
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect cross-linking of silyl-functionalized cage siloxanes via nonhydrolytic siloxane bond formation for preparing nanoporous materials†
Miharu
Kikuchi‡
a,
Taiki
Hayashi‡
 a,
Takamichi
Matsuno
abc,
Kazuyuki
Kuroda
ab and
Atsushi
Shimojima
*abc
a,
Takamichi
Matsuno
abc,
Kazuyuki
Kuroda
ab and
Atsushi
Shimojima
*abc
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: shimojima@waseda.jp
bKagami Memorial Research Institute for Materials Science and Technology, Waseda University, 2-8-26 Nishiwaseda, Shinjuku-ku, Tokyo 169-0051, Japan
cWaseda Research Institute for Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
First published on 5th March 2024
Abstract
Bottom-up synthesis of siloxane-based nanoporous materials from siloxane oligomers is promising for constructing well-defined structures at a molecular level. Herein, we report the synthesis of nanoporous materials consisting of cage-type siloxanes through the nonhydrolytic siloxane bond formation reaction. Cage siloxanes with double-n-ring geometries (n = 4 or 6) modified with dimethylsilyl and dimethylethoxysilyl groups are synthesized and directly cross-linked using a B(C6F5)3 catalyst, resulting in the formation of porous networks composed of alternating cage siloxane nodes and tetramethyldisiloxane (–SiMe2OSiMe2–) linkers. Compared with conventional hydrolysis and polycondensation reactions of alkoxysilyl-modified cage siloxanes under acid conditions, the non-hydrolytic condensation reaction was found favorable for the formation of porous siloxane networks without unwanted cleavage of the siloxane bonds.
Introduction
Siloxane-based nanoporous materials have various applications, including separation, thermal insulation, optics, electronics, and drug delivery, because of their high thermal and chemical stability, controlled pore size, large pore volume, high surface area, and low toxicity.1 Bottom-up assembly of well-defined siloxane oligomers as building blocks is one of the promising methods for precisely controlling the framework structures at the molecular level.2 Cage-type siloxanes with double-n-ring (DnR, n = 3–6) structures3 are especially useful as building blocks because of their simple synthesis, framework rigidity, high symmetry, and multiple reactive sites that are available for cross-linking and chemical modification.2d,e Various functional groups and reactions have been employed for the intermolecular linking of cage siloxanes via Si–O–Si,4 Si–C,5a,b C–C,5c–h and Si–O–C6 bond formations. Among them, the inorganic Si–O–Si bond is crucial to impart high thermal and chemical stabilities.One of the most frequently studied reactions for the siloxane bond formation is the hydrolysis and condensation of silyl groups. Hydrolysis and polycondensation of D4R siloxanes containing various terminal functional groups such as –OMe,4a,b –OSiMe(OEt)2,4d and –OSiMe2Br4c resulted in the formation of cross-linked porous materials. A problem of such hydrolytic condensation reactions is the possible cleavage of the siloxane bonds within the cage frameworks and/or between the silyl groups and cage frameworks. Self-polymerization of D4R siloxanes modified with dimethylsilyl groups (Si8O12(OSiMe2H)8, D4R-SiMe2H) in the presence of a fluoride ion catalyst7 has also been reported. However, it is still difficult to completely prevent the cleavage of the cage framework and the elimination of terminal silyl groups caused by the presence of water in the systems. Cross-linking of D4R silicate hydrates with dichlorodimethylsilane was also reported;8 however, structural disorder occurred due to hydrolysis and subsequent self-condensation of chlorosilane.
Nonhydrolytic siloxane bond formation reactions catalyzed by Lewis acids are useful for the precise synthesis of siloxane oligomers and polymers without the cleavage of siloxane bonds.9 Tris(pentafluorophenyl)borane (B(C6F5)3)-catalyzed condensation between hydrosilanes and alkoxysilanes, the so-called Piers–Rubinsztajn (P–R) reaction,10 has attracted considerable attention because of the fast reaction rates, prevention of hydrolytic Si–O–Si bond cleavage, and easy removal of gaseous hydrocarbon byproducts. These features are quite suitable for the three-dimensional (3D) cross-linking of cage siloxanes to form porous networks with well-defined framework structures. Pan et al. prepared porous materials via the P–R reactions between D4R-SiMe2H and alkoxysilanes such as tetraethoxysilane (TEOS) and bis(triethoxysilyl)benzene.11 However, the P–R reaction generally involves a side reaction, that is, functional group exchange reactions between SiH and SiOR (R = Me, Et, etc.) groups,12 which should cause condensation in random order due to homo-condensation between the cage siloxanes or between the alkoxysilane linkers.
In this study, we report the synthesis of siloxane-based nanoporous materials by the direct cross-linking of cage siloxanes modified with dimethylsilyl (–SiMe2H) and dimethylethoxysilyl (–SiMe2OEt) groups via the P–R reactions (Scheme 1). The absence of linker molecules such as hydrosilanes and alkoxysilanes allows for the formation of well-defined 3D network structures with alternating arrangements of cage siloxanes and tetramethyldisiloxanes without the structural disorder triggered by the functional group exchange reactions. Two types of cage siloxanes with different frameworks (D4R and D6R structures) were used as the model precursors to obtain the porous materials, D4R-PR (Scheme 1(a)) and D6R-PR (Scheme 1(b)), respectively. The degree of cross-linking, degree of cleavage of siloxane bonds, structural periodicity, and pore characteristics of D4R-PR and D6R-PR were compared with the xerogels obtained by conventional hydrolysis and polycondensation (the so-called sol–gel process) of the corresponding cage siloxanes modified with –SiMe2OEt groups.
 | ||
| Scheme 1 Synthetic scheme for preparing cage siloxane-based porous materials through Piers–Rubinsztajn reactions of (a) D4R siloxanes and (b) D6R siloxanes. | ||
Experimental section
Materials
The following reagents were used as received without purification: chlorodimethylsilane (DMSCl, Tokyo Chemical Industry Co., Ltd, >95.0%), ethanol (super-dehydrated, FUJIFILM Wako Pure Chemical Corporation, >99.5%), toluene (super-dehydrated, FUJIFILM Wako Pure Chemical Corporation, >99.5%), platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex solution in xylene (Karstedt's catalyst solution, Pt ∼2%, Sigma-Aldrich), and tris(pentafluorophenyl)borane (B(C6F5)3, Tokyo Chemical Industry Co., Ltd, >98.0%). Tetramethylammonium (TMA) silicate with a D4R structure (TMA8Si8O20·xH2O, TMA-D4R) was prepared using TEOS (Kishida Chemical Co., Ltd, >98.0%) (see the ESI, Procedure S1†).Synthesis of D4R-type cage siloxane modified with dimethylethoxysilyl groups (D4R-SiMe2OEt)
First, TMA-D4R was silylated with DMSCl to obtain D4R-SiMe2H (ESI, Procedure S2†). Then, D4R-SiMe2OEt was synthesized by conversion of the SiH groups of D4R-SiMe2H into SiOEt groups (ESI, Fig. S1†) according to the previous report13 with slight modifications. D4R-SiMe2H (0.60 g, 0.59 mmol), ethanol (1.4 mL, 24 mmol) and a Karstedt's catalyst solution (35.1 μL) were mixed in dehydrated toluene (6 mL) under a nitrogen atmosphere. After stirring at room temperature for 1 d, the volatile components were removed at 40 °C under reduced pressure to obtain a brown viscous solid (0.87 g). The solid (0.39 g) was dissolved in chloroform, and the catalyst and a small amount of partially condensed products formed via the oxidation of the SiH groups were removed by gel permeation chromatography (GPC) to obtain a colorless viscous solid (0.24 g, yield: 68%). 1H nuclear magnetic resonance (NMR) (500.16 MHz, chloroform-d): δ (ppm) = 0.16, 0.17 (s, 48H; SiCH3), 1.21 (t, J = 6.9 Hz, 24H, OCH2CH3), 3.77 (q, J = 6.9 Hz, 16H, OCH2CH3); 13C NMR (125.77 MHz, chloroform-d): δ (ppm) = −1.59, −1.54, 18.28, 58.04, 58.23; 29Si NMR (99.37 MHz): δ (ppm) = −8.90 ((SiO)SiMe2OH), −9.85 ((SiO)SiMe2OEt), −109.67, −110.01 (Si(OSi)4). HR-MS (electrospray ionization) calcd for Si16O28C32H88Na+: 1391.1652; found: 1391.1663.Synthesis of D4R-PR
D4R-SiMe2H (0.10 g, 0.10 mmol) and D4R-SiMe2OEt (0.14 g, 0.10 mmol) were dissolved in dehydrated toluene (4 mL) in a two-neck flask under a nitrogen atmosphere. Upon addition of a toluene solution of B(C6F5)3 (0.02 M, 1.11 mL) to the flask, gas generation was observed followed by the formation of a gel within a few minutes. The wet gel was recovered by filtration, washed with hexane, and dried under reduced pressure to give a white solid.Synthesis of D6R-type cage siloxane modified with dimethylethoxysilyl groups (D6R-SiMe2OEt)
The complex of D6R-type potassium silicate and α-cyclodextrin (αCD) (K12Si12O30·2αCD·xH2O, D6R-αCD) was silylated with DMSCl to obtain D6R siloxanes modified with –SiMe2H groups (Si12O18(OSiMe2H)12, D6R-SiMe2H) (ESI, Procedures S3 and S4†).14 Then, D6R-SiMe2OEt was synthesized by conversion of the SiH groups of D6R-SiMe2H into SiOEt groups (ESI, Fig. S2†). D6R-SiMe2H (0.60 g, 0.39 mmol), ethanol (1.8 mL, 47 mmol) and a Karstedt's catalyst solution (70.2 μL) were mixed in dehydrated toluene (6 mL) under a nitrogen atmosphere. After stirring at room temperature for 1 d, the solvent was removed at 40 °C under reduced pressure to obtain a brown viscous solid. After GPC separation, a colorless viscous solid (0.35 g, yield 44%) was obtained. 1H NMR (500.16 MHz, chloroform-d): δ (ppm) = 0.16, 0.18 (s, 72H; SiCH3), 1.20 (t, J = 6.9 Hz, 36H, OCH2CH3), 3.76 (q, J = 6.9 Hz, 24H, OCH2CH3); 13C NMR (125.77 MHz, chloroform-d): δ (ppm) = −1.39, 18.14, 19.31, 57.92, 58.31; 29Si NMR (119.22 MHz): δ (ppm) = −7.80 to −9.81 (br, (SiO)SiMe2OH), −10.34 ((SiO)SiMe2OEt), −110.03, −111.21, −112.02 (Si(OSi)4). HR-MS (electrospray ionization) calcd for Si24O42C48H132Na+: 2075.2548; found: 2075.2544.Synthesis of D6R-PR
D6R-SiMe2H (0.077 g, 0.05 mmol) and D6R-SiMe2OEt (0.10 g, 0.05 mmol) were dissolved in dehydrated toluene (2 mL) in a two-neck flask under a nitrogen atmosphere. Upon addition of a toluene solution of B(C6F5)3 (0.02 M, 0.53 mL) to the flask, gas generation was observed followed by the formation of a gel within a few minutes. The wet gel was recovered by filtration, washed with hexane, and dried under reduced pressure to give a white solid.Synthesis of a xerogel (D6R-SG) by hydrolysis and polycondensation of D6R-SiMe2OEt
Hydrolysis and polycondensation of D6R-SiMe2OEt were conducted according to the previous report with a slight modification.4dD6R-SiMe2OEt (0.21 g, 0.10 mmol) was dissolved in ethanol (0.42 mL), followed by the addition of H2O (0.043 mL) and 6 mol L−1 hydrochloric acid (0.013 mL). The molar ratio of D6R-SiMe2OEt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol:H2O:HCl was 1:72:30:0.795. The mixture was stirred at room temperature for 4 h to form a wet gel. The gel was dried under reduced pressure to give a xerogel (D6R-SG) as a white solid.
:ethanol:H2O:HCl was 1:72:30:0.795. The mixture was stirred at room temperature for 4 h to form a wet gel. The gel was dried under reduced pressure to give a xerogel (D6R-SG) as a white solid.
Characterization
Liquid-state 1H and 13C NMR spectra were recorded on a JEOL JNM ECZ 500 spectrometer at resonance frequencies of 500.16 and 125.77 MHz, respectively, at ambient temperature using 5 mm glass tubes. Liquid-state 29Si NMR spectra of TMA-D4R, D4R-SiMe2H, D4R-SiMe2OEt, and D6R-SiMe2H were recorded on a JEOL JNM-ECZ500R spectrometer at a resonance frequency of 99.37 MHz at ambient temperature using 5 mm glass tubes. The liquid-state 29Si NMR spectrum of D6R-SiMe2OEt was recorded on a Bruker AVANCE 600 NEO spectrometer at a resonance frequency of 119.22 MHz at 25 °C using a 5 mm glass tube. Chloroform-d was used to obtain lock signals. Tetramethylsilane (δ = 0 ppm) was used as the internal reference for 1H, 13C, and 29Si NMR spectroscopy, and the signals were indicated with an asterisk (*). A small amount of Cr(acac)3 (acac = acetylacetonate) was used as a relaxation agent for the 29Si nuclei. The 29Si NMR spectra were measured with a 45° pulse and a recycle delay of 10 s. Solid-state 29Si magic angle spinning (MAS) NMR spectra were recorded on a JEOL JNM-ECA400 spectrometer at a resonance frequency of 79.43 MHz with a 90° pulse and a recycle delay of 500 s at ambient temperature. The samples for solid-state NMR analysis were placed in a 5 mm zirconia rotor and spun at 8 kHz. Poly(dimethylsilane) was used as an external reference (δ = −33.8 ppm) for solid-state 29Si NMR spectroscopy. High-resolution electrospray ionization mass analysis was conducted using a Thermo Fisher Scientific Exactive Plus spectrometer. Fourier transform infrared (FT-IR) spectroscopy was performed using a JASCO FT/IR-6100 spectrometer. The FT-IR spectra of D4R-SiMe2H, D4R-PR, D6R-SiMe2H, and D6R-PR were obtained using the KBr method. The FT-IR spectra of D4R-SiMe2OEt and D6R-SiMe2OEt were obtained using the attenuated total reflection (ATR) method with an ATR accessory (JASCO ATR PRO ONE) with a diamond prism. Powder X-ray diffraction (XRD) patterns were obtained using a RIGAKU SmartLab diffractometer with Bragg–Brentano geometry equipped with a HyPix-3000 2D X-ray detector using Cu Kα radiation (40 kV, 30 mA). GPC was carried out using a LaboACE LC-7080 recycling preparative HPLC system (Japan Analytical Industry Co., Ltd) equipped with a refractive index detector. JAIGEL-1HR (exclusion limits of 1000, theoretical plate of ≥24000) and JAIGEL-2HR Plus (exclusion limits of 5000, theoretical plate of ≥30000) columns were used with chloroform as the eluent at a flow rate of 7.0 mL min−1. N2 adsorption–desorption measurements were performed with a Quantachrome Autosorb-iQ instrument at −196 °C. The samples were heated at 120 °C for 4.5 h under reduced pressure before the measurement. Brunauer–Emmett–Teller (BET) areas were calculated using the Rouquerol method. Scanning electron microscopy (SEM) images were obtained on a Hitachi S5500 electron microscope with an accelerating voltage of 5 kV. The transmission electron microscopy (TEM) images were obtained on a JEOL JEM-2010 microscope with an accelerating voltage of 200 kV. The samples dispersed in ethanol were dropcast onto carbon-coated Cu mesh grids (Okenshoji Co., Ltd) and dried for SEM and TEM observations.
Results and discussion
D4R-SiMe2OEt was synthesized by the reaction of D4R-SiMe2H with ethanol in the presence of a Pt catalyst. The 29Si NMR spectrum of D4R-SiMe2OEt (Fig. 1A(b)) showed main signals corresponding to the D1 ((SiO)SiMe2OEt: −9.85 ppm) units and the Q4 (Si(OSi)4: −110.01 ppm) units comprising the D4R cage. No M1 signal of (SiO)SiMe2H groups (−1.39 ppm, Fig. 1A(a)) was observed. The downfield-shifted D1 signal (−8.90 ppm) and Q4 signal (−109.67 ppm) indicated that oxidation of the –SiMe2H groups and/or hydrolysis of the –SiMe2OEt groups slightly occurred by a trace of water contained in the reaction solvent and in the eluent for the GPC separation. The integral ratio indicated that 6.5% of these silyl groups were converted to –SiMe2OH groups. Because SiOH groups can also be condensed with the SiH groups in the presence of the B(C6F5)3 catalyst,15 no further purification was performed. | ||
| Fig. 1 (A) Liquid-state 29Si NMR spectra of (a) D4R-SiMe2H and (b) D4R-SiMe2OEt and (c) solid-state 29Si MAS NMR spectrum of D4R-PR. (B) Liquid-state 29Si NMR spectra of (a) D6R-SiMe2H and (b) D6R-SiMe2OEt and (c) solid-state 29Si MAS NMR spectrum of D6R-PR. | ||
The two organosilylated derivatives, D4R-SiMe2H and D4R-SiMe2OEt, were alternately cross-linked via the P–R reaction to form D4R-PR. Gelation began within one minute and the gas generation was completed after several minutes (see the photographs in Scheme 1), suggesting that the condensation reactions between the silyl groups proceeded rapidly. The FT-IR spectrum of D4R-PR (ESI, Fig. S3A†) showed a decrease in the bands of the SiH groups (νSiH at 2145 cm−1) and the SiOEt groups (νCH2 at 2882 and 2928 cm−1), suggesting the progress of the P–R reaction. The band at 564 cm−1, typically observed for the siloxane-based materials containing the D4R units,16 was retained after the reaction. The solid-state 29Si MAS NMR spectrum of D4R-PR (Fig. 1A(c)) showed mainly the D2 (SiMe2(OSi)2: −17.5 ppm) and Q4 (Si(OSi)4: −108.6 ppm) signals, indicating the formation of disiloxane linkers between the D4R cages. The minor signals of uncondensed silyl groups, i.e., M1 ((SiO)SiMe2H: −1.2 ppm) and D1 ((SiO)SiMe2(OH or OEt): around −8 ppm) units, were also observed. A slight Q3 signal (Si(OSi)3OH: −100.0 ppm) indicated the cleavage of the Si–O–Si bonds. The Q3/(Q3 + Q4) integral ratio suggested that 2.6% of the Si(OSi)4 units underwent cleavage, probably due to hydrolysis with a trace of water in the reaction system.17 The integral ratio of the 29Si signals of D2/(M1 + D1 + D2) was 0.91, indicating that the percentage of the condensed silyl groups (Pcond) was 91% (Table 1). Although the P–R reaction is strongly exothermic,9 the FT-IR and 29Si MAS NMR analyses confirmed that the reaction proceeded while retaining the D4R cage structure.
| Sample |
P
conda (%) |
P
cleavageb (%) |
d valuec/nm | S BET /m2 g−1 | V total /cm3 g−1 |
|---|---|---|---|---|---|
| a Percentage of condensed silyl groups calculated from the 29Si MAS NMR results (D2/(M1 + D1 + D2) ratio). b Percentage of hydrolyzed Q4 Si units calculated from the 29Si NMR results (Q3/(Q3 + Q4) ratio). c d value of the peak appearing at the lowest angle in the XRD pattern. d Surface area calculated from N2 adsorption–desorption isotherms using the BET method. e Total pore volume calculated at P/P0 = 0.95. f Numbers in parentheses denote standard deviations for three measurements of the same sample. g The values were not reported in ref. 4d. | |||||
| D4R-PR | 91 | 2.6 | 1.24 | 520 (±80)f | 0.33 (±0.04)f |
| D4R-SG 4d | 964d | —g | 1.14d | —g | —g |
| D6R-PR | 84 | ≤1 | 1.31 | 489 (±23)f | 0.35 (±0.02)f |
| D6R-SG | 90 | 8.4 | 1.26 | 23 | 0.03 |
It is noted that intramolecular condensation between the adjacent silyl groups on the cage via functional group exchange does not occur due to the relatively large distance between the silyl groups on the rigid and strained D4R framework (ESI, Fig. S4(a)†). In fact, no splitting or shouldering of the D2 signal due to the formation of tetrasiloxane rings was observed in the 29Si MAS NMR spectrum. In the P–R reactions involving oligo- or polysiloxanes with terminal –OSiMe2H groups, “dimethylsilane elimination” is also known to occur as a side reaction.18 However, in the case of D4R-PR, the (M1 + D1 + D2)/(Q3 + Q4) integral ratio was 1.0, indicating that this side reaction was negligible. This is probably due to the steric hindrance caused by the adjacent –OSiMe2H groups on the D4R cage.
The P–R reaction was also conducted using D6R siloxanes, which have a larger number of reactive sites (SiH and SiOEt groups) per molecule than the D4R siloxanes. We recently prepared organically bridged cage siloxane-based porous materials via hydrosilylation reactions of D4R siloxanes or D6R siloxanes, and revealed that the D6R-based materials exhibited the higher BET area, larger pore volume, and higher degree of cross-linking.5h,19 To achieve cross-linking of the D6R siloxanes by the P–R reactions, D6R-SiMe2OEt was synthesized by the reaction of D6R-SiMe2H with ethanol in the presence of a Pt catalyst. The 29Si NMR spectrum of D6R-SiMe2OEt (Fig. 1B(b)) confirmed the absence of the (SiO)SiMe2H signals observed for D6R-SiMe2H (Fig. 1B(a)), and showed mainly the D1 signal ((SiO)SiMe2OEt: −10.34 ppm) and the Q4 signal (Si(OSi)4: −111.21 ppm). The slightly shifted D1 (between −7.80 and −9.81 ppm) and Q4 (−110.03 and −112.02 ppm) signals suggested the formation of (SiO)SiMe2OH groups (∼2.0%). Similar to D4R-SiMe2OEt, no further purification was performed.
D6R-PR was prepared in the same way to D4R-PR (see the Experimental section). The FT-IR spectrum of D6R-PR (ESI, Fig. S3B†) showed a decrease in the bands of the SiH and SiOEt groups, while the band assignable to the D6R framework (501 cm−1) remained intact.5h,20 The 29Si MAS NMR spectrum of D6R-PR (Fig. 1B(c)) showed the D2 (SiMe2(OSi)2: −16.8, −20.1 ppm) and the Q4 (Si(OSi)4: −108.1, −110.5 ppm) signals, indicating the progress of the P–R reactions while retaining the silylated D6R units. The minor signals of uncondensed silyl groups (−1.7 and −9.6 ppm) and a Q3 signal (Si(OSi)3OH: −96.4 ppm) were also observed. The Q3 signal, indicative of the cleavage of the Si–O–Si bonds, was only less than 1% relative to the Q4 signal. The integral ratio suggested that 84% of silyl groups were condensed to form siloxane bonds. The (M1 + D1 + D2)/(Q3 + Q4) integral ratio was 1.0, confirming that dimethylsilane elimination did not occur. However, the shoulders of the D2 and Q4 signals might be due to the formation of tetrasiloxane rings by intramolecular condensation between the adjacent silyl groups via functional group exchange. The molecular model of the D6R siloxane suggested that tetrasiloxane rings could be formed because of the angle between the adjacent silyl groups is narrower than that for the D4R siloxane (ESI, Fig. S4(b)†).
The powder XRD patterns of D4R-PR and D6R-PR are shown in Fig. 2A. Both of them exhibit broad diffraction peaks, indicating the presence of short-range structural order. The d value of the peak at the lowest angle in the XRD patterns of D4R-PR (2θ = 7.1°, d = 1.24 nm) and D6R-PR (2θ = 6.7°, d = 1.31 nm) may correspond to the spacing between the cross-linked cage units. The larger periodicity of D6R-PR is attributable to the larger size of the D6R siloxane than D4R siloxane, as we have shown in the previous reports.5h,19 The SEM images of D4R-PR and D6R-PR (Fig. 2B) showed aggregates consisting of irregularly shaped particles with interparticle mesopores. The TEM images of D4R-PR and D6R-PR (Fig. 2C) showed no periodic structures with long-range order.
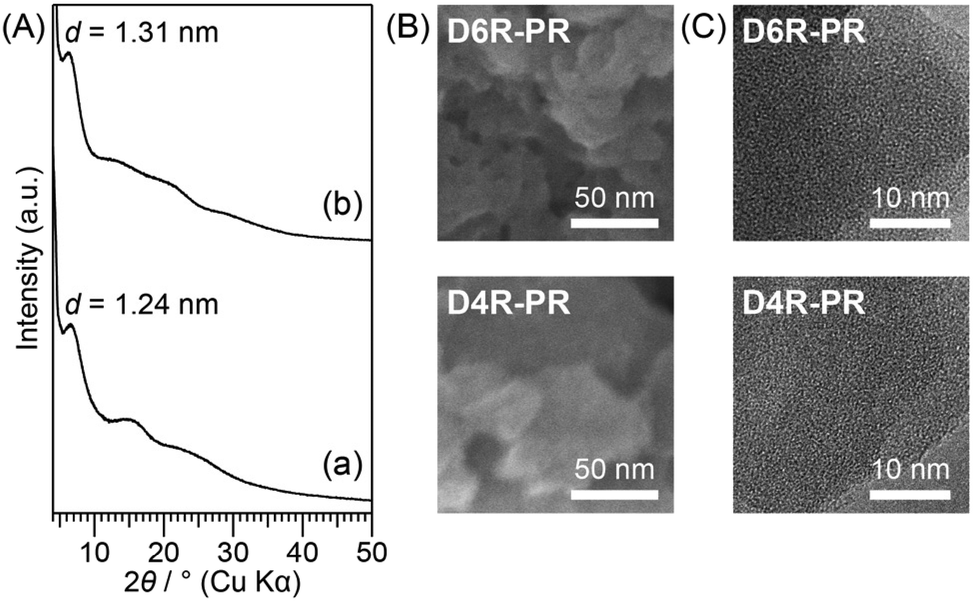 | ||
| Fig. 2 (A) Powder XRD patterns of (a) D4R-PR and (b) D6R-PR. (B) SEM images and (C) TEM images of D4R-PR and D6R-PR. | ||
The N2 adsorption–desorption isotherms of D4R-PR and D6R-PR (Fig. 3) showed uptake at low relative pressures (P/P0 < 0.1) and gradual uptake at higher relative pressures, suggesting the presence of both micropores and mesopores. The BET area (SBET) and the total pore volume (Vtotal) were calculated to be 520 (±80) m2 g−1 and 0.33 (±0.04) cm3 g−1 for D4R-PR, and 489 (±23) m2 g−1 and 0.35 (±0.02) cm3 g−1 for D6R-PR. The hystereses of the isotherms were extended to low relative pressure ranges, which was also observed for porous materials composed of cage siloxanes cross-linked by Si–C linkages through hydrosilylation reactions,5h,19,21 by Si–O–Si linkages through hydrolysis and polycondensation of terminal –Si(OiPr)2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 groups,22 by the cross-linking of D4R silicate with dichlorodimethylsilane,8b and by the P–R reactions of D4R-SiMe2H with tetraethoxysilane;11 however, it is rare that significant cavitation is not observed at around P/P0 = 0.42. This N2 adsorption–desorption behavior should have occurred due to the following reasons: (i) gas desorption was suppressed due to the presence of small pores, whose size was similar to N2, connecting larger pores; and/or (ii) insufficient time was allowed for the system to attain equilibrium.23
CH2 groups,22 by the cross-linking of D4R silicate with dichlorodimethylsilane,8b and by the P–R reactions of D4R-SiMe2H with tetraethoxysilane;11 however, it is rare that significant cavitation is not observed at around P/P0 = 0.42. This N2 adsorption–desorption behavior should have occurred due to the following reasons: (i) gas desorption was suppressed due to the presence of small pores, whose size was similar to N2, connecting larger pores; and/or (ii) insufficient time was allowed for the system to attain equilibrium.23
 | ||
| Fig. 3 N2 adsorption–desorption isotherms of (a) D4R-PR and (b) D6R-PR. The open and filled symbols denote adsorption and desorption, respectively. | ||
In the cross-linking system of cage siloxanes by hydrosilylation reactions,5h,19 D6R siloxane-based material showed higher BET area (701 m2 g−1) and larger pore volume (0.51 cm3 g−1) compared to the D4R siloxane-based material (573 m2 g−1 and 0.40 cm3 g−1, respectively), whereas no significant difference was observed for D4R-PR and D6R-PR. Here, we discuss these differences based on the degree of the intramolecular condensation of the cage siloxanes. In the D6R siloxane system, functional group exchange and subsequent intramolecular condensation might occur, as suggested by 29Si MAS NMR (Fig. 1B(c)), resulting in a decrease in the degree of intermolecular cross-linking. The less cross-linked and hence more flexible network of D6R-PR might undergo shrinkage, and the pore characteristics of D6R-PR become comparable or slightly inferior to those of D4R-PR.
Siloxane network structures similar to those of D4R-PR and D6R-PR can also be formed by the hydrolysis and polycondensation of D4R-SiMe2OEt and D6R-SiMe2OEt, respectively. We previously reported the hydrolysis and polycondensation of D4R-SiMe2OEt under acidic conditions to form a xerogel (hereafter denoted as D4R-SG).4d According to this report, we prepared a xerogel (D6R-SG) from D6R-SiMe2OEt. The 29Si MAS NMR spectrum of D6R-SG (Fig. 4(a)) showed the D1 ((SiO)SiMe2OH: −7.9 ppm), D2 (SiMe2(OSi)2: −15.8 ppm), and Q4 (Si(OSi)4: −107.5, −110.2 ppm) signals, indicating the progress of the hydrolysis and polycondensation reactions. The degree of condensation was calculated to be 90%. The Q3 signal (Si(OSi)3OH: −100.6 ppm) was also observed. From the Q3/(Q3 + Q4) ratio, the degree of the cleavage was calculated to be 8.4%.
 | ||
| Fig. 4 (a) 29Si MAS NMR spectrum, (b) N2 adsorption–desorption isotherms (the open and filled symbols denote adsorption and desorption, respectively), (c) powder XRD pattern, and (d) SEM image of D6R-SG. | ||
The N2 adsorption–desorption measurement confirmed that the product was almost non-porous (Fig. 4(b)) similar to D4R-SG.4d The powder XRD pattern of D6R-SG (Fig. 4(c)) showed broad diffraction peaks. The d value of the peak at the lowest angle was 1.26 nm, which was smaller than that of D6R-PR. No clear differences in the macroscopic morphologies of D6R-PR and D6R-SG could be observed in the SEM image (Fig. 4(d)).
The degrees of cross-linking, degrees of cleavage, periodicities, and pore characteristics of the D4R and D6R-based siloxane networks prepared by the P–R reactions and hydrolysis and polycondensation reactions are summarized in Table 1. The differences are as follows: (i) the periodicity estimated by the d values of the XRD pattern of the samples obtained by the P–R reactions is larger, and (ii) the samples obtained by the P–R reactions are porous solids whereas the samples obtained by the hydrolysis and polycondensation are non-porous.
In the case of hydrolysis and polycondensation of alkoxysilanes under acidic conditions, polycondensation not only occurs in the solution but also continues during the drying process, and significant shrinkage of the gel generally occurs due to the high surface tension of water contained in the siloxane networks.24 Because D4R-SG and D6R-SG have relatively flexible disiloxane linkages between the cages, large shrinkage might occur upon drying. In addition, rearrangement of the siloxane networks can occur via the hydrolytic cleavage of the siloxane bonds between the cages. These drawbacks of the “hydrolytic” siloxane bond formation reactions likely resulted in the formation of non-porous materials.
On the other hand, in the case of the P–R reactions, siloxane bond formation fully proceeded in the solution due to the fast reaction rates. In fact, the values of Pcond for D4R-PR and D6R-PR were high (91% and 84%, respectively), even though the wet gels were washed before drying to remove the catalyst. Therefore, highly cross-linked networks containing solvent molecules were possibly formed. The evaporation of the anhydrous organic solvent with relatively low surface tension should facilitate the retention of the intermolecular spaces between the cages. This is consistent with the fact that the d spacings of D4R-PR and D6R-PR were larger than those obtained by hydrolysis and polycondensation.
The P–R reaction has been used for preparing siloxane-based copolymers, dendrimers, and 3D networks.10 We recently reported the P–R reactions between cyclododecasiloxanes modified with –SiMe2H and –SiMe2OEt groups.25 Gelation did not occur in that system, and subsequent solvent evaporation was necessary to obtain a solid sample, which is in contrast to the rapid gelation by the P–R reactions of the cage siloxanes. It was likely that dimethylsilane eliminations and/or functional group exchange reactions and subsequent intramolecular condensation inhibited the formation of well-cross-linked networks. In the system of cage siloxanes, the rigid siloxane frameworks allowed for the dominant progress of intermolecular cross-linking rather than intramolecular condensation.
Conclusions
We demonstrated the synthesis of nanoporous materials via nonhydrolytic siloxane bond formations between cage siloxanes modified with –SiMe2H and –SiMe2OEt groups. By using rigid cage siloxanes as building blocks without any other linker molecules, the formation of well-defined network structures was achieved, while suppressing the side reactions that compete with the condensation reactions. Furthermore, compared with the conventional hydrolysis and polycondensation reactions, the present method was found advantageous for the suppression of the cleavage of the siloxane bonds and for the formation of porous networks. We expect that the dimethylsiloxane (–SiMe2O–) linkers impart not only flexibility to the porous networks,26 but also high hydrophobicity in conjunction with the presence of few uncondensed silanol groups. The latter feature is particularly effective for applications in the adsorption/separation of hydrophobic molecules and in low-k dielectrics, where water adsorption would degrade the performance.Author contributions
M. K. and T. H. contributed equally to this work. M. K. and T. H. designed the experiments and M. K. performed the experiments and materials characterization. T. M. and K. K. provided scientific and technical support. A. S. supervised the research. M. K. and T. H. wrote the draft of the paper and T. M., K. K., and A. S. substantially contributed to the preparation of the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr Toshimichi Shibue (MCCL, Waseda University)27 and Mr Masashi Yatomi (Waseda University) for the solid-state NMR analysis, Mr Ryoma Uchida for electron microscopic analyses, and Mr Takuya Hikino, Ms Yuka Kawakubo, and Ms Hikaru Mochizuki (Waseda University) for fruitful discussions. This work is based on results obtained from a project commissioned by the New Energy and Industrial Technology Development Organization (NEDO) (JPNP06046). This work was also supported in part by JSPS KAKENHI (grant no. 23H02051). This work was the result of using research equipment (JEOL JNM-ECZ500R (C1028), JEOL JNM-ECA400 (C1026), Thermo Fisher Scientific Exactive Plus (C1057), Rigaku SmartLab (G1009), Quantachrome Autosorb-iQ (G1038), HITACHI S5500 (G1028), and JEOL JEM-2010 (G1024)) shared in MEXT Project for promoting public utilization of advanced research infrastructure (program for supporting construction of core facilities) (grant no. JPMXS0440500023). T. H. is grateful for JST SPRING (grant no. JPMJSP2128) and JSPS Research Fellowships for Young Scientists (grant no. 23KJ2037).Notes and references
- (a) G. J. de A. A. Soler-Illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093 CrossRef PubMed; (b) K. Kanamori and K. Nakanishi, Chem. Soc. Rev., 2011, 40, 754 RSC; (c) Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li, J. Liu, E. Che, L. Hu, Q. Zhang, T. Jiang and S. Wang, Nanomedicine, 2015, 11, 313 CrossRef CAS PubMed.
- (a) Y. Liu, T. Chaiprasert, A. Ouali and M. Unno, Dalton Trans., 2022, 51, 4227 RSC; (b) M. Unno and T. Matsumoto, Russ. Chem. Rev., 2013, 82, 289 CrossRef; (c) S. Kinoshita, S. Watase, K. Matsukawa and Y. Kaneko, J. Am. Chem. Soc., 2015, 137, 5061 CrossRef CAS PubMed; (d) R. M. Laine, J. Mater. Chem., 2005, 15, 3725 RSC; (e) D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081 CrossRef CAS PubMed; (f) A. Shimojima and K. Kuroda, Chem. Rec., 2006, 6, 53 CrossRef CAS PubMed; (g) J. Suzuki, A. Shimojima, Y. Fujimoto and K. Kuroda, Chem. – Eur. J., 2008, 14, 973 CrossRef CAS PubMed.
- (a) D. Hoebbel and W. Wieker, Z. Anorg. Allg. Chem., 1971, 384, 43 CrossRef CAS; (b) D. Hoebbel, W. Wieker, P. Franke and A. Otto, Z. Anorg. Allg. Chem., 1975, 418, 35 CrossRef CAS; (c) D. Hoebbel, G. Garzó, G. Engelhardt, R. Ebert, E. Lippmaa and M. Alla, Z. Anorg. Allg. Chem., 1980, 465, 15 CrossRef CAS; (d) K. Benner, P. Klüfers and J. Schuhmacher, Angew. Chem., Int. Ed. Engl., 1997, 36, 743 CrossRef CAS; (e) M. Haouas, C. Falaise, C. Martineau-Corcos and E. Cadot, Crystals, 2018, 8, 12 CrossRef.
- (a) W. G. Klemperer, V. V. Mainz and D. M. Millar, Mater. Res. Soc. Symp. Proc., 1986, 73, 3 CrossRef CAS; (b) P. C. Cagle, W. G. Klemperer and C. A. Simmons, Mater. Res. Soc. Symp. Proc., 1990, 180, 29 CrossRef CAS; (c) P. G. Harrison and R. Kannengiesser, Chem. Commun., 1996, 415 RSC; (d) Y. Hagiwara, A. Shimojima and K. Kuroda, Chem. Mater., 2008, 20, 1147 CrossRef CAS; (e) A. Shimojima, R. Goto, N. Atsumi and K. Kuroda, Chem. – Eur. J., 2008, 14, 8500 CrossRef CAS PubMed; (f) R. Goto, A. Shimojima, H. Kuge and K. Kuroda, Chem. Commun., 2008, 6152 RSC; (g) A. Shimojima, H. Kuge and K. Kuroda, J. Sol-Gel Sci. Technol., 2011, 57, 263 CrossRef CAS; (h) K. Kawahara, H. Tachibana, Y. Hagiwara and K. Kuroda, New J. Chem., 2012, 36, 1210 RSC; (i) K. Iyoki, A. Sugawara-Narutaki, A. Shimojima and T. Okubo, J. Mater. Chem. A, 2013, 1, 671 RSC.
- (a) C. Zhang, F. Babonneau, C. Bonhomme, R. M. Laine, C. L. Soles, H. A. Hristov and A. F. Yee, J. Am. Chem. Soc., 1998, 120, 8380 CrossRef CAS; (b) T. Hayashi, M. Kikuchi, N. Murase, T. Matsuno, N. Sugimura, K. Kuroda and A. Shimojima, Chem. – Eur. J., 2024, e202304080 CrossRef PubMed; (c) W. Chaikittisilp, A. Sugawara, A. Shimojima and T. Okubo, Chem. – Eur. J., 2010, 16, 6006 CrossRef CAS PubMed; (d) W. Chaikittisilp, A. Sugawara, A. Shimojima and T. Okubo, Chem. Mater., 2010, 22, 4841 CrossRef CAS; (e) W. Chaikittisilp, M. Kubo, T. Moteki, A. Sugawara-Narutaki, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2011, 133, 13832 CrossRef CAS PubMed; (f) Y. Kim, K. Koh, M. F. Roll, R. M. Laine and A. J. Matzger, Macromolecules, 2010, 43, 6995 CrossRef CAS; (g) Y. Peng, T. Ben, J. Xu, M. Xue, X. Jing, F. Deng, S. Qiu and G. Zhu, Dalton Trans., 2011, 40, 2720 RSC; (h) D. Wang, W. Yang, L. Li, X. Zhao, S. Feng and H. Liu, J. Mater. Chem. A, 2013, 1, 13549 RSC.
- Y. Wada, K. Iyoki, A. Sugawara-Narutaki, T. Okubo and A. Shimojima, Chem. – Eur. J., 2013, 19, 1700 CrossRef CAS PubMed.
- N.-h. Hu, C. B. Sims, T. V. Schrand, K. M. Haver, H. E. Armenta and J. C. Furgal, Chem. Commun., 2022, 58, 10008 RSC.
- (a) I. Hasegawa, J. Sol-Gel Sci. Technol., 1995, 5, 93 CrossRef CAS; (b) S. Smet, P. Verlooy, S. Pulinthanathu, C. E. A. Kirschhock, F. Taulelle, E. Breynaert and J. A. Martens, Chem. – Eur. J., 2019, 25, 12957 CrossRef CAS PubMed.
- R. Wakabayashi and K. Kuroda, ChemPlusChem, 2013, 78, 764 CrossRef CAS PubMed.
- S. Rubinsztajn, J. Chojnowski and U. Mizerska, Molecules, 2023, 28, 5941 CrossRef CAS PubMed.
- D. Pan, E. Yi, P. H. Doan, J. C. Furgal, M. Schwartz, S. Clark, T. Goodson and R. M. Laine, J. Ceram. Soc. Jpn., 2015, 123, 756 CrossRef CAS.
- J. Chojnowski, S. Rubinsztajn, J. A. Cella, W. Fortuniak, M. Cypryk, J. Kurjata and K. Kaźmierski, Organometallics, 2005, 24, 6077 CrossRef CAS.
- A. Jurásková, A. L. Skov and M. A. Brook, Ind. Eng. Chem. Res., 2020, 59, 18412 CrossRef.
- N. Sato, H. Wada, K. Kuroda and A. Shimojima, Chem. Lett., 2024, 53, upad048 CrossRef.
- N. Moitra, S. Ichii, T. Kamei, K. Kanamori, Y. Zhu, K. Takeda, K. Nakanishi and T. Shimada, J. Am. Chem. Soc., 2014, 136, 11570 CrossRef CAS PubMed.
- C. S. Blackwell, J. Phys. Chem., 1979, 83, 3251 CrossRef CAS.
- K. M. Rabanzo-Castillo, V. B. Kumar, T. Söhnel and E. M. Leitao, Front. Chem., 2020, 8, 477 CrossRef CAS PubMed.
- J. Chojnowski, W. Fortuniak, J. Kurjata, S. Rubinsztajn and J. A. Cella, Macromolecules, 2006, 39, 3802 CrossRef CAS.
- T. Hayashi, T. Matsuno, K. Kuroda and A. Shimojima, Chem. Lett., 2024, upae025 CrossRef.
- (a) C. S. Blackwell, J. Phys. Chem., 1979, 83, 3257 CrossRef CAS; (b) W. Mozgawa, W. Jastrzębski and M. Handke, J. Mol. Struct., 2005, 744–747, 663 CrossRef CAS.
- J. J. Morrison, C. J. Love, B. W. Manson, I. J. Shannon and R. E. Morris, J. Mater. Chem., 2002, 12, 3208 RSC.
- Y. Hagiwara, A. Shimojima and K. Kuroda, Bull. Chem. Soc. Jpn., 2010, 83, 424 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS.
- (a) C. J. Brinker and G. W. Scherer, Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing, Academic Press, Inc., San Diego, 1990 Search PubMed; (b) U. Schubert, Chemistry and Fundamentals of the Sol–Gel Process, in The Sol–Gel Handbook, ed. D. Levy and Z. Marcos, Wiley-VCH, 2015, pp. 1–28 Search PubMed.
- M. Yoshikawa, H. Shiba, H. Wada, A. Shimojima and K. Kuroda, Bull. Chem. Soc. Jpn., 2018, 91, 747 CrossRef CAS.
- J. Shen, K. Fujita, T. Matsumoto, C. Hongo, M. Misaki, K. Ishida, A. Mori and T. Nishino, Macromol. Chem. Phys., 2017, 218, 1700197 CrossRef.
- C. Izutani, D. Fukagawa, M. Miyasita, M. Ito, N. Sugimura, R. Aoyama, T. Gotoh, T. Shibue, Y. Igarashi and H. Oshio, J. Chem. Educ., 2016, 93, 1667 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental details, 1H NMR spectra, FT-IR spectra, and molecular models. See DOI: https://doi.org/10.1039/d4dt00215f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |