
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the anticancer potential of 4-phenylthiazole derived Ru(II) and Os(II) metalacycles†
Paul
Getreuer
 ab,
Laura
Marretta
c,
Emine
Toyoglu
a,
Orsolya
Dömötör
d,
Michaela
Hejl
a,
Alexander
Prado-Roller
a,
Klaudia
Cseh
a,
Anton A.
Legin
a,
Michael A.
Jakupec
ae,
Giampaolo
Barone
c,
Alessio
Terenzi
c,
Bernhard K.
Keppler
ae and
Wolfgang
Kandioller
*ae
ab,
Laura
Marretta
c,
Emine
Toyoglu
a,
Orsolya
Dömötör
d,
Michaela
Hejl
a,
Alexander
Prado-Roller
a,
Klaudia
Cseh
a,
Anton A.
Legin
a,
Michael A.
Jakupec
ae,
Giampaolo
Barone
c,
Alessio
Terenzi
c,
Bernhard K.
Keppler
ae and
Wolfgang
Kandioller
*ae
aInstitute of Inorganic Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Straße 42, 1090 Vienna, Austria. E-mail: wolfgang.kandioller@univie.ac.at
bVienna Doctoral School in Chemistry (DoSChem), Faculty of Chemistry, University of Vienna, Waehringer Straße 42, 1090 Vienna, Austria
cSTEBICEF-Department, University of Palermo, Viale delle Scienze, Ed. 17, 90128 Palermo, Italy
dDepartment of Molecular and Analytical Chemistry, University of Szeged, Dóm tér 7-8, 6720 Szeged, Hungary
eResearch Cluster “Translational Cancer Therapy Research”, University of Vienna, Waehringer Straße 42, 1090 Vienna, Austria
First published on 1st March 2024
Abstract
In this contribution we report the synthesis, characterization and in vitro anticancer activity of novel cyclometalated 4-phenylthiazole-derived ruthenium(II) (2a–e) and osmium(II) (3a–e) complexes. Formation and sufficient purity of the complexes were unambigiously confirmed by 1H-, 13C- and 2D-NMR techniques, X-ray diffractometry, HRMS and elemental analysis. The binding preferences of these cyclometalates to selected amino acids and to DNA models including G-quadruplex structures were analyzed. Additionally, their stability and behaviour in aqueous solutions was determined by UV-Vis spectroscopy. Their cellular accumulation, their ability of inducing apoptosis, as well as their interference in the cell cycle were studied in SW480 colon cancer cells. The anticancer potencies were investigated in three human cancer cell lines and revealed IC50 values in the low micromolar range, in contrast to the biologically inactive ligands.
Introduction
Since the landmark discovery of cisplatin,1 platinum-based anticancer agents have been effectively utilized for the treatment of numerous cancer types on a global scale.2,3 However, dose-dependent side effects, such as nephrotoxicity, neurotoxicity, hepatotoxicity, and resistances severely hamper their efficacy.4 This implies a strong need for novel metal-based drugs, which more selectively target tumor cells and overcome resistance problems, while featuring unique mechanisms of action.5,6Within the class of metallodrugs, ruthenium-based compounds have garnered attention as promising alternative anticancer agents. Notably, ruthenium(III) complexes, specifically sodium trans-[tetrachlorido-bis(1H-indazole) ruthenate(III)] (BOLD-100, Fig. 1) and imidazolium trans-[Ru(N-imidazole)(S-DMSO)Cl4] (NAMI-A), have shown effective inhibition of tumor metastasis and angiogenesis. BOLD-100, the sodium analog of indazolium trans-[tetrachlorido-bis(1H-indazole)ruthenate(III)] (KP1019, Fig. 1), features enhanced aqueous solubility compared to its indazolium counterpart. Successful clinical phase I trials were conducted, making BOLD-100 the second ruthenium-based anticancer drug to move on to a phase II trial. Throughout phase I trials, it has already demonstrated therapeutic activity in solid tumors, such as non-small cell lung cancer, colorectal carcinoma and particularly in gastrointestinal neuroendocrine tumors.7–9 Furthermore, recent reports of an ongoing phase IIa trial showed promising results.10
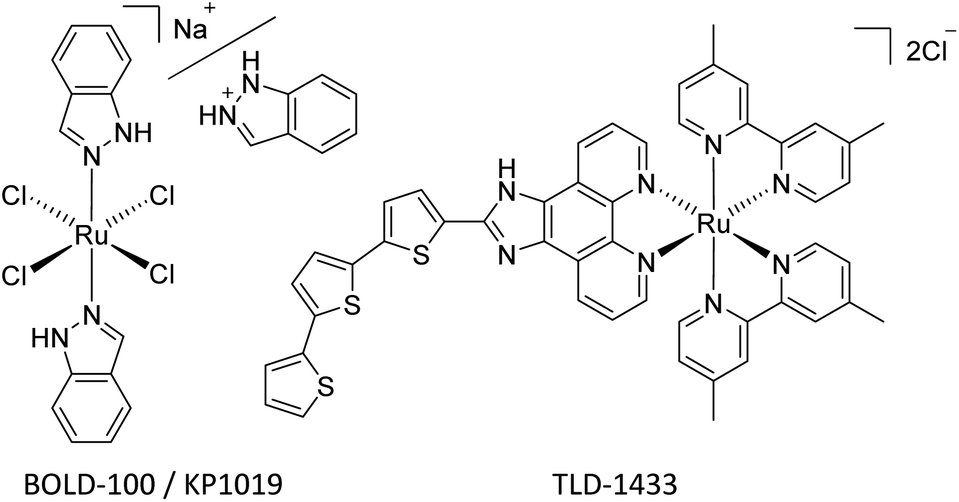 | ||
| Fig. 1 Investigational Ru-based anticancer drugs BOLD 100 (Na+)/KP1019 (indazolium) and TLD1433. | ||
Activation by reduction of the Ru(III) center to the more reactive Ru(II) species is assumed to be an essential step in the mechanism of action.11 Hence, Ru(II) compounds moved to the spotlight of ruthenium-based anticancer research. TLD-1433 (or [Ru(4,4′-dmb)2(IP-3T)](Cl)2 [where: 4,4′-dmb = 4,4′-dimethyl-2,2′-bipyridine; IP = imidazo[4,5-f]-[1,10]phenanthroline; 3T = α-terthiophene], Fig. 1) is currently the furthest developed Ru(II) antitumor drug, undergoing a clinical phase II trial for its potential in photodynamic therapy of nonmuscle-invasive bladder cancer.12–14
Stabilization of the Ru(II) center under normal physiological conditions can be achieved by the coordination of π-bonded arenes, which affords the class of highly stable pseudo-octahedral “piano-stool” complexes. These Ru(II)-arene organometallics provide a versatile framework that paves the way for straightforward refinement of antitumor properties through ligand variation.15–17 Sadler's and Dyson's groups investigated organo-ruthenium complexes extensively, which resulted in the development of the two most prominent and advanced representatives of this substance class, namely, Sadler's RAED organometallics and Dyson's RAPTA complexes.15,18–21
Most Ru(II) arene compounds feature a halido leaving group, priming the complex for biological interactions via substitution of the halido moiety by a water molecule. This process is presumed to be pivotal in the mechanism of action22 and emphasized by the activation-by-aquation hypothesis.23
Due to limited stability of conventional bidentate motifs (e.g. O,O; N,O; S,O), the application of the respective organometallics as anticancer drug candidates remains severely restricted.24 Consequently, scientists emphasized alternatives with improved stability under physiological conditions, which led us to the investigation of C,N-coordination motifs in bidentate ligands for cytotoxic organometallics.15,17,25 Over the last years, especially C,N-coordinated Ru(II) metalacycles featuring 2-arylthiazole,26 2-phenylindole,27 2-phenylpyridin,28 benzimidazole,29,30 4-phenyltriazole15,17 and 2-phenylbenzothiazole25 scaffolds have been reported. In contrast, the biological properties and the cytotoxicity of osma(II)cycles of the general form [Os(arene)LCΛNCl] remain nearly unexplored and were only investigated by Boff et al.,31 Riedl et al.15 and Mokesch et al.25 In every case, promising cytotoxicities in the low micromolar range were reported.
To extend the library of cyclometalated antitumor agents, the present contribution introduces the very robust and versatile 4-phenylthiazole scaffold. We report on the straightforward synthesis and characterization of Ru(II) and Os(II) arene complexes bearing 4-phenylthiazoles as C,N-chelates. We show the cytotoxicity of the synthesized compounds in human lung adenocarcinoma (A549), colon adenocarcinoma (SW480), and ovarian teratocarcinoma (CH1/PA-1) cells. Furthermore, we explore the impact of five 4-phenylthiazole derivatives as C,N bidentate ligands and the difference between Ru(II) and Os(II) metal centers on the biological activity. To elucidate the mechanism of action, we also investigated the cellular accumulation, impact on cell cycle distribution and the compounds’ potential to induce apoptosis in SW480 cells. In addition to these findings, the complexes’ ability to interact with G-quadruplex (G4) structures is also subject of discussion.
Results and discussion
Synthesis
Five 4-phenylthiazole ligands were synthesized based on the procedure of Lee et al.32 utilizing a straightforward Suzuki cross-coupling reaction. 4-Bromothiazole was coupled with the appropriate arylboronic acid or ester, using tetrakis(triphenylphosphine)Pd(0) as catalyst and after workup the desired ligands were obtained in moderate to good yields (42%–89%, Scheme 1).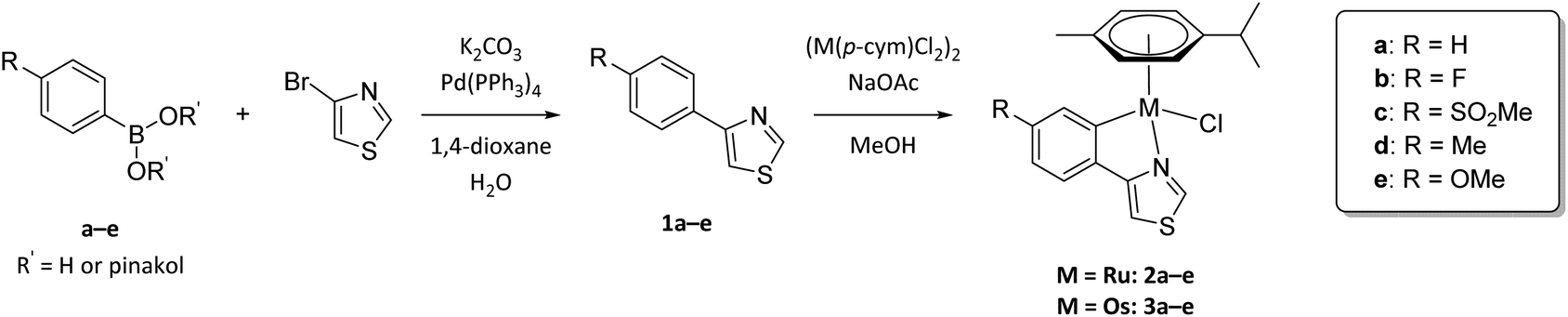 | ||
| Scheme 1 Synthesis of substituted 4-phenylthiazole ligands 1a–e and their Ru(II) (2a–e) and Os(II) (3a–e) metalacycles. | ||
Cyclometalation was achieved via sp2 C–H bond activation. The dimeric precursor [Ru(p-cym)Cl2]2 or [Os(p-cym)Cl2]2 (p-cym = p-cymene) is thereby converted to M(p-cym)(OAc)2 using an excess of sodium acetate. The activated organometallic species was then converted with the ligand, where in the first step the thiazole nitrogen binds coordinatively to the metal center. A subsequent deprotonation of the ortho hydrogen of the phenyl residue by uncoordinated acetate via an intramolecular SE3-mechanism and simultaneous coordination concludes the cyclometalation reaction.33 Lastly, the acetato-complex [M(p-cym)(LCΛN)(OAc)] undergoes transition to the desired chlorido products. However, with high acetate catalyst loadings a fraction of acetato-complex remains in the reaction mixture. To improve the conversion to the chlorido-complexes, a novel step in the work-up of cyclometalation procedures was introduced and exploited. In fact, the dissolution of the crude product in CH2Cl2 and subsequent vigorous stirring with brine grants sufficient chloride ions to convert residual acetato-derivatives.
While precipitation from CH2Cl2 solution with n-hexane can be utilized to purify the target complexes, column chromatography proves to be much more efficient, affording the desired metalacycles in elemental analysis purity and moderate to good yields (46%–86%).
Characterization
The synthesized compounds were characterized by means of standard analytical methods such as 1H-, 13C- and 2D-NMR, high-resolution electrospray ionization mass spectrometry (ESI-MS), X-ray crystallography, and elemental analysis.1H- and 13C-NMR confirmed the formation of the desired cyclometalates. Prior studies with related triazole organometallics showed that the chlorido leaving group is readily replaced by DMSO and due to similar behavior of the complex series CDCl3 was used as NMR solvent.15 Metal coordination breaks the symmetry of the substituted phenyl moiety, resulting in various splitting motifs depending on the substituent in position 4 as can be seen in the 1H spectra of the formed complexes (Fig. S1–S15†). Due to the electron withdrawing effect, the SO2Me residue shifts the protons para and meta to the metalated carbon to lower fields. Lastly, the proton signals of the 4-fluorophenyl ring were easily assignable due to the presence of additional (H,F) couplings.
Thiazole protons always appeared as two doublets, whereas H-2 was shifted downfield (0.2–0.3 ppm) upon metal coordination, due to the electron withdrawing effect of the metal center. Interestingly, the thiazole H-4 proton is considerably shifted upfield (0.2–0.3 ppm) and found around 7.3–7.5 ppm. Furthermore, the arene protons of p-cymene can be observed as four separate doublets in contrast to the free dimeric precursor where the four aromatic cymene protons were found as two doublets. In case of mesyl bearing 2c, ethyl acetate traces (0.25 eq.) from the purification via column chromatography remained in the sample even after drying for several days at 50 °C in vacuo.
High-resolution mass spectrometry of the cyclometalates revealed the presence of the chlorido abstracted species [M − Cl]+, as well as sodium adducts [M + Na]+ in some cases. Additionally, the Os(II)-metalacycles were observed as dimeric sodium adducts [2M + Na]+ (Fig. S16–S25†).
Single crystals of 2a, 2b, 2d and 2e as well as 3a, 3b and 3d, suitable for X-ray diffraction analysis, were obtained by slow diffusion of n-hexane into DCM or CHCl3 complex solutions. Detailed crystal data, data collection parameters, structure refinement details and CCDC-codes and be inferred from the ESI (Fig. S26–S32 and Tables S1–S8†). As depicted in the crystal structure of 2a (Fig. 2) the complexes adopt the so-called piano-stool configuration. Therein, the π-bonded p-cym represents the seat and the mono- and bidentate ligands the three legs of the stool. The investigated complexes are representatives of the monoclinic space groups P21/n (2a, 2e, 3a, 3b) or P21/c (2b, 2d, 3d). Both enantiomers were found in the elemental unit of the complexes. Within the pseudo-octahedral configuration of the compounds, a new distorted five-membered ring arises from the chelating ligand and the metal center. Thus, the phenyl and thiazole rings are locked in plane, featuring low torsion angles of 0.4(4)–4.8(3)° along the thiazole-phenyl bond. The coordination bonds exhibit remarkable similarity between the two metal counterparts, as anticipated due to a mere 0.01 Å difference in their atomic radii (Ru: 1.34 Å, Os: 1.35 Å). Os–N (2.079(3)–2.101(7) Å) and Os–C (2.069(7)–2.081(4) Å) bonds were found to be slightly longer than their Ru analogs (2.065(4)–2.079(2) Å and 2.061(2)–2.070(3) Å, respectively). On the other hand, the osmium metal center is marginally closer to the arene ring (1.6820(2)–1.6847(3) Å) compared to Ru analogs (1.6905(3)–1.6956(3) Å), with ring slippages of 0.062–0.095 Å. A similar trend was observed with the related 4-phenyltriazole based Ru(II)- and Os(II)-arene compounds by Riedl et al.15 The metal-chlorido bond lengths (2.4124(11)–2.4232(6) Å) are consistent with anticipated values.15,25,34–36
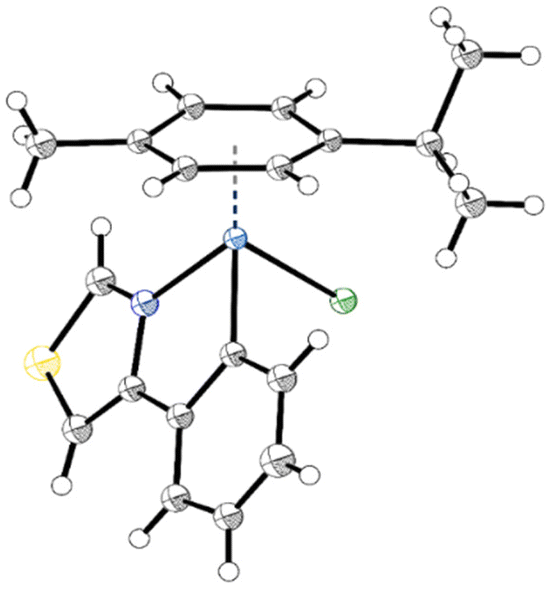 | ||
| Fig. 2 Crystal structure of ruthenacycle 2a. | ||
Stability in aqueous solution
The aqueous stability of the metalacyles was determined via UV-Vis spectroscopy under pseudo-physiological conditions over 24 h at 20 °C using 1% DMF as solubilizer in PBS (40 μM compound concentration, pH 7.4, Fig. S33–S42†). DMF was used instead of DMSO to avoid the formation of solvent adducts.15 Based on the obtained spectra, the ruthenium(II) and osmium(II) compounds undergo (micro)precipitation (except complex 2c), which results in a slight decrease of absorption over time; however, no new peaks, isosbestic points or shifts of peak maxima were observed over 24 h.Chloride ion affinity
The chloride-ion affinity of 2a and 2c was investigated via UV-Vis spectroscopy at various KCl concentrations at 25 °C using 1% DMF as solubilizer in water. The hydrolysis of the M–Cl bond was found to be relatively fast, the equilibrium was reached within ca. 10 min, therefore batch samples were prepared and the H2O/Cl− exchange constants were calculated based on the obtained UV-Vis spectra (Fig. S43 and S44†). The determined constants are log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K(H2O/Cl−) = 1.16 ± 0.01 for 2a and 1.26 ± 0.01 for 2c. Accordingly, the chlorido ligand dissociates in >99% when the complex is dissolved in pure water. In the presence of 100 mM chloride ion concentration (corresponds to the chloride ion level of blood plasm) the original chlorido complexes are present in 60% (2a) and 65% (2c), which are in very good agreement with the results of 1H NMR experiments (Fig. S45†).
K(H2O/Cl−) = 1.16 ± 0.01 for 2a and 1.26 ± 0.01 for 2c. Accordingly, the chlorido ligand dissociates in >99% when the complex is dissolved in pure water. In the presence of 100 mM chloride ion concentration (corresponds to the chloride ion level of blood plasm) the original chlorido complexes are present in 60% (2a) and 65% (2c), which are in very good agreement with the results of 1H NMR experiments (Fig. S45†).
Antiproliferative activity and cellular accumulation
The ruthenium(II) compounds (2a–e), the osmium(II) analogs (3a–e), as well as the corresponding ligands (1a–e) were evaluated for their antiproliferative activity in human lung adenocarcinoma (A549), colon adenocarcinoma (SW480), and ovarian teratocarcinoma (CH1/PA-1) cell lines by means of the colorimetric MTT assay (Table 1 and Fig. S46, S47†). Unlike the free 4-phenylthiazole ligands, for which IC50 values were not reached with the tested concentrations of up to 200 μM, the metalacycles 2a–e and 3a–e demonstrated significant cytotoxic effects with IC50 values in the low micromolar range. In general, all tested complexes exhibit their highest activity in the broadly chemosensitive CH1/PA-1 cell line and the lowest in the multidrug-resistant A549 cell line. Previously published antiproliferative activity of comparable ruthenium(II) based metalacycles of the general form [RuII(arene)LCΛN] was found in the same range as the investigated 4-phenylthiazole derived ruthenacycles (2-aryldiazole (10–150 μM),26 2-phenylindole (0.45–5.4 μM),27 2-phenylpyridine (3–100 μM),28 benzimidazole (1–150 μM),29,30 4-phenyl-1,2,3-triazole (0.65–109 μM),15,17 2-phenylbenzothiazole (2.7–16 μM)25). In contrast to ruthenacycles, osmium analogs [OsII(arene)LCΛN] are relatively sparse in literature. Nevertheless, their antiproliferative activity corresponds well with Riedl's 4-phenyl-1,2,3-triazole (0.98–34 μM)15 and Mokesch's 2-phenylbenzothiazole (1.2–8.7 μM)25 scaffolds. Remarkably, they exhibit slightly higher cytotoxicity than the ruthenium counterparts. Within this compound series, mesyl bearing metalacycles 2c and 3c show the most potent cytotoxic effect. On the other hand, the methyl analogs, 2d and 3d, exhibit the lowest antiproliferative activity within the tested cell lines. The differences between IC50 values of the least and most potent representative of each series generally indicate a moderate impact of the variable substituent.| Compound | A549 [μM] | SW480 [μM] | CH1/PA-1 [μM] |
|---|---|---|---|
| 1a–e | >200 | >200 | >200 |
| 2a | 24 ± 4 | 9.5 ± 0.7 | 3.0 ± 0.9 |
| 2b | 16 ± 1 | 8.6 ± 0.9 | 3.4 ± 0.2 |
| 2c | 12 ± 1 | 5.1 ± 0.4 | 1.6 ± 0.4 |
| 2d | 34 ± 3 | 19 ± 2 | 14 ± 2 |
| 2e | 27 ± 1 | 17 ± 1 | 7.3 ± 0.4 |
| 3a | 17 ± 1 | 9.3 ± 1.1 | 3.0 ± 0.2 |
| 3b | 10 ± 1 | 7.1 ± 0.3 | 2.0 ± 0.4 |
| 3c | 10 ± 1 | 4.4 ± 0.5 | 0.83 ± 0.14 |
| 3d | 17 ± 1 | 9.3 ± 1.6 | 3.7 ± 0.6 |
| 3e | 14 ± 1 | 7.1 ± 1.2 | 2.2 ± 0.3 |
To further elucidate the effects of the varying substituents of the metalacycles, the cellular accumulation of the ruthenium series 2a–e in SW480 cells was assessed. Additionally, the calculated lipophilicity of the unbound ligand (clogP), which serves as a representative measure for the corresponding complexes, was determined using molinspiration (Table 2). Surprisingly, cellular accumulation is most pronounced for both the least lipophilic mesyl derivative 2c and the very lipophilic fluoro derivative 2b. Except for the comparatively low IC50 value of 2a, a clear trend of higher compound cytotoxicities at higher cellular accumulation was found. A direct comparison of IC50 values and drug accumulation is illustrated in Fig. S48.†
P (clogP) values of the corresponding free ligands
| Compound | Ru/cell [fg] | clogP (free ligand) |
|---|---|---|
| 2a | 183 ± 13 | 2.54 |
| 2b | 326 ± 39 | 2.71 |
| 2c | 326 ± 14 | 1.41 |
| 2d | 183 ± 14 | 2.99 |
| 2e | 263 ± 11 | 2.60 |
Mechanism of action
The rather high cytotoxicity of the metalacycles compelled us to conduct further investigations into the mechanism of action of this compound class.Concerning the FRET assay, compounds 2a, 2c, and 2d were incubated at different concentrations with selected oligonucleotides (Tables S9 and S10†) folded as G4s (c-KIT1 and c-MYC) or duplex B-DNA (dsDNA). c-KIT1 and c-MYC were selected considering that these DNA structures form in the promoters of the corresponding oncogenes c-kit and c-myc and have a key role in regulating their expression.44 The ability of the cyclometalates to stabilize such structures was quantified by the increase of the oligonucleotides’ melting temperatures (ΔT1/2). Interestingly, no discernible stabilization with any of the compounds was observed with dsDNA up to a 1/20 [DNA]/[compound] ratio (Fig. S49†). In contrast, a concentration dependent effect was observed when the metal-based compounds were incubated with the G4 motifs (Fig. S50 and Table S10†). The most effective stabilization profile was achieved with c-MYC and compound 2c, which induced a ΔT1/2 of approximately 15 °C at the 1/20 ratio, reaching 18 °C at 1/30 (Fig. 3a). In contrast, lower ΔT1/2 values for c-KIT1 were obtained and only at the highest ratios for 2a and 2d. In comparison to other metal complexes that induce higher ΔT1/2 values at lower concentration,45 the tested compounds can be regarded as modest G4 stabilizers. Despite this, they exhibit a promising preference for G4 over B-DNA and show a clear concentration dependent G4 stabilization. No perturbation of the c-KIT1 and c-MYC structures was observed as a consequence of the interaction with the metalacycles. Their circular dichroism signatures, typical of parallel G-quadruplexes (a positive and a negative band centered at 263 and 241 nm, respectively), remain unaltered after titration of the oligos with increasing amounts of 2a, 2c and 2d (Fig. S51†). As a control, titrations were performed using calf thymus DNA (ct-DNA) selected as B-DNA model, and its structure also remained unaffected (Fig. S52†).
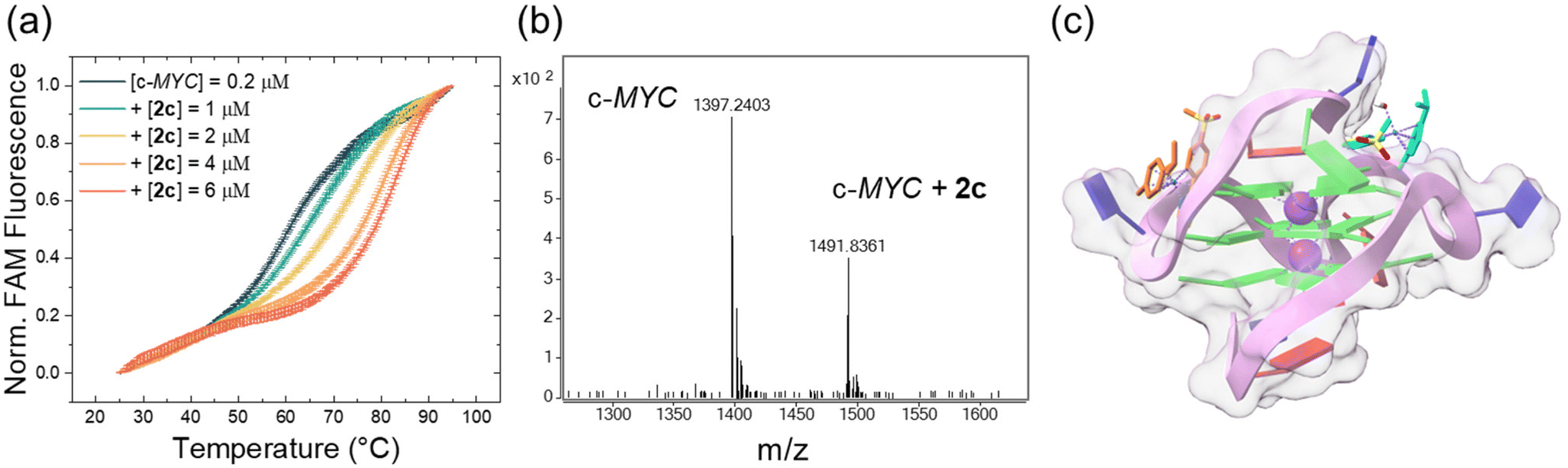 | ||
| Fig. 3 (a) FRET melting profiles of c-MYC (0.2 μM) upon interaction with 2c at the indicated concentration values. (b) MS of 2c incubated with c-MYC for 2 h at room temperature. Five charged masses of c-MYC: found: 1397.2403, calced: 1397.2318 (M-H− for C220H270O131N95P21.) Five charged masses of 2c-c-MYC adduct: found: 1491.8361, calced: 1491.8332 (M − H− for C220H270 O131 N95P21C20H21NO2RuS2) (c) cartoon showing possible binding sites of 2c (R) with c-MYC G4 (PDB id: 1XAV). | ||
As mentioned, the synthesized Ru(II) arene compounds feature a halido leaving group, which after activation-by-aquation, can be substituted by a DNA base. Hence, mass spectrometry was employed to study the adduct formation of 2a, 2c, 2d with 9-ethylguanine. When the cyclometalates (0.2 mM) were incubated with an excess of 9-ethylguanine (0.6 mM) in H2O (4% DMF as solubilizer) for 2 h at room temperature they all readily formed adducts with the selected DNA base model (Fig. S53–S55†). Given such a result and effective stabilization observed in FRET experiments, adduct formation of ruthenacycle 2c with the oligonucleotide c-MYC was also investigated by MS (Fig. 3b). Peaks corresponding to the five charged mass of c-MYC (found: 1397.2403, calcd: 1397.2318) and of the 2c-c-MYC adduct (found: 1491.8361, calcd: 1491.8332) were found (Fig. S56†), confirming a tight interaction between the two entities.
The interaction between the selected G4 structures and compounds 2a, 2c and 2d (R and S enantiomers) was investigated by molecular docking calculations. The NMR resolved structures of c-MYC (PDB id: 1XAV) and c-KIT1 (PDB id: 2O3M) served as G4 models. The structures of both R and S enantiomers of 2a, 2c and 2d were optimized by DFT calculations. Out of 50 possible poses, the most representative ones of each cluster are reported in Fig. S57 and S58† for c-MYC and c-KIT1, respectively.
Overall, the interaction of the metalacycles with the G4 structures takes place within the DNA grooves rather than with the G-tetrads in contrast to what is observed with classic planar G4 binders which interact with the top/end guanine tetrads. Interestingly, the docking free binding energy values (Tables S11 and S12†) suggest that the R enantiomers non-covalently interact better with both c-KIT1 and c-MYC G4s, with compound 2c (R) being the most effective c-MYC binder, corroborating FRET results (Fig. 3c and Fig. S49, S50†). It is worth mentioning that docking results were obtained using the aqua complexes. The reported poses are driven by electrostatic forces and are the step before substitution of the water molecule by a DNA base.
:1:1) of Ac-Met-OMe, Ac-His-OMe and Ac-Cys-OMe for 24 h at rt and analyzed via1H-NMR (1 mM; 10% d-DMF in D2O) and HRMS (5 μM; 1% DMF in 400 μM NH4OAc-solution). Additionally, the ruthenacycles were incubated with equimolar amounts of each amino acid individually and analyzed via the above described 1H-NMR method.
Overall, unsubstituted 2a reacted quantitatively with L-methionine and L-cysteine and only in the case of L-histidine small amounts of unreacted complex were present after 24 h according to NMR, whereby the L-methionine and L-histidine adducts were formed. However, due to the strong trans effect of thiols the complex decomposed after incubation with L-cysteine, releasing bidentate ligand 1a (Fig. S59†). The mass spectrum validated that argument, as only the L-methionine-2a and L-histidine-2a adducts were observed (Fig. 4). Mesyl bearing 2c proved to be less reactive since only traces of the L-methionine-2c and L-histidine-2c adducts were found by MS. This trend was also observed in the performed NMR experiments, where substantial amounts of 2c remained unaltered after 24 h of incubation with each amino acid. Here again, only L-methionine and L-histidine adducts were recorded, while interaction with L-cysteine led to the mentioned loss of bidentate ligand 1c (Fig. S60†). The competitive NMR experiments showed that for both complexes (2a and 2c) the L-methionine adducts are preferably formed. In case of 2a, a substantial amount of free bidentate ligand 1a was observed.
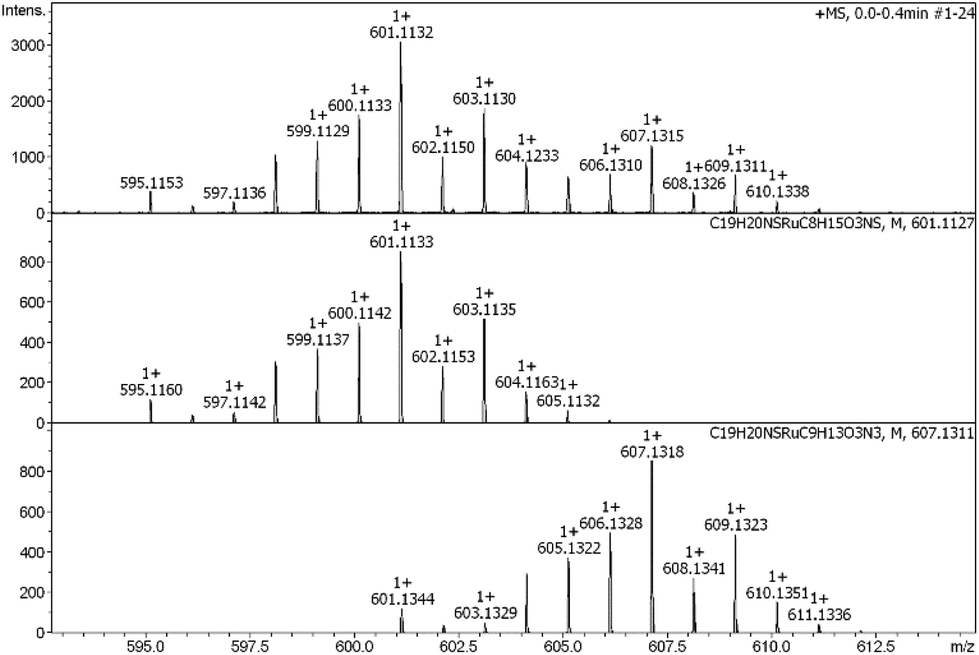 | ||
| Fig. 4 Mass spectrum of amino acid adducts (Ac-Met-OMe, Ac-His-OMe) of 2a. First row: observed mass; second row: calculated mass for Met-adduct; third row: calculated mass for His-adduct. | ||
In tested concentrations, the compounds exhibit minor to moderate effects on the cell cycle of SW480 cells (Table S13†). No clear concentration-effect pattern can be observed. However, there is a tendency of redistribution from the resting (G1/G0) phase towards the synthetic (S) and division (G2/M) phases, which might indicate a modest (ca. 10 ± 5%) but visible inhibition of the cell cycle in the latter two phases (Fig. S61 and S62†). Altogether, the mesyl bearing complexes 2c and 3c induce stronger cell cycle perturbations at IC50 levels, resulting in a 10–16% decrease of cells in G1/G0 phase. Interestingly, the Ru complex 2c induces a redistribution of cells to G2/M phase (by ca. 12%), while the Os analog 3c induces an S-phase inhibition (by ca. 14%) relative to the untreated control (Fig. S63†).
Surprisingly, the compounds only exhibit slight apoptotic effects, except for excessively high concentrations (≥100 μM). Osmium compounds 3c and 3d show the strongest induction of apoptosis after 48 h with 25% and 14%, respectively, at concentrations twice as high as their individual IC50 values. Apoptotic potencies decrease in the following order: 3c > 3d > 2c > 2d, with the ruthenacycle 2d only showing 5% of induced cell death at the stated concentration and incubation time. Generally, induction of cell death occurred slowly, as the fraction of early apoptotic cells is the largest, even after 48 h of incubation.
Experimental part
Materials and methods
Methanol was distilled from Mg/I2 and stored over molecular sieve (3 Å). 4-Bromothiazole (98%, Ambeed), (4-fluorophenyl)boronic acid (TCI Europe), 4,4,5,5-tetramethyl-2-phenyl-1,3,2-dioxaborolane (98%, TCI Europe), 2-(4-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (97%, Ambeed), 4,4,5,5-tetramethyl-2-(p-tolyl)-1,3,2-dioxaborolane (98%, Ambeed), (4-(methyl-(methylene)sulfinyl)phenyl)boronic acid (98%, Ambeed), potassium carbonate (Acros Fisher), tetrakis(triphenylphosphine)palladium(0) (>97%, TCI Europe), 1,4-dioxane (99.5%, Acros Fisher), ruthenium(III) chloride hydrate (38–41% Ru, Johnson Matthey), osmium tetroxide (Johnson Matthey), hydrazine dihydrochloride (≥98.0%, Sigma), α-terpinene (90%, Acros Fisher), sodium acetate anhydrous (≥98.5%, Fluka), potassium chloride (puriss, Molar Chemicals), disodium hydrogen phosphate (puriss, Molar Chemicals), sodium dihydrogen phosphate (puriss, Molar Chemicals), D2O (99.9%, Sigma Aldrich), molecular sieve (3 Å, beads, 4–8 mesh), phosphate buffered saline (pH 7.4, 10×, Gibco), Ac-L-Met-OMe (97%, Ambeed), Ac-L-His-OMe (95%, Ambeed), Ac-L-Cys-OMe (97%, Ambeed) were used without further purification. The dimeric metal precursors [Ru(p-cymene)Cl2]2 and [Os(p-cymene)Cl2]2 were synthesized as described by Geisler et al.46 Ligand 1a was synthesized according to literature and ligands 1b–e were obtained similarly. Acquired spectroscopic data of the ligands 1a–e was in agreement with reported values.47Purification via flash column chromatography was conducted with Biotage® Isolera system and silica gel (VWR, mesh 40–63 μm). 1H-, 13C- and 2D-NMR spectra of the complexes were recorded on a Bruker FT-NMR spectrometer Avance III™ HD 700.40 MHz or on a Bruker FT-NMR spectrometer Avance III™ 600.25 MHz, 1H-NMR spectra of the ligands and amino acid interaction studies were recorded on a Bruker FT-NMR spectrometer AV NEO 500.10 MHz in CDCl3 or D2O and referenced to the residual solvent signals. High resolution ESI mass spectra of the metalacycles and amino acid interaction studies were recorded at the Mass Spectrometry Center of the University of Vienna (Faculty of Chemistry) on a Bruker maXis ESI-Qq-TOF Mass Spectrometer. High resolution mass spectra of 9-ethylguanine and oligonucleotide interactions were recorded on an Agilent 6540 QTOF LC/MS. Single crystal X-ray diffraction data were collected with a STOE STADIVARI Eulerian 4-circle diffractometer (STOE & CIE GmbH, Germany) equipped with an EIGER2 R500 detector (Dectris Ltd, Switzerland). Data were integrated with X-Area Integrate 2.5.3.0 (STOE, 2021) and scaled with X-Area LANA 2.7.5.0 (STOE, 2022). Structures were solved with SHELXS or SHELXT48 and refined with SHELXL49 in the GUI of Olex2.50 Model building was done with Olex2 or ShelXle.51 The structure were validated with CHECKCIF (https://checkcif.iucr.org/). Experimental data and CCDC-Codes Experimental data (available online: https://www.ccdc.cam.ac.uk/conts/retrieving.html) can be found in Table S1.† Crystal data, data collection parameters, and structure refinement details are reported in Tables S2–S8.† Structures, packing, interactions, and data are visualized in Fig. S1–S8.† Elemental analyses were performed by the Microanalytical Laboratory of the University of Vienna with a Eurovector EA 3000(2009) equipped with a high temperature pyrolysis furnace (HT, Hekatech, Germany, 2009). Elemental analyses samples were weighed on a Sartorius SEC 2 ultra-micro balance with ±0.1 μg resolution. Sample weights from 1–3 mg were used. For calibration two NIST-certified reference materials were used: sulfanilamide (C6H8N2O2S) and BBOT (2,5-bis-(5-tert-butyl-2-benzoxazol-2-yl)-thiophenone, C26H26N2O2S). The limit of quantification (LOQ) was 0.05 wt% for C, H, N and 0.02 wt% for S. The presented values are the average of determinations in triplicate. UV-Vis data were recorded on a PerkinElmer Lambda 650 UV-Vis Spectrophotometer with a Peltier element for temperature control. FRET experiments were performed at the AteN Center–Università di Palermo using a Applied Biosystems™ QuantStudio 6 PCR cycler.
K′(H2O/Cl−) constants.53
ESI-MS. Stock solutions in DMF were prepared and diluted with 400 μM NH4OAc solution (pH 7.4) to achieve a final concentration of 5 μM (1% DMF content). The complex solution was subjected to incubation with a balanced mixture (1
:1:1) of protected amino acids Ac-Cys-OMe, Ac-His-OMe and Ac-Met-OMe in 400 μM NH4OAc (5 μM each). Samples were measured after 0, 3 and 24 h of incubation at rt. Mass spectra were recorded on a Bruker maXis ESI-Qq-TOF Mass Spectrometer by direct infusion.
NMR. Stock solutions in DMF-d7 were prepared and diluted with D2O to achieve a final concentration of 1 mM (10% DMF-d7 content). The complex solution was subjected to incubation with an equimolar amount of protected amino acids Ac-Cys-OMe, Ac-His-OMe and Ac-Met-OMe, each individually and with a balanced mixture (1
:1:1).
9-Ethylguanine. DMF stock solutions of metal complexes were prepared, mixed with a stock solution of 9-ethylguanine in H2O and diluted with H2O to achieve a final concentration of 0.2 mM complex and 0.6 mM 9-ethylguanine (4% DMF content). After 2 h of stirring, the samples were analyzed via mass spectrometry. Mass spectra were recorded on an Agilent 6540 QTOF LC/MS Mass Spectrometer by direct infusion.
Oligonucleotide. Oligonucleotide was dissolved in 100 mM NH4OAc solution, heated up to 95 °C for 5 min and cooled down to room temperature slowly. Complex stock solutions in DMF were prepared and diluted with 100 mM NH4OAc solution to a final concentration of 30 μM. The compound solution was subjected to incubation with equimolar amounts of the oligonucleotide solution (30 μM each) for 2 h under constant stirring before a mass spectrum was recorded. Mass spectra were recorded on an Agilent 6540 QTOF LC/MS Mass Spectrometer by direct infusion.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) P calculation.
The octanol–water partition coefficient LogP was calculated for the free ligands with molinspiration (v2014.11). cLogP values allow the comparison of relative lipophilicities within a series of osmium(II)- or ruthenium(II)-arene metalacycles, as the metal-arene fragments remain unchanged.
P calculation.
The octanol–water partition coefficient LogP was calculated for the free ligands with molinspiration (v2014.11). cLogP values allow the comparison of relative lipophilicities within a series of osmium(II)- or ruthenium(II)-arene metalacycles, as the metal-arene fragments remain unchanged.
Conclusions
Within this work, the synthesis of ten 4-phenylthiazole derived ruthena(II)- and osma(II)cycles is described. The obtained complexes were characterized by means of standard analytical techniques and their aqueous stability and interaction with biological molecules were investigated. Amino acid interaction studies showed a stronger affinity towards L-methionine compared to L-histidine and L-cysteine. DNA interaction studies revealed that compounds 2a, 2c and 2d induced a stabilizing effect on G-quadruplex structures with a clear preference of compound 2c for G4 forming in the promoter of the oncogene c-myc. While docking calculations suggested that the R enantiomer interacts tighter with the mentioned DNA motif, mass analysis proved the formation of a 2c-c-MYC adduct. Additionally, the IC50 values of all metalacycles and the corresponding free ligands were determined in three human cancer cell lines (A549, SW480 and CH1/PA-1). While the ligands show no cytotoxic effect in the tested concentration range, the respective complexes proved to be highly active, with IC50 values in the low micromolar range. Except for 2a, higher cellular accumulation is in alignment with higher cytotoxicity. Surprisingly, no clear correlation between lipophilicity and drug accumulation was found. Selected compounds only exhibit a slow and weak induction of apoptosis and only minor cell cycle perturbations were observed.Author contributions
Conceptualization, A. T., B.K. K. and W.K.; data curation, P. G., A. T., O. D., M. A. J. and W. K.; formal analysis, P. G., L. M., E. T., A. P.-R., M. H., K. C., A. A. L., O. D. and G. B.; funding acquisition, O. D., A. T., B. K. K. and W. K.; investigation, P. G., L. M., E. T., O. D., K. C., M. H., A. P.-R., and A. A. L.; methodology, G. B., A. T., M. A. J., B. K. K. and W. K.; project administration, A. T., B. K. K. and W. K.; resources, O. D., M. A. J., G. B., A. T., B. K. K. and W. K.; software, G. B.; supervision, M. A. J., A. T., B. K. K. and W. K.; validation, O. D., M. A. J., G. B., A. T., B. K. K. and W. K.; visualization, P. G., O. D., K. C., A. P.-R., A. A. L., M. A. J., A. T., G. B. and W. K.; writing–original draft preparation, P. G., L. M., O. D., A. A. L, M. A. J., A. T. and W. K.; writing–review and editing, P. G., M. A. J., A. T. and W. K.; All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Vienna, the Doctoral School of Chemistry and Università Degli Studi di Palermo for financial support. We also thank the Centre for X-Ray Structure Analysis (University of Vienna) for determination of the crystal structures, the NMR Centre (University of Vienna) for 2D NMR spectra and the Mass Spectrometry Centre (University of Vienna) for measurement of the MS-spectra. O.D. gratefully acknowledges the financial support from TKP-2021-EGA-32 project of the Development and Innovation Office-NKFIA (Hungary). Finally, we thank AteN Center (University of Palermo) for FRET melting measurements.References
- B. Rosenberg, New Agents for the Control of Tumours, 1971, 15, 42–51 CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F. X. Wang, W. Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- M. A. Jakupec, M. S. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- J. P. C. Coverdale, T. Laroiya-McCarron and I. Romero-Canelón, Inorganics, 2019, 7, 31 CrossRef CAS.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- L. S. Flocke, R. Trondl, M. A. Jakupec and B. K. Keppler, Investig. New Drugs., 2016, 34, 261–268 CrossRef CAS.
- C. Chen, C. Xu, T. Li, S. Lu, F. Luo and H. Wang, Eur. J. Med. Chem., 2020, 203, 112605 CrossRef CAS PubMed.
- G. M. O'Kane, J. L. Spratlin, D.-Y. Oh, S. Y. Rha, E. Elimova, P. Kavan, M. K. Choi, R. A. Goodwin, S. T. Kim, D.-H. Koo, K. Halani, E. R. McAllister, M. Jones, M. Snow, Y. Lemmerick, G. Spera and J. Pankovich, J. Clin. Oncol., 2023, 41, 4098 CrossRef.
- M. A. Jakupec, E. Reisner, A. Eichinger, M. Pongratz, V. B. Arion, M. Galanski, C. G. Hartinger and B. K. Keppler, J. Med. Chem., 2005, 48, 2831–2837 CrossRef CAS PubMed.
- J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef CAS PubMed.
- G. S. Kulkarni, K. A. Richards, P. C. Black, R. A. Rendon, J. Chin, N. D. Shore, G. Jayram, E. V. Kramolowsky, D. Saltzstein, P. K. Agarwal, L. Belkoff, M. A. O'Donnell, A. M. Kamat, M. A. S. Jewett, D. L. Lamm, V. DeGruttola, A. Mandel, R. Dumoulin-White and W. Kassouf, J. Clin. Oncol., 2023, 41, 528 CrossRef.
- A. Chettri, T. Yang, H. D. Cole, G. Shi, C. G. Cameron, S. A. McFarland and B. Dietzek-Ivanšić, Angew. Chem., Int. Ed., 2023, 62, e202301452 CrossRef CAS PubMed.
- C. A. Riedl, L. S. Flocke, M. Hejl, A. Roller, M. H. M. Klose, M. A. Jakupec, W. Kandioller and B. K. Keppler, Inorg. Chem., 2017, 56, 528–541 CrossRef CAS PubMed.
- W. D. J. Tremlett, D. M. Goodman, T. R. Steel, S. Kumar, A. Wieczorek-Błauż, F. P. Walsh, M. P. Sullivan, M. Hanif and C. G. Hartinger, Coord. Chem. Rev., 2021, 445, 213950 CrossRef CAS.
- C. A. Riedl, M. Hejl, M. H. M. Klose, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, Dalton Trans., 2018, 47, 4625–4638 RSC.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- F. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernández, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2007, 13, 2601–2613 CrossRef CAS PubMed.
- M. Schmidlehner, V. Pichler, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, J. Organomet. Chem., 2015, 782, 69–76 CrossRef CAS.
- S. Mokesch, K. Cseh, H. Geisler, M. Hejl, M. H. M. Klose, A. Roller, S. M. Meier-Menches, M. A. Jakupec, W. Kandioller and B. K. Keppler, Front. Chem., 2020, 8, 1–15 CrossRef PubMed.
- M. Martínez-Alonso, N. Busto, F. A. Jalón, B. R. Manzano, J. M. Leal, A. M. Rodríguez, B. García and G. Espino, Inorg. Chem., 2014, 53, 11274–11288 CrossRef PubMed.
- L. Belsa, C. López, A. González, M. Font-Bardía, T. Calvet, C. Calvis and R. Messeguer, Organometallics, 2013, 32, 7264–7267 CrossRef CAS.
- J. Ruiz, V. Rodríguez, N. Cutillas, A. Espinosa and M. J. Hannon, Inorg. Chem., 2011, 50, 9164–9171 CrossRef CAS.
- G. S. Yellol, A. Donaire, J. G. Yellol, V. Vasylyeva, C. Janiak and J. Ruiz, Chem. Commun., 2013, 49, 11533–11535 RSC.
- J. Yellol, S. A. Pérez, A. Buceta, G. Yellol, A. Donaire, P. Szumlas, P. J. Bednarski, G. Makhloufi, C. Janiak, A. Espinosa and J. Ruiz, J. Med. Chem., 2015, 58, 7310–7327 CrossRef CAS PubMed.
- B. Boff, C. Gaiddon and M. Pfeffer, Inorg. Chem., 2013, 52, 2705–2715 CrossRef CAS PubMed.
- W. Lee, C. Shin, S. E. Park and J. M. Joo, J. Org. Chem., 2019, 84, 12913–12924 CrossRef CAS PubMed.
- E. Ferrer Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161–10170 CrossRef PubMed.
- A. F. A. Peacock, A. Habtemariam, R. Fernández, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739–1748 CrossRef CAS PubMed.
- S. Mokesch, M. S. Novak, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, Organometallics, 2015, 34, 848–857 CrossRef CAS.
- W. Kandioller, C. G. Hartinger, A. A. Nazarov, C. Bartel, M. Skocic, M. A. Jakupec, V. B. Arion and B. K. Keppler, Chem. – Eur. J., 2009, 15, 12283–12291 CrossRef CAS PubMed.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 RSC.
- J. Qian, R. Liu, N. Liu, C. Yuan, Q. Wu, Y. Chen, W. Tan and W. Mei, Molecules, 2022, 27, 3046 CrossRef CAS PubMed.
- Q. Wu, K. Zheng, S. Liao, Y. Ding, Y. Li and W. Mei, Organometallics, 2016, 35, 317–326 CrossRef CAS.
- W. Streciwilk, A. Terenzi, R. Misgeld, C. Frias, P. G. Jones, A. Prokop, B. K. Keppler and I. Ott, ChemMedChem, 2017, 12, 214–225 CrossRef CAS PubMed.
- W. Streciwilk, A. Terenzi, F. Lo Nardo, P. Prochnow, J. E. Bandow, B. K. Keppler and I. Ott, Eur. J. Inorg. Chem., 2018, 2018, 3104–3112 CrossRef CAS.
- L. A. Hager, S. Mokesch, C. Kieler, S. Alonso-de Castro, D. Baier, A. Roller, W. Kandioller, B. K. Keppler, W. Berger, L. Salassa and A. Terenzi, Dalton Trans., 2019, 48, 12040–12049 RSC.
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474 CrossRef CAS PubMed.
- G. W. Collie, G. N. Parkinson, S. Neidle, F. Rosu, E. De Pauw and V. Gabelica, J. Am. Chem. Soc., 2010, 132, 9328–9334 CrossRef CAS PubMed.
- T. Kench and R. Vilar, in Annual Reports in Medicinal Chemistry, Academic Press Inc., 2020, vol. 54, pp. 485–515 Search PubMed.
- H. Geisler, S. Harringer, D. Wenisch, R. Urban, M. A. Jakupec, W. Kandioller and B. K. Keppler, ChemistryOpen, 2022, 11, e202200019 CrossRef CAS.
- S. Tani, T. N. Uehara, J. Yamaguchi and K. Itami, Chem. Sci., 2014, 5, 123–135 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- O. Dömötör, S. Aicher, M. Schmidlehner, M. S. Novak, A. Roller, M. A. Jakupec, W. Kandioller, C. G. Hartinger, B. K. Keppler and É. A. Enyedy, J. Inorg. Biochem., 2014, 134, 57–65 CrossRef PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- A. E. Egger, C. Rappel, M. A. Jakupec, C. G. Hartinger, P. Heffeter and B. K. Keppler, J. Anal. At. Spectrom., 2009, 24, 51–61 RSC.
- K. Cseh, H. Geisler, K. Stanojkovska, J. Westermayr, P. Brunmayr, D. Wenisch, N. Gajic, M. Hejl, M. Schaier, G. Koellensperger, M. A. Jakupec, P. Marquetand and W. Kandioller, Pharmaceutics, 2022, 14, 2644 CrossRef PubMed.
- L. D'Anna, S. Rubino, C. Pipitone, G. Serio, C. Gentile, A. Palumbo Piccionello, F. Giannici, G. Barone and A. Terenzi, Dalton Trans., 2022, 52, 2966–2975 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc, Wallingford CT, 2016.
- Y. Wang, X. Jin, H. S. Yu, D. G. Truhlar and X. He, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8487–8492 CrossRef CAS PubMed.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 2008, 72, 650–654 CrossRef.
- G. M. Morris, H. Ruth, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 70–82 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Ligand synthesis, 1H and 13C NMR spectra, mass spectra, X-ray diffraction analysis, stability in aqueous solution, MTT-data, G-quadruplex interaction studies, amino acid interaction data, apoptosis and cell cycle investigations. CCDC 2296745–2296747, 2296749–2296751 and 2296753. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00245h |
| This journal is © The Royal Society of Chemistry 2024 |