zinna, G. Pescitelli and A. Escuer, Chem. – Eur. J., 2018, 24, 7653–7663 CrossRef CAS PubMed.
- N. Kelly, J. Schulz, K. Gloe, T. Doert, K. Gloe, M. Wenzel, M. Acker and J. J. Weigand, Z. Anorg. Allg. Chem., 2015, 641, 2215–2221 CrossRef CAS.
- P. Levín, D. Escudero, N. Díaz, A. Oliver, A. G. Lappin, G. Ferraudi and L. Lemus, Inorg. Chem., 2020, 59, 1660–1674 CrossRef.
- L. Fabbrizzi, J. Org. Chem., 2020, 85, 12212–12226 CrossRef CAS PubMed.
- M. J. Romero, R. Carballido, L. Rodríguez-Silva, M. Maneiro, G. Zaragoza, A. M. González-Noya and R. Pedrido, Dalton Trans., 2016, 45, 16162–16165 RSC.
- C. A. J. Hooper, L. Cardo, J. S. Craig, L. Melidis, A. Garai, R. T. Egan, V. Sadovnikova, F. Burkert, L. Male, N. J. Hodges, D. F. Browning, R. Rosas, F. Liu, F. V. Rocha, M. A. Lima, S. Liu, D. Bardelang and M. J. Hannon, J. Am. Chem. Soc., 2020, 142, 20651–20660 CrossRef CAS PubMed.
- L. Cardo, I. Nawroth, P. J. Cail, J. A. McKeating and M. J. Hannon, Sci. Rep., 2018, 8, 2–8 CrossRef.
- L. J. Childs, M. Pascu, A. J. Clarke, N. W. Alcock and M. J. Hannon, Chem. – Eur. J., 2004, 10, 4291–4300 CrossRef CAS.
- P. E. Kruger, N. Martin and M. Nieuwenhuyzen, J. Chem. Soc., Dalton Trans., 2001, 1966–1970 RSC.
- F. Habib, J. Long, P. H. Lin, I. Korobkov, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, Chem. Sci., 2012, 3, 2158–2164 RSC.
- G. Novitchi, J. P. Costes, J. P. Tuchagues, L. Vendier and W. Wernsdorfer, New J. Chem., 2008, 32, 197–200 RSC.
- S. Fernández-Fariña, I. Velo-Heleno, M. Martínez-Calvo, M. Maneiro, R. Pedrido and A. M. González-Noya, Int. J. Mol. Sci., 2023, 24, 8654 CrossRef PubMed.
- G. M. Sheldrick, Program for Scaling and Correction of Area Detector Data, University, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- I. Usón and G. M. Sheldrick, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 106–116 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. Mccabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van De Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 466–470 CrossRef CAS.
- G. Han, Y. Zhou, Y. Yao, Z. Cheng, T. Gao, H. Li and P. Yan, Dalton Trans., 2020, 49, 3312–3320 RSC.
- G. A. Jeffrey, An introduction to hydrogen bonding, Oxford University Press, 1997 Search PubMed.
- S. H. Sumrra, A. U. Hassan, M. Imran, M. Khalid, E. U. Mughal, M. N. Zafar, M. N. Tahir, M. A. Raza and A. A. C. Braga, Appl. Organomet. Chem., 2020, e5623 CrossRef CAS.
- H. Ünver, C. T. Zeyrek, B. Boyacioglu, M. Yıldız, N. Demir and A. Elmali, J. Chem. Crystallogr., 2019, 49, 232–244 CrossRef.
- L. D. S. Mariano, I. M. L. Rosa, N. R. De Campos, A. C. Doriguetto, D. F. Dias, W. D. Do Pim, A. K. S. M. Valdo, F. T. Martins, M. A. Ribeiro, E. E. B. De Paula, E. F. Pedroso, H. O. Stumpf, J. Cano, F. Lloret, M. Julve and M. V. Marinho, Cryst. Growth Des., 2020, 20, 2462–2476 CrossRef CAS.
- J. Hildebrandt, N. Häfner, H. Görls, M. C. Barth, M. Dürst, I. B. Runnebaum and W. Weigand, Int. J. Mol. Sci., 2022, 23, 6669 CrossRef CAS PubMed.
- I. V. Ershova, I. V. Smolyaninov, A. S. Bogomyakov, M. V. Fedin, A. G. Starikov, A. V. Cherkasov, G. K. Fukin and A. V. Piskunov, Dalton Trans., 2019, 48, 10723–10732 RSC.
- B. Ivanova and M. Spiteller, J. Mol. Struct., 2022, 1248, 131488 CrossRef CAS.
- S. Fernández-Fariña, I. Velo-Heleno, M. Martínez-Calvo, R. Barcia, Ò. Palacios, M. Capdevila, A. M. González-Noya and R. Pedrido, Int. J. Mol. Sci., 2023, 24, 2246 CrossRef PubMed.
- J. Zhao, S. Wang and W. Zhang, J. Chem. Crystallogr., 2022, 52, 34–42 CrossRef CAS.
- P. Cucos, F. Tuna, L. Sorace, I. Matei, C. Maxim, S. Shova, R. Gheorghe, A. Caneschi, M. Hillebrand and M. Andruh, Inorg. Chem., 2014, 53, 7738–7747 CrossRef CAS PubMed.
- (a) M. Vázquez, A. Taglietti, D. Gatteschi, L. Sorace, C. Sangregorio, A. M. González, M. Maneiro, R. M. Pedrido and M. R. Bermejo, Chem. Commun., 2003, 3, 1840–1841 RSC; (b) R. A. Tigaa and A. de Bettencourt-Dias, J. Chem. Crystallogr., 2017, 47, 233–240 CrossRef CAS; (c) C. Puigjaner, A. Portell, A. Blasco, M. Font-Bardia and O. Vallcorba, Crystals, 2021, 11, 342 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, compound characterization and synthetic and crystallographic data; Fig. S1–S14 and Tables S1–S8. CCDC 2321087–2321094. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00279b |
| This journal is © The Royal Society of Chemistry 2024 |