
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to long-lived room temperature phosphorescence through auration of 2,1,3-benzothiadiazole†
Mauricio
Posada Urrutia‡
 a,
Nidhi
Kaul‡
b,
Tobias
Kaper
a,
Dustin
Hurrell
c,
Linus
Chiang
c,
Jordann A. L.
Wells
b,
Andreas
Orthaber
b,
Leif
Hammarström
b,
Lukasz T.
Pilarski
a and
Christine
Dyrager
*a
a,
Nidhi
Kaul‡
b,
Tobias
Kaper
a,
Dustin
Hurrell
c,
Linus
Chiang
c,
Jordann A. L.
Wells
b,
Andreas
Orthaber
b,
Leif
Hammarström
b,
Lukasz T.
Pilarski
a and
Christine
Dyrager
*a
aDepartment of Chemistry-BMC, Uppsala University, Box 576 751 23, Uppsala, Sweden. E-mail: christine.dyrager@kemi.uu.se
bDepartment of Chemistry-Ångström, Uppsala University, Box 523, 751 20 Uppsala, Sweden
cDepartment of Chemistry, University of the Fraser Valley, V2S7M8, Abbotsford, BC, Canada
First published on 28th February 2024
Abstract
A series of 2,1,3-benzothiadiazole–Au(I)–L complexes have been synthesised, structurally characterised and investigated for their photophysical properties. These are the first organometallic Au(I) complexes containing a C–Au bond on the highly electron-deficient benzothiadiazole unit. The complexes exhibit solution-phase phosphorescence at room temperature, assigned to the intrinsic triplet state of the benzothiadiazole unit that is efficently populated through its attachment to gold. Comparison with routinely reported Au(I) complexes, which include intervening alkenyl linkers, suggests that previous assignments of their phosphorescence as 1π → π*(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR) might be incomplete. Our observations affirm that, in addition to the heavy atom effect, breaking symmetry in the involved aryl motif may be of importance in controlling the luminescence properties.
CR) might be incomplete. Our observations affirm that, in addition to the heavy atom effect, breaking symmetry in the involved aryl motif may be of importance in controlling the luminescence properties.
Introduction
Organometallic Au(I) complexes have emerged as an attractive class of luminescent emitters with prospects for various applications, including OLED materials and chemical sensing.1–5 Their luminescent behaviour is usually ascribed to the heavy-atom effect of gold that induces large spin–orbit coupling (SOC), which results in efficient intersystem crossing (ISC) from the singlet to the triplet excited state.6–8 However, the presence of an Au atom does not always dramatically increase the intersystem crossing rate (kISC);9 many emissive organometallic Au(I) structures display very weak phosphorescence or solely exhibit prompt or delayed fluorescence.4,9 The two-coordinate linear geometry of Au(I) complexes also enables close aurophilic contacts (2.7–3.5 Å) between gold centres (Au⋯Au), which may affect the nature of the emission.6The most extensively investigated class of luminescent Au(I) complexes are acetylene-based derivatives (exemplified by R1–CC–Au–R2).4,10,11 Their emission has been attributed to the triplet excited state manifold of the alkynyl fragment.4,9 The photophysics of aryl–Au(I)–L complexes (i.e., with a direct aryl C–Au bond), however, has not been explored to the same extent or compared with the former in great detail. Also, the structural diversity of such emitters is relatively unexplored, especially species based on (hetero)aryl units beyond simple polycyclic aromatic hydrocarbons (PAHs).12–16 An early report by Gray and co-workers described Au(I) pyrenyls exhibiting pyrene-based π → π* phosphorescence at 77 K.13 More recently, Thompson and co-workers observed both inter-ligand charge transfer (ICT) and metal-to-ligand charge transfer (MLCT) emissions in aryl–Au(I)–NHC complexes at room temperature,17,18 where changing the donor strength of the aryl group allowed for tuning between the two emissive states. Therefore, the organic unit in aryl–Au(I) complexes is not limited to acting merely as a relativistic spectator; the origin and characteristics of observed emissions can, in fact, largely depend on the aryl motifs, together with the position of auration. The development of aryl–Au(I) complexes that contain structurally and electronically diverse aromatic units is, therefore, a promising strategy for the discovery of new Au(I)-based luminophores.
As part of our ongoing research into harnessing the photophysical properties of BTD derivatives,19–22 we sought to investigate BTD–Au(I) complexes as promising luminophores. Here, we report a series of emissive aryl–Au(I)–L complexes in which the gold centre is directly attached to the highly electron-deficient 2,1,3-benzothiadiazole (BTD) unit via a C–Au bond (Scheme 1). Hitherto, organometallic Au(I) luminophores based on fluorescent heteroaromatic chromophores (e.g., coumarin, BODIPY, and BTD) have only been acetylides.4,9,11,23 Among these are BTD–CC–Au–PCy3 (reported by Che and co-workers in 2017, see Table 3), which exhibit prompt and delayed fluorescence.9 We found that the absence of the acetylene linker precludes delayed fluorescence and instead grants access to long-lived (∼100 μs) room temperature phosphorescence in solution. This feature arises due to the direct attachment of gold, which significantly enhances ISC to the triplet state and thereby amplifies the weak intrinsic phosphorescence of the parent BTD unit.24 In this study, we gained important insight into the origins of this phosphorescence, as well as reconsidered the previous reported assignments for related acetylide-based luminophores.
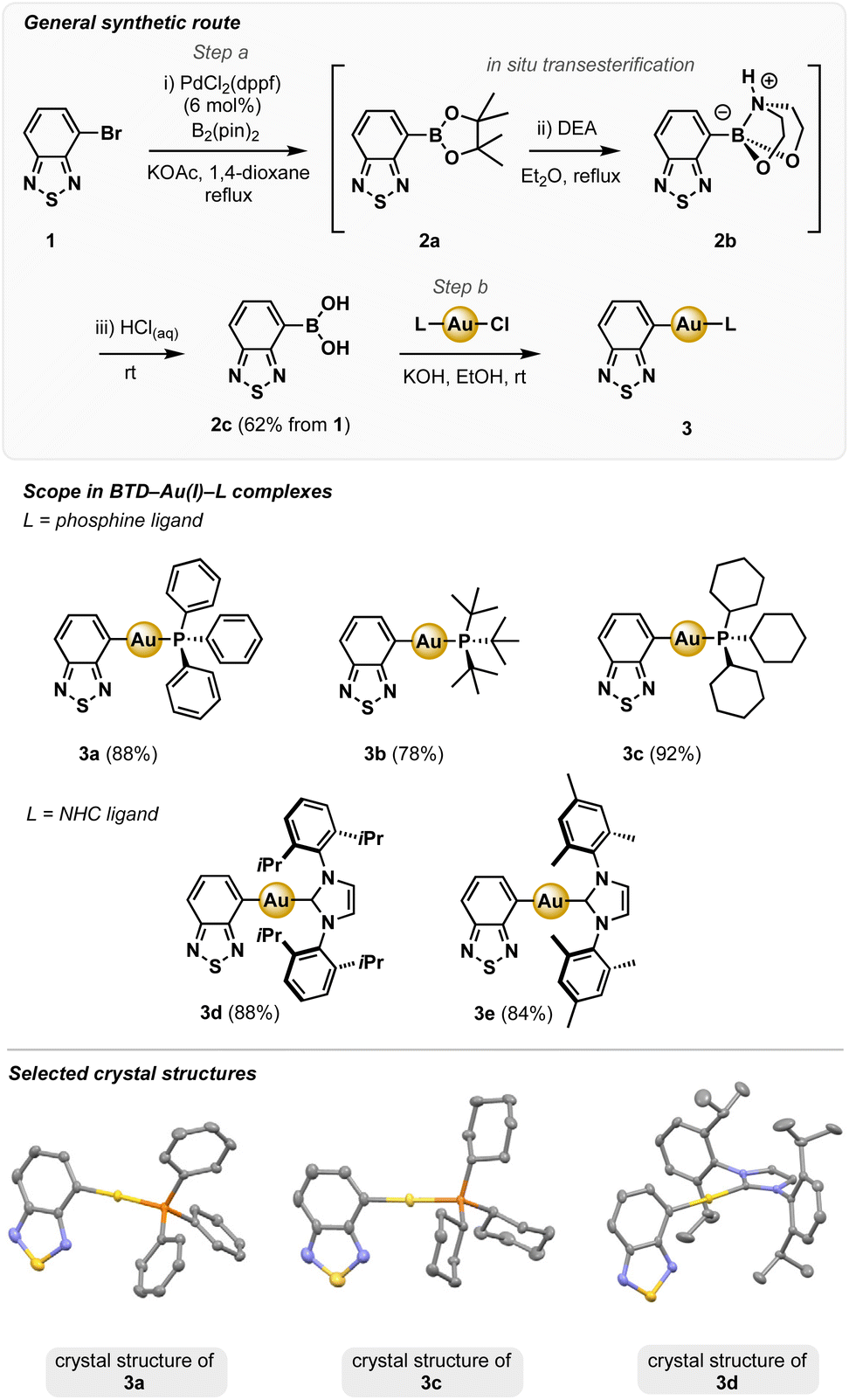 | ||
| Scheme 1 Top and middle: synthesis and scope of BTD–Au(I)–L complexes 3a–3e. Conditions: (a) (i) B2(pin)2, KOAc, PdCl2(dppf) (6 mol%), 1,4-dioxane, reflux, 90 min; (ii) diethanolamine (DEA), Et2O, reflux, 30 min; (iii) 4 M HCl(aq), r.t., 5 min. 62% over 3 steps. (b) L–Au–Cl, KOH, EtOH, r.t., 24 h. Bottom: selected crystal structures of 3a, 3c and 3d. | ||
Results and discussion
Synthesis and characterisation
Novel BTD–Au(I) complexes 3a–e were synthesised from 1via Miyaura borylation (Scheme 1).25In situ deprotection of the corresponding pinacol boronate ester 2a, via transesterification with diethanolamine (DEA) and acid hydrolysis of adduct 2b, gave boronic acid 2c in 62% overall yield.26,27 The corresponding Au(I) complexes 3a–e were generated in good to excellent yields (78–92%) via transmetalation between 2c and L–Au(I)–Cl salts (L = phosphine, NHC) in EtOH at r.t.,13,28,29 and were obtained as analytically pure, air-stable colourless solids that exhibit solid-state emission (ESI, Fig. S3†).Complexes 3a–e were characterised by 1H, 13C and, where applicable 31P NMR spectroscopy, and high-resolution mass spectrometry (HRMS). X-ray diffraction (XRD) data for all complexes 3a–e is given in the ESI† and selected crystal structures of 3a, 3c and 3d are shown in Scheme 1 (bottom panel). Importantly, none of the crystal structures show close intermolecular Au⋯Au interactions; the shortest distances range between 5.6–8.6 Å, which is beyond the limit for aurophilic contacts.6
Electrochemical analysis
The electrochemical properties of THF solutions of 3a–e were investigated using cyclic voltammetry. All the complexes exhibited irreversible redox waves at ca. 1 V vs. Fc/Fc+, and no redox waves in the cathodic region of the voltammogram within the electrochemical window of the experiments (Table 1; ESI, Fig. S12–16 and Tables S8–12†). While a few BTD–M compounds (M = Pd,30 Zn,31 Ag,32 Sn33) have been reported previously, their electrochemical or photophysical properties have not. As such, there is a lack of directly comparable examples for the electrochemical studies presented in this work. Conversely, the electrochemical properties of the alkyne linked analogue BTD–CC–Au–PCy3 in CH2Cl2 have been reported, where a BTD-acetylide-based reduction wave was observed at −1.94 V vs. Fc/Fc+, without a redox wave in the anodic region.9 The lack of cathodic waves for 3a–e suggests their reduction occurs at more negative potentials, likely outside the measured electrochemical window, implying the LUMO of complexes 3a–e is higher in energy that of their alkynyl-bridged congeners. On the other hand, anodic waves were observed for 3a–e, while none were observed for their alkyne-containing analogue in CH2Cl2. This suggests the HOMO of 3a–e is higher in energy in comparison to BTD–CC–Au–PCy3. The origins of these frontier molecular orbitals were further investigated by density functional theory (DFT) computations (vide infra).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| BTD–Au(I)–L complex | E 1 (V) | E 2 (V) |
|---|---|---|
| a Values were determined in THF at 298 K, referenced to a ferrocene standard. Supporting electrolyte: 0.1 M [nBu4N]PF6, scan rate: 100 mV s−1. | ||
| 3a | 0.95 | 1.05 |
| 3b | 1.13 | — |
| 3c | 1.11 | — |
| 3d | 1.15 | — |
| 3e | 0.93 | 1.09 |
Steady-state and time resolved photophysics
The photophysical properties of compound 3a–e were investigated in 2-Me-THF (Table 2 and Fig. 1). All complexes absorb in the blue region (below ∼400 nm) with two characteristic absorption bands; one centred at ∼300 nm with some fine structure and a broader band at ∼350 nm (Fig. 1A). Extinction coefficients for the latter range from 2800–5500 M−1 cm−1. Comparing the absorption spectra of 3a–e with the spectrum of unsubstituted BTD suggests that the lowest energy band is a direct consequence of the auration. This observation differs from the explanation invoked for the alkynyl-bridged analogue BTD–CC–Au–PCy3, where the lowest energy band was attributed to dipole-allowed intraligand transitions of the alkynyl motif, 1π → π*(CCR), with some charge transfer character.9 Furthermore, ligation of gold in BTD–CC–Au–PCy3 was suggested to result in a bathochromic shift of the 1IL band. However, given that the bridging alkynyl is entirely absent in 3a–e, its involvement can be ruled out.
 | ||
| Fig. 1 Photophysical data in 2-Me-THF. (A) UV-Vis absorption spectra for 3a–3e at room temperature (conc. 15 μM). (B) Emission spectra for 3a–3e (conc. 15 μM) recorded between 540–800 nm. λex = 355 nm (C) UV-Vis absorption spectrum, excitation spectrum (monitored at 620 nm) and emission spectrum (degassed solution, λex = 355 nm) for 3d at room temperature plotted together with the emission spectrum of 3d at 77 K (glassy matrix, λex = 355 nm D) compound 3d in 2-Me-THF. Left vial: at room temperature, 298 K, in the presence of air; middle vial: degassed (N2 sparged) solution at 298 K; right vial: glassy matrix at 77 K in the presence of air. λex = 365 nm. | ||
| BTD–Au(I)–L complex | UV/Vis absorption | Emission | ||||
|---|---|---|---|---|---|---|
| λ max/nm (ε/M−1 cm−1) |
λ
em, λphosb (nm) |
λ
phos at 77 Kc (nm) |
τ
PFd (ns) |
τ
phose (μs) |
Φ
phosf |
|
| a Data were obtained from steady-state measurements using degassed 2-Me-THF solutions of 3a–e (15μM) at 298 K unless otherwise specified. b λ ex = 355 nm. c Measurements at 77 K were performed in the presence of air. d Emission lifetimes of prompt fluorescence (τPF) were determined using time-correlated single photon counting (TCSPC) measurements, see ESI† for details. e Phosphorescence lifetimes (τphos) were determined using time resolved emission measurements. f Phosphorescence quantum (Φphos) yields were obtained using [Ru(bpy)3][PF6]2 in aerated acetonitrile (Φphos = 0.018)36 as the standard. Φphos values are rounded to the nearest centesimal. | ||||||
| 3a | 306 (13600), 313 (13400), 346 (5500) |
415, 621 | 622 | 2 | 96 | 0.03 |
| 3b | 305 (11900), 312 (12000), 349 (4300) |
416, 624 | 624 | 2 | 88 | 0.03 |
| 3c | 305 (11900), 312 (10900), 349 (3900) |
416, 625 | 624 | 2 | 104 | 0.04 |
| 3d | 305 (9800), 312 (7200), 356 (3800) | 416, 631 | 629 | 2 | 102 | 0.04 |
| 3e | 305 (7300), 312 (7200), 356 (2800) | 415, 630 | 629 | 2 | 100 | 0.06 |
A better understanding into the origin of the lowest energy band for 3a–e can be obtained by considering the detailed spectroscopic analysis of BTD's transitions (which has been reported in the literature).24,34,35 Accordingly, the first two electronically allowed 1ππ* transitions at 306 nm and 328 nm have been assigned as 1A1 ← 1A1 and 1B2 ← 1A1, respectively, with about a fifth of the latter's intensity being sourced in 1A1 ← 1A1 as well. The former band is strong and does not shift upon auration. The latter band is weak and hard to resolve in the solution spectra of the unsubstituted BTD. The dominant vibration, characterizing this weak intensity band, is a fully symmetric (a1) C4C5C9 angle bend with a ring contraction (ESI, Fig. S2†). The B2 vibrations also contribute and indulge in Herzberg–Teller intensity stealing, however, presumably from the 1A1 ← 1A1 band.35 Since the Au(I) centre is directly ligated in the C4-position of 3a–e, it can be rationalised that this band becomes more allowed and is bathochromically shifted due to the auration. Moreover, changing the auxiliary ligand from phosphine to carbene (NHC) resulted in a small but discernible redshift of the lowest energy absorption band, which indicates some minimal charge transfer (CT), considering that NHCs are generally stronger σ-donors than phosphine ligands.
At room temperature, excitation at 355 nm into the lowest energy absorption band of 3a–e results in bright orange-red phosphorescence, in both the solid phase and degassed solutions. The latter has emission maxima between 621 nm and 630 nm (Fig. 1B). Phosphorescent quantum yields (Φphos) range between 0.03 and 0.06, which are notable for mononuclear Au(I) complexes in solution at room temperature. Lifetimes close to a hundred microseconds (ESI, Fig. S5†) also support the triplet nature of the red emission (Stokes shift of ca. 12500 cm−1).
We note that some recently reported Au(I)–carbene complexes show no or very low phosphorescence at room temperature in solution (Φ ≤ 0.01), and that the phosphorescence lifetimes of these reached at best 41 μs under such conditions.18 Further work from Thompson and co-workers shows emissive Au(I)–carbene complexes with lifetimes up to 3.3 μs in solution, though the emission was not ascribed primarily to ligand centred phosphorescence.17 The photoluminescence of these complexes originated from a variety of states including ICT and MLCT. The comparatively large phosphorescence quantum yield for the compounds presented here, as well as their long lifetime, is again reinforced. Previous reports have described phosphorescent BTD derivatives in the solid state,37,38 at 77 K,24,37 and in solution.37,39 However, for the solution-based phosphors the quantum yields have peaked at only 0.7%.39 The longest lifetime that has been previously observed for a BTD derivative at room temperature solution has also remained modest, at 5.4 μs. To date and to the best of our knowledge, the phosphorescence quantum yield and phosphorescence lifetime described in this study remain the largest for BTD-based phosphors in solution at room temperature.
The vibrational spacings seen for 3a–e are similar to those observed for the very weak phosphorescence of the parent BTD (observed only when deuterated or in halogenated solvents at 77 K)24 suggesting an orbital parentage with 3π → π* character from the BTD unit. Furthermore, the emission intensity of 3a–e in solution is significantly higher at 77 K (glassy matrix) than at room temperature (cf.3d, Fig. 1D). Changing the auxiliary ligand, from phosphine to NHC, also results in a small but discernible redshift of the emission as well as the absorption;28 some charge transfer (CT) is thus indicated. The excitation spectra monitored at 620 nm unambiguously traces the ground state absorption spectrum, including the lowest energy band, which is characteristic of the complexes (Fig. 1C and ESI, Fig. S4†).
Complexes 3a–e also exhibit prompt fluorescence (λmax ∼ 415 nm), with lifetimes longer than unsubstituted BTD37 (τ ∼ 2 ns; ESI, Fig. S6†). It can therefore be inferred that auration alters the intrinsic photophysics. At higher laser excitation power(s), a low degree of photodegradation (<1% from the absorption data) was observed. This was evident by the appearance of strong emission signals at approximately 475 nm with lifetimes of a few tens of nanoseconds. Notably, these signals are identical to those reported for highly emissive Au nanoparticles/clusters40 and should therefore not be mistaken for delayed fluorescence. The phosphorescence in our study is spectrally distinct from Au nanoparticle luminescence. Based on the prompt fluorescence lifetime of 2 ns (kobs = 5 × 108 s−1) in the complexes, only the upper limit of the kISC can be estimated to 108 s−1 since the observed rate constant is the sum of all accessible decay pathways, which includes intersystem crossing. This would make it potentially an order of magnitude faster than the values reported for the alkynyl-bridged counterparts, which exclusively exhibit prompt and delayed fluorescence in solution.9 It is worthwhile to note that the lifetimes and yields alone also do not permit for an evaluation of the intersystem crossing efficiency. Since Φphos = ΦISC[kr/(kr + knr)] (where 1/(kr + knr) is the observed phosphorescence lifetime), kr and ΦISC may not be disentangled.
Overall, the data suggest that a suitable photophysical descriptor for 3a–3e is perturbation of the intrinsic emission behaviour of the heteroarene moiety by the Au(I) centre. Direct linkage to the gold, without the alkynyl linker in this case, can be thought to impact intersystem crossing rates favourably and enhance phosphorescence quantum yields in BTD. Support for such an analysis is found by comparing the aurated BTD units presented in this study with Br-substituted BTD derivatives very recently reported in the literature,37 as well as with the alkynyl bridged analogue reported by Che and co-workers.9 The relevant structures, together with critical photophysical data at room temperature, is tabulated in Table 3. For BTD–CC–Au–PCy3, phosphorescence is exclusively observed at 77 K,9 with very similar emission maxima and lifetime as for the complexes reported here (3a–e). On observing the trend in Table 3, it can be clearly seen that direct linkage of a heavy atom at the 4-position of BTD would make the weak 1B2 ← 1A1 transition more allowed due to broken symmetry, in conjunction with SOC. In fact, 4,7-dibromination is more effective for achieving BTD-based phosphorescence than introducing Au with an alkynyl linker (cf.A and B with BTD–CC–Au–PCy3). For 4,7-dibromo-BTD (A), phosphorescence is only observed in the solid state and the yields remain well over an order of magnitude smaller than the solution phase yields of the complexes in the present work. Thus, the photophysical picture of accessing the triplet manifold of the heteroarene, via heavy atom substitution at the C4-position, is reinforced via direct auration of BTD.
| 3a–e (this work)a | BTD 37 | A 37 | B 37 |
BTD–CC–Au–PCy39 |
|
|---|---|---|---|---|---|
| a Values represent 3d in 2-Me-THF. Table 2 contains photophysical data all complexes (3a–e). b Phosphorescence quantum yield, Φphos. Values are rounded to the nearest centesimal or indicated as <0.01 if appliable. c Emission maxima, λmax (nm). d Phosphorescence lifetimes, τphos (μs). e Not determined. f In THF. | |||||
| Structure |

|

|

|

|

|
| Phosphorescence (Φphos,bλmax,cτphosd) in: |
|||||
| Solution, r.t. | 0.04, 629 nm, 102 μs | Not observed | Not observed | <0.01,f 670 nm,f n.d.e | Not observed |
| Solid state, r.t. | n.d.e | <0.01, 685 nm, 37 μs | 0.04, 648 nm, 585 μs | ||
Computational studies
The absorption properties of 3a–e were investigated computationally via DFT and TD-DFT calculations. The phenyl, tert-butyl and cyclohexyl moieties of 3a–c were truncated to methyl groups, giving BTD–Au–PMe3, to expedite computations. For comparison, the same molecular truncation and computational parameters were applied to the previously reported acetylide analogue, giving BTD–CC–Au–PMe3, which resulted in frontier molecular orbitals (ESI, Fig. S8†) and vertical excitation energies (ESI, Tables S3 and 4†) that are near identical to those described in the literature.9 Using this methodology, the frontier molecular orbitals of BTD–Au–PMe3 (representing 3a–3c) and BTD–Au–NHCs (3d and 3e) were found to be similar to BTD–CC–Au–PMe3, comprising of the antibonding combinations of an Au d-orbital and a π-system of the BTD fragment (ESI, Fig. S9–11†). These orbitals are predominantly localised on the BTD fragment with minimal contribution from the PMe3 or the NHC ligands (Fig. 2). This is consistent with both the experimental observations of the auxiliary ligand showing a minimal contribution to the observed transitions, and the photophysics of the BTD–Au(I)–L complexes 3a–3e being best described as a perturbation of that of the heteroarene moiety. Further, the TD-DFT calculated vertical excitation transitions of BTD, BTD–Au–PMe3 and the BTD–Au–NHCs (3d and 3e) originating from their respective HOMO and LUMO, reflects the observed trends, even if not the precise transition energies. The calculated orbitals for the HOMO and LUMO are consistent with symmetry labels A (symmetric with respect to the principal rotation axis) and B (anti-symmetric with respect to the principal rotation axis) for the C2v point group of BTD. It can therefore represent the weaker symmetry-forbidden band at ca. 325 nm seen in experiments. Meanwhile, the stronger band at ca. 300 nm is between the symmetric HOMO−1 and LUMO, i.e., 1A1 ← 1A1. In unsubstituted BTD, the computed oscillator strengths are consistent with such an analysis. Upon auration, the relative oscillator strengths of the two bands are reversed, reflecting the more allowed nature of the weaker band in 3a–e, which is qualitatively captured at this level of theory. Finally, the predicted transitions of BTD–Au–PMe3 are blue-shifted by 4000 cm−1 in comparison to BTD–CC–Au–PMe3, consistent with their respective experimental absorption values (Table 2 and ESI, Tables S3–7†).9
 | ||
| Fig. 2 HOMO and LUMO of (A) truncated BTD–AuPMe3 and (B) BTD–Au–NHC. | ||
The singlet (Sn) and triplet (Tn) excited state energies of BTD–Au(I)–L (L = truncated PMe3, or NHCs: IPr and IMes) were investigated via TD-DFT calculations at their corresponding optimised singlet ground state (S0) geometries. For BTD–Au–PMe3, three triplet excited states (T1–T3) were found to be lower in energy than the lowest singlet excited state (S1), with the energy difference between the highest energy triplet (T1) and lowest energy singlet (S1) excited states (ΔEST) being 12300 cm−1, which falls in the proximity of the experimentally observed difference between the fluorescence and phosphorescence spectral maxima (ca. 7500 cm−1). Similarly, both 3d and 3e exhibit five triplet excited states (T1–T5) lower in energy than the lowest singlet excited state (S1) and a small ΔEST (3d: 12000 cm−1; 3e: 12100 cm−1). Notably, while exclusively fluorescence or delayed fluorescence properties were reported for BTD–CC–Au–PCy3, 3a–c exhibit phosphorescence. The observation of delayed fluorescence indicates that the triplet is also populated in the alkynyl-bridged complexes, but the radiative rate constant is not competitive. It can be hypothesised that direct coordination to Au results in a comparatively stronger SOC for 3a–3e, resulting in a higher radiative rate constant, and the observed phosphorescent properties.
Conclusions
A series of BTD–Au(I) complexes was synthesised, characterised (NMR, HRMS, CV and XRD) and investigated for their photophysical behaviour. These are the first isolated coordination complexes containing a C–Au bond on the BTD unit and the first aryl–Au(I) complexes that comprise a highly electron-deficient heterocyclic unit. In solution the complexes display long-lived room temperature phosphorescence (τ ∼ 100 μs) with appreciable quantum yields (Φphos = 0.03–0.06). The origin of the phosphorescence was traced to the triplet manifold of BTD (3LC emission), which is efficiently populated specifically due to its direct auration. The strong phosphorescence compared to acetylene-linked Au(I) complexes suggest a larger radiative rate constant for the T1–S0 transition, which can presumably be rationalised by a stronger SOC coupling with Au(I) directly linked on the BTD unit. In addition, the unsymmetrical substitution on BTD increases the oscillator strength of the lowest, symmetry-forbidden singlet–singlet transition of BTD and should also increase the rate constant for phosphorescence. This reveals the tantalising prospect that otherwise weak luminophores can form the basis of efficient organo–Au(I) phosphors with compact, simple, and robust motifs. We anticipate that this strategy may be generalisable for other weak luminophore/heavy atom combinations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council, Dnr: 2018-03524 (to CD) and a NSERC Discovery Grant, RGPIN-2018-06744 (to L. C.). Compute Canada is acknowledged for access to computational resources. This study made use of the NMR Uppsala infrastructure, which is funded by the Department of Chemistry – BMC and the Disciplinary Domain of Medicine and Pharmacy.References
- X. He and V. W.-W. Yam, Coord. Chem. Rev., 2011, 255, 2111–2123 CrossRef CAS.
- E. E. Langdon-Jones and S. J. A. Pope, Chem. Commun., 2014, 50, 10343–10354 RSC.
- J. M. López-de-Luzuriaga, M. Monge and M. E. Olmos, Dalton Trans., 2017, 46, 2046–2067 RSC.
- M. Pujadas and L. Rodríguez, Coord. Chem. Rev., 2020, 408, 213179 CrossRef CAS.
- X. Li, Y. Xie and Z. Li, Chem. – Asian J., 2021, 16, 2817–2829 CrossRef CAS PubMed.
- H. Schmidbaur, Gold Bull., 2000, 33, 3–10 CrossRef CAS.
- A. Vogler and H. Kunkely, Coord. Chem. Rev., 2001, 219–221, 489–507 CrossRef CAS.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- K. T. Chan, G. S. M. Tong, W.-P. To, C. Yang, L. Du, D. L. Phillips and C.-M. Che, Chem. Sci., 2017, 8, 2352–2364 RSC.
- L. Gao, D. V. Partyka, J. B. Updegraff Iii, N. Deligonul and T. G. Gray, Eur. J. Inorg. Chem., 2009, 2009, 2711–2719 CrossRef.
- A. Möller, P. Bleckenwegner, U. Monkowius and F. Mohr, J. Organomet. Chem., 2016, 813, 1–6 CrossRef.
- M. Osawa, M. Hoshino and D. Hashizume, Chem. Phys. Lett., 2007, 436, 89–93 CrossRef CAS.
- D. V. Partyka, A. J. Esswein, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2007, 26, 3279–3282 CrossRef CAS.
- W. Y. Heng, J. Hu and J. H. K. Yip, Organometallics, 2007, 26, 6760–6768 CrossRef CAS.
- L. Gao, M. A. Peay, D. V. Partyka, J. B. Updegraff III, T. S. Teets, A. J. Esswein, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2009, 28, 5669–5681 CrossRef CAS.
- R. A. Vogt, M. A. Peay, T. G. Gray and C. E. Crespo-Hernández, J. Phys. Chem. Lett., 2010, 1, 1205–1211 CrossRef CAS.
- T.-y. Li, D. S. Muthiah Ravinson, R. Haiges, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2020, 142, 6158–6172 CrossRef PubMed.
- T.-y. Li, P. I. Djurovich and M. E. Thompson, Inorg. Chim. Acta, 2021, 517, 120188 CrossRef CAS.
- H. Appelqvist, K. Stranius, K. Borjesson, K. P. R. Nilsson and C. Dyrager, Bioconjugate Chem., 2017, 28, 1363–1370 CrossRef CAS PubMed.
- K. Colas, S. Doloczki, A. Kesidou, L. Sainero-Alcolado, A. Rodriguez-Garcia, M. Arsenian-Henriksson and C. Dyrager, ChemPhotoChem, 2021, 5, 632–643 CrossRef CAS.
- K. Colas, K. O. Holmberg, L. Chiang, S. Doloczki, F. J. Swartling and C. Dyrager, RSC Adv., 2021, 11, 23960–23967 RSC.
- S. Doloczki, K. O. Holmberg, I. Fdez Galvan, F. J. Swartling and C. Dyrager, RSC Adv., 2022, 12, 14544–14550 RSC.
- A. Pinto, M. Echeverri, B. Gómez-Lor and L. Rodríguez, Dalton Trans., 2022, 51, 8340–8349 RSC.
- B. R. Henry and J. D. Morrison, J. Mol. Spectrosc., 1975, 55, 311–318 CrossRef CAS.
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508–7510 CrossRef CAS.
- J. Sun, M. T. Perfetti and W. L. Santos, J. Org. Chem., 2011, 76, 3571–3575 CrossRef CAS.
- P. A. Inglesby, L. R. Agnew, H. L. Carter and O. T. Ring, Org. Process Res. Dev., 2020, 24, 1683–1689 CrossRef CAS.
- F. J. L. Ingner, A.-C. Schmitt, A. Orthaber, P. J. Gates and L. T. Pilarski, ChemSusChem, 2020, 13, 2032–2037 CrossRef CAS.
- For recent solvent-free approaches to the synthesis of organo-Au(I) complexes, see: (a) F. J. L. Ingner, Z. X. Giustra, S. Novosedlik, A. Orthaber, P. J. Gates, C. Dyrager and L. T. Pilarski, Green Chem., 2020, 22, 5648–5655 RSC; (b) G. Pisanò and C. S. J. Cazin, ACS Sustainable Chem. Eng., 2021, 9, 9625–9631 CrossRef.
- K. Kubota, R. Takahashi and H. Ito, Chem. Sci., 2019, 10, 5837–5842 RSC.
- H. Langhals, P. Knochel, A. Walter and S. Zimdars, Synthesis, 2012, 3465–3476 CrossRef CAS.
- T. S. Sukhikh, D. A. Bashirov, S. Shuvaev, V. Y. Komarov, N. V. Kuratieva, S. N. Konchenko and E. Benassi, Polyhedron, 2018, 141, 77–86 CrossRef CAS.
- Y. Farré, M. Raissi, A. Fihey, Y. Pellegrin, E. Blart, D. Jacquemin and F. Odobel, Dyes Pigm., 2018, 148, 154–166 CrossRef.
- J. M. Hollas and R. A. Wright, Spectrochim. Acta, Part A, 1969, 25, 1211–1225 CrossRef CAS.
- R. D. Gordon and R. F. Yang, J. Mol. Spectrosc., 1971, 39, 295–320 CrossRef CAS.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449–2458 CrossRef CAS.
- T. Ishi-i, R. Kichise, I. S. Park, T. Yasuda and T. Matsumoto, J. Mater. Chem. C, 2023, 11, 3003–3009 RSC.
- H. Shi, L. Zou, K. Huang, H. Wang, C. Sun, S. Wang, H. Ma, Y. He, J. Wang, H. Yu, W. Yao, Z. An, Q. Zhao and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 18103–18110 CrossRef CAS PubMed.
- G. D. Gutierrez, G. T. Sazama, T. Wu, M. A. Baldo and T. M. Swager, J. Org. Chem., 2016, 81, 4789–4796 CrossRef CAS PubMed.
- H. Kawasaki, K. Hamaguchi, I. Osaka and R. Arakawa, Adv. Funct. Mater., 2011, 21, 3508–3515 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2026788 and 2125287–2125290. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00238e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |