
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu(dppf) complexes can be synthesized from Cu-exchanged solids and enable a quantification of the Cu-accessibility by 31P MAS NMR spectroscopy†
Elif
Kaya
,
Daniel
Dittmann
 ,
Maximilian
Schmidt
and
Michael
Dyballa
*
,
Maximilian
Schmidt
and
Michael
Dyballa
*
Institute of Technical Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: michael.dyballa@itc.uni-stuttgart.de
First published on 22nd March 2024
Abstract
Herein, we apply three different copper-exchanged materials (Na–[Al]SBA-15, silica, Na–MCM-22) as hosts for a direct synthesis of CuI(1,1′-bis(diphenylphosphino)ferrocene = dppf) complexes in cationic ion exchange position. Using 31P MAS NMR spectroscopy, we show that identical complexes as after ion exchange are generated if the solids are applied as reactants directly. The homogeneity of copper exchanges is evaluated by EDX spectroscopy. Both CuI and CuII result in the formation of complexes, thereby oxidizing dppf. Cu-particles were not reactive. Optimized conditions for a maximized complex formation are identified applying quantitative 31P MAS NMR spectroscopy and ICP-OES. Only accessible copper in cationic position of the solids forms the complexes. This enables a quantification of the amount of copper in mesopores vs. the total copper amount. Thus, besides a new synthesis of the complex a suitable method for quantitative elucidation of the location of copper cations is demonstrated herein.
Introduction
Most industrially relevant large-scale conversions are conducted catalytically. Usually, catalyst and reactant and/or products are thereby present in different phases. The support in such heterogeneous catalysis systems is usually a solid, as this can cost-efficiently be separated from the reaction mixture.1 Typical solid catalysts are porous in order to enlarge the surface area for an efficient contact between reaction medium and catalyst. Exemplary supports are zeolites, metal–organic frameworks, and mesoporous materials.2–5 The diameter of the pores determines the dimension of the species that can be hosted, which leads to shape-selectivity effects during catalysis.6 The support must furthermore stabilize the catalytically active sites in the solid. Copper is a frequently investigated active site, and a variety of ways is applied to support active copper in form of a heterogeneous catalyst.One form of stabilization is ion exchange into the cation position of bridging Si(OX)Al groups (with X = monovalent cation) that are found in silicoaluminates.2,7 For catalyst synthesis, the CuII ions are typically ion exchanged into porous silicoaluminates prior to the conversion of the hydrocarbons. This metal cations catalyse, for example, the selective oxidation of methane and ethane by solid catalysts.8–14 In contrast to heterogeneous catalysis, where ion exchange is applied frequently, this is a seldom investigated technique for metalorganic complex immobilization. Copper complexes can be (1) adsorbed onto the surface, (2) bound to the surface using it as ligand (referred to as surface organometallic chemistry, SOMC), (3) bound to the surface using a linker (molecular heterogeneous catalysts) and finally (4) charged complexes can be immobilized by ion exchange.15–18 In the case of surface organometallic chemistry (SOMC), the complex formation takes place inside the pores with the surface as a ligand.15,16,19 A potential application of SOMC is also the formation of defined nanoparticles20 or the better understanding of processes occurring on often ill-defined heterogeneous catalysts.21,22 Counter-intuitively also for covalently bound molecular heterogeneous catalysts a strong interaction with the surface can be present.15,16 Such strong surface interactions impact on the catalytic performance of the materials. Complexes formed by methods of SOMC and likewise molecular heterogeneous catalysts form part of the surface or are covalently bound to the surface, respectively, and used for catalytic applications. As synthesis of complexes on a solid support means also removing these complexes in a second step, thus, only ion exchange and adsorption remain as industrially feasible pathways. Herein we synthesize complexes in ion exchange position of a solid support to enable their later release.
In catalytic reactions, interactions between copper and surface play an important role. For example, copper-based catalysts immobilized on Al2O3 were shown to outperform their homogeneous counterparts in terms of selectivity in atom-transfer radical cyclizations.23 Also location-dependent selectivity changes during catalysis by Rh-complexes in 1,2-additions were found, indicating that the location of the active site within the solid is important for the application as catalyst.24 Unfortunately, if immobilized in porous materials, such complexes remain often in the outer parts of the catalysts.24,25 The reason is the large diameter of the complexes and the strong interactions with the surface. Conclusively, it was also shown that ion exchange can lead to a very strong binding between CuI(dppf) (dppf = 1,1′-bis(diphenylphosphino)ferrocene) complexes and surfaces.26 This motif was previously identified to be interesting for electrochemical or anticancer applications.27,28 In this favourable cases, metalorganic complexes bear a phosphorus nucleus (I = ½) which can be investigated quantitatively using 31P MAS NMR spectroscopy.29–34 Finally it can be summarized that techniques to finely distribute such complexes in pores are of interest for the community. We herein thus also focus on how synthesized cationic complexes as counter ions in ion exchange position can be applied to characterize the support.
The typical way to place a cationic complex in counter ion position of an inorganic support is as follows: first the metalorganic complex, in this case consisting of CuI and dppf, is synthesized. Then the cationic complex is ion-exchanged into the solid. This procedure is visualized in route I in Scheme 1.26 In this work, it shall thus be investigated weather a direct CuI(dppf) complex formation in an ion exchanger is possible. This new, alternative route involves a direct synthesis of the copper-exchanged solid and then the dppf is reacted directly with the cations. A schematic presentation of this procedure is shown in route II in Scheme 1. To maximize the surface and the amount of ion exchange sites, the copper-exchanged aluminated [Al]SBA-15 is used as support. This amorphous silicoaluminate has a large surface area and is capable of a quantitative ion exchange inside its mesopores of usually 6 nm and above, which is large enough to host metalorganic complexes inside the pores.35 Typical heterogeneous catalysts contain copper in its highest oxidation state (CuII). It should be noted that this cation might form a variety of oxidizing copper-oxo species, for example CuII(OH), depending on the surface of the support and the conditions.36–40 Thus it shall be tested if the complex formation occurs also with a precursor containing this cation. In route II the complex forms inside a porous solid. It is important to note that a complex formation is only possible if (1) the dppf has access to the copper and if (2) there is enough space available to form a complex.32 A similar picture is observed if noble metals react with phosphines in inorganic complexes as triphenylphosphines and higher analogues. Also in this case a complex forms directly inside a mesopore that is large enough to host the resulting complex.41,42 The sensitivity of synthesis route II for space and accessibility determined by the solid can be used as tool for characterization. In other words, the direct complex synthesis enables information about the location of the formerly present ions inside the solid. This makes route II interesting for quantitative elucidating the spatial distribution of ion exchanged copper.
 | ||
| Scheme 1 Reaction scheme for visualizing the synthesis of CuI(dppf) complexes in counter ion position synthesized by ion exchange (route I) or by reaction with CuI/II counter ions (route II). | ||
Results and discussion
Physicochemical characterization of supports
In this work three different supports for copper are applied. The standard characterization of these materials is found in Table 1. For checking the presence of regular mesoporosity, small-angle X-ray powder diffraction patterns (XRD) were recorded (see Fig. S1 in the ESI†). Likewise, the crystalline zeolite Cu–MCM-22 was investigated by wide-angle XRD (see Fig. S2 in the ESI†). Both patterns agree with those reported in literature for SBA-15 and MCM-22.35,43,44 Thus, we conclude that our materials have an intact meso- or micropore structure. The Cu–[Al]SBA-15 synthesized with a Si/Al ratio of 14 shows in comparison to literature a higher BET surface area.35 This is a result of the large surface area of the siliceous parent SBA-15 (1160 m2 g−1) before alumination. In accordance with literature,35 no micropores were detected after alumination while a large mesopore volume Vmeso of 1.34 ml g−1 was maintained. Thus, the support can not only host cations but also larger complexes within its mesopores. The silica A200 is a commercial, fumed silica support with negligible aluminum impurities, a BET surface area of 185 m2 g−1, and secondary mesopores as described previously.45 The state of aluminum in Cu–[Al]SBA-15 and Cu–MCM-22 was investigated by 27Al MAS NMR spectroscopy (see Fig. S3 in the ESI†). The spectrum of Na–[Al]SBA-15 contains a single peak at a chemical shift of δ27Al = 53 ppm. This peak is assigned to a tetrahedral aluminum within the framework and proves a maximum in well-exchangeable cationic sites.35 In silica A200, we find no aluminum, neither by ICP-OES nor by 27Al MAS NMR. The 27Al MAS NMR spectrum of MCM-22 shows two peaks. A peak at δ27Al = 56 ppm can again be assigned to aluminum in the framework. The peak at δ27Al = 0 ppm indicates presence of extra-framework aluminum, which agrees well with previous findings on the aluminum state in this zeolite structure.44| Material | Si/Ala | Cua [mmol g−1] | BET surfaceb [m2 g−1] | V pore [ml g−1] | V micro [ml g−1] | V meso [ml g−1] | Si(OH) densityc [mmol g−1] | BAS content H-formd [mmol g−1] |
|---|---|---|---|---|---|---|---|---|
| a Determined by ICP-OES, error ±1. b From N2 physisorption. c From 1H MAS NMR after subtracting BAS density. d From 1H MAS NMR after NH3 adsorption. | ||||||||
| Cu–[Al]SBA-15 | 14 | 0.27 | 850 | 1.34 | — | 1.34 | 1.19 | — |
| Cu@silica (A200) | >1700 | 1.26 | 185 | 0.24 | — | 0.24 | 0.39 | — |
| Cu–MCM-22 | 19 | 1.02 | 630 | 0.79 | 0.18 | 0.61 | 0.18 | 0.17 |
The copper content of the materials was investigated by ICP-OES (see Table 1). Cu–[Al]SBA-15 has thus 0.27 mmol g−1 copper cations. In addition to the directly evaluation of copper, we also cross-checked by ICP-OES that the Na-content of the material decreased after ion exchange, from 1.07 to 0.34 mmol g−1 Na+. The larger decrease in mmol g−1 is reasonable, as the 2-fold charged copper cations will exchange cations stoichiometrically. Furthermore, an incomplete ion exchange indicates that some Na-cations could not be exchanged. For a better understanding, despite Na-cations are still present, the resulting material is labeled Cu–[Al]SBA-15. The copper content of impregnated silica A200 (labeled Cu@silica) was 1.26 mmol g−1 while the copper content of the ion-exchanged zeolite Cu–MCM-22 was 1.02 mmol g−1. SEM images of the support after ion exchange with copper are provided in Fig. 1. The elongated structures of SBA-15 particles are typical for this mesoporous material.35 To prove a homogeneous alumination of the SBA-15 material and a homogeneous exchange with copper, EDX mappings were performed. The Si–EDX mapping resembles the structures of the SEM as this element constitutes the backbone of the material. The Al–EDX mapping shows identical structures, in lower intensity due to the lower abundance of aluminum. Absence of bright spots indicates absence of aluminum deposits and conclusively, a homogeneous alumination distribution was achieved by alumination, as expected from tests in previous work.35 Also the Cu–EDX mapping resembles the structures from the SEM pictures. Again, an absence of bright spots indicates a homogeneous distribution of ion exchanged copper and absence of copper particles. As further proof of a homogeneous copper distribution, the backscattered electron detector (BSE-detector) was applied. No bright spots appeared and thus no copper particles formed. We conclude that the copper cations are homogeneously stabilized at ion exchange sites associated with aluminum and that no copper deposits were formed on Cu–[Al]SBA-15. Comparable figures of EDX on Cu@silica and Cu–MCM-22 are found in Fig. S4 and S5 in the ESI.† Thereby, for Cu–MCM-22 a similar picture as for Cu–[Al]SBA-15 is observed as also here all copper is introduced by ion exchange. In contrast, bright spots occur on Cu@silica when using the BSE-detector. Thus, Cu-particles form on the silica surface as result of the impregnation and the absence of stabilizing ion exchange sites. These Cu-particles give characteristic bright spots in the BSE-detector and indicate a bad stabilization of cationic copper.46
 | ||
| Fig. 1 SEM and EDX screenings on copper-exchanged [Al]SBA-15 (top). Characteristic Cu-particles formed on Cu@silica are revealed by the BSE-detector. | ||
The density of Si(OH) groups on the material surfaces was quantified applying 1H MAS NMR spectroscopy. A density of 1.19 mmol g−1 was found for Na–[Al]SBA-15 and agrees well with the 1.14 mmol g−1 reported previously.45 Thus, this surface can be considered polar, compared to the other two material surfaces. A lower Si(OH) density of 0.24 mmol g−1 was found for silica A200, in agreement with a literature value of 0.39 mmol g−1.45 The Si(OH) density of the zeolite H–MCM-22 is, with 0.18 mmol g−1, comparable. The higher Si(OH) density on [Al]SBA-15 results from the amorphous surface structure. In contrast, on the crystalline structure of MCM-22 or on the heat-treated fumed silica surface more Si(OH) groups formed siloxane bridges upon water release. MCM-22 and [Al]SBA-15 are aluminum-doted, which results in Brønsted acid site (BAS) formation on the material H-form (after ion exchange).7 Quantitative 1H MAS NMR spectroscopy was thus applied to evaluate the acidity of the respective H-forms (see Fig. S6 in the ESI†). All materials show peaks of Si(OH) groups at a chemical shift of δ1H = 1.8 ppm., while broad peaks at ∼2.5 ppm are assigned to interacting Si(OH) groups. The latter peak broadens for Na–[Al]SBA-15 after NH3-loading as a result of the heat treatment applied during NH3-desorption.35 The H–MCM-22 shows peaks at 3.9 ppm and above, caused by Brønsted acidic bridging Si(OH)Al groups in free or disturbed state.47–49 These groups react with NH3 to form NH4+ which gives a quantifiable peak at 6.4 ppm.32,50 The peak intensity indicates the accessibility of 0.17 mmol g−1 BAS. This is low compared to the ∼0.8 mmol g−1 BAS expected from the Si/Al ratio. However, this is reasonable due to the formation of EFAl, as it was indicated by 27Al MAS NMR spectra (vide supra). This is in-line with previous findings for similar H–MCM-22.44 Note that MCM-22 was copper ion exchanged from its Na-form and that all copper-bearing materials applied in this study for complex formation experiments were free of detectable BAS.
Reaction of Cu–[Al]SBA-15 with dppf
The formed CuI(dppf) complexes are in the following investigated using 31P MAS NMR spectroscopy (see Fig. 2). The neat dppf results in a slim signal at a chemical shift of δ31P = −15 ppm. From literature it is known that adsorption of phosphines at silicoalumina surfaces usually leads to downfield shifts of the peaks.24,41 In seldom cases also for dppf, an additional peak at −9 ppm, caused by dppf in surface interaction, is found herein. In Fig. 1, below the spectrum of neat dppf, the benchmark in form of an ion exchanged complex Cu–[Al]SBA-15, synthesized according to route I in Scheme 1, is shown. The spectrum contains a multitude of lines, since each resonance is split into 2I + 1 = 4 individual lines due to 1J coupling of the 31P nuclei of the dppf ligand with the 63Cu/65Cu (I = 3/2) cations. We see that there are two different spins associated with the central copper and, due to a higher natural abundance of 63Cu, the lines belonging to 65Cu are less intense. Due to a γ(65Cu)/γ(63Cu) = 1.07 larger 1J coupling, the lines belonging to 65Cu complexes are furthermore found outside of the lines belonging to 63Cu complexes.51 As preciously shown, both 31P nuclei present in the dppf become inequivalent upon ion exchange.26 Thus, a characteristic splitting into 8 overlapping lines is observed (4 lines for each 31P nucleus in dppf). The outer lines of the coupling pattern are found at chemical shifts of about δ31P = −7 and −37 ppm, respectively. This supports the conclusion, that the herein investigated complexes are cations in ion exchange position and agrees with literature findings, 1H–31P FSLG HETCOR spectra provided herein (vide infra), and the spectra of similar ion exchanged materials provided previously.26 Next, the reaction of CuI–[Al]SBA-15 with dppf was investigated. For the reaction, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry between CuI and dppf was applied. An identical spectrum as for the benchmark is received and it will later become important to remark that no additional peaks are found at higher or lower field, respectively. This clarifies, that exclusively CuI(dppf) complexes, but no by-products were formed upon reaction of CuI in cationic ion exchange position with dppf. The complexes can be quantified with an external standard applying 31P MAS NMR spectroscopy. This results in amounts of 0.10 to 0.05 mmol g−1 complexes within the mesoporous [Al]SBA-15 support after ion exchange of reaction with CuI, respectively (see also quantitative indications in Fig. 2).
:1 stoichiometry between CuI and dppf was applied. An identical spectrum as for the benchmark is received and it will later become important to remark that no additional peaks are found at higher or lower field, respectively. This clarifies, that exclusively CuI(dppf) complexes, but no by-products were formed upon reaction of CuI in cationic ion exchange position with dppf. The complexes can be quantified with an external standard applying 31P MAS NMR spectroscopy. This results in amounts of 0.10 to 0.05 mmol g−1 complexes within the mesoporous [Al]SBA-15 support after ion exchange of reaction with CuI, respectively (see also quantitative indications in Fig. 2).
 | ||
| Fig. 2
31P MAS NMR spectra of (from top to bottom) neat dppf, CuI(dppf) ion exchanged into Na–[Al]SBA-15, dppf reacted with CuI–[Al]SBA-15, and dppf reacted with CuII–[Al]SBA-15. The stoichiometry 1:1 and 1:10 indicates the molar rations between copper and dppf, while the loading amounts in mmol g−1 indicate the amount of CuI(dppf) derived from quantitative 31P MAS NMR spectroscopy. | ||
In a next step, the reaction of dppf with CuII is addressed. This formation of CuI(dppf) requires a reduction of the CuII cations present. The species responsible for oxygen-transfer could be a Cu(OH)+ unit that forms upon ion exchange in aqueous media to balance the charge of CuII on an ion exchange site with single negative charge. Furthermore, similar sites were identified by XAS in copper-exchanged zeolite catalysts.39,52 Additional peaks at higher chemical shift at δ31P of ∼68 ppm appear. Typically, peaks at such low-field chemical shift of δ31P ≈ 68 ppm originate from the oxidation of phosphines to the corresponding phosphine oxide, which is supported by previous work on a variety of phosphines.24,53–56 Unfortunately, it was herein not possible to isolate this oxidation product and to further characterize it. As this happens only in case of CuII presence, oxidation due to oxygen impurities during sample handling can be excluded. Also a previous calcination at 823 K after copper ion exchange of Cu–[Al]SBA-15 leads to identical 31P MAS NMR spectra. This excludes that any surface impurity caused this peak. It is thus associated with dppf oxidation to the corresponding oxide. The 31P MAS NMR spectrum of the resulting CuI(dppf) is identical to the spectra recorded after ion exchange or reaction with CuI (vide supra). In accordance with the reactivity of other phosphines we conclude that herein the dppf reduces the CuII to CuI. The latter subsequently forms a complex identical to those received after ion exchange of CuI(dppf) into the material or after reaction with CuI–[Al]SBA-15. Quantification shows that a reaction in a 1:1 stoichiometry between CuII and dppf leads to an amount of 0.22 mmol g−1 CuI(dppf) complex formed. This is a reasonable quantity with respect to the 0.27 mmol g−1 copper initially present on the Cu–[Al]SBA-15 material. Note that not all copper has reacted to form complexes. Furthermore, the reaction with dppf was also tested in absence of solvent to check if solvent is necessary. This was done according to the solid state loading approach previously applied for loading a variety of other phosphines and phosphine oxides on solids.35,42,57–59 Briefly, the Cu–[Al]SBA-15 solid was mixed with neat, solid dppf and the mixture was heated over 2 h with a heating rate of 1 K min−1 at 333 K under N2. However, on Cu–[Al]SBA-15 no CuI(dppf) complex formed and only the slim 31P MAS NMR peak associated with the neat dppf was found. We conclude that the formation of CuI(dppf) complexes from cationic copper requires a solvent. It is not relevant if the solvent is acetonitrile or ethanol, however, a larger quantity of the complex forms if ethanol is applied.
1H–31P FSLG HETCOR was performed to further clarify the identity of complexes synthesized by the two routes indicated in Scheme 1. For the 2D spectra in Fig. 3 it is immediately noted that similar correlations exist for both synthesis routes I and II. Correlation peaks at δ1H ≈ 4, 5 and 7 ppm are associated with the protons of the cyclopentadienyl and phenyl rings, respectively. The splitting of the cyclopentadienyl ring protons into two individual signals with 4 lines each is caused by magnetic inequivalence of the 31P nuclei of dppf upon ion exchange (vide supra and elsewhere26). As result, 1J(63/65Cu, 31P) scalar couplings of 1200 and 1340 Hz are found, in agreement with previous findings.26 A proton at δ1H ≈ 10 ppm is found in both 2D spectra and indicates an interaction between complexes and Si(OH) on the surface. Thus, it was verified that identical structures have formed within mesoporous [Al]SBA-15 also after reaction of the copper-exchanged Cu–[Al]SBA-15 with dppf. This is the first proof that both route I and route II indicated in Scheme 1 are applicable to generate cationic CuI(dppf) complexes.
 | ||
| Fig. 3
1H–31P FSLG HETCOR spectra on (top) Na–[Al]SBA-15 after ion exchange of CuI(dppf) complexes and (bottom) after reaction of dppf with CuII–[Al]SBA-15 with indicated loading stoichiometry 1:2 Cu:dppf. | ||
Quantitative study of the complex formation
Quantitative 31P MAS NMR spectroscopy applied on (neat) metalorganic complexes is hampered by usually long T1-times.29–31,33,34,51,60 However, if the phosphine is tightly bound to the surface the T1-relaxation of the 31P nuclei is significantly decreased.33,42 In order to approximate the T1-time we performed a delay time variation (see Fig. S7 and S8 in the ESI†). For the neat CuI(dppf) complex, an increasing 31P signal intensity is found for increasing delay time between scans from 5 to 240 s. In other words, even 240 s delay yields no quantitative spectrum of the pure complex under our measurement conditions (see Fig. S7 in the ESI†). Also for neat dppf, the peak intensity at δ31P = −15 ppm is not constant after 240 s delay time. However, the relaxation behavior changes dramatically if the complex is ion exchanged into the support. In our case, a delay between scans of 10 s or longer is sufficient, to yield spectra of similar intensity (see Fig. S8 in the ESI†). The shorter T1-time is rationalized by the strong interaction between complex and surface (see cross-peak with Si(OH) δ1H ≈ 10 ppm in Fig. 3) and the close proximity to 27Al in the ion exchange position. Conclusively, delays above 10 s were quantitative. However, to include a safety threshold, herein 60 s delay were applied for quantitative evaluation of the 31P spectra. We furthermore verified that for each individual sample that indeed quantitative spectra were gained. Therefore, a second measurement with a delay time of 40 s was performed. Only if peak intensities after applying 40 and 60 s delay were identical, a quantification was performed. It is fair to note that this was the case for all herein investigated complexes. To reveal potential pitfalls, it is worth showing 1H MAS NMR spectra of materials (see Fig. S9 in the ESI†). Peaks of ethanol appear at δ1H = 3.6 and 1.2 ppm and indicate its adsorption on the surface. The peak intensity, and thus the quantity of adsorbed ethanol, varies from sample to sample. However, for a quantification the dry material mass needs to be determined. This makes a treatment in vacuum, to remove adsorbed ethanol prior a mass determination, necessary. The successful ethanol removal can, in reverse, be checked by 1H MAS NMR.In Fig. 2 it was found that the stoichiometry between copper and dppf ligand impacts the amount of finally formed complexes. Because, counter-intuitively, fewer CuI(dppf) complexes (0.03 mmol g−1) were formed after reaction in 1:10 Cu:dppf stoichiometry than in 1:1 stoichiometry (0.22 mmol g−1). Furthermore, a strong and slim peak at δ31P = −15 ppm is present after probing in 1:10 Cu:dppf stoichiometry. This peak belongs to neat dppf, which indicates that previously formed CuI(dppf) complex decomposed over time. To further investigate this decomposition, 31P MAS NMR measurements on the sample (1:10 stoichiometry) were performed over a period of several days (see Fig. 4). Thereby, a decreasing intensity of the peaks associated with the CuI(dppf) complex and in parallel an increasing intensity of the peak of neat dppf at δ31P = −15 ppm is observed. This supports a decomposition of the initially formed CuI(dppf) complexes within the time span of 43 h. Such a decomposition is only observed, if dppf is used in large excess. Conversely, in case of a 1:1 stoichiometry, samples are often stable over days without loss in signal intensity and thus complex decomposition. Of course, this requires storage in a closed, air-free container. The stability of the complex in this case proves that the dppf is stable against oxidation over time and supports that it is only oxidized in presence of CuII cations (vide supra).
 | ||
| Fig. 4
31P MAS NMR after different storage time t on a CuII–[Al]SBA-15 sample after reaction with dppf in 1:10 stoichiometry. The intensity of the peak at −15 ppm increases over time. | ||
Next, optimized formation conditions for a maximized CuI(dppf) complex formation were identified. First the stoichiometry between dppf and CuII ions during reaction was addressed (see Fig. 5a). A 1:1 stoichiometry between CuII and dppf results in the largest complex amount formed. Conversely, more dppf applied during reaction leads to a strong decrease of the number of complexes finally observed. Apart from complex decomposition, proven in Fig. 4, leaching of copper cations could potentially be a reason for a decreased complex amount. The copper-content of the samples after loading was thus cross-checked by ICP-OES (see Fig. 5(b)). After 2 h reaction time in a 1:1 stoichiometry the initial copper content is maintained (0.27 mmol g−1). As, according to 31P MAS NMR spectroscopy, 0.22 mmol g−1 complex was formed, it is calculated that 81% of the initially available copper reacted to CuI(dppf) complexes. This shows that the present copper cations reacted nearly quantitatively to the complex. The 19% discrepancy is rationalized by inaccessible copper cations, not reachable by dppf for a reaction. Potentially, also the space available around these cations inside the SBA-15 pores is insufficient for a CuI(dppf) complex formation. If larger dppf quantities are used for the reaction, a strong decrease of the copper-content of the samples is found by the ICP-OES measurements. This decrease is supported by 31P MAS NMR spectroscopy. Thus, leaching of the copper in higher CuII:dppf stoichiometry is the reason for a lower quantity of cationic CuI(dppf) complexes observed.
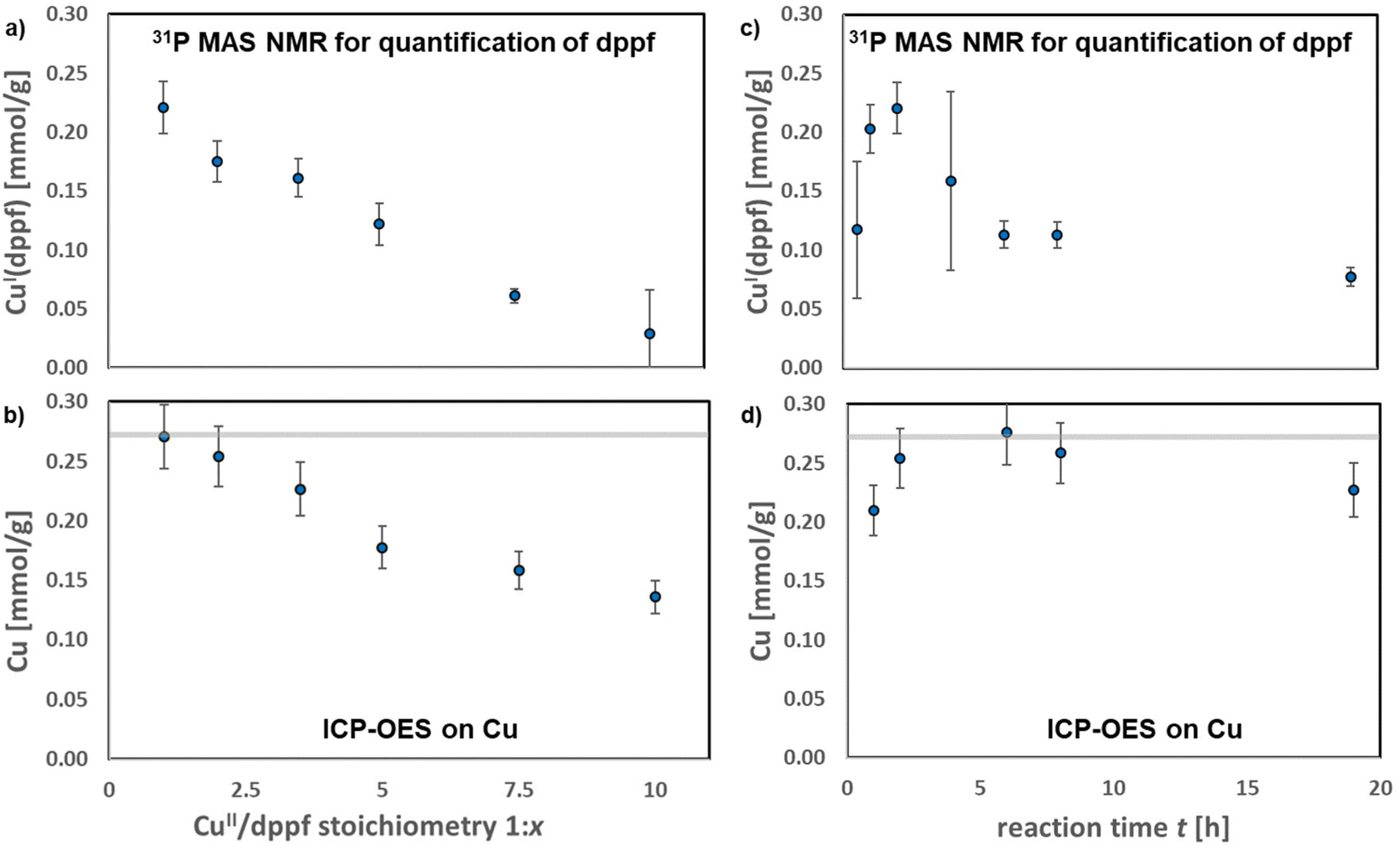 | ||
| Fig. 5 Quantitative screening of CuI(dppf) complex formation conditions. In the first column a varied CuII/dppf stoichiometry after 2 h reaction time was investigated by (a) 31P MAS NMR and (b) by ICP-OES on the Cu-content. On the right hand side, a reaction time screening in a 1:1 CuII:dppf stoichiometry was investigated by (c) 31P MAS NMR and (d) by ICP-OES on the Cu-content. For the evaluation of 31P MAS NMR spectra a minimum error of ±10% was assumed, even if smaller errors were obtained. Likewise, for ICP-OES measurements an error of ±10% is indicated. The initial Cu-content of the samples prior reaction is indicated by a grey bar. | ||
Also the reaction time influences the quantified amount of CuI(dppf) complexes (see Fig. 5(c) and (d)). For a maximized amount of complexes an intermediate reaction time of 1 to 2 h is beneficial. After longer reaction times, the quantity of complexes decreases remarkably. The reason for the lower quantity of CuI(dppf) formed is not leaching, as clarified by the ICP-OES measurements and a maintained Cu-content. As discussed previously for larger dppf quantities (see Fig. 4), the initially formed complexes decompose. Summarizing, in order to maximized the amount of CuI(dppf) complexes, a CuII/dppf stoichiometry of 1:1 and a reaction time of 2 h is suggested. These are the conditions that should be applied if the copper cation accessibility is investigated using dppf.
Now the accessibility of copper cations in various supports is compared using dppf. The until here applied solid, Cu–[Al]SBA-15, contains large mesopores of 6.7 nm diameter. However zeolites, frequently used heterogeneous catalysts, have micropores with a diameter far below 1 nm.2,43 As the reaction with dppf probes the available copper cations almost quantitatively, this reaction is suited to track accessible copper cations on zeolite surfaces. In particular Cu–MCM-22 (MWW structure) is applied herein, as this material was recently shown to be active in the oxidation of methane to methanol.13 The amount of external ion exchange sites was investigated using triphenylphosphine, which was applied on the H-form of the material according to literature.44 In the spectrum in Fig. 6, top, a broad peak at 12 ppm indicates the formation of protonated TPP (<0.01 mmol g−1). Thus, only negligible amounts of ion exchange sites are located on the external surface. The copper cations introduced by ion exchange will thus be located nearly exclusively inside the micropores or pore mouths. There, they should not be accessible for the large dppf ligand. Indeed, the 31P MAS NMR spectrum (see Fig. 6, middle) after reaction with dppf shows only the peak of neat dppf at δ31P = −15 ppm. No peaks of cationic CuI(dppf) complexes appear and thus no such complexes formed. It is concluded that both methods, loading with dppf and TPP probe molecules, lead to similar results. Namely, the copper cations are located exclusively in MCM-22 micropores. Finally, the question arises if dppf is also reacting with Cu-particles, commonly observed side products of a copper exchange into zeolites.46 Thus, Cu@silica synthesized by impregnation was also investigated in the reaction with dppf (see Fig. 6, bottom). This material contains exclusively Cu-particles, as verified by the BSE detector in Fig. 1. After reaction with dppf, the 31P MAS NMR spectrum contains only the slim peak of neat dppf and no peaks associated with formed complexes. Thus, the quantitative evaluation of formed CuI(dppf) complexes is a valuable tool to quantify the accessibility of copper cations. The reaction does not occur if the copper is not accessible and the formation of CuI(dppf) complexes is not observed if the copper is not in cationic position.
 | ||
| Fig. 6 31P MAS NMR spectra after TPP loading on H–MCM-22 (top) and after reaction of CuII–MCM-22 (middle) and CuII@silica (bottom) with dppf. | ||
Conclusion
Herein a new pathway to synthesize CuI(dppf) complexes (dppf = 1,1′-bis(diphenylphosphino)ferrocene) as cations within mesoporous [Al]SBA-15 is introduced. Therefore, ion exchanged copper cations were directly reacted with dppf. These complexes can later be released from the solid by ion exchange to enable their industrial production. 31P MAS NMR proves the identity of CuI(dppf) complexes after conventional ion exchange and after reaction with the solid. This is supported by 1H–31P FSLG HETCOR measurements. CuII cations are in situ reduced to CuI upon oxidation of dppf to the respective phosphine oxide. Maximized amounts of CuI(dppf) are received if a CuII/dppf stoichiometry of 1:1 and a reaction time of 2 h is applied during synthesis. On our Cu–[Al]SBA-15 material 81% of the initially present copper cations formed the complex. Copper leaching can occur if dppf is applied in too high stoichiometry. Also a decomposition of CuI(dppf) complexes to dppf over multiple hours is observed if unfavorable reaction conditions are applied. Cu-particles or copper located inside zeolite micropores does not contribute to the CuI(dppf) complex formation. Thus, the quantitative evaluation of peaks caused by in situ formed CuI(dppf) complexes is a potential tool to evaluate the amount of accessible cationic copper. The method introduced herein is thus not only of potential use for synthesizing complexes, but also for determining the accessibility of supported copper cations.
Experimental
Material preparation
The synthesis of [Al]SBA-15 by synthesis of SBA-15 and subsequent alumination was performed according to literature.4,35 Briefly, Pluronic® P123 (16.0 g, 2.76 mmol) was dissolved in a solution of demineralized water (520 mL) and 37 wt% hydrochloric acid (80 mL) at room temperature. After addition of tetraethyl orthosilicate (36.6 mL, 165 mmol) the solution was stirred at 318 K for 7.5 h and aged at 353 K for 15.5 h under static conditions. For the synthesis of Na–[Al]SBA-15, sodium aluminate (0.12 g) was added to calcined SBA-15 (1.0 g) in water (200 mL) and the reaction mixture was stirred at room temperature for 16 h. Na–[Al]SBA-15 was obtained after calcination at 823 K for 5 h.For the copper ion exchange, 0.45 mmol metal salt (copper(I) chloride for CuI–[Al]SBA-15 or copper(II) acetate for CuII–[Al]SBA-15) was diluted in 100 mL water, then 1 g Na–[Al]SBA-15 was added and stirred at room temperature for 24 h. After ion exchange, Cu–[Al]SBA-15 was thoroughly washed. MCM-22 was synthesized according to literature61 and subsequently calcined at 813 K for 48 h. It was 2-fold ion exchanged with 1 M aqueous NaNO3 solution and washed nitrate-free before copper ion exchange was performed, as described above. Fumed silica A200 was purchased from Evonik Industries AG, Germany. Cu@silica was obtained by wet impregnation (0.045 mmol metal salt, 10 mL demin. Water, 1 g A200) and calcined at 823 K for 5 h. The reaction with 1,1′-bis(diphenylphosphino)ferrocene (dppf) was performed by stirring a reaction mixture of Cu–[Al]SBA-15 (0.075 g) and dppf (0.3–3.0 mmol) at room temperature in ethanol or acetonitrile (50 mL) for x h (x = 0.5 to 24 h, if not otherwise stated 2 h). After measurement, the weight of the solid was determined after desorbing solvent molecules (ethanol) for 6 h at 333 K in vacuum. A complete removal of the solvent checked by 1H MAS NMR spectroscopy. The reaction of dppf with Cu@silica and Cu–MCM-22 was performed analogously. Also a solid state approach for reaction between dppf and copper was applied, by mixing the respective Cu-bearing solid with neat dppf in a 1:2 stoichiometry (Cu:dppf) and heating it 2 h at 333 K (heating rate of 1 K min−1) under N2.
Characterization
Chemical analysis was conducted using inductively coupled plasma optical emission spectrometry (ICP-OES) on an IRIS Advantage instrument. The structure of the samples was investigated by X-ray diffraction (XRD). Therefore, a Bruker D8 diffractometer equipped with an X-ray tube for CuKα radiation (λ = 1.5418 Å) was used. The XRD patterns were recorded in a 2θ range of 0.7–5° for mesoporous and 5–55° for microporous samples. Nitrogen physisorption was performed at 77 K using a Quantachrome Autosorb 3B. Prior to measurements, the samples were activated at 623 K for 16 h. The surface of materials was evaluated using the Brunauer–Emmett–Teller (BET) equation. The micropore volume was determined according to the V–t method (deBoer) and the mesopore volume was determined as difference to the total pore volume of p/p0 = 0.99. SEM images using SE and BSE-detectors were recorded on a Tescan Vega3 equipped with an energy dispersive X-ray spectroscopy (EDX) detector using APEX software for evaluation.1H, 27Al, and 31P MAS NMR spectroscopy was performed on a Bruker Avance III 400 WB spectrometer with a magnetic field of 9.4 T. The resonance frequencies were 400.1 MHz (1H), 104.2 MHz (27Al), and 161.9 MHz (31P). 1H and 31P measurements were performed after π/2 excitation, whereas for qualitative 27Al π/8 excitation was used. The samples for the 27Al MAS NMR measurements were in a fully hydrated state. 4 mm rotor spinning rates of 8 kHz were applied, if not stated otherwise. Typical recycle delays were 5 s (qualitative 1H), 20 s (quantitative 1H), 5 s (31P CP), and 0.5 s (27Al). 31P MAS NMR direct excitation measurements were recorded using high-power proton decoupling (HPDEC) with recycle delays of 40 and 60 s, if not stated otherwise. For quantification of 31P MAS NMR spectra, hydrated VPI-5 was used as an external standard, as described elsewhere.621H–31P FSLG HETCOR spectra were collected at 11 kHz spinning rate with a repetition time of 5 s. The indirect (1H) dimension was referenced using the phenyl protons as internal standard.
For a determination of the BAS density, an ammonia loading with 60 mbar ammonia gas (Westfalen, Germany) was performed through a vacuum line. To desorb excess ammonia, a subsequent evacuation at 453 K for 2 h was performed. Activation of the samples before quantitative 1H measurements was done at elevated temperature in vacuum, with a heating rate 1 K min−1 applying 723 K for 12 h. For quantification of 1H MAS NMR spectra a dehydrated zeolite H,Na–Y (35% ammonium exchanged) was used as an external standard. The amount of external ion exchange sites was performed after activating the NH4-form of the respective zeolites and evaluating the triphenylphosphine (TPP) loaded material by quantitative 31P MAS NMR spectroscopy as described elsewhere.44 Briefly, under N2 the material was combined with pre-calculated amounts of TPP, then 0.8–1 mL of dried dichloromethane (DCM) was added, before the mixture was stirred 1 h and the vessel was finally opened under N2 until complete evaporation of the solvent. NMR spectra were evaluated using TopSpin and Dmfit.63
Conflicts of interest
There is nothing to declare.Acknowledgements
The authors want to thank Heike Fingerle for ICP-OES measurements, Faeze Tari, and Nagme Ay for N2-physisorption measurements.References
- P. B. Weisz, Heterogeneous Catalysis, Annu. Rev. Phys. Chem., 1970, 21, 175–196 CrossRef CAS.
- J. Weitkamp, Zeolites and catalysis, Solid State Ionics, 2000, 131, 175–188 CrossRef CAS.
- B. M. Weckhuysen and J. Yu, Recent advances in zeolite chemistry and catalysis, Chem. Soc. Rev., 2015, 44, 7022–7024 RSC.
- V. Meynen, P. Cool and E. F. Vansant, Verified syntheses of mesoporous materials, Microporous Mesoporous Mater., 2009, 125, 170–223 CrossRef CAS.
- V. V. Butova, M. A. Soldatov, A. A. Guda, K. A. Lomachenko and C. Lamberti, Metal-organic frameworks: structure, properties, methods of synthesis and characterization, Russ. Chem. Rev., 2016, 85, 280–307 CrossRef CAS.
- M. Stöcker, Gas phase catalysis by zeolites, Microporous Mesoporous Mater., 2005, 82, 257–292 CrossRef.
- M. Hunger, Br⊘nsted Acid Sites in Zeolites Characterized by Multinuclear Solid-State NMR Spectroscopy, Catal. Rev., 1997, 39, 345–393 CrossRef CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- K. Kvande, S. Prodinger, B. G. Solemsli, S. Bordiga, E. Borfecchia, U. Olsbye, P. Beato and S. Svelle, Cu-loaded zeolites enable the selective activation of ethane to ethylene at low temperatures and pressure, Chem. Commun., 2023, 59, 6052–6055 RSC.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- P. Vanelderen, B. E. R. Snyder, M.-L. Tsai, R. G. Hadt, J. Vancauwenbergh, O. Coussens, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Spectroscopic Definition of the Copper Active Sites in Mordenite: Selective Methane Oxidation, J. Am. Chem. Soc., 2015, 137, 6383–6392 CrossRef CAS PubMed.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, Reaction Conditions of Methane-to-Methanol Conversion Affect the Structure of Active Copper Sites, ACS Catal., 2014, 4, 16–22 CrossRef CAS.
- K. Kvande, M. Mawanga, S. Prodinger, B. G. Solemsli, J. Yang, U. Olsbye, P. Beato, E. A. Blekkan and S. Svelle, Microcalorimetry on Cu-MCM-22 Reveals Structure–Activity Relationships for the Methane-to-Methanol Reaction, Ind. Eng. Chem. Res., 2023, 62, 10939–10950 CrossRef CAS.
- M. B. Park, S. H. Ahn, A. Mansouri, M. Ranocchiari and J. A. van Bokhoven, Comparative Study of Diverse Copper Zeolites for the Conversion of Methane into Methanol, ChemCatChem, 2017, 9, 3705–3713 CrossRef CAS.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Surface Organometallic and Coordination Chemistry toward Single-Site Heterogeneous Catalysts: Strategies, Methods, Structures, and Activities, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- M. K. Samantaray, S. K. Mishra, A. Saidi and J.-M. Basset, Surface organometallic chemistry: A sustainable approach in modern catalysis, J. Organomet. Chem., 2021, 945, 121864 CrossRef CAS.
- R. Augustine, S. Tanielyan, S. Anderson and H. Yang, A new technique for anchoring homogeneous catalysts, Chem. Commun., 1999, 1257–1258, 10.1039/A903205C.
- D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, Heterogenization of Homogeneous Catalysts in Metal–Organic Frameworks via Cation Exchange, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed.
- M. K. Samantaray, E. Pump, A. Bendjeriou-Sedjerari, V. D'Elia, J. D. A. Pelletier, M. Guidotti, R. Psaro and J.-M. Basset, Surface organometallic chemistry in heterogeneous catalysis, Chem. Soc. Rev., 2018, 47, 8403–8437 RSC.
- C. Copéret, Single-Sites and Nanoparticles at Tailored Interfaces Prepared via Surface Organometallic Chemistry from Thermolytic Molecular Precursors, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef PubMed.
- S. R. Docherty and C. Copéret, Deciphering Metal–Oxide and Metal–Metal Interplay via Surface Organometallic Chemistry: A Case Study with CO2 Hydrogenation to Methanol, J. Am. Chem. Soc., 2021, 143, 6767–6780 CrossRef CAS PubMed.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Bridging the Gap between Industrial and Well-Defined Supported Catalysts, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef PubMed.
- S. E. Maier, O. Bunjaku, E. Kaya, M. Dyballa, W. Frey and D. P. Estes, Surface immobilized Cu-1,10-phenanthroline complexes with α-aminophosphonate groups in the 5-position as heterogenous catalysts for efficient atom-transfer radical cyclizations, Dalton Trans., 2023, 52, 8442–8448 RSC.
- C. Rieg, M. Kirchhof, K. Gugeler, A.-K. Beurer, L. Stein, K. Dirnberger, W. Frey, J. R. Bruckner, Y. Traa, J. Kästner, S. Ludwigs, S. Laschat and M. Dyballa, Determination of accessibility and spatial distribution of chiral Rh diene complexes immobilized on SBA-15 via phosphine-based solid-state NMR probe molecules, Catal. Sci. Technol., 2023, 13, 410–425 RSC.
- S. R. Kousik, F. Ziegler, D. Sipp, A. Rodríguez-Camargo, H. Solodenko, W. Gassner, G. Schmitz, B. V. Lotsch, M. R. Buchmeiser, K. Koynov and P. Atanasova, Imaging the Permeability and Passivation Susceptibility of SiO2 Nanosphere-Based Mesoporous Supports for Molecular Heterogeneous Catalysis under Confinement, ACS Appl. Nano Mater., 2022, 5, 14733–14745 CrossRef CAS.
- M. Schnierle, S. Klostermann, E. Kaya, Z. Li, D. Dittmann, C. Rieg, D. P. Estes, J. Kästner, M. R. Ringenberg and M. Dyballa, How Solid Surfaces Control Stability and Interactions of Supported Cationic CuI(dppf) Complexes—A Solid-State NMR Study, Inorg. Chem., 2023, 62, 7283–7295 CrossRef CAS PubMed.
- D. Schweinfurth, N. Büttner, S. Hohloch, N. Deibel, J. Klein and B. Sarkar, Heterobimetallic Cu–dppf (dppf = 1,1′-Bis(diphenylphosphino)ferrocene) Complexes with “Click” Derived Ligands: A Combined Structural, Electrochemical, Spectroelectrochemical, and Theoretical Study, Organometallics, 2013, 32, 5834–5842 CrossRef CAS.
- C. Bravo, M. P. Robalo, F. Marques, A. R. Fernandes, D. A. Sequeira, M. F. M. Piedade, M. H. Garcia, M. J. V. de Brito and T. S. Morais, First heterobimetallic Cu(i)–dppf complexes designed for anticancer applications: synthesis, structural characterization and cytotoxicity, New J. Chem., 2019, 43, 12308–12317 RSC.
- L. Bemi, H. C. Clark, J. A. Davies, C. A. Fyfe and R. E. Wasylishen, Studies of phosphorus(III) ligands and their complexes of nickel(II), palladium(II), and platinum(II) immobilized on insoluble supports by high-resolution solid-state phosphorus-31 NMR using magic-angle spinning techniques, J. Am. Chem. Soc., 1982, 104, 438–445 CrossRef CAS.
- G. A. Bowmaker, J. V. Hanna, R. D. Hart, P. C. Healy, S. P. King, F. Marchetti, C. Pettinari, B. W. Skelton, A. Tabacaru and A. H. White, Mechanochemical and solution synthesis, X-ray structure and IR and 31P solid state NMR spectroscopic studies of copper(i) thiocyanate adducts with bulky monodentate tertiary phosphine ligands, Dalton Trans., 2012, 41, 7513–7525 RSC.
- G. A. Bowmaker, J. V. Hanna, S. P. King, F. Marchetti, C. Pettinari, A. Pizzabiocca, B. W. Skelton, A. N. Sobolev, A. Tăbăcaru and A. H. White, Complexes of Copper(I) Thiocyanate with Monodentate Phosphine and Pyridine Ligands and the P(,N)-Donor Diphenyl(2-pyridyl)phosphine, Eur. J. Inorg. Chem., 2014, 2014, 6104–6116 CrossRef CAS.
- M. Dyballa, Solid-State NMR Probe Molecules for Catalysts and Adsorbents: Concepts, Quantification, Accessibility, and Spatial Distribution, Energy Fuels, 2023, 37, 18517–18559 CrossRef CAS.
- D. S. Zasukhin, I. A. Kasyanov, Y. G. Kolyagin, A. I. Bulygina, K. C. Kharas and I. I. Ivanova, Evaluation of Zeolite Acidity by 31P MAS NMR Spectroscopy of Adsorbed Phosphine Oxides: Quantitative or Not?, ACS Omega, 2022, 7, 12318–12328 CrossRef CAS PubMed.
- A. Zheng, S.-B. Liu and F. Deng, 31P NMR Chemical Shifts of Phosphorus Probes as Reliable and Practical Acidity Scales for Solid and Liquid Catalysts, Chem. Rev., 2017, 117, 12475–12531 CrossRef CAS PubMed.
- Z. Li, M. Benz, C. Rieg, D. Dittmann, A.-K. Beurer, D. Häussermann, B. Arstad and M. Dyballa, The alumination mechanism of porous silica materials and properties of derived ion exchangers and acid catalysts, Mater. Chem. Front., 2021, 5, 4254–4271 RSC.
- V. L. Sushkevich, R. Verel and J. A. van Bokhoven, Pathways of Methane Transformation over Copper-Exchanged Mordenite as Revealed by In Situ NMR and IR Spectroscopy, Angew. Chem., Int. Ed., 2020, 59, 910–918 CrossRef CAS PubMed.
- N. F. Dummer, D. J. Willock, Q. He, M. J. Howard, R. J. Lewis, G. Qi, S. H. Taylor, J. Xu, D. Bethell, C. J. Kiely and G. J. Hutchings, Methane Oxidation to Methanol, Chem. Rev., 2023, 123, 6359–6411 CrossRef CAS PubMed.
- M. Dyballa, K. Thorshaug, D. K. Pappas, E. Borfecchia, K. Kvande, S. Bordiga, G. Berlier, A. Lazzarini, U. Olsbye, P. Beato, S. Svelle and B. Arstad, Zeolite Surface Methoxy Groups as Key Intermediates in the Stepwise Conversion of Methane to Methanol, ChemCatChem, 2019, 11, 5022–5026 CrossRef CAS.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, The Cu-CHA deNOx Catalyst in Action: Temperature-Dependent NH3-Assisted Selective Catalytic Reduction Monitored by Operando XAS and XES, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- A. A. Kolganov, A. A. Gabrienko, S. A. Yashnik, E. A. Pidko and A. G. Stepanov, Nature of the Surface Intermediates Formed from Methane on Cu-ZSM-5 Zeolite: A Combined Solid-State Nuclear Magnetic Resonance and Density Functional Theory Study, J. Phys. Chem. C, 2020, 124, 6242–6252 CrossRef CAS.
- C. Rieg, D. Dittmann, Z. Li, A. Kurtz, I. Lorenz, D. P. Estes, M. Buchmeiser, M. Dyballa and M. Hunger, Noble metal location in porous supports determined by reaction with phosphines, Microporous Mesoporous Mater., 2021, 310, 110594 CrossRef CAS.
- C. Rieg, D. Dittmann, Z. Li, R. Lawitzki, K. Gugeler, S. Maier, G. Schmitz, J. Kastner, D. P. Estes and M. Dyballa, Quantitative Distinction between Noble Metals Located in Mesopores from Those on the External Surface, Chem. – Eur. J., 2021, 27, 17012–17023 CrossRef CAS PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures: https://www.iza-structure.org/databases/, (accessed 08.09., 2023).
- C. Rieg, Z. Li, A. Kurtz, M. Schmidt, D. Dittmann, M. Benz and M. Dyballa, A Method for the Selective Quantification of Brønsted Acid Sites on External Surfaces and in Mesopores of Hierarchical Zeolites, J. Phys. Chem. C, 2021, 125, 515–525 CrossRef CAS.
- Z. Li, D. Dittmann, C. Rieg, M. Benz and M. Dyballa, Confinement and surface sites control methanol adsorbate stability on MFI zeolites, SBA-15, and a silica-supported heteropoly acid, Catal. Sci. Technol., 2022, 12, 2265–2277 RSC.
- M. Dyballa, D. K. Pappas, E. Borfecchia, P. Beato, U. Olsbye, K. P. Lillerud, B. Arstad and S. Svelle, Tuning the material and catalytic properties of SUZ-4 zeolites for the conversion of methanol or methane, Microporous Mesoporous Mater., 2018, 265, 112–122 CrossRef CAS.
- M. Hunger, Multinuclear solid-state NMR studies of acidic and non-acidic hydroxyl protons in zeolites, Solid State Nucl. Magn. Reson., 1996, 6, 1–29 CrossRef CAS PubMed.
- Y. Jiang, J. Huang, W. Dai and M. Hunger, Solid-state nuclear magnetic resonance investigations of the nature, property, and activity of acid sites on solid catalysts, Solid State Nucl. Magn. Reson., 2011, 39, 116–141 CrossRef CAS PubMed.
- W. Yang, Z. Wang, J. Huang and Y. Jiang, Qualitative and Quantitative Analysis of Acid Properties for Solid Acids by Solid-State Nuclear Magnetic Resonance Spectroscopy, J. Phys. Chem. C, 2021, 125, 10179–10197 CrossRef CAS.
- D. Coster, A. L. Blumenfeld and J. J. Fripiat, Lewis Acid Sites and Surface Aluminum in Aluminas and Zeolites: A High-Resolution NMR Study, J. Phys. Chem., 1994, 98, 6201–6211 CrossRef CAS.
- H. Yu, X. Tan, G. M. Bernard, V. V. Terskikh, J. Chen and R. E. Wasylishen, Solid-State 63Cu, 65Cu, and 31P NMR Spectroscopy of Photoluminescent Copper(I) Triazole Phosphine Complexes, J. Phys. Chem. A, 2015, 119, 8279–8293 CrossRef CAS PubMed.
- E. Borfecchia, D. K. Pappas, M. Dyballa, K. A. Lomachenko, C. Negri, M. Signorile and G. Berlier, Evolution of active sites during selective oxidation of methane to methanol over Cu-CHA and Cu-MOR zeolites as monitored by operando XAS, Catal. Today, 2019, 333, 17–27 CrossRef CAS.
- L. Peng, P. J. Chupas and C. P. Grey, Measuring Brønsted Acid Densites in Zeolite HY with Diphosphine Molecules and Solid State NMR Spectroscopy, J. Am. Chem. Soc., 2004, 126, 12254–12255 CrossRef CAS PubMed.
- L. Peng and C. P. Grey, Diphosphine probe molecules and solid-state NMR investigations of proximity between acidic sites in zeolite HY, Microporous Mesoporous Mater., 2008, 116, 277–283 CrossRef CAS.
- D. J. Zalewski, P. J. Chu, P. N. Tutunjian and J. H. Lunsford, The oxidation of trimethylphosphine in zeolite Y: a solid-state NMR study, Langmuir, 1989, 5, 1026–1030 CrossRef CAS.
- C. Rieg, D. Dittmann, Z. Li, A. Kurtz, E. Kaya, S. Peters, B. Kunkel, M. Parlinska-Wojtan, S. Wohlrab, A. M. Abdel-Mageed and M. Dyballa, Introducing a Novel Method for Probing Accessibility, Local Environment, and Spatial Distribution of Oxidative Sites on Solid Catalysts Using Trimethylphosphine, J. Phys. Chem. C, 2022, 126, 13213–13223 CrossRef CAS.
- B. Hu and I. D. Gay, Probing Surface Acidity by 31P Nuclear Magnetic Resonance Spectroscopy of Arylphosphines, Langmuir, 1999, 15, 477–481 CrossRef CAS.
- B. Hu and I. D. Gay, Acid Sites on SiO2–Al2O3 Monolayer Catalysts: 31P NMR Probes of Strength and Accessibility, J. Phys. Chem. B, 2001, 105, 217–219 CrossRef CAS.
- C. Bornes, D. Stosic, C. F. G. C. Geraldes, S. Mintova, J. Rocha and L. Mafra, Elucidating the Nature of the External Acid Sites of ZSM-5 Zeolites Using NMR Probe Molecules, Chem. – Eur. J., 2022, 28, e202201795 CrossRef CAS PubMed.
- G. A. Bowmaker, S. E. Boyd, J. V. Hanna, R. D. Hart, P. C. Healy, B. W. Skelton and A. H. White, Structural and spectroscopic studies on three-coordinate complexes of copper(i) halides with tricyclohexylphosphine, J. Chem. Soc., Dalton Trans., 2002, 2722–2730, 10.1039/B111121N.
- S. Unverricht, M. Hunger, S. Ernst, H. G. Karge and J. Weitkamp, in Studies in Surface Science and Catalysis, ed. J. Weitkamp, H. G. Karge, H. Pfeifer and W. Hölderich, Elsevier, 1994, vol. 84, pp. 37–44 Search PubMed.
- M. Dyballa, C. Rieg, D. Dittmann, Z. Li, M. Buchmeiser, B. Plietker and M. Hunger, Potential of triphenylphosphine as solid-state NMR probe for studying the noble metal distribution on porous supports, Microporous Mesoporous Mater., 2020, 293, 109778 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Modelling one- and two-dimensional solid-state NMR spectra, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt00147h |
| This journal is © The Royal Society of Chemistry 2024 |