
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimising the acid–base ratio of Mg–Al layered double oxides to enhance CO2 capture performance: the critical role of calcination conditions†
D. W. Justin
Leung
,
Katherine R.
Laney
 *,
Philip
Kenyon
,
Nicholas H.
Rees
,
Jean-Charles
Buffet
,
Chunping
Chen
* and
Dermot
O'Hare
*
*,
Philip
Kenyon
,
Nicholas H.
Rees
,
Jean-Charles
Buffet
,
Chunping
Chen
* and
Dermot
O'Hare
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: dermot.ohare@chem.ox.ac.uk; chunping.chen@chem.ox.ac.uk; Tel: +44 (0)1865 27268
First published on 14th March 2024
Abstract
The effect of calcination conditions (ramp rate, calcination temperature and time) on the formation of Mg2Al layered double oxides (Mg2Al LDOs) as well as their CO2 capture performance, has been systematically investigated. This study explores novel insights into the intricate relationship between these calcination conditions and the resulting surface characteristics, which play a vital role in CO2 capture efficiency. Notably, it is revealed that a rapid ramp rate (100 °C min−1) significantly increases surface area and hydroxyl concentration, leading to a 69% increase in CO2 capture efficiency compared to slower ramp rate. Conversely, short calcination times (1 h) and fast ramp rates (100 °C min−1) are observed to compromise CO2 adsorption due to the presence of dehydrated LDHs. A critical acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :base ratio of 0.37, achieved from a fast ramp rate (100 °C min−1) at 400 °C for 2 h, was found as a key threshold for optimising surface properties, effectively balancing favourable hydroxyl and less favourable strong acid sites, thereby maximizing CO2 capture performance.
:base ratio of 0.37, achieved from a fast ramp rate (100 °C min−1) at 400 °C for 2 h, was found as a key threshold for optimising surface properties, effectively balancing favourable hydroxyl and less favourable strong acid sites, thereby maximizing CO2 capture performance.
Introduction
Layered double hydroxides (LDHs) are a family of anionic layered material with a general formula of [Mz+1−xM′y+x(OH)2]a+[(An−)a/n]·mH2O wherein M and M′ are different cations with charges z (typically 1 or 2) and y (typically 3 or 4), respectively;1 A is the anion in the interlayer galleries to balance the net positive charge of metal hydroxide layers; x is the ratio of M′ to total metals (M + M′), and it has been demonstrated that MgAl-based LDHs exhibit pure crystalline phares when 0.33 ≥ x ≥ 0.18.2 Water is incorporated into the interlayer region, establishing hydrogen bonds with the hydroxyl groups of the cationic layers and interlayer anions. This interaction stabilises the layered structure, connecting individual layers while preserving translational mobility.3 At elevated temperatures, LDHs undergo thermal decomposition. Initially, they release surface and interlayer water molecules, leading to the formation of dehydrated LDHs when heated up to 200 °C. Subsequently, they undergo transformation into Layered Double Oxides (LDOs) through dehydroxylation and the removal of anions (such as CO32−) in the temperature range of 300 to 750 °C. When temperature is higher than 750 °C, it forms a crystalline spinel phase which cannot be reconstructed to the LDH structure.2,4Both LDHs and their thermal derivatives (LDOs) have found successful applications in a wide range of areas, including catalysis, anionic adsorbents, flame retardants, gas barrier membrane and biomaterials.5–11 In particular, MgAl–CO3 LDOs have been demonstrated as promising CO2 adsorbents due to their inherent basicity, low adsorption heat, rapid adsorption/desorption kinetics, simple regenerability and good stability.7,8,12,13 Considerable efforts have been made to enhance the CO2 adsorption capacity including the optimisation of adsorption conditions, the control of calcination parameters (temperature and time), the design of LDHs/LDOs synthesis methods, as well as the incorporation of alkaline metal doping or other hybrid materials.12,14–17 However, the influence of the calcination process, including the parameters such as ramp rate, temperature and calcination duration time, on the surface chemistry of the material and its subsequent effects on CO2 capture through both chemisorption and physisorption has not been fully explored yet. Being able to optimise the calcination parameters for the LDHs materials and fundamentally understand their influence on the surface physio-chemical properties holds the key to unlock the full potential of LDHs/LDOs as efficient CO2 sorption materials.
Herein, we report a systematic study to investigate the impact of calcination conditions of Mg2Al LDHs ([Mg0.66Al0.34(OH)2](CO3)0.17(H2O)0.95) on the formation of Mg2Al LDOs. This study includes varying ramp rate (5–100 °C min−1), calcination time (1–6 h) and calcination temperature (100–600 °C). Additionally, we access the CO2 capture performance of the resulting calcined products. The physical property and surface chemistry of each calcined material was characterised extensively using N2 adsorption isotherms, the titration of surface hydroxyl groups, solid state nuclear magnetic resonance (ssNMR) and temperature programmed desorption (TPD).
Experimental
Synthesis of materials
[Mg0.66Al0.34(OH)2](CO3)0.17(H2O)0.95 (Mg2Al LDHs) was synthesised using the co-precipitation method.13 Solution A was made by dissolving Mg(NO3)2·6H2O (17.09 g, 0.067 mol) and Al(NO3)3·9H2O (12.50 g, 0.033 mol) in 100 mL deionised (DI) water. Na2CO3 (5.30 g, 0.050 mol) was dissolved in another 100 mL DI water to make solution B. Solution A was added to solution B dropwise for an hour while stirring, with the solution mixture kept at pH 10 by the addition of 4 M NaOH solution. After aging for 24 h with stirring at room temperature, the solid was filtrated and washed with DI water until pH 7. The final solid product was then dried overnight in a vacuum oven at 30 °C.The LDOs were obtained from the calcination at various conditions. The nomenclature for the obtained calcined LDH samples is as follows: calcination temperature (°C) – calcination time (h) – ramp rate (°C min−1). For example, 400-2 h-5 stands for a sample calcined at 400 °C for 2 h with a ramp rate of 5 °C min−1. For CO2 capture and TPD measurements, LDOs was generated by in situ calcination of LDHs. All other calcinations were carried out in a box furnace and the LDOs used immediately to prevent reconstruction.
Hydroxyl titration
The OH titrations were carried out using a procedure adapted from literature.18 5 mg samples and 10 mg Mg(CH2Ph)2(THF)2 were mixed in 500 μL d6-benzene containing Si(SiMe3)4 as an internal standard (c = 6.23 mmol dm−3) in a Young's tap NMR tube. Production of toluene resulting from protonation of the benzyl by the surface hydroxyl groups was monitored by 1H NMR spectroscopy and the toluene methyl resonance integrated against that of Si(SiMe3)4.Material characterisation
The powder X-ray diffraction (XRD) data were collected using a PANAnalytical X'Pert Pro diffractometer in refection mode at 40 kV and 40 mA using Cu Kα radiation. Surface area analysis was carried out using the BET method on a Micromeritics Tristar II plus. Nitrogen adsorption and desorption isotherms were collected at 77 k and ∼100 mg of sample were degassed at 110 °C overnight prior to analysis. Acidity and basicity were measured on a Micromeritics AutoChem II 2920 Chemisorption Analyser equipped with a thermal conductivity detector (TCD). The sample (100 mg) was loaded in a quartz U-tube and first purged in a flow of He (50 cm3 min−1) at room temperature for 10 min to remove impurities. The temperature was then raised to 400 °C at a ramp rate of 10 °C min−1 and held at this temperature for 1 h to remove water and any other impurities. The temperature was then reduced to 100 °C. For basicity tests, CO2 in He (30 cm3 min−1 CO2, 50 cm3 min−1 He) flown over the sample for 1 h. For acidity tests, a 10% NH3 in He gas flow was used instead. Physically adsorbed CO2/NH3 was then removed by changing the gas flow to He (50 cm3 min−1) for 1 h. For the desorption step, the sample was heated from 100 to 400 °C under He flow (50 cm3 min−1). Samples with a ramp rate of <90 °C min−1 were calcined in situ under the specified conditions. Samples with a ramp rate of >90 °C min−1 were calcined ex situ first and then transferred into the machine for an extra pre-treatment to 400 °C at a ramp of 10 °C min−1 to clean the surface.CO2 capture measurement
CO2 capture was carried out on a Micrometritics 3Flex with a chemisorption attachment. 100 mg of sample was calcined in situ under N2 atmosphere under the calcination conditions described. The sample was cooled to 40 °C at a ramp of 30 °C min−1 and left to evacuate under vacuum at 40 °C for 3 h. The CO2 measurements were then carried out twice at 40 °C. The first value provides the total adsorption, the second gives physisorption. The difference between the two values provides the chemisorption value.Results and discussion
Ramp rate
The ramp rate leading to the desired temperature for LDH calcination, is typically reported within the range of 1–20 °C min−1. However, the exploration into higher ramp rates and their effects on the materials has been limited. In this work, we investigated different ramp rates (5, 20, 50 and 100 °C min−1) reaching 400 °C to understand their impact on the physical and chemical properties of calcined products derived from Mg2Al LDHs. Additionally, we assessed their CO2 capture performance and their relationship between surface properties and CO2 adsorption. The calcination temperature was maintained at 400 °C for 2 h. After calcination, all samples exhibit the same typical powder X-ray diffraction (XRD) pattern of Mg2Al LDOs regardless of the ramp rates, displaying the characteristic (111), (200) and (220) Bragg reflections of MgO (Fig. S1†). Similarly, all samples present similar total pore volume (ca. 0.9 cm3 g−1) across all ramp rates (Fig. 1a). However, it was surprising to find that the surface area increases from 147.2 to 200.1 m2 g−1 as the ramp rate increases from 5 to 100 °C min−1 (Fig. 1a). This phenomenon is likely attributed to the thermal decomposition response of materials under varying ramp rates. As evident from the SEM images (Fig. S8†), the particles exhibit an approximate average diameter size of 144 nm at a ramp rate of 5 °C min−1. However, the diameter reduces to 101 and 85 nm when the ramp rate increases to 50 and 100 °C min−1 respectively. Concurrently, there is a decrease in platelet thickness from 27.4 to 18 nm. The accelerated ramp rate seems to induce a more vigorous breakdown of the cluster, likely due to the faster transitions through the distinct stages of thermal decomposition.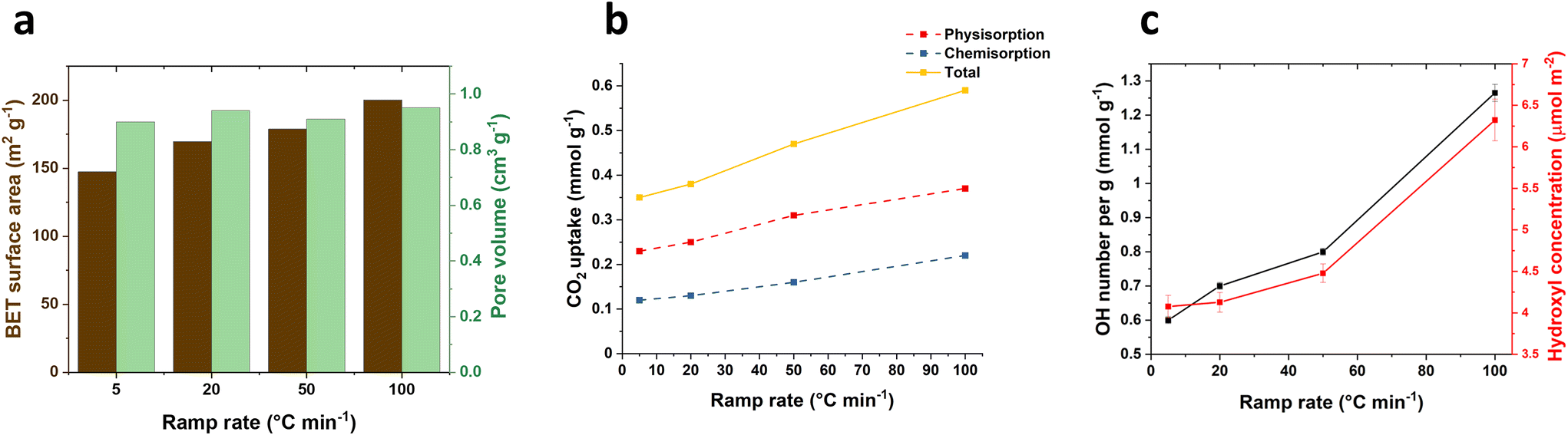 | ||
| Fig. 1 (a) BET surface area and pore volume for 400-2 h-X samples, (b) CO2 adsorption for 400-2 h-X samples at 40 °C and 100 kPa and (c) surface hydroxyl numbers for 400-2 h-X samples. | ||
CO2 adsorption measurements were carried out in 100% CO2 atmosphere at 40 °C and 100 kPa. In parallel with the observed trend in surface area, the total CO2 adsorption (Fig. 1b) increases from 0.35 to 0.59 mmol g−1 as the ramp rate increases from 5 to 100 °C min−1, displaying a significant 69% increase. Interestingly, this increase occurred despite the samples being calcined at the same temperature (400 °C). For ramp rates below 50 °C min−1, 34% of the CO2 was chemically adsorbed on the LDOs samples and this proportion further rose to 37% when the ramp rate reached 100 °C min−1. This suggests that the surface of the calcined samples might undergo changes with the rapid temperature increase, facilitating CO2 chemical adsorption. Furthermore, we noted that there is still an increase in CO2 uptake capacity when normalised to the surface area (Fig. S3†), implying that the enhanced CO2 capture is not solely attributed to an increase in surface area.
The surface hydroxyl number of the calcined samples was examed using the previously reported method.18 The results as shown in Fig. 1c reveal a significant increase in the surface hydroxyl number, rising from 0.60 to 1.26 mmol g−1 (twice as high) with the ramp rate increasing from 5 to 100 °C min−1. This notable shift in the hydroxyl numbers indicates a responsive change in surface chemistry of the calcined materials to varying ramp rates. The accelerated decomposition at a fast ramp rate, particularly at 100 °C min−1, likely leads to an incomplete dehydroxylation process. This results in a greater presence of surface hydroxyl groups remaining in the calcined samples, even after calcination at 400 °C for 2 h. Notably, this increase in the hydroxyl number remains consistent even when the values are normalised to surface area. This suggests that the observed changes are also not solely due to alternations in the material's surface area but indicative of a more complex and nuanced kinetically-controlled modification on the surface of the calcined materials. This trend in surface hydroxyl number aligns well with that of CO2 uptake capacity (Fig. 1b), indicating the positive interconnected relationship between the surface chemistry and the CO2 adsorption, where the surface hydroxyl number may play a key role in facilitating CO2 adsorption on the calcined samples.
The surface chemistry of the calcined samples was further investigated through probe molecule adsorption and temperature-programmed desorption (TPD) techniques. Ammonia gas (NH3) served as the probe for the surface acid sites, while CO2 probes the Lewis basic sites of the calcined samples. As shown in Fig. 2a, the profiles of the 400-2 h-X samples presents a broad desorption peak of NH3 from 100–400 °C and this distinct desorption pattern provides clear evidence that the material calcined with rapid ramp rate could generate significantly more surface acid sites. The total number of acid sites and the acid site density were summarised in Table 1. We found that both total number and the density of the acid sites increase with increasing the ramp rate. The highest total acid site number can reach 1.34 mmol g−1 when a fast the ramp rate 100 °C min−1 was applied, marking a more than sevenfold increase compared to the sample prepared at 5 °C min−1 (0.17 mmol g−1). To further investigate the different bonding modes to the surface of the calcined samples, the NH3 TPD profiles were deconvoluted into 2 groups based on the analysis reported by Prinetto et al. as shown in Scheme S1.†19 The first group, centred at around 150 °C, corresponds to the hydrogen bonding of NH3 to surface hydroxyl groups or the basic surface oxygens. The other group, at higher temperature around 230 °C, is attributed to the bonding motifs between nitrogen and surface metals. The proportion of the first group increases from 15% to 40% when the ramp rate increases from 5 to 100 °C min−1, a trend is consistent with what we found in surface hydroxyl number in Fig. 1c. A similar phenomenon was observed in the total number of basic sites and the basic site density, as obtained from CO2 TPD Fig. 2b. Both total number and density increase with increasing the ramp rate. The sample treated at 100 °C min−1 displayed triple the number of basic sites compared to that at 5 °C min−1 (3.65 and 1.21 mmol g−1, respectively). As previous reported, there are three types of CO2 bonding modes: bicarbonate, bidentate and monodentate as illustrated in Scheme S2.†20 In our study, the bicarbonate bonding mode from the lower desorption temperature (below 100 °C) was not measured. Therefore, the desorption profiles in Fig. 2b were deconvoluted into bidentate around 155 °C and monodentate modes at a higher temperature around 225 °C. With the slower ramps, the strong basic sites account for ca. 70% of the total measured basicity while at 100 °C min−1, it comprises only 65%. This indicates that even though there is a significant overall increase in number of basic sites, the predominant contribution comes from an increase in weaker bidentate nature. Additionally, we noted that CO2 desorption is completed at lower temperature (350 °C) for the sample subjected to 100 °C min−1 ramp rate. This finding implies that some strongest monodentate basic sites may be absent or less predominant when using a fast ramp rate (100 °C min−1).
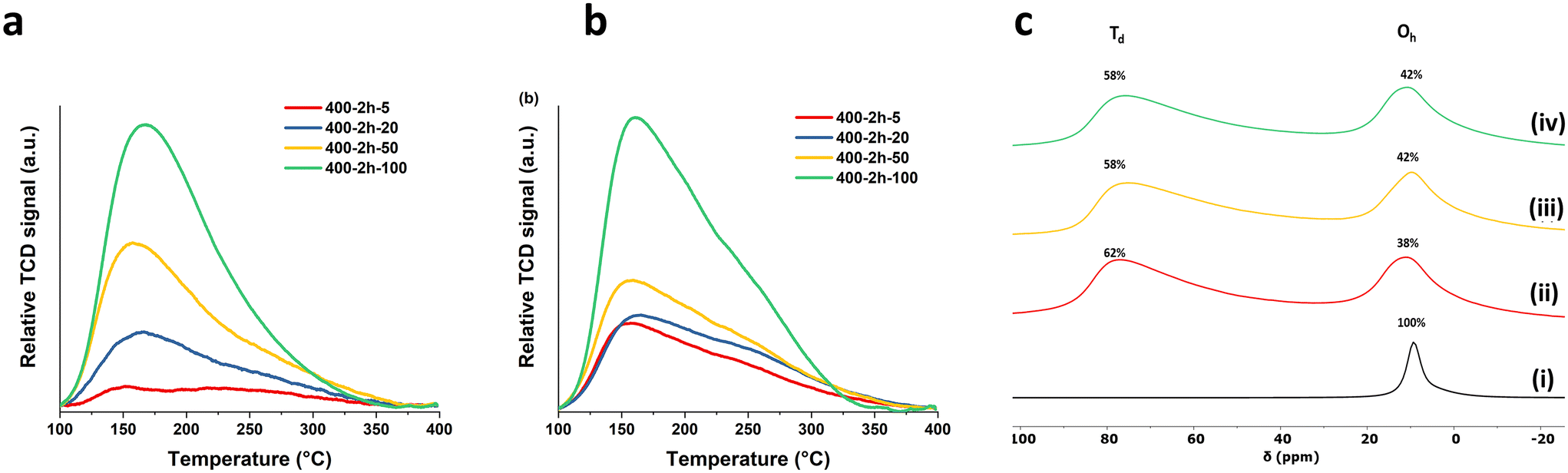 | ||
| Fig. 2 (a) NH3 TPD, (b) CO2 TPD of 400-2 h-X samples and (c) 27Al DPMAS ssNMR of (i) Mg2Al LDH, (ii) 400-2 h-5, (iii) 400-2 h-50 and (iv) 400-2 h-100. | ||
| Total basic sites (mmol g−1) | Basic site density (μmmol m−2) | Total acid sites (mmol g−1) | Acid site density (μmol m−2) | Total acid sites/total basic sites | CO2 uptake (mmol g−1) | |
|---|---|---|---|---|---|---|
| 400-2 h-5 | 1.21 | 8.22 | 0.17 | 1.15 | 0.14 | 0.35 |
| 400-2 h-20 | 1.46 | 8.61 | 0.40 | 2.36 | 0.27 | 0.38 |
| 400-2 h-50 | 1.89 | 10.58 | 0.84 | 4.70 | 0.44 | 0.47 |
| 400-2 h-100 | 3.65 | 18.24 | 1.34 | 6.70 | 0.37 | 0.59 |
| 400-1 h-100 | 2.14 | 17.22 | 1.10 | 8.85 | 0.51 | 0.38 |
| 400-3 h-100 | 3.56 | 17.57 | 1.11 | 5.48 | 0.31 | 0.41 |
| 400-6 h-100 | 2.86 | 11.87 | 1.31 | 5.44 | 0.46 | 0.4 |
The 27Al DPMAS ssNMR spectroscopy experiments were carried out to investigate the changes in Al environment corresponding to variations in ramp rates. As shown in Fig. 2c, Mg2Al W LDHs present a single distinct resonance peak at lower field which corresponds to the six-coordinated (octahedral) Al species in the LDHs layers. After calcination, an additional resonance at around 78 ppm was observed in all calcined samples. This is attributed to four-coordinated (tetrahedra) Al sites, indicating the Al atoms have diffused out of the octahedral structure of brucite layer during the calcination.21,22 The proportion of tetrahedral sites (Td%) reaches to the highest value (62%) at a ramp rate of 5 °C min−1, followed by a slight decrease to 58% when ramp rates increase to 50 and 100 °C min−1. This is likely due to the shorter calcination time associated with higher ramp rate, leading to an incomplete migration of Al out from octahedral structure of MgO. The 1H NMR spectroscopy (Fig. S9†) displays the characteristic proton resonances of LDH: the surface Mg2Al–OH at δ = 1.2 ppm and the interlayer water molecules at δ = 4.8 ppm.23 When calcined, several resonances arise between δ = 0–3 ppm, which correspond to the OH resonances in the bulk MgO structure with different neighbouring metals.24 The 13C CPMAS NMR spectroscopy (Fig. S10†) shows a slight change in the CO32− environment from δ = 171 ppm to δ = 167 ppm after post calcination. This is due to the change in environment resulting from the dehydration and dehydroxylation during the calcination process.
Calcination temperature
The calcination temperatures (100, 200, 300, 400, 500, 600 °C) were investigated using a fast ramp rate (100 °C min−1) and hold at the desired temperature for 2 h. The structure decomposition behaviour along with increasing temperature is similar to that observed with a slower ramp rate, as previously report.25,26 As shown in Fig. 3a, at 100 °C, the Mg2Al LDHs maintain a well-defined crystalline structure with characteristic harmonic (00l) Bragg reflections and in-plane (110) and (113) Bragg reflections. The (00l) Bragg reflections shift to higher angle with the increasing temperature up to 300 °C, primarily attributed to the dehydration process of LDH involving the loss of the interlayer water, resulting in a decrease of interlayer space. This is consistent with the observations from thermal gravity analysis (TGA) (Fig. S2†). Upon further temperature increase, the samples displayed a typical Mg2Al LDOs diffraction patterns, featuring characteristic (111), (200) and (220) Bragg reflections from MgO. The surface area and the pore volume (Fig. 3b) remain relatively constant when the temperature below 300 °C. However, a significant increase is observed as the temperature rises further, attributed to the removal of water and carbonates during the calcination, creating pores and channels in the materials. The highest surface area reaches up to 294 m2 g−1 and the pore volume can peak at 1.21 cm3 g−1 when the calcination temperature reaches 500 °C. Continued temperature elevation results in a decline in both surface area and pore volume due to the further growth of mixed metal oxides particles.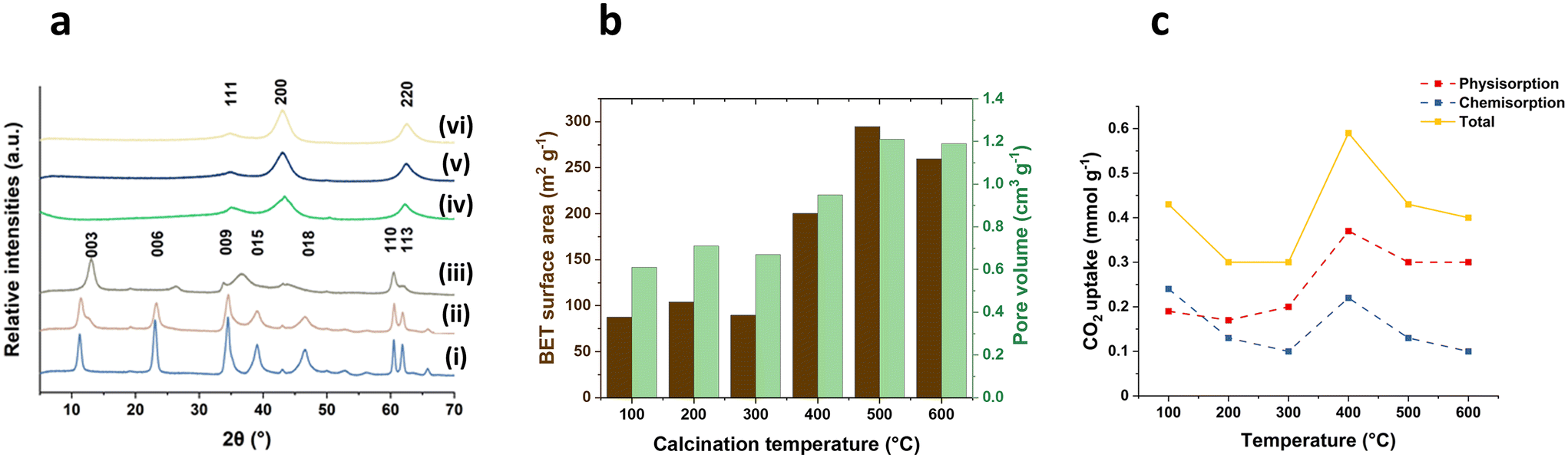 | ||
| Fig. 3 (a) XRD of X-2 h-100 samples ((i) 100-2 h-100, (ii) 200-2 h-100, (iii) 300-2 h-100, (iv) 400-2 h-100, (v) 500-2 h-100 and (vi) 600-2 h-100), (b) BET surface area and pore volume of X-2 h-100 samples, and (c) CO2 adsorption for X-2 h-100 samples at 40 °C and 100 kPa. | ||
The CO2 uptake at 40 °C and 100 kPa using X-2 h-100 samples are presented in Fig. 3c. The maximum CO2 capture value (0.59 mmol g−1) was obtained at a calcination temperature of 400 °C. This trend aligns with previous literature even though using different ramp rates.22,27 However, it is surprising to find that the sample treated at 100 °C displayed a relatively high CO2 uptake, notably exhibiting the highest chemisorption value (0.24 mmol g−1) among all the tested samples. The unexpected results could be attributed to the dissolution of CO2 in the interlayer water molecules of LDHs in 100-2 h-100 sample. A similar enhancement in CO2 capture has been reported in the literature through addition of H2O.8,28
Calcination time
The impact of calcination times (1 h, 2 h, 3 h and 6 h) at a rapid ramp rate (100 °C min−1) was also explored and the calcination temperature was fixed at 400 °C. The powder XRD patterns of the resulting calcined samples (400-X-100) are shown in Fig. 4a. Interestingly, with a fast ramp rate (100 °C min−1) and a short calcination time (1 h) at 400 °C, the material still contains the Bragg reflections of LDHs ((003), (006) and (110) at 2θ = 12.5°, 26.9° and 61.0° respectively), indicating that the LDHs is not fully transformed into an LDOs structure within such a rapid and short calcination process. Extending the calcination time to 2 h or more, the Bragg reflections indicate that LDHs has been decomposed and transferred into a LDOs structure with (111), (200) and (220) Bragg reflections at 2θ = 35°, 43° and 63° respectively. The surface area increased from 124 to 240 m2 g−1 as the calcination time increases from 1 to 6 h (Fig. 4b). Similarly, the total hydroxyl number exhibited a nearly linear increase with increasing calcination time (Fig. S6†). However, the hydroxyl concentration per surface area remained relatively consistent when calcination time is longer than 2 h, indicating that the increased hydroxyl number is primarily attributed to the greater exposed surface area. Slightly higher hydroxyl concentration at 1 h calcination time is mainly due to the presence of LDHs.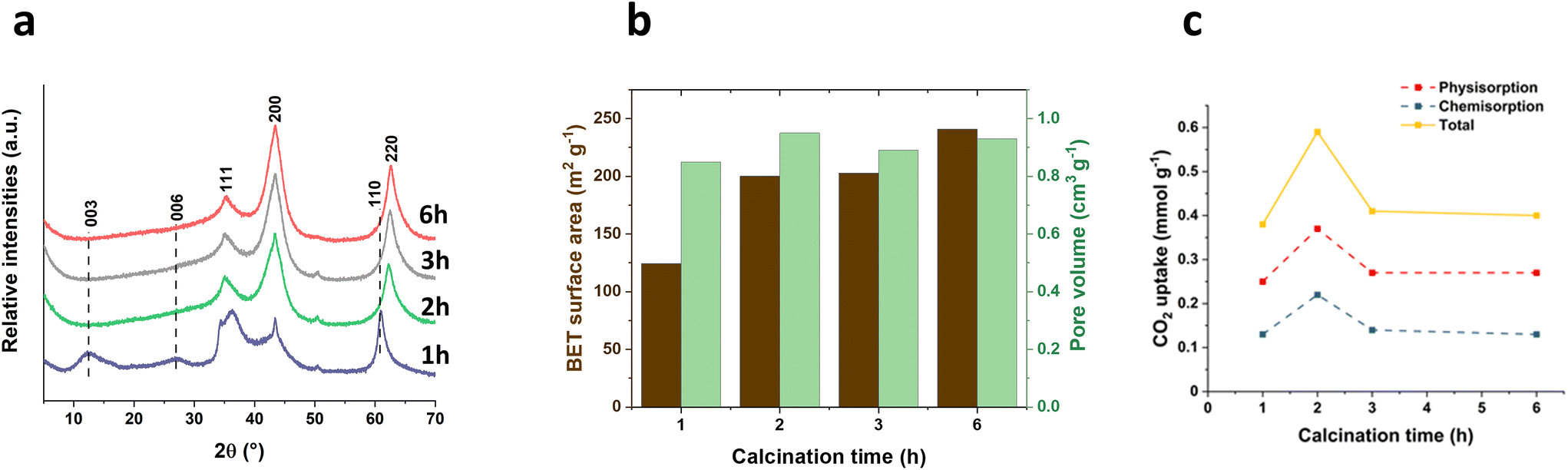 | ||
| Fig. 4 (a) XRD of 400-X-100 samples, (b) BET surface area and pore volume of 400-X-100 samples, and (c) CO2 adsorption for 400-X-100 samples at 40 °C and 100 kPa. | ||
CO2 uptake capacity (Fig. 4c) of the subsequent LDOs increases with calcination time up to 2 h, followed by a decrease in capacity at longer calcination times (3 h and 6 h). The sample calcined for 2 h (400-2 h-100) displayed a highest CO2 capture value of 0.59 mmol g−1, making an almost 50% increase compared with the value (ca. 0.4 mmol g−1) observed in samples calcined for 3 or 6 h. However, too short a calcination time and fast ramp rate may result in lower CO2 capture value due to part of the material containing a dehydrated LDHs structure which is less active as a CO2 capture material compared to LDOs.
The impact of calcination time on CO2 capture performance is linked to the surface changes in their acidity and basicity. As shown in Table 1 and Fig. S7,† the sample calcined for 2 h (400-2 h-100) possessed the highest total number of acid sites (1.34 mmol g−1) while 1 h calcination time led to the lowest acidity of 1.10 mmol g−1. The acid site density decreases with increasing calcination time, from 8.85 μmol m−2 for 1 h to 5.44 μmol m−2 for 6 h. Interestingly, 400-2 h-100 also displayed the highest number of basic sites (3.65 mmol g−1) as determined by CO2 TPD. The total number of basicity increases sharply between calcination times of 1 and 2 h, then steadily decreases with prolonged calcination, reflecting the trend observed in their CO2 capture performance. Basic site densities of these samples also follow a similar trend. However, the sample (400-6 h-100) displayed significantly less basicity sites and basicity density. The deconvolution of CO2–TPD profile (Table S1†) indicates that the sample 400-6 h-100 has a lower proportion of monodentate binding mode centred at around 215 °C (63%) compared to the shorter calcination times of 1–3 h (65%). Chizallet et al. suggested that the difference in the basic strength of MgO correlates to their coordination environment: a lower oxygen coordination number leads to an increase in basicity due to a higher concentration of the lone pair density.29 This lower coordination numbers are generated by surface defects which create steps, edges, corners or holes. The decrease in basicity sites and basicity strength of the sample (400-6 h-100) is likely due to the prolonged calcination time, which could lead to increase the surface ordering of the LDOs, allowing more oxygens to migrate into more coordinationally saturated environments.
Considering the overall impact of acid and base sites on the CO2 capture, our previous findings revealed a positive correlation between CO2 capture and acid:base ratio within a limited range of acid:base ratio (<0.2).13 In this study, we have expanded the scope by presenting a wider range of acid:base ratio obtained from all the samples calcined at 400 °C. As shown in Fig. 5, our results indicate that when the acid:base ratio is below 0.37, the CO2 uptake increases with the ratio which is consistent with our earlier observations. However, at higher acid:base ratio (>0.37), the CO2 uptake capability decreases. This decline is attributed to the acidic nature of CO2, an excessive concentration of acidic sites creates a less favourable surface for the CO2 interaction. The optimal ratio of 0.37 strikes a balance between the number of acid and base sites, facilitating sufficient hydroxyl participation in the bicarbonate bonding mode without oversaturating the surface with less favourable interactions between the CO2 molecules and acidic sites. This balance is crucial for achieving enhanced CO2 capture efficiency.
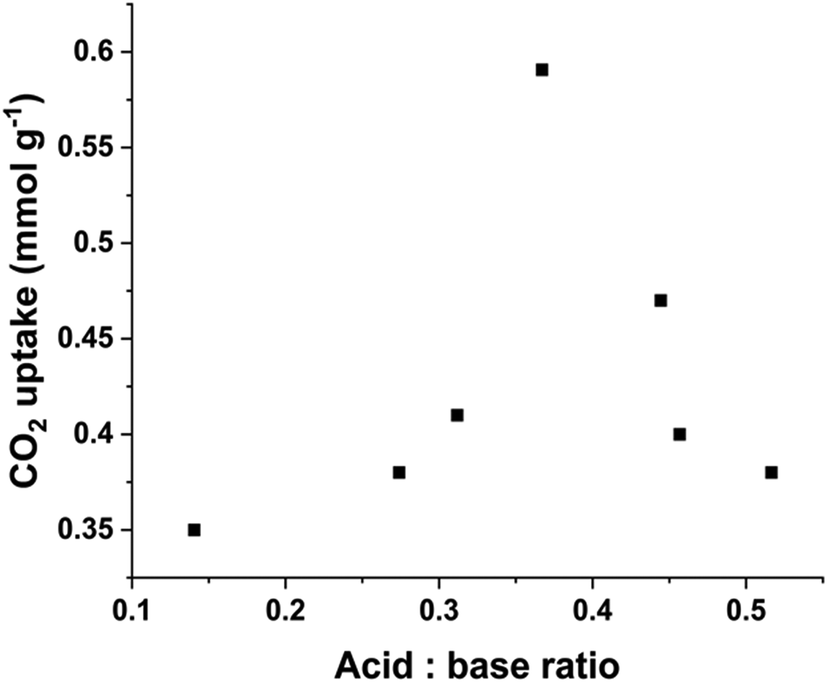 | ||
| Fig. 5 CO2 capture as a function of acid:base ratio of LDOs calcined at 400 °C. | ||
Conclusions
A comprehensive investigation into the surface chemistry and CO2 capture performance of LDO samples prepared following calcination under varying conditions reveals important relationships between the calcination parameters and ultimate CO2 capture performance. A 100 °C min−1 ramp rate to 400 °C, significantly enhances the formation of surface acid and basic sites. The resulting material (400-2 h-100) exhibits a remarkable increase in surface area, hydroxyl numbers and acid/basic site numbers, leading to superior CO2 capture efficiency (69% increase than others). On the other hand, excessively short calcination times and fast ramp rate may result in the presence of a hydrated LDH structure, compromising CO2 capture effectiveness. Furthermore, we discovered the importance of balancing acid and base sites for maximise CO2 capture performance. A critical acid:base ratio of 0.37 emerges as the key threshold, ensuring a favourable surface for CO2 interaction. This study expands on prior knowledge by presenting a broader range of acid:base ratios and underscores the complex interaction between surface functionality and CO2 capture efficiency.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank SCG Chemicals Public Co., Ltd. (Thailand) for funding.References
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- G. Marcelin, N. J. Stockhausen, J. F. M. Post and A. Schutz, J. Phys. Chem., 1989, 93, 4646–4650 CrossRef CAS.
- J. Rocha, M. Del Arco, V. Rives and M. Ulibarri, J. Mater. Chem., 1999, 9, 2499–2503 RSC.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- M. M.-J. Li, C. Chen, T. C. E. Ayvalı, H. Suo, J. Zheng, I. F. Teixeira, L. Ye, H. Zou, D. O'Hare and S. C. E. Tsang, ACS Catal., 2018, 8, 4390–4401 CrossRef CAS.
- X. Zhu, C. Chen, Q. Wang, Y. Shi, D. O'Hare and N. Cai, Chem. Eng. J., 2019, 366, 181–191 CrossRef CAS.
- X. Zhu, T. Ge, F. Yang, M. Lyu, C. Chen, D. O'Hare and R. Wang, J. Mater. Chem. A, 2020, 8, 16421–16428 RSC.
- C. Chen, P. Gunawan, X. W. D. Lou and R. Xu, Adv. Funct. Mater., 2012, 22, 780–787 CrossRef CAS.
- J. Yu, C. Chen, J. B. Gilchrist, J.-C. Buffet, Z. Wu, G. Mo, F. Xie and D. O'Hare, Mater. Horiz., 2021, 8, 2823–2833 RSC.
- J. Yu, K. Ruengkajorn, D.-G. Crivoi, C. Chen, J.-C. Buffet and D. O'Hare, Nat. Commun., 2019, 10, 2398 CrossRef PubMed.
- N. N. Meis, J. H. Bitter and K. P. de Jong, Ind. Eng. Chem. Res., 2010, 49, 1229–1235 CrossRef CAS.
- D. J. Leung, C. Chen, J.-C. Buffet and D. O'Hare, Dalton Trans., 2020, 49, 9306–9311 RSC.
- H. Du, A. D. Ebner and J. A. Ritter, Ind. Eng. Chem. Res., 2011, 50, 412–418 CrossRef CAS.
- Q. Wang, Z. Wu, H. H. Tay, L. Chen, Y. Liu, J. Chang, Z. Zhong, J. Luo and A. Borgna, Catal. Today, 2011, 164, 198–203 CrossRef CAS.
- J. M. Silva, R. Trujillano, V. Rives, M. Soria and L. M. Madeira, Chem. Eng. J., 2017, 325, 25–34 CrossRef CAS.
- T. Taher, Y. Gusti Wibowo, S. Maulana, N. Rahayu Palapa, A. Rianjanu and A. Lesbani, Mater. Lett., 2023, 338, 134068 CrossRef CAS.
- C. M. Wright, K. Ruengkajorn, A. F. Kilpatrick, J.-C. Buffet and D. O'Hare, Inorg. Chem., 2017, 56, 7842–7850 CrossRef CAS PubMed.
- F. Prinetto, G. Ghiotti, R. Durand and D. Tichit, J. Phys. Chem. B, 2000, 104, 11117–11126 CrossRef CAS.
- J. Di Cosimo, V. Dıez, M. Xu, E. Iglesia and C. Apesteguıa, J. Catal., 1998, 178, 499–510 CrossRef CAS.
- M. Bellotto, B. Rebours, O. Clause, J. Lynch, D. Bazin and E. Elkaïm, J. Phys. Chem., 1996, 100, 8535–8542 CrossRef CAS.
- Y. Gao, Z. Zhang, J. Wu, X. Yi, A. Zheng, A. Umar, D. O'Hare and Q. Wang, J. Mater. Chem. A, 2013, 1, 12782–12790 RSC.
- P. J. Sideris, U. G. Nielsen, Z. Gan and C. P. Grey, Science, 2008, 321, 113–117 CrossRef CAS PubMed.
- J. M. Rimsza, E. G. Sorte and T. M. Alam, ACS Omega, 2019, 4, 1033–1044 CrossRef CAS PubMed.
- W. Yang, Y. Kim, P. K. T. Liu, M. Sahimi and T. T. Tsotsis, Chem. Eng. Sci., 2002, 57, 2945–2953 CrossRef CAS.
- E. Kanezaki, Solid State Ionics, 1998, 106, 279–284 CrossRef CAS.
- M. K. Ram Reddy, Z. P. Xu, G. Q. Lu and J. C. Diniz da Costa, Ind. Eng. Chem. Res., 2006, 45, 7504–7509 CrossRef CAS.
- K. Coenen, F. Gallucci, G. Pio, P. Cobden, E. van Dijk, E. Hensen and M. van Sint Annaland, Chem. Eng. J., 2017, 314, 554–569 CrossRef CAS.
- C. Chizallet, G. Costentin, M. Che, F. Delbecq and P. Sautet, J. Phys. Chem. B, 2006, 110, 15878–15886 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation technique details, XRDs, BET surface area, TGA, hydroxyl numbers, SEM images, NMR, TPD data. See DOI: https://doi.org/10.1039/d4dt00270a |
| This journal is © The Royal Society of Chemistry 2024 |