
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-based carbon monoxide releasing molecules with promising cytotoxic properties
Ahmed M.
Mansour†
 *a,
Rabaa M.
Khaled†
b,
Giarita
Ferraro
c,
Ola R.
Shehab
b and
Antonello
Merlino
*c
*a,
Rabaa M.
Khaled†
b,
Giarita
Ferraro
c,
Ola R.
Shehab
b and
Antonello
Merlino
*c
aDepartment of Chemistry, United Arab Emirates University, Al-Ain, United Arab Emirates. E-mail: Mansour_am@uaeu.ac.ae; inorganic_am@yahoo.com
bDepartment of Chemistry, Faculty of Science, Cairo University, Gamma Street, 12613, Egypt. E-mail: Olashehab@sci.cu.edu.eg
cDepartment of Chemical Sciences, University of Naples Federico II, Napoli, Italy. E-mail: antonello.merlino@unina.it
First published on 16th May 2024
Abstract
Carbon monoxide, the “silent killer” gas, is increasingly recognised as an important signalling molecule in human physiology, which has beneficial biological properties. A particular way of achieving controlled CO administration is based on the use of biocompatible molecules that only release CO when triggered by internal or external factors. These approaches include the development of pharmacologically effective prodrugs known as CO releasing molecules (CORMs), which can supply biological systems with CO in well-regulated doses. An overview of transition metal-based CORMs with cytotoxic properties is here reported. The mechanisms at the basis of the biological activities of these molecules and their potential therapeutical applications with respect to their stability and CO releasing properties have been discussed. The activation of metal-based CORMs is determined by the type of metal and by the nature and features of the auxiliary ligands, which affect the metal core electronic density and therefore the prodrug resistance towards oxidation and CO release ability. A major role in regulating the cytotoxic properties of these CORMs is played by CO and/or CO-depleted species. However, several mysteries concerning the cytotoxicity of CORMs remain as intriguing questions for scientists.
1. Introduction
Carbon monoxide is a colourless and odourless gas that is often formed as a result of partial oxidation of carbon-based molecules. High concentrations of CO are hazardous to humans as it has affinity to haemoglobin (HbA) 210–250 times > than O2. As a consequence, the resulting carboxyhaemoglobin (COHb) impairs O2 storage and delivery in the body.1,2 CO starts with unselective binding and unequal distribution in the case of the inhalation route causing coma, convulsions, respiratory depression, and even more fatal consequences. In 1949, Sjöstrand discovered that haemoglobin breakdown in vivo generated CO.3 Elevated haem levels, following erythrocyte destruction, enhances endogenous CO generation, as evidenced by an increase in COHb levels.4 Tenhunen and co-workers identified haem degradation via the haem oxygenase (HO) as the main mechanism for endogenous CO generation.5 Enzymatic haem metabolism produces the vast bulk of CO in the human body. HOs (HO-1 and HO-2) catalyse this metabolism, which takes place in the liver and spleen reticuloendothelial system.6 HO-1 produces approximately 16 mL h−1 of CO in the human body. However, the cellular CO amount is in the nanomolar range. Iron-dependent lipid peroxidation7 and cytochrome P-450 self-inactivation8 produce a limited amount of this distinct metabolite.CO is not always harmful to humans. CO works as a signalling molecule in the neural system, and it has been shown to have vasorelaxant,9 and heart protective properties.10 Numerous papers have also demonstrated the significance of the CO activities in the immunological,11 reproductive,12 respiratory,13 gastrointestinal,14 liver,15 and kidney16 systems. The CO deficiency, on the other hand, has been linked to diabetes, sepsis, colitis, and vascular complications.17,18 The development of prodrugs that can deliver CO steadily and measurably may offer a new way to directly administrate this therapeutic gas and tackle the drawback of unselective binding and distribution. It has been, and still is, difficult for pharmaceutical chemists to come up with safe, workable methods for administering therapeutic doses of CO. Among the methods used to supply potentially effective doses of carbon monoxide to specific organs and tissues, the creation of pharmacologically effective prodrugs known as CO releasing molecules (CORMs) is particularly interesting. Several CORMs have been synthetized and studied in the last years.19 These molecules can distribute CO into biological systems in a secure and controlled manner. CORMs can be generally classified in five groups: boroncarboxylates,19 oxalates,20 α,α-dialkyl-aldehydes, silacarboxylates,21 and metal carbonyl complexes (MCCs). Metal-free CO donors could offer advantages when compared to MCCs,22,23 but their application is in part limited by the conditions needed for CO release or by the low kinetics of gas release.24,25 The structures, CO releasing activity and some biological properties of CORMs have been critically discussed in previous reviews.26–28 MCCs possess antimicrobial properties,20,21,29 and bactericidal activity against different species including Neisseria gonorrhoeae,30Pseudomonas aeruginosa,31Helicobacter pylori,32 and Salmonella enterica serova typhimurium.33 Their role as modulators of inflammation in multiple pathological conditions is well documented. In addition, they contribute to the balance of the redox equilibrium acting as antioxidant molecules. Being reservoirs of CO, a known neurotransmitter, MCCs are involved in neuroprotection and neuronal differentiation processes.34 These properties have been also summarized, together with CORM carrier conjugate systems.35 However, a comprehensive examination of the cytotoxic properties of MCCs is missing. In this respect, it should be underlined that not all the MCCs reported in the literature were examined for cytotoxic properties. Here, we focused our attention on MCCs. In particular, an overview of the anticancer features of metal-based CORMs is reported,35 with the intention of defining the mechanism of action of these potential drugs and discussing their potential therapeutical applications in relation to their biocompatibility and CO releasing properties. The review begins with a brief overview of early-generation CORMs, mechanisms of CO release, CO detection techniques, and it continues with the discussion of the anticancer properties of transition metal-based CORMs, including photoinduced CORMs. The discussion focusses on many cancer hallmarks, including proliferation, apoptosis, angiogenesis, inflammation, etc.
2. Metal-based CORMs
MCCs are able to directly release CO upon internal or external activation. To act as a CORM, a MCC needs to be readily soluble in water and ideally sufficiently stable when held at room temperature (r.t.). Furthermore, it should survive in the blood, remaining active up to the desired targets and should produce only non-toxic fragments after the CO release.24 The properties of the auxiliary ligands in MCCs influence the metal core electron density (e.d.) and hence its stability toward oxidation. Also, the strength of the metal–ligand bonds could stabilize the metal ligand field, slowing down the possible ligand exchange reactions or, oppositely, speeding up CO substitution. As a result, the structure of the first coordination sphere is critical for tuning the chemistry and stability of a certain MCC to withstand the plasma proteins, react to a precise trigger, and develop a certain CO release profile.25,36 The first transition MCCs were examined by Motterlini's group.37 They showed that when Fe(CO)5 and Mn2(CO)10 (denominated CORM-1) (Fig. 1) are exposed to a cold light, a CO release is promoted and that these simple metal carbonyls can deliver controlled quantities of CO causing potent vasorelaxant effects in rat aortic rings pre-contracted with phenylephrine, similarly to endogenous CO.37 Afterward, the first-generation CORMs were quickly replaced by Ru(II) carbonyl complexes due to their low bioavailability as well as the necessity for the photoactivation to produce substantial CO release. The first Ru(II) based complex to be investigated as CORM is CORM-2 ([RuCl2(CO)3]2, Fig. 1). All the previously mentioned CORMs have poor solubility in water. Then, the scientists sought out MCCs with glycinate as an auxiliary ligand, such as [Ru(CO)3Cl(glycinate)], namely CORM-3 (Fig. 1). The CO release from this molecule is prompted by solvent-assisted ligand exchange. When administered into the body, CORMs are exposed to high concentrations of biomolecules, which promote ligand exchange reactions that result in the spontaneous release of CO. The half-life, t1/2, value of CORM-3, for example, is 98 h at ambient temperature in water; yet, this value decreases to 3.6 min in human plasma, when it encounters glutathione and other biomolecules. Since the distribution of such CORMs in tissues largely depends on both t1/2 value in a specific medium and the required time to reach their desired target, this could have an adverse effect on the ability to control site-specific delivery.38 Several triggers were utilized to promote the CO release including changes in pH,39 oxidation state,40 and temperature.41 In addition, the use of enzymes that cleave bonds at the ligand periphery in enzyme-triggered CORMs has been also explored.42 Furthermore, it is now simple to control the time, location, and dose of CO release using light. The essential characteristics of the well-known classes of CORMs are outlined in the next sections.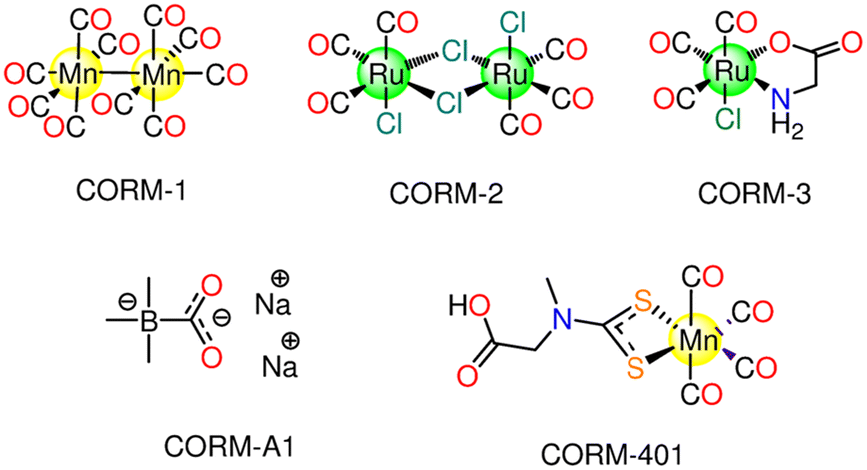 | ||
| Fig. 1 First-generation CORMs: CORM-1, CORM-2, CORM-3 and CORM-401. The structure of the non-metallic CORM-A1 is also reported. | ||
3. CO release mechanisms
3.1. Thermally activated CORMs
The ligand exchange reaction with the medium is one of the mechanisms that promote the CO release from CORMs. For example, CORM-2 and CORM-3 are able to thermally release CO via the ligand exchange mechanism.37,43 Several research groups presented a series of iron(0) CORMs featuring norbornadiene ligands that thermally released CO via the loss of norbornadiene and/or substitution with solvent molecules.44 Motterlini and co-workers proposed the water-soluble non-metallic CORM-A1 (Fig. 1). CORM-A1 is one of the most utilised CORMs in CO biology research because of its commercial availability, high water solubility, and fast CO release kinetics (t1/2 = 2–21 min in phosphate buffered saline, PBS, solutions at pH 5.5 and 7.4 at 37 °C). The CO release mechanism of CORM-A1 is based on a protonation-induced decomposition, which leads to a spontaneous CO liberation consequent to the generation of an unstable borane carbonyl intermediate (Scheme 1).45,46 The release mechanism was firstly proposed by Motterlini in 2005; he measured the CO released by the means of the myoglobin (Mb) assay.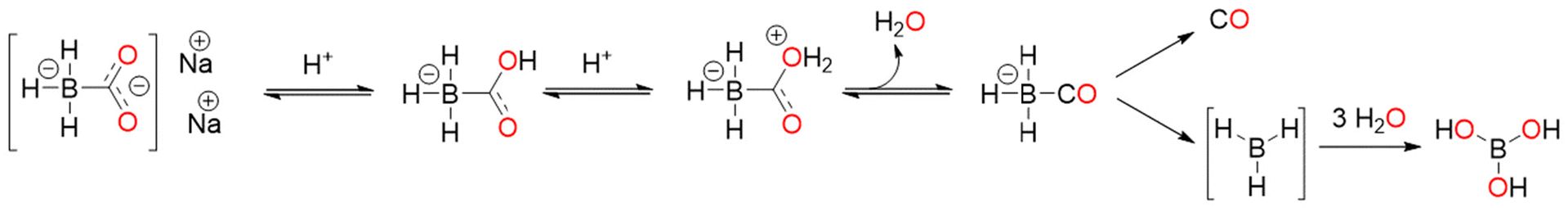 | ||
| Scheme 1 Possible CO release mechanism of CORM-A1. | ||
In 2016 Klein and coworkers used gas phase FTIR to confirm that the rate of CO release was dependent on the rate of formation of the intermediate, which released CO at a constant rate with a half-life of 33 min, and that the pH of buffered solutions increased the intermediate rate formation.47 This peculiar mechanism, different from the one of other CORMs, allows CO release from CORM-A1 with tunable rate through adjustments of pH and temperature, by changing the rate of protonation and decomposition, respectively. The CO release from this non-metallic compound is strongly influenced by the presence of other molecules. Bauer and coworkers identified NAD+ and NADP+ as accelerators of CO release, while H2O2 seemed to diminish and, when present in high excess, abolish its liberation.19 CORM-A1 induces a gradual, significant dose-dependent vasorelaxation over time in isolated aortic rings. Besides, the in vivo treatment with this compound caused a moderate drop in arterial pressure. The CO-depleted form of CORM-A1 is inactive and does not exhibit the same impact, suggesting that CO is the mediator of the reported effects.45
3.2. Enzyme-triggered CORMs
At first, Schmalz and coworkers developed a novel idea known as “enzyme-triggered CO-releasing molecules (ET-CORMs)”.48 Dienol-Fe(CO)3 compounds, featuring acycloxybutadiene ligands, easily decompose under mildly oxidative environments, which are triggered through enzymatic cleavage of the ester moiety by the intracellular esterases.48,49 The ancillary ligands (cyclohexenone or cyclohexanedione) and the ester group that they include have a significant impact on the CO release activity and, consequently, on their biological activity. On the contrary, the equivalent enones in cyclohexanedione ET-CORM have no effect on the biological behaviour. The mechanism at the basis of the CO release from ET-CORMs typically involves two steps: first, hydrolysis of the ester, then oxidation of the resultant dienol-Fe(CO)3 moiety to release CO, Fe-ions and the associated ligand (Scheme 2).50 The variable biological activity was thought to be a reflection of how easily dienol-Fe(CO)3 intermediates are oxidised.42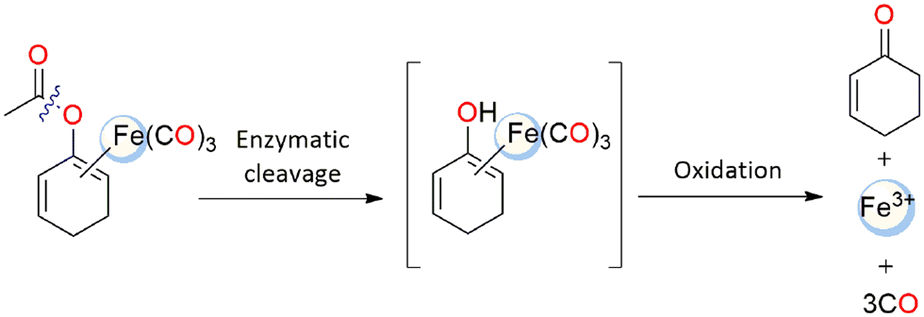 | ||
| Scheme 2 Possible enzyme-triggered CO release mechanism from an ET-CORM. | ||
3.3. Redox
It has been reported that some Mo(0) and Fe(0) carbonyls can release carbon monoxide when exposed to ambient oxygen, changing the oxidation state of Mn+. Na[Mo(CO)3(histidinate)], ALF186, can easily release all its three CO molecules in vitro and in vivo under biological conditions. Decarbonylation of ALF186 occurs after metal oxidation by O2, the event that causes CO delivery and quick diffusion into the blood stream after injection in vivo. In the dark at 37 °C, ALF186 easily releases 75% of its total CO content (2.26 equivalents) in 2 h and one equivalent after about 30 min. Additionally, no CO is emitted under the comparable anaerobic conditions. The complex is not haemolytic and has minimal cytotoxicity.51 Also, CORM-401 (Fig. 1) was found to interact with reactive oxygen species (ROS), which are widely acknowledged as essential mediators in CO signalling actions.523.4. Photoinduced CORMs
If CORM-2 spontaneously releases CO when injected into living tissues, other MCCs have been studied as photochemical CO-releasing agents since they do not release CO spontaneously but only when irradiated. In this respect, it should be underlined that the use of light as an external stimulus is advantageous, since light beam is non-invasive and can be easily changed in terms of energy, frequency, and spatial location. As a result of the fact that some MCCs release CO when lighted, these molecules also can regulate CO delivery. From a biological standpoint, a photoinduced CORM, photoCORM as it was called by Ford et al.,53 and its photoproducts, or iCORM, should be safe. Given that CO-depleted species have to be non-toxic, they should be identified and characterized and their reaction/interaction with the medium, oxygen or biomolecules should be studied. Besides, photoCORMs ought to be stable and soluble in aqueous solution at r.t. or, at the very least, soluble in mixed solvents like aqueous/dimethyl sulfoxide (DMSO) solutions, which are often used for testing the biological activity of metallodrugs. PhotoCORMs must release CO only after stimulation with a proper wavelength. Different excitation wavelengths can be used, depending on the MCC features, i.e., the stability and the strength of the carbonyl-metal bond. It should be desirable the use of red light so that it can be irradiated through skin. The ability of the photoCORM to absorb visible light is vital for photo delivery to biological targets and stability.54 An important limit of photoCORMs is their difficult localization after administration due to the rapid diffusion; the conjugation or functionalization with peptides, polymeric matrices or other supramolecular structures is a common strategy to get over toxicity to untargeted healthy tissues.55Kinetic investigations have been conducted to unveil details of CO release mechanism and possible formation of intermediates during the illumination of photoCORMs, in particular using Mn(I) tricarbonyl complexes.56–59 Quantum chemical calculations, in solution IR and EPR spectroscopy demonstrated the formation of Mn(CO)2 species, upon illumination, which were easily oxidized in subsequent dark processes.56,57 From Mn(I) tricarbonyl complexes, only one CO molecule is released photolytically; the remaining CO molecules require a second dark process. One CO molecule was photochemically released on very short timescales, according to femtosecond transient absorption UV pump/mid-IR probe spectroscopic studies; however, a portion of the excited molecules were shown to undergo geminate recombination.59 It was confirmed by Lynam and Fairlamb60 that illumination of tricarbonyl Mn(I) complex at 400 nm, in acetonitrile, resulted in loss of one CO and in the formation of a triplet dicarbonyl Mn(I) analogue, where the vacant coordination position is occupied by a solvent molecule. The lifetime of the dicarbonyl species was 20 ps. This species does not change during the experiment (800 μs), so any additional CO thermal loss must occur more slowly than 800 μs.
4. First-generation CORMs
Several studies have been done on the first-generation CORMs (CORM-1, CORM-2, and CORM-3), CORM-A1 and CORM-401. Therefore, it is essential to get an insight into the main features of these compounds and their biological impact.4.1. CORM-1
In 2002, Motterlini and co-workers revealed that CORM-1 could release CO upon activation by cold light source. CORM-1 releases CO via dissociation and not by Mn–Mn cleavage.61 Mimicking endogenously generated CO, CORM-1 triggers vasodilation on rat aortic rings contracted with phenylephrine, decreases coronary vasoconstriction ex vivo and acute hypertension in animal models. It was also noted that CORM-1 does not exhibit cytotoxic effects.37In 2004, the effects of 50–600 μM CORM-1, genuine CO and non-adrenergic noncholinergic nerve stimulation on the internal anal sphincter (IAS) were compared. CORM-1 causes relaxation to the rat IAS, in a dose-dependent fashion, and its influence is not affected by neurohumoral antagonists as propranolol, hexamethonium, guanethidine, indomethacin and atropine. Also, the HO inhibitor Tin-protoporphyrin IX (SnPP-IX) used to show non-adrenergic noncholinergic relaxation suppression does not influence the effects of CORM-1. Conversely, the guanylate cyclase inhibitor ODQ decreases the relaxation of IAS caused by CORM-1.62 Following the light stimulation, CORM-1 was found to have concentration-dependent vasodilatation in isolated porcine cerebral arterioles.63,64 The CO released by CORM-1 elevates Ca2+-activated potassium channels (KCa) activity by 4.9 and 3.5 times, respectively, in new born pig cerebral arteriole smooth muscle cells. In such cells, the KCa channels have poor Ca2+ sensitivity, and CO stimulates KCa channels via increasing Ca2+ sensitivity.65 In Sprague-Dawley rats, intrarenal administration of CORM-1 increases renal blood flow, COHb levels, glomerular filtration rate, and subsequent urinary cGMP excretion. The renal protective role of HO is suppressed by the HO-inhibitor Co(III) Protoporphyrin (CoPP). Co-treatment with CORM-1 and CoPP reverses the inhibition effect on HO activity that could cause renal failure. In addition, CORM-1 elevates renal NO levels and nitrates/nitrites excretion, implying that alterations in NO release could contribute to the HO–CO system's renal effects.66 To enhance the water solubility and bioavailability, Schiller and co-workers embedded for example CORM-1 into poly(L-lactide-co-D/L-lactide) fibres to produce nonporous non-woven. The CO release rate depends on frequency since the complex released CO four times faster after irradiation at 365 nm than at 480 nm. The non-woven released 3.4 μmol of CO per mg. The hybrid complex has no cytotoxic activity in the dark against 3T3 mouse fibroblasts but exhibits strong photo-cytotoxicity when irradiated at 365 nm.67 The hydrophobicity of CORM-1 also encouraged the design and synthesis of a drug delivery system able to transport hydrophobic substances, including polymeric micelles and microbubbles (MB). In this frame, CORM-1-containing polymeric microbubbles (CO-MBs) have been developed. This has allowed for the ultrasound and magnetic resonance imaging-driven light-activated CO release. CO-MBs release one CO mole per mole of loaded CORM-1 upon irradiation. The results show that CO-MBs are promising theragnostic agents for reducing hypoxia-related and ROS-mediated damage to cells and tissues in cardiovascular disease.68
4.2. CORM-2
Motterlini and co-workers showed how CORM-2 instantly releases CO into organisms with a reported yield of 0.7 mol of CO per mole of CORM in DMSO/PBS solutions (pH 6.8). This process is favoured by CORM-2 ligands exchange with solvent molecules like DMSO, as reported in Scheme 3. In fact, CORM-2 spontaneously liberates CO in contrast to CORM-1, which needed cold light as a trigger.37 According to 13C NMR spectroscopy, the freshly dissolved CORM-2 does not appear as a dimer in DMSO, since two distinct peaks corresponding to di-carbonyl and tri-carbonyl monomers were observed. It is probable that during the dissolution stage, DMSO behaves as a metal coordinating ligand encouraging the formation of monomers (Scheme 3). The appearance of di-carbonyl monomers may be taken as evidence that CO has been released.37 In DMSO, the CO release from CORM-2 is slow with several reaction routes, that are involved in the formation of mono-, di- and tricarbonyl Ru(II) species.69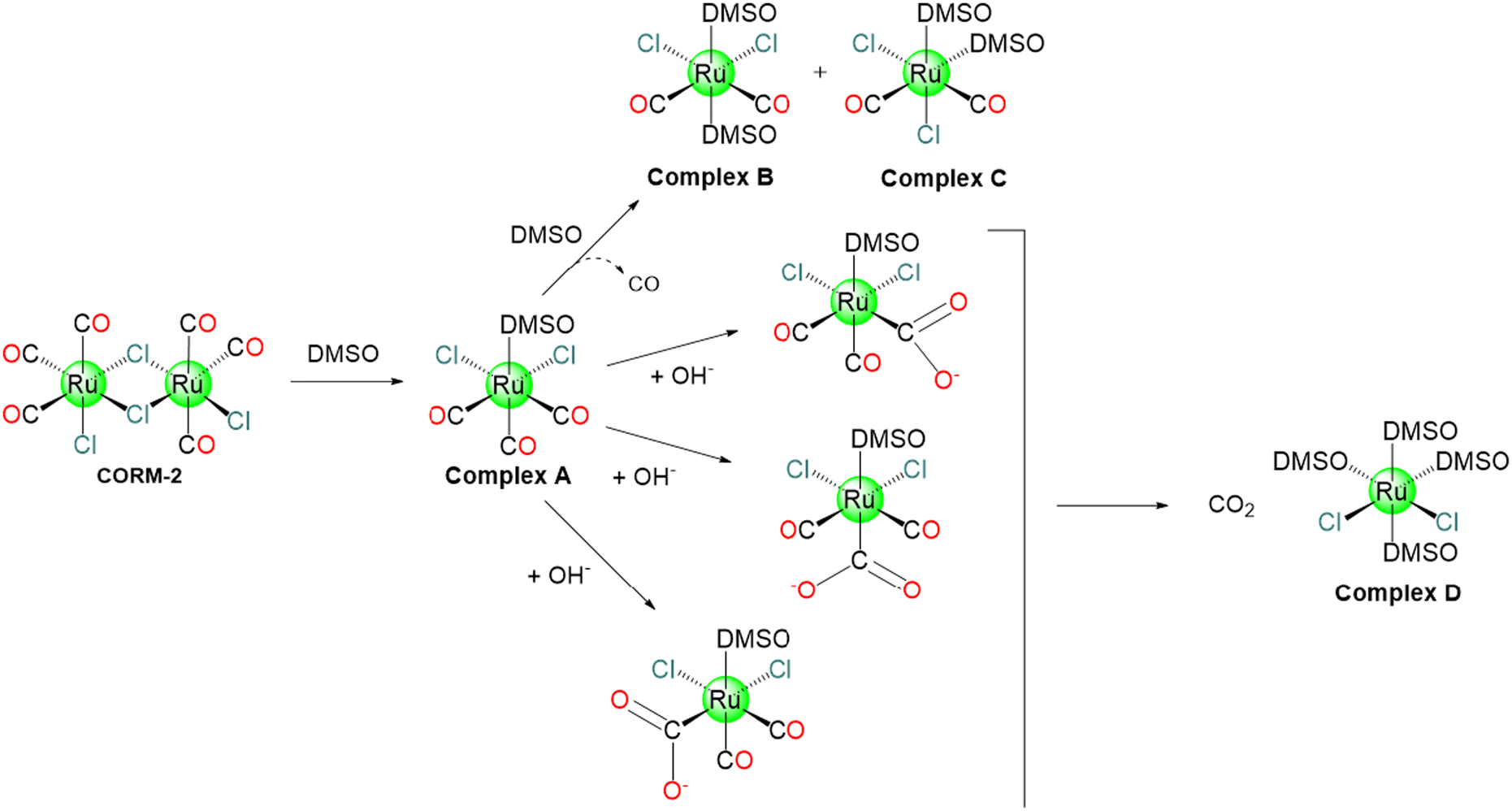 | ||
| Scheme 3 Possible CO release mechanism of CORM-2. | ||
At r.t., the dimeric CORM-2 easily releases CO in murine serum, whereas the monomeric Ru(II) species do not. This indicates that a significant portion of CORM-2 in the presence of DMSO is inactive. Photochemical stimulation causes CO release from the inactive molecules. For this reason, the combination of thermal and photochemical techniques can significantly enhance CO delivery yield.70 CORM-2 interferes with coagulation factors and enhances thrombus formation. In fibrinogen-deficient plasma, the modification of fibrinogen concentration with CORM-2 dramatically increases clot formation velocity (30–50%).71 20 μM of CORM-2 persuaded hydrogen peroxide induces cell damage as determined by lactate dehydrogenase (LDH) release from rat cardiomyocytes. On the other hand, the LDH activity is directly suppressed by 400 μM CORM-2. CORM-2 and its CO depleted form (iCORM-2) as well as the CO gas reduce the cisplatin-stimulated caspase-3 activity in Madin–Darby canine kidney MDCK and HeK cells, implying an anti-apoptotic action. Alternatively, CORM-2 and iCORM-2 cause considerable cell damage, including reduced viability, aberrant cytology, elevated apoptosis and necrosis, cell cycle arrest, and decrease mitochondrial enzyme activity. Low doses of CO, released from CORM-2, display cytoprotective properties. These findings indicate that iCORM-2 is cytotoxic, and that its build up would severely restrict its potential clinical use.72 CORM-2 (10–100 μM), and not iCORM-2, inhibits the lipopolysaccharide-induced inflammation in murine RAW264.7 macrophages in a dose-dependent fashion.73,74 CORM-2 reduces liver inflammation in septic mice,75 LPS (lipopolysaccharide)- or CLP (cecal ligation and puncture)-induced endotoxemia and sepsis,76 and has anti-inflammatory actions on the progression of intestinal ischemia–reperfusion injury (IRI) in rats suffering haemorrhagic shock.77 Also, CORM-2 possesses CO-mediated reduction in leukocyte infiltration in harmed mice intestines via the interference with activation of nuclear factor κB (NF-κB), which is important for cellular proteins expression via a CO-regulated signalling pathway,78 and intercellular adhesion molecule 1 protein expression, hence decreasing endothelial cells’ pro-adhesive character.79 CORM-2 can regulate a variety of genes with roles in intestinal inflammation and tumour development.80
CORM-2 has also other potential medicinal applications, including cardioprotective,81 antimicrobial,82–86 analgesic, and anti-nociceptive actions,87 anti-apoptotic,88 and angiogenic capabilities.89 Also, it possesses CO-mediated protective effect to the kidney against ischemic injury.90 After being dissolved in 10% (v/v) DMSO, CORM-2 was included in a basic cosmetic oil-in-water emulsion to produce a topical lotion. Topical CORM-2 treatment diminishes the chronic acute inflammatory erythema and epidermal hyperplasia following tumour effects in albino Skh-1 hairless mice with UVB-induced photo-carcinogenesis. CORM-2 provides a significant dose-dependent medium suppression of early tumour appearance.91 Additionally, CORM-2 inhibits the abnormal growth of pulmonary artery smooth muscle cells in humans, accompanied with pulmonary hypertension.92
The therapeutic potential and inflammatory action of CORM-2 on a murine orthotopic lung cancer model was assessed in vivo. 80 mice were divided into two sets, control set and orthotopic lung cancer set. The tumour set was either left untreated or treated with DMSO or CORM-2. The body weight of the control group increased over time, whereas it greatly decreased in the tumour group. Administration of CORM-2 significantly reversed this negative effect and increased body weight significantly. It also increased the thymus and spleen indices. Treated mice showed no evident cancer emboli formation. CORM-2 inhibited local inflammation reaction as well as the central intracellular protein synthesis signalling, which in turn restricted abnormal cell proliferation and cancer.93 100 μM of CORM-2 and iCORM-2 reduce the viability of human primate peripheral blood mononuclear cells (PBMCs) and human leukaemia HL-60 cells. Also, CORM-2 and iCORM-2, in the concentration range of 0.01–100 μM, cause DNA damage. CORM-2 significantly decreases H2O2-induced oxidative stress in normal and cancer cells, while iCORM-2 increases the free radical levels of HL-60 cells in the presence of H2O2. Both CORM-2 and iCORM-2 exhibit geno- and cytotoxicity, antioxidant actions and the potential to induce the HO-1 gene. These effects could be caused by both the released CO and iCORM-2.94
Nanocomposites of ferritin (Fr) with CORM-2 have been prepared and tested for their ability to release CO in vitro and in vivo.95 The X-ray structure of this molecule reveals the existence of 72 Ru binding sites at level of Glu, His and Cys residues. The CO release ability of the nanocomposites can be regulated by changing the metal ligands, using single point Fr mutants. It has been shown that the protein cage improves the performance of the CORM, significantly increasing its uptake and t1/2 value for CO release that is 18-fold higher than CORM-3. Besides, the uptake of the nanocomposite is approximately four times higher than that of CORM-3 itself. Indeed, Fr enters the cell via receptor-mediated endocytosis. The nanocomposites also increase nuclear factor kappa B (NF-κB) activation 10-times more than CORM-3.96 Using CORM-2, Ueno and co-workers also prepared RuII carbonyl-incorporated cross-linked hen egg white lysozyme (HEWL) crystals that release CO and significantly increased NF-κB activity.97
Overall, these data strengthen the potential of CORM-2 to act as a cytotoxic molecule able to trigger and interfere with different biological pathways. However, despite this evidence, many studies have highlighted the issues related to the lack of CO release from CORM-2 assessing that it does not reliably and efficiently deliver CO and questioning its role as donor for studying CO biology. Bauer and coworkers summarized most of the relevant data regarding this point.19 These studies finger-point the most used method to follow CO release, the Mb assay, as responsible of an oversight that has hampered the recognition of the effects of Na2S2O4 (sodium dithionite, used in the Mb assays) in facilitating CO release from CORM-2. The first insight was given by McLean in 2012, who stated that CO released from CORM-2 strictly depends on the sulphite species. Interestingly, it was found that in potassium phosphate buffer, CORM-2 did not release CO in the presence of reduced myoglobin alone, but only when a 0.1% Na-dithionite is added to the reaction mixture. In the same way, other sulphite compounds were also found to promote the release of CO.
4.3. CORM-A1
CORM-A1, Na2[H3BCO2], was proposed in 2005 by Motterlini and co-workers as a water-soluble borane-based CO donor. It releases CO, via the protonation process, slower than CORM-3. Dehydration caused by protonation was suggested to produce an unstable intermediate, a borane-carbonyl complex, which releases CO spontaneously. Based on Mb assay, the t1/2 values of 60 μM CORM-A1 in 0.04 M PBS solutions at r.t. were found to be 2.5 min and 21 min at pH = 5.5 and 7.4, respectively.45 Over the time, in isolated aortic rings, CORM-A1 induced a dose-dependent vasorelaxation that was inhibited by guanylate cyclase inhibitor ODQ and greatly increased by guanylate cyclase stimulator YC-1. Like this, pretreatment with YC-1 significantly enhanced the modest drop in mean arterial pressure that resulted after the in vivo injection of CORM-A1 (30 μmol kg−1 i.v.). iCORM-1 did not increase hypotension or vasorelaxation.19,45 According to Chlopicki and co-workers, CORM-A1 modulated platelet bioenergetics to prevent platelet aggregation. The loss of cytosolic NAD+ was found to be the cause of the antiplatelet action of CORM−A1, which in turn prevented glycolysis and mitochondrial respiration.98 In mice, the release of CO from CORM-A1 results in a decrease in renal vascular resistance and an increase in renal blood flow (RBF).99 CORM-A1 possesses strong antioxidant and antiapoptotic effects100 and could be a promising therapy for non-infectious posterior uveitis.1014.4 CORM-3
CORM-3 was proposed in 2003 by Motterlini's group and introduced as a “quick releaser” to generate one mole equivalent of CO (measured as COMb) (t1/2: 4 to 18 min) at pH 7.4 in PBS and biological fluids (cell culture media, human blood plasma).37 The molecule originates from CORM-2 and glycine.43 CORM-3 is stable in water for more than 24 h at 37 °C and at acidic pH. However, because of the lability of the chloride and glycinate ligands, in biological fluids and physiological solutions, it releases CO43 upon replacement of these ligands with higher metal affinity ligands, e.g., glutathione. Such liability also allows CORM-3 to bind blood proteins. As it happens for CORM-2, CORM-3 mostly produces CO2, not CO, under near-physiological conditions in the absence of a strong nucleophile or a reducing agent (Scheme 4).19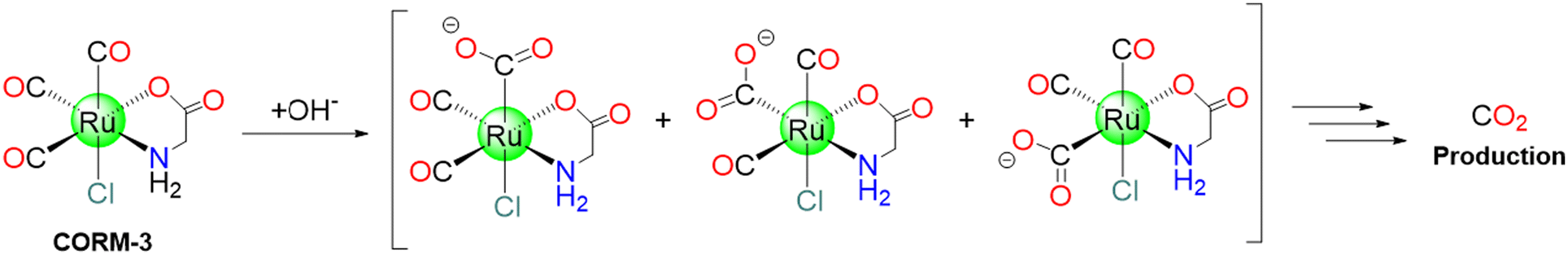 | ||
| Scheme 4 Possible CO2 release mechanism of CORM-3. | ||
CORM-3 exhibits a t1/2 value of 98 h at 37 °C in distilled water, while only 3.6 min in human plasma.102 In solution, CORM-3 exhibits a pH-dependent equilibria. At pH = 3, it is readily produces the [Ru(CO)2(CO2H)Cl(glycinate)]− species. At physiological pH, [Ru(CO)2(CO2H)OH(glycinate)]− and [Ru(CO)2(CO2)Cl(glycinate)]2− coexist in solution.102 In 2011, Santos and coworkers, using gas chromatography (GC) equipped with a thermal conductivity detector (GC-TCD), revealed that when CORM-3 is dissolved in aqueous solutions and closed in a flask, only CO2 can be detected. This can be explained considering this mechanism: one CO is attacked by a water molecule, leading to the formation of CO2. The results imply that plasma proteins, lacking haem, accelerate CORM-3 decomposition by allowing the formation and release of CO2, that is accelerated by covalently binding of cis-RuII(CO)2 moieties to protein residue side chains. The reaction with haem-containing proteins, like Mb, on the other hand, proceeds in a distinct manner because one of the three CO ligands is swiftly delivered to the haem. Also in this case, the remaining cis-RuII(CO)2 moiety binds the protein.103
To probe at molecular level what happens when CORM-3 interacts with proteins, different techniques have been used. Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES), Fourier-transform infrared spectroscopy (FTIR), Liquid-Chromatography Mass Spectrometry (LC-MS)103 and crystallographic studies demonstrated that this molecule is able to bind different human proteins (for example serum albumin (HSA), transferrin and haemoglobin) but also small model proteins, like horse heart Mb and HEWL.104 In this respect, useful information on the structure of the final adduct(s) formed between CORM-3 and proteins have been gained by solving the molecular structure of its HEWL adduct.103 The X-ray structure, refined at 1.67 Å, shows that a Ru(CO)2fragment binds the protein at level of one His (His15) and two Asp (Asp18 and Asp52) side chains (Fig. 2). Thus, the two ancillary ligands and one CO are released upon the protein binding. Water molecules complete the metal coordination sphere.
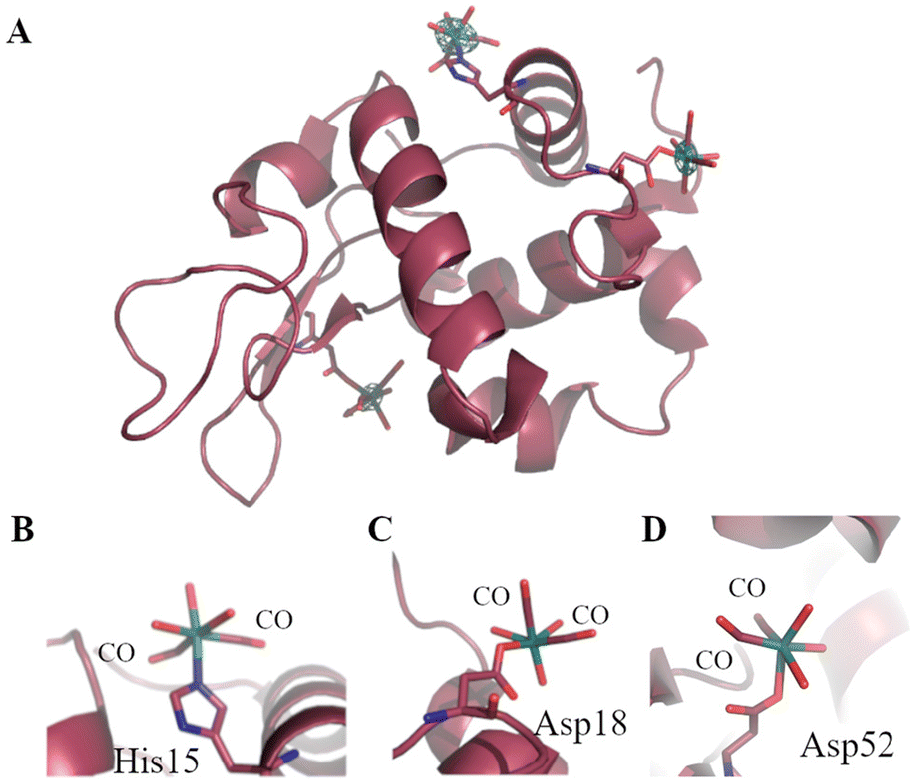 | ||
| Fig. 2 Ribbon model of the adduct formed when CORM-3 reacts with HEWL (A). The Ru(CO)2 fragments bound to His15 (B), Asp18 (C) and Asp52 (D) are highlighted. In panel A, anomalous difference electron density map close to the Ru centres is reported in green at 3.0σ. | ||
These findings indicate that in the CORM-3 protein adducts one CO ligand is lost. In preclinical research, CORM-3 was found to exhibit intriguing biological properties,105–107 such as cardioprotective,61,108 vasodilatory,43,109 antioxidant,110 anti-inflammatory,111–113 antibacterial,31,114–116 anti-ischemic,106,117,118 and anti-apoptotic119–121 activities. A few noteworthy instances of the therapeutic efficacy provided by CORM-3 include the prevention of myocardial infarction and heart failure,107,122,123 kidney protection from cisplatin toxicity124 and the improvement in tissues preservation for transplantation.108 Also, CORM-3 exhibits neuroprotective effect in rats,125 as well as suppression of growth, invasion, and metastasis in tongue squamous cell carcinoma (TSCC) cells.126 CORM-3 (10–100 μM), but not iCORM-3, suppresses LPS-induced inflammation in murine RAW264.7 cells. When CORM-3 is added 3 or 6 h after LPS exposure, it reduces nitrite levels. CORM-3 also significantly lowers the levels of tumour necrosis factor-α (TNF-α), another mediator of inflammatory functions.73 In ischemia-induced acute renal failure (ARF), CORM-3 exhibits CO-mediated protective effect against renal damage.90 The toxicity of CORM-3 is low as cynomolgus monkeys displayed no negative impacts after having a dose of 4 mg kg−1 for one month.127 CORM-3 suppresses both the generation of O2˙− (IC50 = 1.66 μM) and CD11b expression (IC50 = 1.20 μM) in human polymorphonuclear neutrophils (PMNs) and reduces CD54 and CD203 expression as well as histamine release in perivascular mast cells (MCs) with IC50 values of 6.78, 1.18, and 1.15 μM, respectively. CORM-3 has a potent anti-inflammatory effect by suppressing the oxidative burst in PMNs, overexpression of adhesion molecules in PMNs and vascular endothelial cells, histamine release, and MCs overexpression of an activation marker.128 The pharmacologic activities of CORM-3 on porcine aortic endothelial cells (PAEC) and PBMC were evaluated in vitro. High PAEC proliferation was noticed at doses of 300 and 500 μm of CORM-3, but at higher concentration (≥50 μm) it reduced ConA-activated primate lymphocyte proliferation as well as the primate xenogeneic reaction towards pig PBMC. These effects have been shown to be CO dependent.
TNF-α production is considerably suppressed in vivo when several doses of CORM-3 are administered. These findings indicate that CORM-3 possesses anti-inflammatory and immunomodulatory characteristics in primates, which may have clinical implications for allografted and xenografted organs.129 In the concentration range of 1–20 μM, CORM-3, but not iCORM-3, dramatically elevates the mitochondrial oxygen consumption rate. Conversely, 100 μM of CORM-3 inhibit the cytochrome c oxidase and thus the ADP-dependent respiration. In the presence of Mb, the uncoupling action mediated by CORM-3 was blocked. CORM-3, but not iCORM-3, is a regulator of mitochondrial respiration, rising the levels of H2O2 that respiration generates.110 CORM-3 has DNA as biological target, but the mode of action is quite different from cisplatin, since Ru(II) does not generate intramolecular DNA cross-links. It was verified that CORM-3 causes a significant increase in DNA strand breakage in poorly differentiated colon carcinoma RKO cells as evidenced by alkaline comet test.130 A variety of challenges, including the instability of CORM-3 in water, short t1/2 value (see paragraph 4.3) in human plasma, and limited cellular absorption, are impeding its clinical development.36 Due to their instability, poor water solubility for CORM-2, lack of selectivity, and questionable reactivities to biomolecules, the clinical use of CORMs as therapeutic drugs is constrained.24
4.5. CORM-401
CORM-401, [Mn(CO)4{S2CNMe(CH2CO2H)}] (Fig. 1), was introduced by Motterlini and co-workers in 2011.131 CORM-401 releases 3.2 moles of CO molecules with t1/2 = 0.8 min via dissociative reversible mechanism. It is soluble and stable in aqueous media, but CO is rapidly released in the presence of a CO receptor, such as Mb, that hampers the rebinding of CO.131 When CORM-401 is pre-incubated in the presence of Na-dithionite, CO release is detected. The CO release is still detectable when CORM-401 is in buffer alone. As a result, while Na-dithionite is not necessary for CO release from CORM-401, but it does increase the CO release kinetics.132 So, Hb-CO assay is performed as a suitable alternative to the Mb assay. The fact that CORM-401 releases three times more CO than CORM-3 (3.2 equiv. vs. one equivalent) makes it feasible to be administered in much less quantity.131A CO release mechanism by CORM-401 was proposed on the basis of computational data.133 The calculations suggest that CO release occurs via a three-step mechanism, involving: dissociation of an axial CO ligand from Mn; binding of a nucleophile to the vacant position and formation of an intermediate that then dissociates and releases CO (Scheme 5).
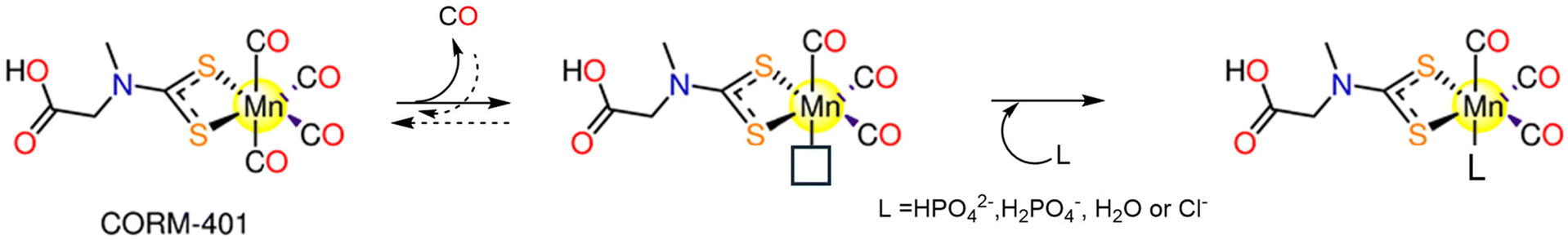 | ||
| Scheme 5 Possible CO release mechanism of CORM-401. | ||
Since its discovery, CORM-401 showed a variety of therapeutic applications, including anti-inflammatory,134–136 antibacterial,136–139 angiogenic,140 anti-carcinogenic,141,142 anti-metastatic, anti-ischemic,143 and cytoprotective properties under the oxidative stress caused by H2O2.144,145 A photodynamic therapy (PDT)-driven, controlled CO release system, was constructed using CORM-401. Exposure to near-IR light triggered fast intracellular CO release from CORM-401 by limiting the H2O2 generated during PDT. In vitro and in vivo, the integration of PDT and CO therapy had strong synergistic anticancer benefits and enhanced therapeutic safety.146,147 For treatment of diabetic wounds, a multifunctional hydrogel dressing containing CORM-401 was designed. The CORM-401 dressing demonstrated blood glucose control, CO dependent anti-oxidative stress, antibacterial and anti-inflammatory activities.148 The cell viability of murine RAW264.7 macrophages was lowered by 25% when 100 μM of CORM-401 was administered, while nitrite generated in response to one μg ml−1 lipopolysaccharide (LPS) treatment decreased by 70%.131 In addition, a two-part metabolic reaction takes place by CO delivered by CORM-401 which are inhibition of glycolysis,148 and uncoupling of mitochondrial respiration.148,149 It was reported that the oral administration of CORM-401 lowers body weight gain and enhances insulin resistance in an obese model.149
Even after considering all the prior successes of first-generation CORMs, there are still unresolved issues that present intriguing challenges for researchers. When it comes to the chemical and physical CORMs features, which molecule has the main role, the CORM or the iCORM? In addition, some reports concluded that, in the absence of a strong nucleophile or reducing agent, CORM-2 and CORM-3 primarily produce CO2 rather than CO, under conditions close to the physiological ones. Furthermore, it is known that such CORMs are extremely reactive towards a variety of biologically relevant molecules and that reaction with an oxidant and/or a nucleophile influences or completely determines the amount of the released CO. The reactivity of such CORMs towards biomolecules has been rarely examined.
5. Detection of carbon monoxide
The CO release rates in CORMs must be measured and quantified using reliable and precise methods. A variety of assays have been developed to measure the release of CO, including colorimetric CO sensing,150 myoglobin assay,132 laser infrared absorption,151 and GC.152 GC-TCD is used as gold standard in the quantification of CO from different sources, providing information also on the amount of other gas species.53 However, this method is not efficient when a continuous monitoring of the reaction is needed.153 In the Mb assay, the formation of CO is detected and quantified spectrophotometrically. The reduced deoxy-Mb interacts with the released CO producing carbonmonoxy-Mb (MbCO). The deoxy-Mb absorption band at 557 nm declines whereas the MbCO absorption bands at 540 and 577 nm rise. Despite myoglobin assay has been and still is widely utilized its efficacy was questioned due to long-term instability, interference with coloured CORMs, turbidity and CO loss from the CORM being reliant on the quantity of Na2S2O4 used as a reducing agent.132,155FTIR analysis was also used to quantify CO release from CORMs,53 because of the distinctive band at 2142 cm−1 of CO gas. It should be noted that FTIR spectra are largely dependent on total gas pressure. For this reason, CO detection in different samples should be carried out at the same total pressure of 1 atm.24 Rimmer and co-workers conjugated FTIR technique with GC equipped with a thermal conductivity detector (GC-TCD).53 This approach can track the CORM breakdown's CO release in real time. This may be carried out immediately in the reaction chamber without gas extraction from the sample. In the case of CORMs that are light-activated, the irradiation can be directly coupled to the setup. Hence, it is possible to measure CO in situ during the irradiation. Moreover, IR absorption spectroscopy may also quantify and identify various gaseous byproducts.153
cis-[Rh2(C6H4PPh2)2(O2CCH3)2](CH3COOH)2 was used as a chromogenic technique for CO detection. The interchange of the axial acetic acid ligands with CO causes colour variation from violet to orange yellow. The probe has remarkable recognition properties, for instance a visible colour shift at concentrations of CO that begin to be harmful (50 ppm). However, the rhodium complex dissolves well only in organic solvents making its utilization for detection and quantification difficult.154
CO can be detected also using fluorescent probes. Some research groups employed a cyclopalladated probe (CO Probe 1, COP-1) to perform palladium-mediated carbonylation process. Through heavy-atom electronic effects, the Pd atom quenches the fluorescence of boron dipyrromethene (BODIPY). Binding of CO to the cyclopalladated probe leads to production of Pd(0) and strongly fluorescent BODIPY dye. The emission increases 10 times in the presence of CO, with a minimum detectable concentration of one μM of CO. The palladium probe is non-toxic and may be used in biological systems.156
Another research group used a haem protein in the fabrication of another fluorescent probe, named COSer. This sensor can bind CO specifically because it contains a circularly permuted yellow fluorescent protein that has been introduced into the regulatory domain of the CO-sensing protein from bacteria. In response to CO, the fluorescence intensity of the probe doubles after 10 min.157 In contrast to COSer, the COP-1 probe amplifies the fluorescence signals more. The irreversible interaction between CO and COP-1 probe adds an advantage to COP-1 compared to the reversible one in the case of COSer. Both COSer and COP-1 serve distinct functions; COSer can be used for real-time detection of CO, while COP-1 is more efficient for detection of low CO concentrations because of its great sensitivity.158 In general, the ability to detect low concentrations of CO quickly and selectively is crucial. So, more specific, sensitive, and quantitative CO detection techniques are required to avoid the drawbacks of the previously mentioned methods.
6. Next generation CORMs
For researchers interested in investigating the potential of using MCCs as CO delivery systems, the complexes by Motterlini and co-workers marked a turning point. They were inspired to continue their trials to develop CORMs based on other metals and co-ligands that could be used to deliver CO clinically. It was rapidly apparent that to develop stable, biocompatible CORMs that could be activated both internally and externally, it was necessary to find specific metal–CO patterns and unique ligand design principles. Metals like Cr, Mn, Co, Ni, W, and Mo interact with CO gas to generate volatile MCCs even when the metals are in their elemental solid state. Metal carbonyls of groups 3–5, 9 and 10, have not been examined as CORMs because of their lability. The most promising metal candidates for CORMs, except for the radioactive element Technetium, are those from Group 6 (Cr and Mo), Group 7 (Mn and Re) and Group 8 (Fe, and Ru), which satisfy the 18-electron rule. Due to their highly regulated substitution chemistry and oxidative stability, Mn(I) carbonyl derivatives can be employed with many auxiliary ligands, including biomolecules. Unfortunately, there is worrying evidence that Mn is toxic in the brain, hence it is highly advised against using drugs that contain Mn.159 Contrarily, ruthenium has been tested on animals in a variety of potential antitumour drugs,160 and NO-scavenging compounds, and has been found to be non-toxic.161 In addition to Mn(I) (Table 1) and Ru(II) ions, metal carbonyls of other elements such as Co(0), Fe(0), and Re(I) have been investigated in the context of CORMs with an emphasis on their CO releasing kinetics and cytotoxic properties (Table 2). In the next section, the ability of these CORMs to release CO under various conditions and their cytotoxic characteristics were discussed.| CORM | Activation wavelength | Solvent used | CO release kinetics | CO equivalents (Myoglobin assay) | Experimental cancer model | Anticancer activity | Ref. |
|---|---|---|---|---|---|---|---|
| a In the dark: IC50 (μM) = 7.4 ± 0.2 (6a), >1 ± 0.1 (6b), 11.4 ± 0.9 (6c), 52 ± 2 (6d) and 9.9 ± 0.7 (6e). b In the dark: IC50 (μM) = 51.93 (7a), 22.89 (7b), 3.22 (7c), 17.32 (7d) and 6.49 (7e). | |||||||
| Manganese(I) CORMs (section 6.1) | |||||||
| 1 | 365 nm | DMSO | 1.96 moles | HT-29 | When irradiated, 1 reduced the cell biomass by 30%. | 162 | |
| 2 | ≥520 nm | CH2Cl2, CH3CN and 20% (v/v) CH3CN/H2O | k CO (min−1): 21.94 ± 0.01 (CH2Cl2) | HeLa and MDA-MB-231 | About 60% reduction in cell viability upon illumination. | 165 | |
| 11.216 ± 0.01 (φ550: 0.48 ± 0.01) (CH3CN) | |||||||
| 4.987 ± 0.01 (CH3CN/H2O) | |||||||
| 3 | ≥520 nm | CH2Cl2 | k CO: 15.28 ± 0.01 min−1 | 165 | |||
| 4 | 10–15 mW visible light | CH2Cl2, CH3CN, 20% (v/v) DMSO/H2O and 40% (v/v) CH3CN/H2O | k CO (min−1): 4.32 ± 0.01 (CH2Cl2) | MDA-MB-231 | 4 decreased the cell viability by 50% upon illumination via CO-triggered apoptosis. | 166 | |
| 1.05 ± 0.01 (CH3CN) | |||||||
| 0.23 ± 0.01 (CH3CN/H2O) | |||||||
| 0.61 ± 0.01 (DMSO/H2O) | |||||||
| 5 | 365 nm | CH2Cl2 DMSO | 1.3 moles | A549, HeLa, HCT-15 and normal PBMCs | Suppression to colon (7.15 ± 0.24 μM), lung (12.5 ± 1.33 μM), and cervical (20.7 ± 0.94 μM) cells upon illumination. Nontoxic to PBMCs. | 167 | |
| 6a–6e | 365 nm | DMSO | t 1/2 (min) = 5.7, 6.4, 4.8, 9.5 and 6.9 in that order. | 6a–6e: 2.4, 1.0, 2.1, 1.4, and 2.0 equiv. | MCF-7 | IC50/UV (μM) = 3.1 ± 0.1, 21 ± 1, 10 ± 1, 2.91 ± 0.07 and 9.7 ± 0.6 in that order.a | 168 |
| 7a–7e | 366 nm | DMSO PBS | t 1/2 (min) = 9.5, 11.4, 13.9, 3.9 and 8.7 in that order. | 7a–7e: 1.4, 1.4, 1.5, 1.7, and 2.2 equiv. | MCF-7 | 7a–7e are cytotoxic under dark and light conditions. | 169 |
| IC50/UV (μM) = 2.91, 12.25, 1.79, 1.43 and <1 in that order.b | |||||||
| 8a–8b | Low-power visible light | PBS, and CH3CN. | k CO (min−1): PBS: 1.54 ± 0.02 (8a), 0.44 ± 0.02 (8b) | MDA-MB-231 | The cell viability is decreased by 50% upon illumination. | 170 | |
| CH3CN: 0.91 ± 0.02 (8a), 0.51 ± 0.02 (8b) | |||||||
| 9 | 365 nm | DMSO | HeLa, A549, and HCT-15 | IC50 (μM): 15.7 ± 0.98 (A549) and 28.7 ± 0.16 (HCT-15). | 171 | ||
| 10 | 365 nm | DMSO | 1.8 equivalents | A549, MDA-MB-231, HeLa, HCT-15 and normal HEK-293 cells | IC50 (μM): 15.4 ± 0.67 (HeLa), 15.8 ± 1.75 (A549) and 14.5 ± 0.97 (HCT-15).171 | 171 and 172 | |
| IC50 (μM): 24.12 ± 1.03 (HeLa), 21.37 ± 1.72 (A549), 13.69 ± 0.91 (HCT-15) and 21.89 ± 0.59 (MDA MB-231). 10 showed low toxicity against normal HEK-293 cells (>50 μM).145 | |||||||
| 11 | Low power visible light | CH3CN and 2% (v/v) CH3CN/PBS | k CO (min−1) = 2% CH3CN/PBS: 0.13 (φ = 0.39 ± 0.03) | HT29 and normal HEK-293 cells | - The viability of HT-29 cells decreased by 47%. | 173 | |
| CH3CN: 10.5 ± 0.02 (φ = 0.35 ± 0.03) | Negligible effect on HEK-293 cells. | ||||||
| 12a–12d | 400–700 nm and 350 nm | Aqueous DMF 10% (v/v) and CH3CN | 12a–12d: kCO (s−1) in CH3CN = 1.92 × 10−3, 2.76 × 10−3, 3.30 × 10−3 and 4.46 × 10−3. | HeLa | Reduced the viability of HeLa cells after 30 min of illumination, with IC50 = 7.29–36.05 μM. | 174 | |
| 13a–13d | 365, 405, 435 nm | CH3CN | t 1/2 (s) = 365 nm: 11.03, 12.47, 12.69 and 44.54 | (equivalents) 365 nm: 2.04, 2.43, 2.17, 2.05 | - HEK-293T and A549. | - The photoCORMs and their iCORMs, exhibited dose-dependent cytotoxicity. Irradiation with blue or purple light showed no influence on the cytotoxicity. | 175 |
| 405 nm: 8.46, 17.20, 46.28 and 59.59 | 405 nm: 1.64, 2.23, 2.01, 1.55 | ||||||
| 435 nm: 92.31, 95.75, 56.81 and 65.90. | 435 nm: 1.08, 1.24, 1.85, 1.32 | ||||||
| k CO (s −1 ) = 365 nm: 2.62 × 10−2, 1.21 × 10−2, 1.84 × 10−2 and 1.95 × 10−2. | |||||||
| 405 nm: 1.39 × 10−2, 1.38 × 10−2, 1.45 × 10−2 and 5.70 × 10−2 | |||||||
| 435 nm: 6.66 × 10−3, 1.81 × 10−2, 4.88 × 10−3 and 6.40 × 10−3 | |||||||
| 14–15 | 15 mW visible light | 2% (v/v) CH3CN aqueous solution and CH2Cl2 | k CO min−1 in CH2Cl2 = 1.03 (φ380 = 0.35 ± 0.02) (14) and 0.66 (φ380 = 0.23 ± 0.02) (15). | HT-29 | Dose-dependent viability suppression upon irradiation, IC50 = 40 and 70 μM for 14 and 15. | 176 | |
| 16a–16e | 365 nm | DMSO | t 1/2 (min) of 16a–16e = 5 (φ: 0.36), 13 (φ: 0.10), 6 (φ: 0.28), 15 (φ: 0.12) and 10 (φ: 0.16), respectively. kCO mol−1 min−1 = 10.8, 0.8, 8.0, 1.9 and 3.4 | Normal HL-7702 and SK-Hep1 | -16b: the lowest cytotoxicity due to its poor solubility. | 177 and 178 | |
| - 16c has high cytotoxicity against SK-Hep1 cells. | |||||||
| 17a–17d | 365 nm | HCT-15, A549, HeLa and normal HEK-293 cells. | - 17b: cytotoxic against A549 but had a little effect on HCT-15 (13.18 ± 2.57 μM) and HeLa (12.05 ± 3.12 μM) cells. It showed cytotoxicity against normal HEK-293 cells. | 179 | |||
| - 17c: cytotoxic against HCT-15 (33.12 ± 5.03 μM), A549 (929.83 ± 3.25 μM) and HeLa (11.41 ± 2.61 μM) cells. Not toxic against HEK-293. | |||||||
| - 17d: less toxic against all tested cell lines | |||||||
| 18a–18g | 365 nm | 18a: 2.65 ± 0.13 18c: (spontaneous release): 3.61 ± 0.089 (equivalents) | HCT-15, A549, HeLa and normal HEK-293 cells | - 18c: highly active even at small doses, IC50 = 6.74 ± 1.77 (A549), 2.54 ± 0.579 (HeLa), and 4.92 ± 0.89 (HCT-15) μM | 180 | ||
| - 18c showed no cytotoxicity against normal HEK-293 cells. | |||||||
| 19 | Low-power visible light | CH3CN | In CH3CN: kCO = 0.13 min−1 | HT29 and normal HEK-293 cells | 10%–48% decrease in viability upon illumination but no cytotoxicity against HEK-293 cells. | 181 | |
| 20a–20d | 420 nm | t 1/2 (s) | (equivalents) 20a, 20b and 20d: 1.45 ± 0.03, 2.3 ± 0.02, 2.49 ± 0.02 | MCF-7, A549-LD, A549-HD, HT29-LD, HT29-HD and 16HBE14o- cells. | - The B12-conjugated photoCORMs were less toxic against MCF-7 cells than their free analogues. | 183 | |
| 20a: 2.3 ± 0.1 | |||||||
| 20b: 3.31 ± 2.30 | B12–20a: 2.26 ± 0.06 | - B12–20b and B12–20c showed moderate cytotoxicity (IC50 = 40 and 17 μM). | |||||
| 20c: 5.65 ± 1.0 | B12–20b: 2.74 ± 0.13 | iCORMs of 20b and B12–20b showed more toxicity in a dose-dependent manner than their 20b and B12–20b analogues. | |||||
| 20d: 3.46 ± 0.1 | B12–20c: 2.20 ± 0.05 | - The iCORM of 20b showed maximum effect at 12.5 μM. | |||||
| B12–20a: 12.9 ± 0.6 | B12–20d: 2.39 ± 0.09 | ||||||
| B12–20b: 13.7 ± 1.5 | |||||||
| B12–20c: 15.8 ± 1.9 | |||||||
| B12–20d: 13.3 ± 1.4 | |||||||
| 21a–21d | 525 and 468 nm | DMSO, CH2Cl2 | At 468 nm in DMSO: 21b: t1/2 = 1.91 ± 0.16 min (kCO = (0.60 ± 0.05) × 10−2 s−1) | 21b: 525 nm: one 468 nm: 3 (equivalents) | HepG2 | - At 525 nm, the complexes showed significant cytotoxicity. | 184 |
| - 21b (IC50 = 7.1 μM) was the most phototoxic complex. | |||||||
| 22a–22d | 525 and 468 nm | DMSO, CH2Cl2 | At 468 nm in DMSO: 22b: t1/2 = 2.27 ± 0.27 min (kCO = (0.50 ± 0.05) × 10−2 s−1) | 22b: 525 nm: one equivalents, 468 nm: 3 equivalents | HepG2 | At 525 nm, the complexes showed significant cytotoxicity. | 184 |
| 23 | 400–700 nm | 10% (v/v) DMF/H2O, DMSO/H2O, and CH3CN | k CO of 23-AuNPs = 33.7 × 10−3 s−1 | A549 | - 23: IC50 = 39.2 μM in red light, and 89 μM under dark conditions. | 185 | |
| - 23-AuNPs caused apoptosis that is dependent upon caspase 3/7 in A549 cells with IC50 = 232 μg mL−1 upon illumination. | |||||||
| 24a–24f | 365 nm | DMSO, CH2Cl2 | HepG2 | - In the dark: IC50 = 18.1 (24a) and 11.8 (24c) μM, while 24b and 24d were inactive. | 186 | ||
| - Upon irradiation: IC50 = 7.9 (24a), 6.6 (24c), 5.7 (24b) and 6.7 (24d) μM. | |||||||
| 24g | 625 nm | PBS | k CO and t1/2 = (1.8 ± 0.5) × 10−2 s−1 and 39 ± 10 s | U87, MCF-7 and HeLa | The targeted/localized CO release improved cytotoxicity through apoptosis towards MCF-7, HeLa and U87 cells. | 187 | |
| (φ625) = (4.30 ± 0.03) × 10−2 | |||||||
| 25a–25b | 468 nm | DMSO and H2O | k CO and t1/2 = In H2O: 2.1 × 10−3 s−1 and 5.4 min (25a) and = 2.2 × 10−3 s−1 and 5.2 min (25b) | In H2O: 0.66 (25a) and 1.31 (25b) equivalents | SW-620, MDA-MB-231 and normal HEK-283T cell. | Both complexes showed inactivity towards the tested cells under dark and illumination circumstances up to 50 μM. | 188 |
| In DMSO: 1.1 × 10−3 s−1 and 10.26 min (25) and = 1.3 × 10−3 s−1 and 8.93 min (25b) | |||||||
| 26–27 | 468 nm | DMSO and 25% (v/v) DMSO/H2O | k CO and t1/2 in DMSO = (26.0 ± 0.18) × 10−3 s−1 and 4.54 ± 0.17 min for 26 = (7.0 ± 0.10) × 10−4 s−1 and 17.28 ± 3.6 min for 27. | MDA-MB-231 and HEK-293T cells | - 26: no cytotoxicity in absence and presence of light. | 189 | |
| - 27: concentration-dependent cytotoxic behaviour (IC50 = 19.62 μM in the dark and 11.43 μM) upon irradiation. The same cytotoxic behaviour was against normal HEK 283T cells. | |||||||
| - The viability of cells cotreated with 30 nM paclitaxel and 10 μM 27 was 27%, suggesting that 27 might increase the cytotoxicity of paclitaxel in the context of resistance. | |||||||
| 28 | 468 nm | DMSO and 20% (v/v) DMSO/H2O | k CO and t1/2 in DMSO = 5.05 × 10−4 s−1 and 21.21 min | THP-1 and BM cells | - Under dark conditions: 28 prevented THP-1 from multiplying whereas exhibiting no effect on BM cells. | 190 | |
| - Upon irradiation, 28 demonstrated THP-1-like potency to that observed in the dark and generated a severe effect on BM cells that could be the cause of the photo-released CO. | |||||||
| MnCO-Ferritin | 456 nm | PBS buffer pH 7.4 | t 1/2 in PBS = 2.5 ± 0.2 min | HEK293 cells | - The quantity of discharged CO from MnCO-Ferritin is modulated by the degree of irradiation | 191 | |
| -The light-activated CO-releasing characteristics of MnCO-Ferritin lead to NF-κB activation | |||||||
| Compound | Activation method | Solvent used | CO release kinetics | CO equivalent | Experimental cancer model | Anticancer activity | Ref. |
|---|---|---|---|---|---|---|---|
| a In myoglobin and aqueous plasma solutions. b Using PLE enzyme. c Using LCR enzyme. d At pH 5.8. e At pH 6.3. f At pH 7.4. | |||||||
| - Molybdenum(0) CORMs (Section 6.2) | |||||||
| 29 | Redox | PEG300/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) :4) |
one equivalents after 0.5 h and 2.26 equivalents after 2 h, in the dark | LLC-PK1, RAW264.7 macrophages and HepG2 cells | - LLC-PK1 and RAW264.7 cells: no toxicity | 51 and 192 | |
| Deoxygenated aqueous media | - HepG2 cells: the rate of survival reduced by 30% at 100 μM | ||||||
| 30 | Reaction with FeCl3 | Cell culture medium | - Normal HEK-293 or NIH3T3 cells | Neither the CORM nor the probable byproduct was toxic. OCORS produced and released CO into the stomach of the animals without causing systemic exposure | 194 and 195 | ||
| Aqueous solution | - Pig models | ||||||
| - Cobalt(0) CORMs (section 6.3) | |||||||
| 31a–31g | Direct release | DMSO/H2O CH3OH/H2O | t 1/2 for 31a–31g = 41.8, 58.8, 71.8, 62.7, 34.8, 71.6 and 47.1 min | - HeLa cells | - IC50 for 31a–31g = 36.20, 73.39, 124.88, 36.89, 42.95, 79.29 and 51.56 μM. | 196 | |
| - Rat modelsa | - With low LD50 values, these complexes showed minimal in vivo toxicity against rats. | ||||||
| 32a–32e | Direct release | - DMSO | t 1/2 for 32a–32e in buffered Mb = 55.1, 53.2, 40.1(54.7)a, 41.8(78.5)a, 60.5, 33.5 and 71.6(143.4)a | - HeLa and HepG2. | - IC50 for 32a–32e against HeLa: 83.24 ± 6.2, 110.21 ± 8.6, 40.61 ± 2.7, 36.20 ± 2.5, 85.85 ± 6.4, 78.45 ± 5.9 and 79.29 ± 4.1 μM. | 197 | |
| - Ethanol | - Mice and rat models | -IC50 for 32a–32e against HepG2: 79.54 ± 6.7, 139.04 ± 9.1, 58.79 ± 3.2, 39.25 ± 1.9, 75.04 ± 4.7, 69.85 ± 3.4 and 68.57 ± 2.4. | |||||
| - Buffered Mb solution | - Zebrafish larvae | - Animal tests showed that 32a and 32f had the smallest LD50 (300–500 mg kg−1). The rest of the complexes were less toxic with 32d and 32g having the highest LD50 values of 2500–5000 mg kg−1 and >5000 mg kg−1 | |||||
| - 32d induced developmental toxicity on zebrafish larvae. At 0.5 and 1.0 μM, it showed no toxicity, but at 5.0, 10, and 20.0 μM it was toxic | |||||||
| 33a–33k | Direct release | - H2O | t 1/2 (min) | - HeLa, A549, HT-29, HepG2, and MCF-7. | - Low activity against tested cancer cells when compared to cisplatin. When compared to 5-FU, 34a and 34b showed superior activity and selectivity to the HT-29 (37.9 and 55.8 μM) and MCF-7 (33.6 and 49.3 μM) cells | 198 | |
| 34a–34e | - Culture medium | 33a–33k: 32.8, 21.8, 30.5, 34.7, 38.9, 42.6, 26.4, 16.8, 30.9, 24.8 and 16.9 | - Myocardial H9c2 cells | - Compared to 33d, 34a had a higher ability to down-regulate COX-2 expression. | |||
| - 0.5% Sodium carboxymethyl cellulose:DMSO (3:1 v/v) |
34a–34d: 25.6, 26.8, 16.3 and 15.8 | - SHR rats | - 33a, 33j, and 34a acted as antioxidants to myocardial H9c2 cells exposed to H2O2. | ||||
| - A concentration-dependent antihypertensive impact on SHR rats | |||||||
| 35a–35e | Direct release | - DMSO | HepG2, MDA-MB-231 and HeLa cells | - IC50 = 4.7–548.6 μM. | 199 | ||
| 36a–36e | - 0.1 M PBS at pH = 7.4 | - 35a, but not its iCORM, exhibited notable selectivity towards HepG2 cells (IC50 = 4.7 ± 0.76 μM). At 50 μM, 35a had a cytotoxic effect against HepG2, MDA-MB-231 and HeLa cells with cell viability of 21.21%, 12.14% and 23.99% | |||||
| 37a–37c | |||||||
| - Iron(II) and Iron(0) CORMs (Section 6.4) | |||||||
| 38 (CORM−F3) | Metal oxidation | DMSO, ethanol | t 1/2 ≈ 55 min | 0.25 M mol−1 | Causes vasorelaxation and prevent inflammation in vitro | 200 | |
| 38 (CORM−F3) 39 (CORM−F7) 40 (CORM−F8) 41 (CORM−F11) | Metal oxidation | DMSO | k CO (nmol min−1) = 38: 0.19, 39: 0.007, 40: 0.041, 41: 0.041 | - Thoracic aortic rings of male adult Sprague-Dawley rats | - 100 μM of 38 induced vascular relaxation in isolated aortic segments and suppressed endotoxin-stimulated inflammation reaction of RAW264.7 macrophages in a concentration-dependent manner | 201 | |
| - Murine RAW264.7 macrophages | - 38 and 40 showed less toxicity against RAW246.7 macrophages | ||||||
| 42a–42k | Metal oxidation | H2O | t 1/2 (min) 42a: one | Murine RAW264.7 macrophages | Only 42b exhibited no cytotoxicity even at 100 μM | 203 | |
| Ethanol | 42i: >3000 | ||||||
| 43 (CORM−S1) | > 400 nm | Aqueous solution | 470 nm: two equivalents | Ca2+-and voltage-activated K+ (BK, Slo1) channels | When exposed to light, a higher outward current was generated with similar variation in membrane potential, giving a measurement for released CO | 204 | |
| 44–48 | Enzyme-triggered | Murine RAW264.7 macrophages | - 45 and 48 showed no toxicity against murine RAW267.4 macrophages up to 100 μM | 48, 205 and 206 | |||
| - 44, 46 and 47 had IC20 values in the ranges 11–28 μM and 14–38 μM | |||||||
| - 47 showed the highest inhibition of NO generation. At 15 and 5 μM, 47 suppressed LPS-induced NO production by up to 68 ± 6% and 33 ± 6% | |||||||
| - 5 μM of 46 led to 30 ± 7% suppression of NO production, while 44 (25 μM) reduced NO generation by only 16 ± 10% | |||||||
| - For up to 50 μM, 45 and 48 did not inhibit NO production | |||||||
| 44–56 | Enzyme-triggered | PBS (0.1 M, pH = 7.4) and DMSO mixture (≈ 17% DMSO) | t 1/2 (min) 44: 43b (128),c45: __,b46: 21,b47: 5,b56: 28,b49: __,b50: 133,b51: 25,b52a: 478,b52b: 51,b53: 10,b54: 6119,c55: 108b | 44: 2.2b (2.4),c45: 0.3,b46: 2.4,b47: 3.0,b56: 2.6,b49: 0.1,b50: 1.0,b51: 3.1,b52a: 1.7,b52b: 2.3,b53: 2.1,b54: 0.6,c55: 1.3b | Murine RAW264.7 macrophages | 47 and 51 showed promising suppression of NO-production. The NO-inhibition was shown to be highly influenced by the enone by-products of monoester-bearing complexes but not in the case of diester-containing ones | 49 |
| 44, 46 and 57 | Enzyme-triggered | - DMSO | HUVEC | - EC50 = 8.2 ± 1.5 and 7.22 ± 1.12 μM for 46 and RAMB@46vs. EC50 = 448.9 ± 50.23 and 457.3 ± 8.23 μM for 44 and RAMB@44 | 42 | ||
| - Used as RAMB (randomly methylated-beta-cyclodextrin) complexes | - The 44-derived suppression of VCAM-1 expression decreased over time, and the 57-derived inhibition seemed to rise. Both 44 and 57 prevented NFκB irrespective of IκBα degeneration. Both ET-CORMs stimulated Nrf-2, which in turn caused HO-1 to be expressed | ||||||
| 44, 46 and 52b | Enzyme-triggered | DMSO | Pre-contracted small rat mesenteric arteries. | - 44 and 46 caused significant dilation, while 52b did not cause any effect | 206 | ||
| - 46 did not cause vasodilation in the case of KCl- pre-treated mesenteric arteries | |||||||
| 58 | Direct release | 58–60: | t 1/2 (min) 61: 18 | One mole of CO/mole of each complex | - Pre-contracted rat aortic smooth muscle cells (A7r5) | - 58–60 caused more significant cytotoxic effects on vascular and inflammatory cells and isolated vessels compared to 61 | 207 |
| 59 | DMSO | - Murine RAW264.7 macrophages | - Against macrophages, the IC50 values of 58–60 and 61 were 9.1, 11.9, 23.6 and 797 μM | ||||
| 60 | 61: H2O | - 58 and its iCORM caused vasorelaxation in isolated aortic rings and reached maximum after 60 min. 59 and 60 led to the same results, however their iCORMs had less vasorelaxation effect | |||||
| 61 | - 58–60 caused a total loss in the cell viability of murine smooth muscle cells | ||||||
| - 61 caused fast dose-dependent vasorelaxation that reached maximum after 10 min | |||||||
| - In contrast to 58, the iCORM of 61 showed minimum vasorelaxation. 61 and not its iCORM greatly reduced LPS-induced NO production without any obvious toxicity up to 100 μM | |||||||
| 44–48 | Enzyme-triggered | 44, 45, 46 and 54: mixture of 0.2 mL DMSO to 1.0 mL PBS | - HUVEC | - 46 and 53 showed cytotoxicity at low concentrations. 46 was toxic only against HUVEC cells, while 53 showed cytotoxicity against HUVEC and PTEC cell lines. 52b and 56 had decreased toxicity | 208 | ||
| 52b | - PTEC | - The cell damage caused by cold preservation was decreased in a concentration-dependent manner by 44. Only ET-CORMs containing 2-cyclohexenone reduced the damage caused by cold preservation. | |||||
| 53–56 | - The cell protection was dramatically diminished when acetate in 44 was replaced with pivalate in 45 | ||||||
| - VCAM-1 expression was significantly suppressed by 44, 52b and 56 and to some degree by 45 | |||||||
| 62a–62d | Enzyme-triggered | DMSO | t 1/2 = 5 h | HUVEC | Only when the CORM and PGA were administered together in an in vitro test, the CO-induced suppression of the inflammation reaction and an elevation of the expression of HO-1 become apparent | 209 | |
| 63–66 | Enzyme-triggered and direct release for 63 and 66 | DMSO | Murine bone marrow-derived DCs | - FumET-CORMs caused substantial suppression of LPS-stimulated pro-inflammatory signaling routes and blockage of downstream (IL)-12 or -23 production. 63–66 can change dendritic cells into anti-inflammatory phenotypes. | 210 | ||
| - 64 and 65 were nontoxic below 25 μM | |||||||
| 44, 46, 62c, 62d, 67 and 68 | Enzyme-triggered | PBS (0.1M; pH7.4)/DMSO (5:1) mixture |
(Equivalents) 46: 2.5 | HUVEC | The utilization of certain membrane associated enzymatic activity could allow tissue-targeted CO administration based on the finding that extra- and intracellular CO release generate anti-inflammatory characteristics | 211 | |
| 67–68: >1.5 within 50 h | |||||||
| 69–70 | Enzyme-triggered | HUVEC | 69 and 70 were toxic in a dose-dependent manner, with 69 being more toxic. While both CORMs induced HO-1, they could not decrease TNF-α-mediated expression of VCAM-1 | 213 | |||
| 71a–71i | Enzyme-triggered and Direct release | PBS (0.1M; pH7.4)/DMSO (5:1) mixture |
(equivalents) - Spontaneous release: | HUVEC | - 71e and 71f showed no toxicity up to 500 μM. 71h and 71i were toxic > 50 μM. | 214 | |
| 71a: 1.8 after 2.5 days | - The anti-inflammatory effect of 71h and 71i was stronger than 71e and 71f. This was noticeable for the suppression of the expression of VCAM-1, but there was not a significant variance in HO-1 inhibition among the two Mito-CORM classes. While 71h and 71i suppressed mitochondrial respiration in both basal and stressful settings, glycolysis increased. 71e and 71f elevated both mitochondrial respiration and glycolysis | ||||||
| 71b and 71c: 2 after 2 days. | |||||||
| 71d: 3 after 10 h. | |||||||
| - PLE-triggered: | |||||||
| 71b and 71c: 0.6 after 5 days | |||||||
| 71d: 3 after 10 h. | |||||||
| 71e and 71f: 3 | |||||||
| 71g :1.5 | |||||||
| - Ruthenium( II ) CORMs (Section 6.5) | |||||||
| 72a–72i | Ligand exchange | DMSO | t 1/2 (min) 72a: 4.9 72b: 3.2 | - L929 murine fibroblast cells. | - Very weak antiproliferative activity against murine L929 fibroblasts (IC50 = 62.69–255.48 mg l−1) | 215 | |
| 73a–73d | Methanol | 72c: 1.1 72d: 2.1 | - Mice and rat models. | - Against mice: LD50 values of 800–1000 mg kg−1 (72a and 72h), 1100–1500 mg kg−1 (72g and 74b) and 150–200 mg kg−1 (73a) | |||
| 74a–74b | 72e: 1.6 72f: 1.0 | - On rats in vivo: a little impact on liver function but did cause physiological harm to liver cells. Detrimental effect on the kidney in both functional and physiological approaches | |||||
| 72g: 10.6 72h: 2.6 | - No accumulation in major tissues or organs and are unable to pass through the blood–brain barrier | ||||||
| 72i: 13.2 | |||||||
| 73a: 10.6 73b: 2.5 | |||||||
| 73c: 2.4 73d: 4.3 | |||||||
| 74a: 15.8 74b: 14.2 | |||||||
| 75a–75n | Aqueous solutions (PBS pH 7.4 or H2O) | 75d: 50 min | Murine RAW264.7 macrophages | - Up to 100 μM, no toxicity. | 216 | ||
| - Reduction in NO generation in a concentration-dependent mechanism. 75k was the most efficient at reducing NO generation | |||||||
| 76 | Decom-position | PBS pH = 7.4 | RAW264.7 cells | - Decrease ROS production during the release of CO and had no bactericidal activity | 217 | ||
| HeLa cells | - No toxicity against RAW264.7 cells | ||||||
| - Anti-inflammatory properties | |||||||
| - HeLa cells have been incubated with 50 μM of 76 before being subjected to COP-1, and considerable rise in intracellular fluorescence was observed | |||||||
| - No increase in the amount of COHb in sheep blood when incubated at 37 °C | |||||||
| 77–79 | - DMSO/PBS | - Balb/c mice | - At 2.5 mg, 78 significantly slowed tumour growth of CT-26 cancer cells in Balb/c mice in vivo. 77 had no effect on the tumor | 218 | |||
| - PBS | - Murine CT-26 cells, CH1/PA-1, A549 and SW480 | - 77 and 78, in vitro, showed toxicity against CH1/PA-1 (77: 56 ± 3, 78: 55 ± 1 μM), A549 (77: 212 ± 24, 78: 16 ± 5 μM) and SW480 (77: 48 ± 4, 78: 44 ± 7 μM) cell lines with IC50 values that were nearly the same | |||||
| - Animal experiments: 20% propylene glycol | |||||||
| - Cytotoxicity: DMSO or DMF (maximum 0.5% v/v) | |||||||
| 80–81 | 350 nm | 0.8% (v/v) DMSO/H2O mixture | A431 and HEK-293 cells | - Increased cytotoxicity against A431 cells at 350 nm | 221 | ||
| - 81 was promptly taken up by A431 and HEK-293 cells and distributed throughout the cytoplasm | |||||||
| 82a–82e | 365 nm | DMSO | MCF-7 | Except for 82d (IC50 = 45.08 ± 3.5 μM), the studied complexes showed no toxicity to MCF-7 cells under dark conditions. By light, the complexes developed cytotoxicity based on the type of the substituent (82a (14.32 ± 1.2 μM) > 82d (23.0 ± 2.8 μM) > 82b (23.2 ± 3.2 μM) > 82c (24.3 ± 3.5 μM) > 82e (26.9 ± 3.1 μM)). | 222 | ||
| 83a–83l | 365 nm | DMSO | t 1/2 (s) at 60 μM | - Murine RAW264.7 macrophages | Although it showed minimum toxicity against murine RAW264.7 macrophages, 83a exhibited anticancer action under illumination. At 50 μM, 83a and 83h showed 12.5% and 6.65% loss in cells activity by illumination. | 223 | |
| Ethanol | 83a: 166 83b: 276 | - HT-29 cells | |||||
| 83c: 249 83d: 189 | |||||||
| 83e: 1209 83f: 632 | |||||||
| 83g: 962 83h: 1096 | |||||||
| 83i: 1450 83j: 966 | |||||||
| 83k: 2699 83l: 2472 | |||||||
| - Rhenium( I ) and Rhenium( II ) CORMs (section 6.6) | |||||||
| 84–90 | pH-dependent | CH3OH | t 1/2 (min) Precursor: 1.0,d 2.5e and 5.7f | One mol mol−1 of complex | Neonatal rat ventricular cardiomyocytes (NRCs) | The precursor complex and 88a, 89a and 89b protected NRCs from ischemia-reperfusion stress in vitro | 39 |
| DMSO | 84–86: no CO release | ||||||
| PBS | 87: 8.4,d 12.3e and 14.0f | ||||||
| 88a: 29.8,d 41.3e and 42.3f | |||||||
| 88b: 19.9,d 27.0e and 40.7f | |||||||
| 89a: 9.7,d 10.2e and 17.2f | |||||||
| 89b: 15.2,d 20.3e and 23.6f | |||||||
| 91a–91b | pH-dependent | H2O | 20 min for 91a and 91b | One mol mol−1 within two h | Cardiomyocytes | Non-toxic, even after CO release, and showed cellular protection against ischemia-reperfusion injury. | 225 |
| DMSO | |||||||
| 92 | 365 and 405 nm | CH3CN | Φ at 365 nm = 0.21 ± 0.01 | At 405 nm: one CO | PPC-1 | - Nontoxic to PPC-1 cells | 226 and 227 |
| Aqueous media | Φ at 405 nm = 0.11 | - Built up in the cytoplasm yet did not pass through the nuclear membrane | |||||
| 93–94 | 360 nm | CH3CN | k CO (min−1) | UV-A light: one CO molecule | MDA-MB-231 | 93 was rapidly internalized by the cancerous cells | 170 |
| PBS | In PBS: 0.32 ± 0.02 (93) and 0.27 ± 0.02 (94) | ||||||
| In CH3CN: 0.30 ± 0.02 (93) | |||||||
| 95 | Low-power UV light | CH3CN | k CO = 0.31 min−1 | MDA-MB-231 | A dose-dependent loss in the viability caused by CO-induced apoptosis upon exposure to light | 228 | |
| MTT assay: 2:3 v/v CH3CN/PBS |
|||||||
| 96a–96e | Low-power UV light (5 mW cm−2) | CH3CN | k CO (min−1) 96a: 0.07 ± 0.02 | 96a: three CO | MDA-MB-231 | The luminescent complexes, with auxiliary ligands of varying lipophilicity, exhibited significant cellular uptake and distributed largely throughout the cytoplasm. 96d displayed moderate nuclear accumulation along with cytosolic distribution. | 229 |
| 96c: no CO release | 96d and 96e: one CO | ||||||
| 96d: 1.59 ± 0.02 | |||||||
| 96e: 0.07 ± 0.02 | |||||||
| 97–98 | 365 nm | DMSO | HepG2 and HeLa | 97 against HepG2: IC50 = 14.2 ± 4.8 μM | 171 | ||
| 98 against HeLa: IC50 = 12.4 ± 2.9 μM | |||||||
6.1. Manganese(I) CORMs
In 2008, the research group of Schatzschneider examined the potential of 1 (Fig. 3) to release CO when irradiated with UV light (λ = 365 nm).162,163 About 1.96 moles of CO were released to the Mb solution. In the dark, 1 was inactive against HT29 cells up to 100 μM, however, the complex acquired activity when irradiated, as it reduced the cell biomass by 30%. The activation wavelength, the solvent used, the CO release kinetics and equivalents, based on the Mb solution, the experimental cancer model, and the cytotoxic properties of all the tested Mn(I) PhotoCORMs are presented in Table 1.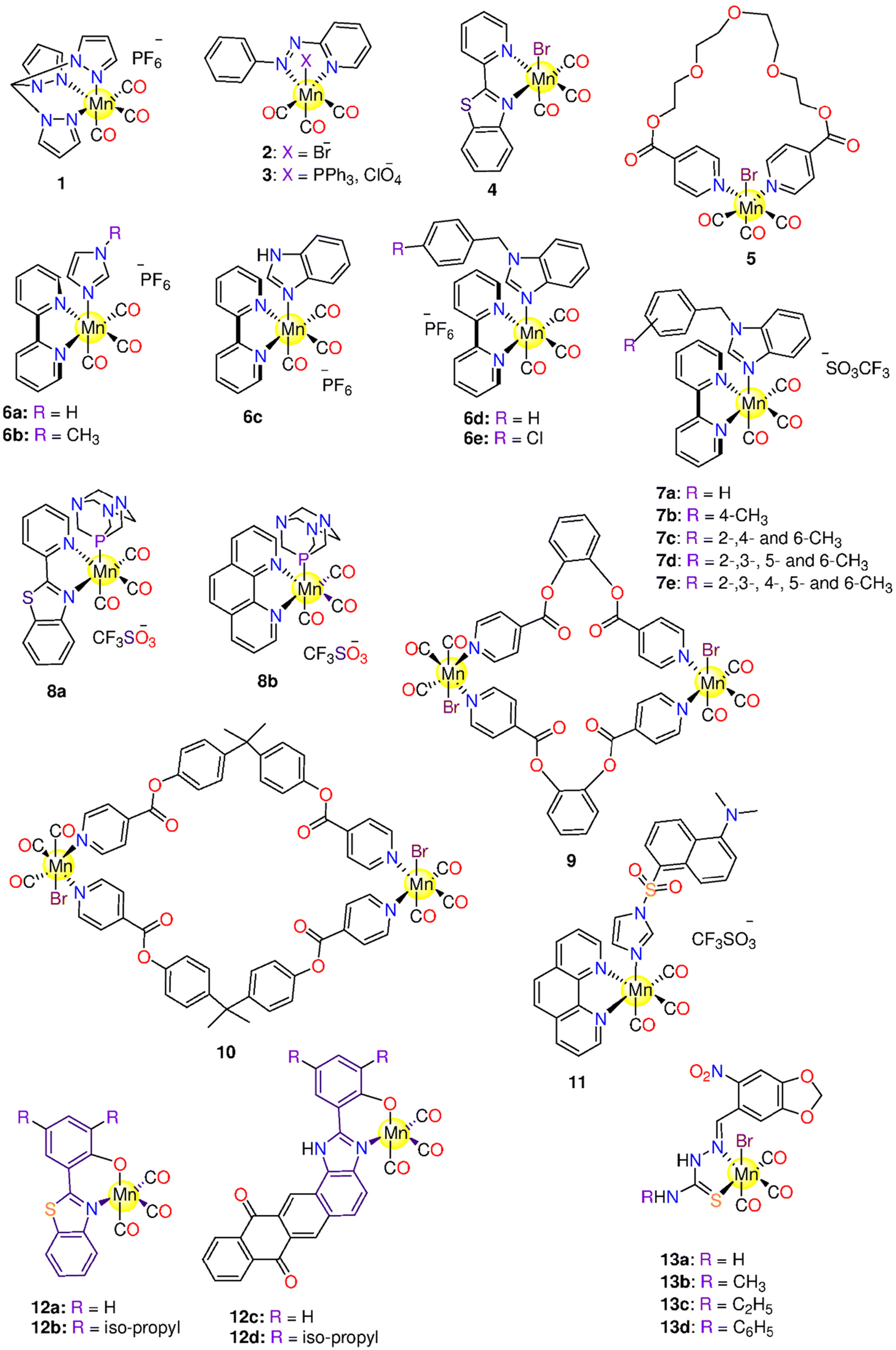 | ||
| Fig. 3 Structures of the Mn(I) photoCORMs 1–13. | ||
Next, Mascharak synthesized photoCORMs 2–4 (Fig. 3), which liberates CO upon illumination with 15 mW visible light.165 These molecules display good stability in some organic solvents under dark conditions. The kCO values of 2–4 in CH2Cl2 are 21.94 ± 0.01, 15.28 ± 0.01 and 4.32 ± 0.01 min−1, respectively. In CH3CN, the kCO value of 4 is 1.05 ± 0.01 min−1. However, a slower CO release was observed in 20% DMSO/H2O and 40% CH3CN/H2O solutions of 4 with kCO values of 0.61 ± 0.01 and 0.23 ± 0.01 min−1, respectively. Complex 2 reduced the viability of HeLa and MDA-MB-231 cell lines by 60% upon illumination.165 The highly fluorescent 2-(2-pyridyl) benzothiazole ligand provides an interesting method for tracking CO distribution within the cells.164 When tested against MBA-MB-231 cells, 4 exhibited 50% decrease in the viability upon illumination via the CO-triggered apoptosis.166
The Mn-based metallo-crown ether 5 (Fig. 3) has good solubility in some organic solvents and releases CO by 365 nm light. Complex 5 was stable in the dark up to 6 h, while upon illumination it releases about 1.3 moles of CO. The complex showed selective suppression to colon, lung, and cervical cancer cells upon the illumination with IC50 values of 7.15 ± 0.24, 12.5 ± 1.33 and 20.7 ± 0.94 μM, respectively. On the other hand, 5 is nontoxic to the normal cells.167
Five 2,2′-bipyridine complexes 6a–6e (Fig. 3), bearing imidazole derivative in the axial position, were examined as CO prodrugs when exposed to 365 nm UV light.168 About 1.0–2.4 CO equivalents were photo-released from 6a–6e with t1/2 of 4.8–9.5 min according to Mb assay. Under the dark conditions, 6a, with imidazole ring, showed the highest cytotoxicity (IC50 = 7.4 ± 0.2 μM) against breast cancer MCF-7 cell line, while the 6b analogue, with methyl-substituted imidazole ring, displayed the lowest cytotoxicity (IC50 > 1 ± 0.1 μM). The presence of a methyl group on an imidazole moiety can result in steric hindrance and impair CO release, or it could change the imidazole nitrogen electronegativity, resulting in decreased reactivity. Complexes 6c–6e showed significant cytotoxicity against MCF-7 cells with IC50 values of 11.4 ± 0.9, 52 ± 2 and 9.9 ± 0.7 μM, respectively. Upon the illumination, 6a, 6c and 6e showed cytotoxicity, however illumination did not seem to induce a touchable enhancement in cytotoxicity. Alternatively, 6b and 6d did not exhibit cytotoxicity upon illumination.168 Afterward, the same research group prepared another series of bipyridine-based Mn photoCORMs (7a–7e) which were also activated by 365 nm light.1697a–7e are stable in the dark for 4 h in DMSO. When dissolved in PBS, these molecules displayed good dark stability over 16 h in the presence of Mb and Na-dithionite. The number of equivalents of released CO increased on going from 7a to 7e (1.4, 1.4, 1.5, 1.7, and 2.2 equivalents, respectively) as the number of methyl groups on the benzyl moiety increased. This could be due to enhanced electron donation via higher methyl groups number, which in turn raised the e.d. on the Mn ion and hence strengthened Mn–CO π-back bonding. However, there is no consistent variation in the t1/2 values based on the number of methyl groups, 9.5 (7a), 11.4 (7b), 13.9 (7c), 3.9 (7d), and 8.7 (7e). When incubated with MCF-7 cells, 7a–7e exhibited cytotoxic effects under both the dark and illumination conditions.
Mascharak and co-workers described the antiproliferative activity of two water-soluble Mn(I) complexes, 8a and 8b (Fig. 3).170 Under dark conditions, for not less than 48 h, 8a and 8b showed good stability in CH3CN, water, and PBS. Via the illumination with broad-band low-power visible light, 8a showed CO release associated with the appearance of fluorescence at about 400 nm due to the de-ligation of 2-(2-pyridyl)benzothiazole ligand. According to Mb assay, the complexes showed kCO values of 1.54 ± 0.02 (8a) and 0.44 ± 0.02 min−1 (8b) in PBS and 0.91 ± 0.02 (8a) and 0.51 ± 0.02 min−1 (8b) in CH3CN. With a concentration of 100 μM, 8a and 8b caused 50% decrease in the cell viability of MDA-MB-231 cells upon illumination.170
Two novel binuclear semi-rigid ester Mn(I)-based carbonyl complexes, 9 and 10 (Fig. 3), were prepared and proved to release CO via illumination at 365 nm.171 The reduced Mb solution of 10 remained stable for 12 h, however, by exposure to the light source, 10 released 1.8 equivalents of CO. Upon illumination, 9 showed cytotoxicity against lung (IC50 = 15.7 ± 0.98 μM) and colon (IC50 = 28.7 ± 0.16 μM) cancer cells only, while 10 showed cytotoxicity against cervical, lung, and colon cancer cell lines with IC50 values of 15.4 ± 0.67, 15.8 ± 1.75 and 14.5 ± 0.97 μM, respectively.
In DMSO, 10 (Fig. 3) showed no CO release in the dark in the presence of Mb and Na-dithionite up to 18 h.172 However, it released CO when exposed to a 365 nm light, according to Mb assay and FTIR studies. Complex 10 showed cytotoxicity toward cervical (HeLa), lung (A549), colon (HCT-15) and breast (MDA MB-231) cancer cell lines with IC50 values of 24.12 ± 1.03, 21.37 ± 1.72, 13.69 ± 0.91 and 21.89 ± 0.59 μM, respectively. With IC50 > 50 μM, 10 showed low toxicity against normal HEK-293 cells. Both iCORM and CO may contribute to the cytotoxicity of 10. The mechanism by which 10 triggered apoptosis was mediated by the production of ROS and consequently the loss of mitochondrial membrane potential.172
To track the photoCORM into the target cells, Mascharak and co-workers introduced the luminescent complex 11 (Fig. 3), which was activated to release CO via illumination with light > 400 nm.173 An equivalent Re(I)-based tricarbonyl complex was prepared but it was activated with UV-light (λ < 315 nm). An equivalent Re(I)-based tricarbonyl complex was prepared but it was activated with UV-light (λ < 315 nm). Complex 11 showed good solubility in CH3CN, CH2Cl2, and CHCl3. In a 2% (v/v) CH3CN/PBS solvent mixture, the kCO value of 11 was 0.13 min−1 (φ = 0.39 ± 0.03), while in CH3CN, a kCO value of 10.5 ± 0.02 min−1 (φ = 0.35 ± 0.03) was observed. In the dark, when human colorectal adenocarcinoma HT-29 cells were co-incubated with 11, no cell damage was seen, even at 100 μM. When illuminated with low power vis light, cells viability decreased by 47%. Interestingly, 11 had a negligible effect on normal HEK-293 cells in the light or under dark conditions. Notably, the low cytotoxicity of the photolyzed solution indicates that CO release may exclusively induce cytotoxicity.173
Musib and co-workers prepared four Mn(I) complexes, 12a–12d (Fig. 3), that liberate CO rapidly via activation with visible light (400–700 nm).174 Photoactivation led to variations in the luminescence features of the complexes, which allowed tracking photo-released CO. 12a–12d were soluble in DMSO, dimethyl formamide (DMF) and CH3CN. In the dark, the solutions of 12a–12d in 10% (DMF/H2O) were stable for up to 72 h. When illuminated at 350 nm, the complexes had less luminescence than the corresponding free ligands. It was seen that the greater the π-accepting character, the greater the trans effect, which resulted in Mn–CO bond lability and so, more CO release. In the dark, the complexes were non-toxic with IC50 > 50 μM. Complexes 12a–12d reduced the cell viability of HeLa after 30 min of the illumination, with IC50 values ranging from 7.29 to 36.05 μM.174
Four Mn(I) photoCORMs, 13a–13d (Fig. 3), containing 6-nitro-piperonal thiosemicarbazone ligands, were found to have both antibacterial and cytotoxic characteristics.175 The photoCORMs were categorized as fast CO releasers. When exposed to a UV/Vis light source (365 (UV), 405 (purple) and 435 (blue) nm), the CO release t1/2 values were between 11 and 95 s. While the purple and blue lights caused the release of around 1.5 and 1.0 equivalents of CO, respectively, the UV-light produced two equivalents of CO. PhotoCORMs 13a–13d and their iCORMs, exhibited dose-dependent cytotoxicity against A549 as well as normal HEK-293T cell lines. Irradiating the cultures with blue or purple light showed no influence on the cytotoxicity of these molecules. The results imply that some CORMs have cytotoxic effects on HEK-293T, but that CO release from these molecules is not the only toxic mechanism.175
As a continuation of their efforts on finding the appropriate Mn(I) photoCORMs with desirable properties for the clinical applications, Mascharak and co-workers synthesized two visible light-activated luminescent photoCORMs, 14 and 15 (Fig. 4), functionalized with diazabutadiene ligand.176 For 24 h, 14 showed good stability in 2% (CH3OH/H2O) mixture as well as CH2Cl2 and CHCl3. The CO release kinetics of 14 and 15, in CH2Cl2, were fast with kCO values of 1.03 and 0.66 min−1 and φ380 = 0.35 ± 0.02 and 0.23 ± 0.02, respectively. In comparison to the hydrophilic complex 14, the lipophilic luminescent complex 15 demonstrated strong membrane permeability, allowing for fast accumulation in the cell and driving CO-triggered cell death more effectively. In HT-29 cells, 14 appeared to accumulate mostly in the cytoplasm due to its hydrophilicity, while 15 accumulated along the cell membrane because of its lipophilicity. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay showed a concentration-dependent inhibition of the viability of HT-29 cells upon the irradiation with IC50 = 40 and 70 μM for 14 and 15, respectively. Interestingly, the ligands and the iCORMs did not show any cytotoxicity in the dark or upon illumination. The activation of Caspase-3/7 confirmed the CO-induced apoptotic cell death.176
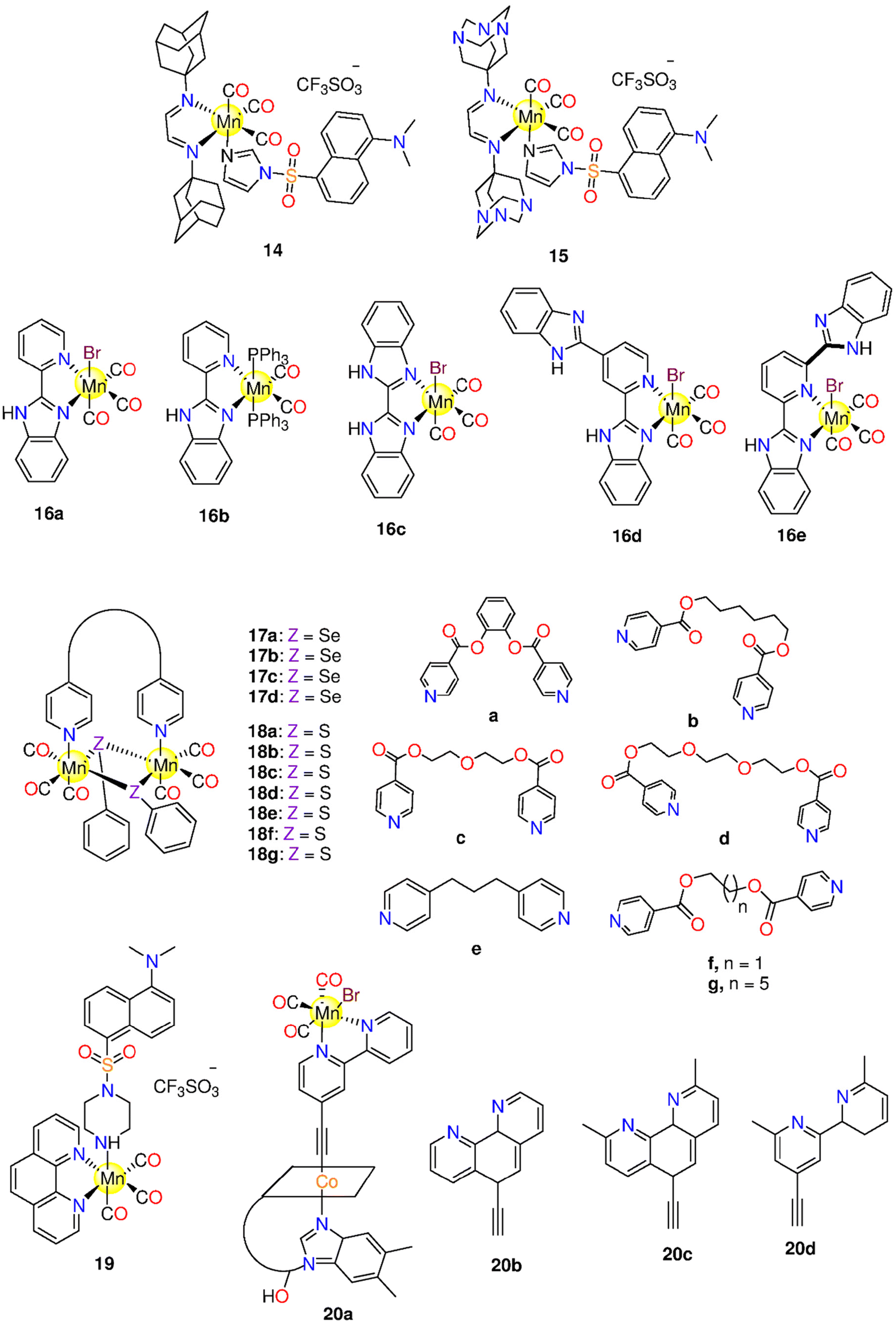 | ||
| Fig. 4 Structures of the Mn(I) photoCORMs 14–20. | ||
Five Mn(I) photoCORMs, 16a–16e (Fig. 4), with benzimidazole ligands, were found to release CO rapidly when illuminated at 365 nm.177 The synthesis and photoactivatable properties of 16a and 16e, upon the exposure to 468 nm LED, were previously reported by our research groups.17816a–16e were soluble in organic solvents, such as tetrahydrofuran (THF), DMSO and CH3CN, however they were insoluble in water. The complexes displayed good stability for 24 h in the dark. By comparing the t1/2 values of the complexes (going from 5 min for 16a to 15 min for 16d), it was observed that extending the degree of conjugation and unsaturation in the ligand framework is helpful for increasing the time of CO release, and so the luminescence intensity of 16a–16e could steadily be increased. The CORMs showed kCO values in the range 1.9 to 10.8 mol−1 min−1 according to the CO sensor data. Since the emission bands of 16a–16e extended to the visible light region, their features of luminescence could satisfy the needs of bioimaging studies. After the incubation of HL-7702 (human normal liver) and SK-Hep1 (hepatic adenocarcinoma) cells with 16a–16e, bright green fluorescence was found. Complex 16c displayed the highest cytotoxicity against SK-Hep1, owing to high lipo-solubility and biocompatibility of the benzimidazolyl moiety. Compound 16b, on the other hand, showed the lowest cytotoxicity due to its restricted solubility.177,178
The binuclear selenolato-bridged Mn(I) metallacycles 17a–17d (Fig. 4) were highly soluble in polar protic and aprotic organic solvents.179 Complex 17c showed instability in the dark and released relatively little amounts of CO as well as liberated its CO molecules upon the illumination at 365 nm. When tested against cancer cell lines, 17c demonstrated toxicity against HCT-15, A549 and HeLa cell lines with IC50 = 33.12 ± 5.03, 29.83 ± 3.25 and 11.41 ± 2.61 μM, respectively. Interestingly, 17c was safe to HEK-293 cells. Complex 17b had a significant cytotoxicity against A549 cancer cells, but it only had little effect on HCT-15 and HeLa cancer cells. It also showed cytotoxicity against normal HEK-293 cells. On the other hand, 17d was less toxic against both normal and cancerous cell lines.179
Similarly, a series of thiolato bridged Mn-based photoCORMs, 18a–18g (Fig. 4), soluble in polar organic solvents, was found to release CO when irradiated at 365 nm.180 When the deoxy-Mb solution of 18a was kept in the dark, no spectral changes were observed. However, when exposed to a 365 nm wavelength a release of about 2.65 ± 0.13 equivalents of CO was found. Alternatively, 18c showed very slow spontaneous CO release (about 3.61 ± 0.089 equivalents) in the dark under the Mb assay conditions. According to MTT assay, 18c was more active even at lower concentrations with IC50 values of 6.74 ± 1.77, 2.54 0.58, and 4.92 ± 0.89 against A549, HeLa and HCT-15 cancer cells, in that order. Excitingly, the same complex was not toxic against HEK-293 cells. The spontaneous ability of 18c to liberate CO in the dark could be the cause of its broad-spectrum cytotoxicity.180
The luminescent photoCORM 19 (Fig. 4) was synthesized to facilitate luminescent tracking in a cellular medium.181 Complex 19 was dark-stable for 24 h in 2% (v/v) (CH3CN/PBS). Myoglobin assay confirmed that, in acetonitrile, 19 released CO upon illumination with kCO value of 0.13 min−1. IR spectroscopy indicated loss of all the CO molecules upon the exhaustive photolysis. Upon the complexation with Mn(I), the pipdansyl ligand emission was reduced. Following photolysis of 19 in solution, the ligand luminescence intensity was totally recovered. Replacing the imidazole linker in 11 with piperazine resulted in a greater luminescence intensity of 19 when compared to 11.173 The strong luminescence of 19 hampers to see the HT-29 cells when their concentration is lower than that of 11.173 The cytotoxicity of 19 against HT-29 cells revealed 10–48% decrease in viability upon illumination, but no significant changes in the absence of light were observed. Complex 19 did not show toxicity against HEK-293 cells even at 100 μM concentration.
In 2020, the CO releasing and cytotoxicity properties of 20a–20d (Fig. 4), bearing ethynyl-α-diimine ligands coupled with vitamin B12, were examined. These photoCORMs were tested on several cancer cells to see whether or not the effect of CO on vitality was cell-type dependent.182 It was observed that the cytotoxicity of 20a–20d was affected by the cell density. The higher the cell confluence, the lower the cytotoxic effect, and vice versa. When the complexes were subjected to 420 nm light source, they quickly released 2.3 equivalents of CO. The type of the α-diimine ligand has no impact on the CO release kinetics. Under dark conditions, the B12-conjugated photoCORMs were less toxic against MCF-7 than their free analogues (<10 μM). Complexes B12–20b and B12–20c showed moderate cytotoxicity against MCF-7 cells with IC50 = 40 and 17 μM, respectively. When the cytotoxicity under illumination and dark conditions were compared, no specific patterns were seen. Complex 20b and its B12 conjugate showed good stability under the physiological conditions. When the iCORMs of 20b and B12–20b were examined, it was surprisingly found that the iCORMs showed more toxicity, in a dose-dependent manner, than their 20b and B12–20b analogues. The iCORM of 20b showed maximum effect at concentration of 12.5 μM. The results indicated that both CO and iCORM might be involved in these photoCORM cytotoxicity.183
In the same year, some of us synthesized two series of visible-light induced photoCORMs, 21a–21d and 22a–22d (Fig. 5), using some N,N-bidentate ligands. In DMSO and CH2Cl2, the complexes were dark-stable. However, when illuminated at 525 nm, these photoCORMs released CO. Both 21b and 22b release one and three CO molecules at 525 and 468 nm, respectively. Upon irradiation at 468 nm, 21b and 22b exhibited t1/2 values of 1.91 ± 0.16 min (kCO = (0.60 ± 0.05) × 10−2 s−1) and 2.27 ± 0.27 min (kCO = (0.50 ± 0.05) × 10−2 s−1), respectively. The compounds did not exhibit cytotoxicity against human hepatocarcinoma HepG2 cells in the dark, but after being exposed to illumination at 525 nm, they showed significant cytotoxicity. The cytotoxicity could be related to the released CO ligands or iCORM, which inhibited the proliferation of the cell line. Complexes 21b and 22b liberated CO upon illumination in the same fashion, but the cytotoxicity of 21b (IC50 = 7.1 μM) was higher than that of 22b. This finding suggested that the iCORM is the primary cause of the acquired cytotoxicity.184
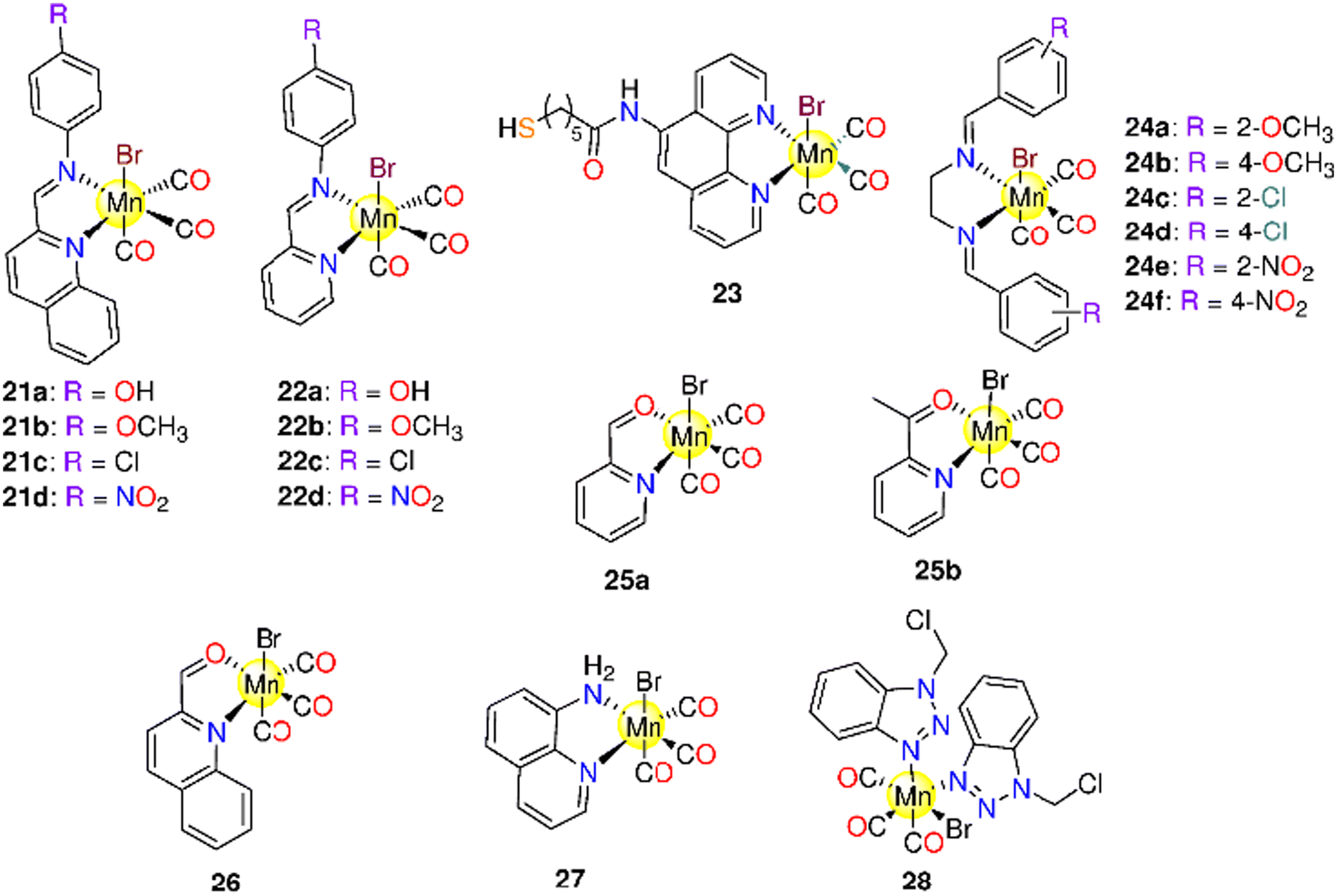 | ||
| Fig. 5 Structures of the Mn(I) photoCORMs 21–28. | ||
Complex 23 (Fig. 5), incorporating 1,10-phenanthroline derivative, was found to be soluble in some organic solvents and in 10% (v/v) DMF/H2O mixture. In the dark, 23 showed good stability for 5 days in 10% (v/v) DMSO/H2O mixture at pH 7.2. Gold nanoparticles (AuNPs) were functionalized with 23 to yield water-soluble 23-AuNPs, which demonstrated good stability at room temperature for 5 days under dark condition. The CO photo release from 23 and 23-AuNPs was assessed using 400–700 nm light. In the dark, the loaded nanoparticles were nontoxic to A549 cells, whereas they caused apoptosis in the same cell line, with IC50 = 232 μg mL−1, upon irradiation with red light. Complex 23 was cytotoxic to cells, with an IC50 of 39.2 μM when exposed to red light and 89 μM in the dark. The primary mechanism underlying the photo-cytotoxicity was dual photosensitization with red light, which resulted in fast, synergistic CO release and singlet oxygen (1O2) production. In A549 cells, photo-triggered CO release led to TURN-ON luminescence.185
In 2021, six Mn(I) photoCORMs, 24a–24f (Fig. 5), containing N,N-bidentate Schiff-base ligands bearing different substituents at the ortho- and para-positions, were also proposed and investigated to study how the substituents affected the kinetics of CO release. The complexes maintained the dark stability in both coordinating and non-coordinating solvents. However, in coordinating solvents, upon the incubation, interchange between the bromo ligand and solvent molecules occurred. When 24a–24f were exposed to a 365 nm light source, they released CO. Complexes 24a and 24c, with o-OCH3 and o-Cl groups, had the highest cytotoxicity in the dark (IC50 = of 18.1 and 11.8 μM) and upon illumination (IC50 = of 7.9 and 6.6 μM) against HepG2. On the other hand, 24b and 24d were inactive in the dark, however, the illumination enhanced their cytotoxicity indicating that iCORM or synergism between CO and the iCORM were responsible for their biological activity.186
The fac-Mn(CO)3 fragment of the peptide azopyridine bioconjugate 24g was capable of being effectively internalized and the local CO release was obtained following red-light illumination without changing the nature of the peptides. The targeted/localized CO release improved cell cytotoxicity via apoptosis against U87 (human malignant glioblastoma), MCF-7, and HeLa cell lines. The kCO and t1/2 values of 24g are (1.8 ± 0.5) × 10−2 s−1 and 39 ± 10 s, respectively.188
Recently, we synthetized two water-soluble Mn-based 468 nm-activated photoCORMs, 25a and 25b (Fig. 5).188 Upon incubation in water for 16 h, both complexes showed a slightly decrease in absorbance, which is due to bromo-DMSO exchange. The fact that complex 25b, which contains 2-acetyl pyridine, is able to liberate CO more quickly than 25a suggests that the methyl group is involved in controlling the kinetics of CO release. The Mb assay indicated that the solvent plays a part in influencing the CO release kinetics by demonstrating that the route for CO release in water (kCO and t1/2 = 2.1 × 10−3 s−1 and 5.4 min (25a) and 2.2 × 10−3 s−1 and 5.2 min (25b)) is slower than in DMSO (kCO and t1/2 = 1.1 × 10−3 s−1 and 10.26 min (25a) and 1.3 × 10−3 s−1 and 8.93 min (25b)). In water, 25a and 25b released about 0.66 and 1.31 equivalents of CO, respectively. Both complexes are not cytotoxic for malignant and normal cells under dark and light conditions. The fact that the tested photoCORMs remained inactive even under illumination proved how crucial the kind of iCORM is for controlling the cytotoxicity of Mn(I) photoCORMs.
Following that, we looked at the dark stability of 26 and 27 (Fig. 5), their CO release kinetics when exposed to 468 nm, and their cytotoxicity against MDA-MB-231 in both dark and light settings.189 Upon incubation for 16 h in 25% (v/v) DMSO/H2O solution, 26 exhibited pronounced change in the metal–ligand charge transfer band. On the other hand, 27 appeared to be stable under the same conditions. The kCO and t1/2 values were found to be (26.0 ± 0.18) × 10−3 s−1 and 4.54 ± 0.17 min for 26 and (7.0 ± 0.10) × 10−4 s−1 and 17.28 ± 3.6 min for 27, respectively. Neither in the presence of light nor without it, complex 26 exhibited any discernible cytotoxicity towards MDA-MB-231 cells. On the other hand, 27 owned a dose-dependent cytotoxic effect with IC50 = 19.62 μM in the dark and 11.43 μM upon illumination. A similar behaviour was seen when the complexes were tested against normal HEK-293T cells. Accordingly, iCORM or synergism between free CO and iCORM play an essential part in controlling the cytotoxicity of these photoCORMs. Complex 27 was combined with the chemo-therapeutic agent paclitaxel and tested against MDA-MB-231 cells. Viability of the cells cotreated with paclitaxel and 27 was 27%. This value is significantly lower than that obtained when the same cells are exposed to only paclitaxel as well as lower than that of the cells treated with 27, suggesting that this compound might increase the cytotoxicity of paclitaxel in the context of resistance.189
Recently, we introduced 28 (Fig. 5), featuring 1-(chloromethyl)-1H-benzotriazole, which released CO at 468 nm.190 The dark stability tests carried out in various solvents and/or in the presence of biomolecules show a significant likelihood of ligand exchange with the coordinating solvents and histidine. For histidine, HEWL, and calf thymus (CT-DNA), the CO release process plateau was reached after 75, 21, and 25 min, respectively. Complex 28 inhibited THP-1 (human acute monocytic leukaemia) proliferation in the dark, but it does not have negative effects on BM cells, contrarily to the free ligand, which was not cytotoxic to THP-1 and normal BM cells. Upon illumination, 28 generated an acute effect on the normal cells that could be the cause of the photo-generated CO. Because the uncoordinated benzotriazole ligand is inactive under identical experimental conditions, the iCORM may be a Mn(II) complex and be less potent than the parent complex.
Fujita and co-workers designed a photoactive ferritin based CO-releasing system,191 able to release CO under light irradiation. In particular, the amount of CO released from the Fr cage can be altered by light exposure. The mutation R52C allowed to stabilize the binding sites of MnCO moieties within the nanocage. Through X-ray crystallography the authors identified 48 Mn binding sites with partial occupancies (0.60–0.80), even if no electron density peaks could be assigned to CO ligands. The presence of CO coordinated to Mn was detected by ATR-IR spectroscopy, following the signal of CO-stretching frequencies. The CO release from the MnCO-loaded nanocages was studied as function of irradiation time: the nanoconstruct t1/2 was 2.5 ± 0.2 min, while than that of CORM-1 was 11.4 ± 0.8 min under the same conditions. As a confirmation of the CO-releasing properties of the Fr construct, HEK-293 cells treated with MnCO-Fr, after 10 min of light exposure, showed a cooperative activation of NF-κB accomplished by CO with TNF-α. These results support the potential of protein cages as carriers for CORMs.
6.2 Molybdenum(0) CORMs
Complex 29 (Na[Mo(CO)3(histidine)], Fig. 6) is a water-soluble oxygen-activated CORM that is stable in the deoxygenated aqueous solutions in inert atmosphere.19229 could be activated by O2 to release CO into the circulatory system and other tissues after oral, intraperitoneal, or intravenous administration. Under aerobic conditions, 29 releases one CO equivalent after 30 min and 2.26 equivalents after 2 h in the dark. However, this behaviour slightly varies with lowering pH, as it slows down by around 50% at pH of 2.5 (the stomach's pH). After 24 h of incubation, 29 is not harmful to LLC-PK1 or RAW264.7 cells, however when 100 M of 29 is given to HepG2 cells, their survival rate drops by about 30%. Mice given daily intraperitoneal injections of 20 mg kg−1 of 29, dissolved in PEG300/water (1:4), for 40 days did not exhibit any evident behavioural abnormalities or issues with their internal or exterior organs. Acute toxicity appeared only at a dose of 500 mg kg−1 that is substantially higher dose than what is required for therapeutic activity in the majority of diseases.5129 interacts with HSA and HEWL. Upon interaction with HEWL at solid state a polyoxomolybdate cluster [PMo12O40]3− is formed.51
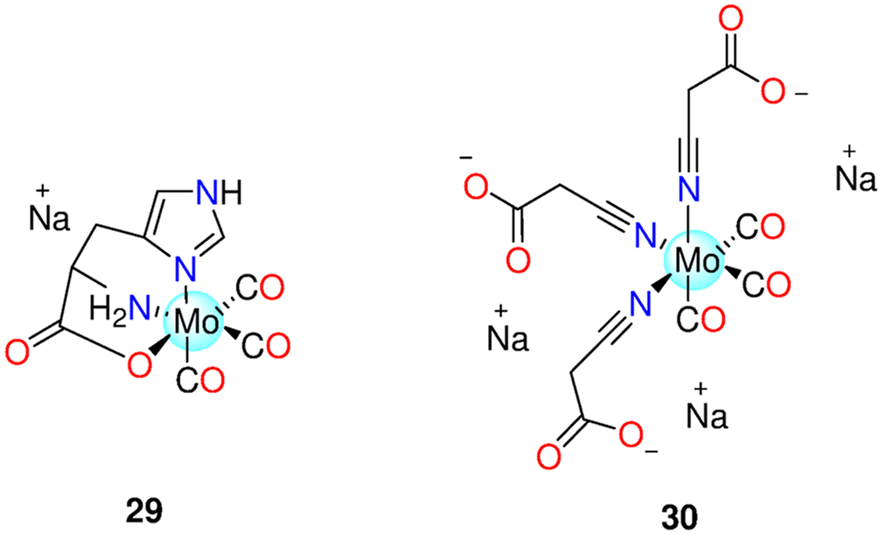 | ||
| Fig. 6 Structures of the Mo(0) CORMs 29 and 30. | ||
The phospholipid phosphatidylcholine was mixed with Mo(CO)5L and Mo(CO)4L2 (L = Ph2P(CH2)6SO3Na) to generate new distinct aggregates. Metallosomes are mixed vesicles with features comparable to liposomes that can be produced using both compounds. FTIR spectroscopy revealed that these mixed systems act as photoCORMs in the presence of UV/Vis light. Toxicity inquiries of various mixed aggregate systems against human dermal fibroblasts have showed that metallosomes have relatively low toxicity like liposomes that are free of metallo-surfactants. Micro-FTIR microscopy, coupled with synchrotron radiation, was utilised to examine the penetration of the studied complexes into cells, revealing metallo-surfactant penetration.193
Recently, researchers developed an oral CO releasing system, based on Na3[Mo(CO)3(N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CH2CO2)3], 30 (Fig. 6),194 that allows for tuneable CO liberation into the gastrointestinal tract, while limiting the release of any other component contained in the device, which could pose a safety risk. The surrounding silicon membranes of the CO release system as well as the load of the Mo(0) complex and iron(III) chloride solution govern CO release rates. Therapeutic CO was generated and released by the reaction of 30 with FeCl3. Neither the CORM nor the probable by-product isocyano-acetate were toxic to human embryonic kidney HEK-293 or mouse embryonic fibroblast NIH3T3 cells. It was demonstrated that the CO releasing system locally produced and released CO into the stomach of the animals without causing systemic exposure.195
C–CH2CO2)3], 30 (Fig. 6),194 that allows for tuneable CO liberation into the gastrointestinal tract, while limiting the release of any other component contained in the device, which could pose a safety risk. The surrounding silicon membranes of the CO release system as well as the load of the Mo(0) complex and iron(III) chloride solution govern CO release rates. Therapeutic CO was generated and released by the reaction of 30 with FeCl3. Neither the CORM nor the probable by-product isocyano-acetate were toxic to human embryonic kidney HEK-293 or mouse embryonic fibroblast NIH3T3 cells. It was demonstrated that the CO releasing system locally produced and released CO into the stomach of the animals without causing systemic exposure.195
6.3 Cobalt(0) CORMs
The binuclear Co-based CORMs, Co2(CO)6HCC–CH2OCOR (31a–31g) (Fig. 7) showed slight solubility in water and good solubility in DMSO, DMF, CH2Cl2, and THF.196 In the pH range of 2–10, the DMSO/H2O or CH3OH/H2O mixtures of the complexes showed stability for 7–10 h. This series of the Co(0) complexes released CO via oxidation. As CO releasers, most of the complexes have t1/2 value of about 60 min. The t1/2 values were affected by the CORM structure. In vivo, these complexes release CO at a comparatively modest rate. Among the entire series, 31a with the acetyl substituent had the maximum cytotoxicity against HeLa cells, with an IC50 value of 36.20 μM. Compound 31b showed less toxicity than 31a revealing the role of the acetyl substituent in controlling the toxicity of this class of Co(0) CORMs. As examples, 31a and 31f were discovered to raise intracellular ROS levels, decrease cell division and proliferation, trigger apoptosis in HeLa cells, and stop the cell cycle in the G2/M phase. Following the release of CO, Co(0) may undergo oxidation to produce the active species Co(II) and Co(III), which are attracted to endogenous substrates like DNA and RNA-containing proteins causing cell damage. The CO ligands in CORMs make it easier for these molecules the membrane penetration, which improves medication cellular absorption.196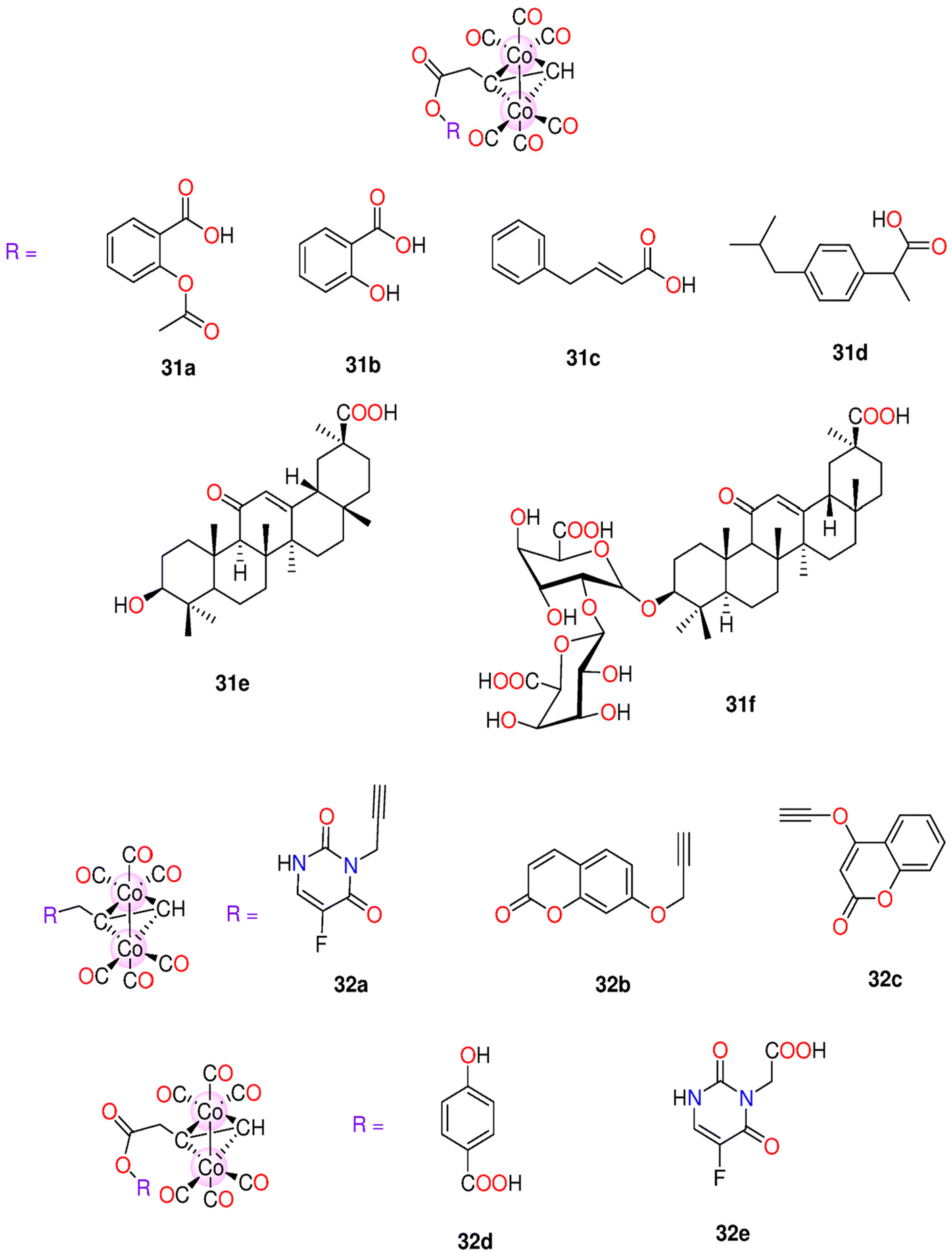 | ||
| Fig. 7 Structures of the Co(0) CORMs 31a–31f and 32a–32e. | ||
Afterward, the same research group prepared another series of Co based CORMs, 32a–32e (Fig. 7), which were categorized as slow CO releasers with t1/2 values in the range 33.5–71.6 min in Mb solution. However, when measured in blood plasma in vivo, the t1/2 values were longer. When tested against HeLa and HepG2 cell lines, the complexes 31a, 31e and 32a–32e had IC50 values in the ranges 36.2–110.21 μM and 39.25–171.34 μM, respectively. Complex 31a showed the highest potency against both cell lines with IC50 = 36.20 ± 2.5 and 39.25 ± 1.9 μM, respectively. Animal tests on rats demonstrated that 31a and 32e had the smallest LD50 values (300–500 mg kg−1). The biodistribution of CORMs in the tissues and organs has been correlated to side chain substituents, which have a substantial impact on the kinetics of CO release, potency, and cell viability. After numerous successive dosages, the CORMs damaged the liver and kidneys both morphologically and functionally. Zebrafish larvae were significantly impacted by developmental toxicity caused by both 31a and 32e. At low concentrations (0.5 and 1.0 μM), they showed no cytotoxicity, but at higher concentrations (5.0–20.0 μM) they are toxic.197
Next, similar binuclear complexes, 33a–33k and 34a–34e (Fig. 8), were synthesized. The t1/2 values were proven to be dependent on the structures of the non-CO ligands similar to 31a–31g and 32a–32e. Complex 33f, with pyridine ring and chloride atom in the 3-position of the phenyl ring, had the longest t1/2 value (42.6 min), while 34d was the fastest CO releaser with t1/2 = 15.8 min. All complexes showed lower activity against the proliferation of HeLa, A549, HT-29, HepG2, and MCF-7 cells when compared to cisplatin. However, when compared to 5-Fluorouracil, 34a and 34b showed superior activity and selectivity to the HT-29 (37.9 and 55.8 μM) and MCF-7 (33.6 and 49.3 μM) cell lines. With respect to 33d, 34a had a higher ability to down-regulate cyclooxygenase-2 (COX-2) expression. This might be due to celecoxib structural fragment, a COX-2 specific inhibitor, being present in 34a. After being exposed to H2O2 for an hour, myocardial H9c2 cells were treated with 33a, 33j, and 34a, which enhanced the survival rate of the cells. This suggested that the complexes have a protective effect against oxidative damage. The efficacy of 33a to increase H9c2 cell viability was the highest, while 34a significantly increased the cell survival rate. However, when the cells experienced harm for 8 h, the protective action was not noticeable. Additionally, the complexes demonstrated a concentration-dependent anti-hypertensive impact on SHR rats.198
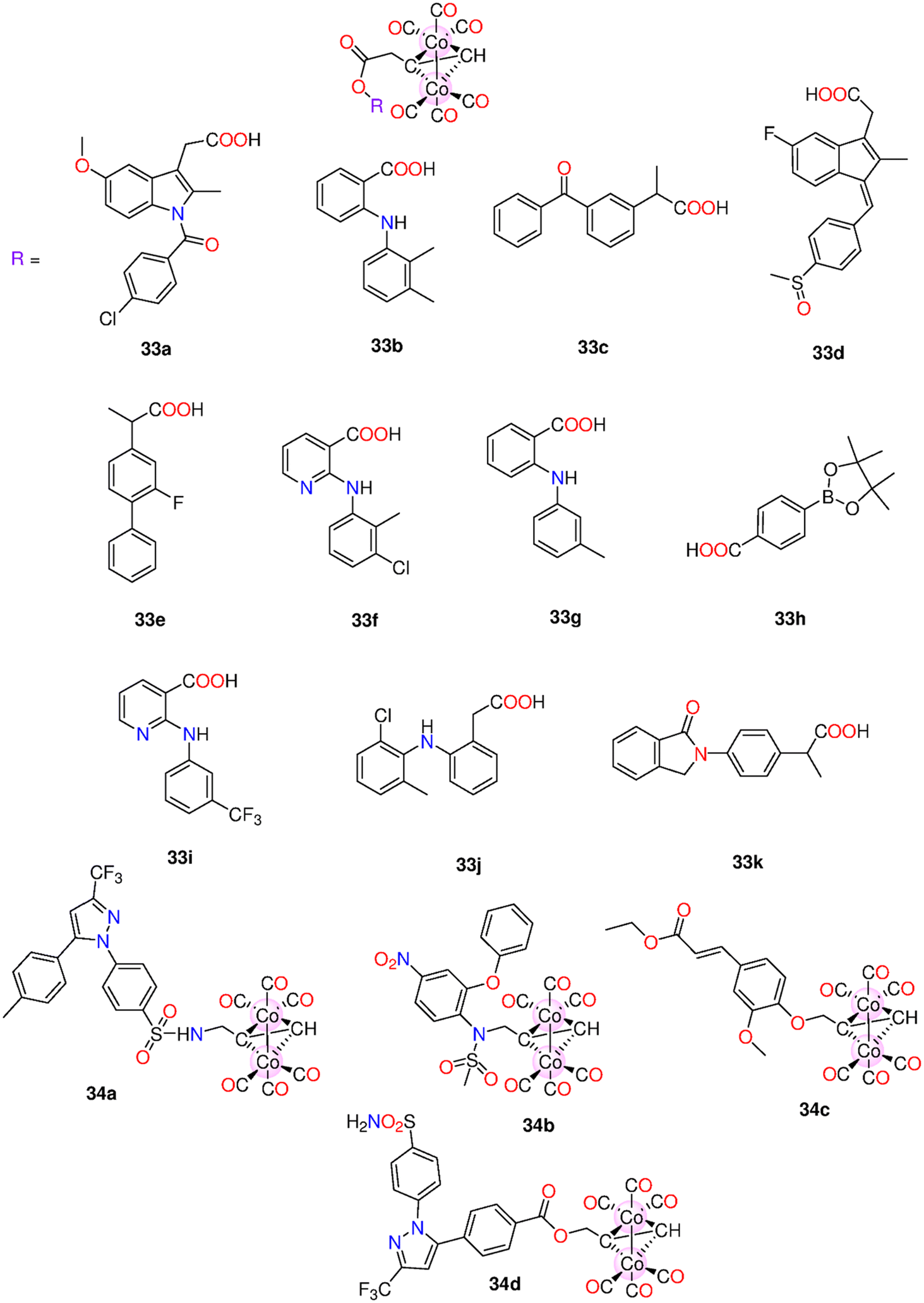 | ||
| Fig. 8 Structures of the Co(0) CORMs 33a–33k and 34a–34d. | ||
The cytotoxicity of two series of Co-based CORMs, 35a−35e, 36a−36e as well as 17α-ethinyl estradiol based complexes 37a–37c (Fig. 9) were examined against HepG2, HeLa, and MDA-MB-231 cells.199 These complexes have good solubility in organic solvents. The complexes showed IC50 values in the range 4.7–548.6 μM against the tested cell lines. Complex 35a exhibited high selectivity to HepG2 with IC50 = 4.7 ± 0.76 μM. At a concentration of 50 μM, 35a had a cytotoxic effect against HepG2, HeLa and MDA-MB231 cells with cell viability of 21.21%, 12.14% and 23.99%, respectively. However, the iCORM of 35a showed minimum effect on the cells. It was suggested that the anticancer action could be the result of combined impacts of CO and CO depleted species. In HepG2 cells, complex 35a increased the amounts of ROS in the mitochondria and decreased the dose-dependent mitochondrial membrane potential. According to a western blot investigation, 35a reduced COX-2 expression. Molecular docking calculations showed that 35a can bind Arg120 in the protein active site through the formation of a hydrogen bond. The COX-2 and mitochondrial pathways may be targeted by 35a to cause apoptosis in HepG2 cells.199
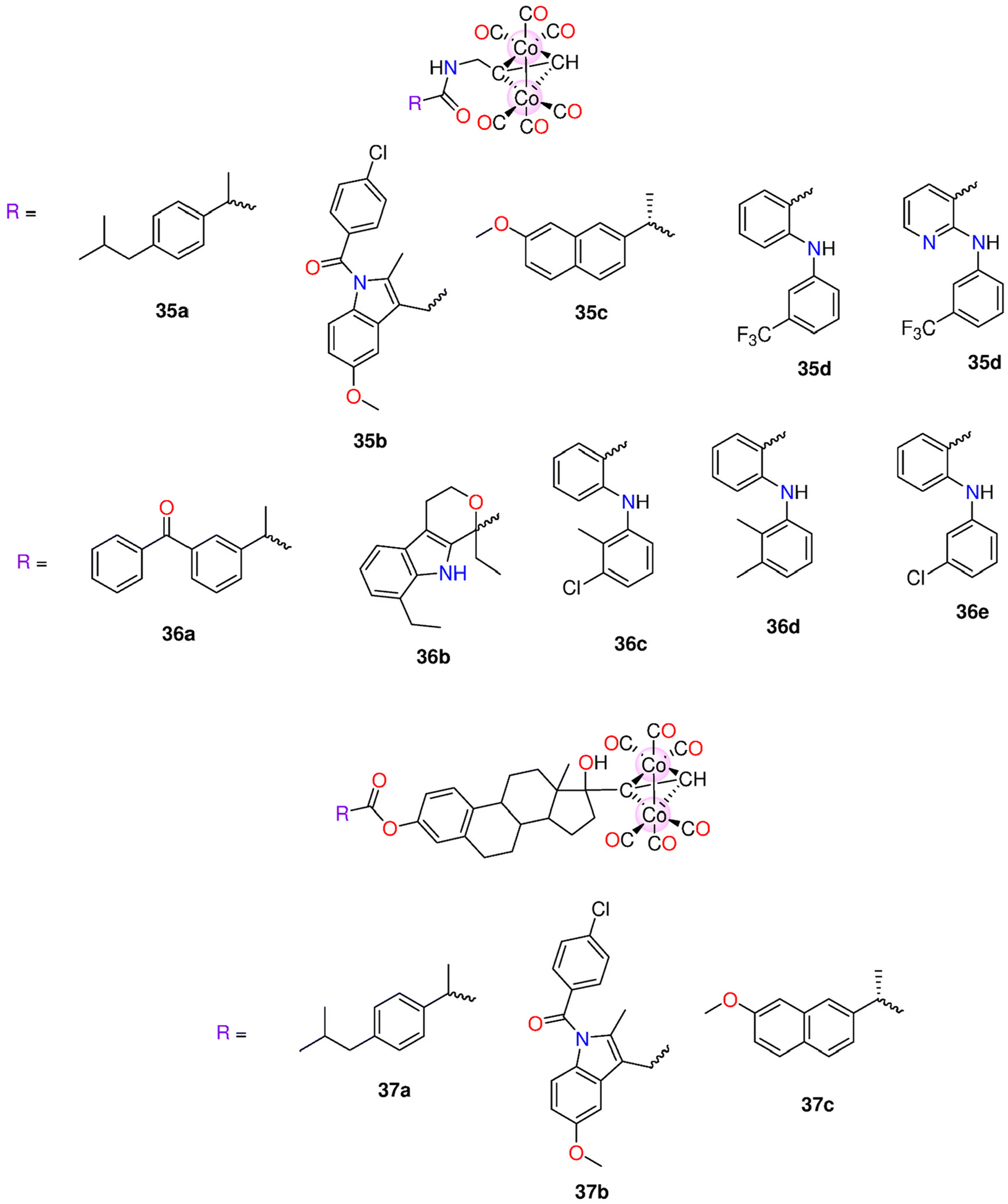 | ||
| Fig. 9 Structures of the Co(0) CORMs 35a–35e, 36a–36e and 37a–37c. | ||
6.4 Iron(II) and iron(0) CORMs
Iron-based CORMs can release CO when triggered by light, redox state changes, and bond breakage caused by an enzyme at the ligand in enzyme-triggered CORMs. In 2005, Motterlini and co-workers presented the iron-containing complex CORM-F3, 38 (Fig. 10).200 Complex 38 steadily released CO via the oxidation processes and has been proven to cause vasorelaxation and prevent inflammation in vitro. Next, three Fe-CORMs have been introduced such as CORM-F7, CORM-F8 and CORM-F11, 39–41, respectively (Fig. 10). Motterlini's group investigated how the structure of CORMs can be altered to preserve CO dependent functions while reducing the cellular toxicity. The rate of CO release was greatly reduced by about 4.5-fold when the Br at the 4-position of the 2-pyrone in 38 (kCO = 0.19 nmol min−1) was replaced with a Cl in 40 (kCO = 0.041 nmol min−1). A further reduction was noticed when the substituent at positions 4- and 6- is a –CH3 group in 41 (kCO = 0.041 nmol min−1) or a hydrogen in 39 (kCO = 0.007 nmol min−1). 100 μM of 38 induced vascular relaxation in detached aortic segments and suppressed endotoxin-stimulated a dose-dependent inflammation reaction in macrophages. Both 38 and 40, with halogens and a –CH3 group meta to each other, showed less toxicity against RAW246.7 macrophages.201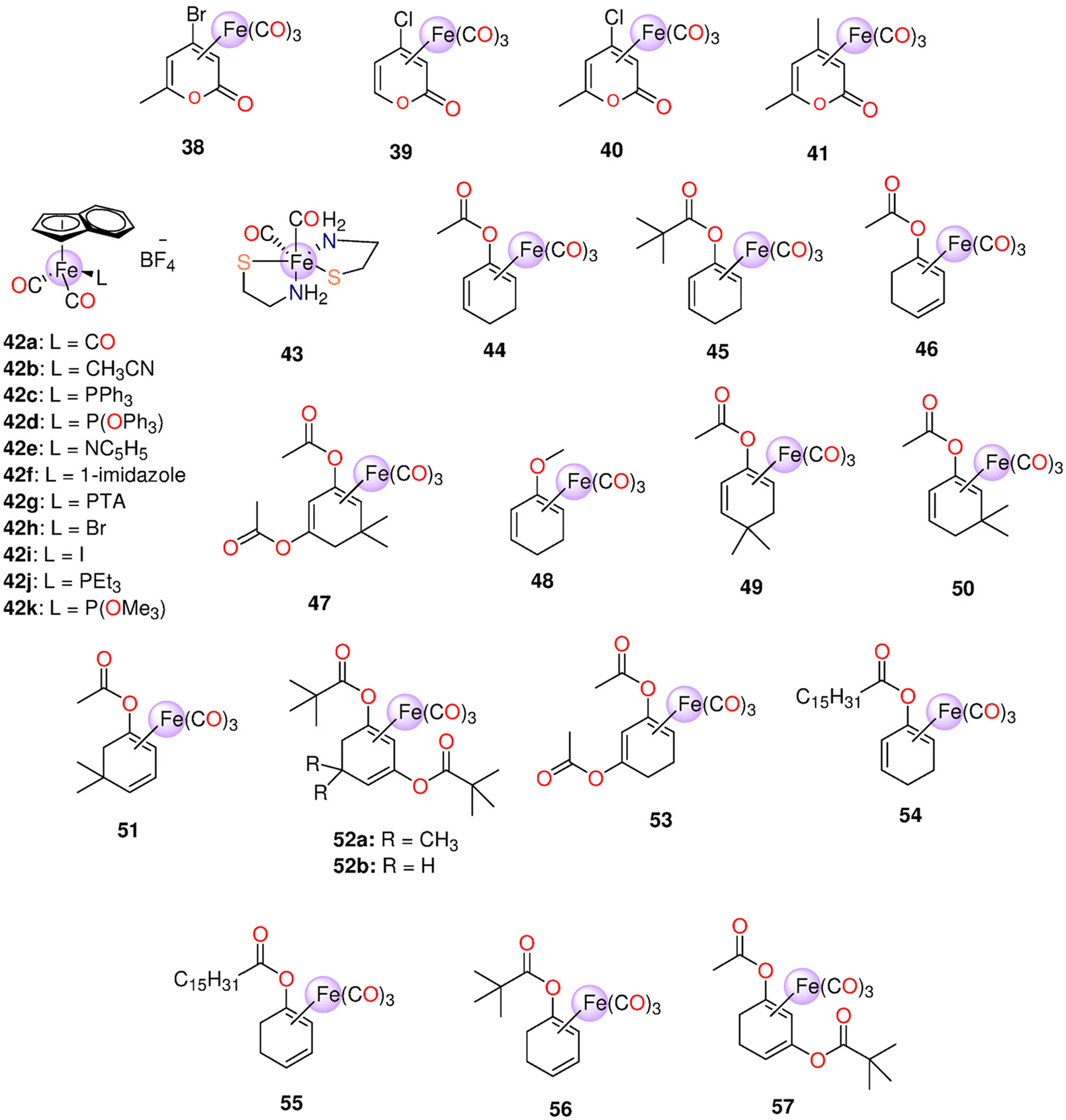 | ||
| Fig. 10 Structures of the Fe(0) CORMs 38–57. | ||
When indenyl ligand replaced cyclopentadienyl ligand, the rates of the substitution processes significantly increased.202 This fact served as the basis for the synthesis of Fe-based CORMs 42a−42k (Fig. 10) with indenyl ligands that may release CO more quickly than their equivalent cyclopentadienyl CORMs. Complex 42a had the shortest t1/2 value of one min, while 42j exhibited the highest t1/2 of >5000 min. Among this series of iron CORMs, 42b exhibited no cytotoxicity even at 100 μM towards rodent RAW264.7 macrophages. 42a was the only compound that released CO more quickly than the comparable cyclopentadienyl molecule. Also, indenyl CORMs are much more cytotoxic than the cyclopentadienyl ones.203
CORM-S1 (dicarbonyl-bis(cysteamine)iron(II)), 43 (Fig. 10), is a water-soluble Fe(II) photoCORM that can release CO via irradiation with visible light source higher than 400 nm.204 The rate of CO release from 43 was proportional to the intensity of the irradiation source. Complex 43 is relatively stable under dark ambient conditions, however the irradiation with a 470 nm light for 15 min led to the release of two CO molecules. In the dark, 43 had no effect on ion currents that crossed voltage-activated potassium channels, and the cell membrane's structure remained unaffected. When exposed to light, a higher outward current was generated with a similar variation in the potential of the membrane, giving a measurement for CO released from 43.204
Acyloxydiene–Fe(CO)344–46, diacetoxydiene-Fe(CO)347, and the methoxy-substituted 48,205 were prepared and are referred to as ET-CORMs. In cells, the complexes are cleaved by the intracellular esterases (e.g.: PLE, pig-liver esterase). An LCR (lipase of Candida rugosa) surpassed PLE for 44 and 45. Complex 45 reacted far more slowly than 44, 46, and 47. Under these conditions, the methoxy 48 did not show any reactivity, as anticipated. When PLE is present, CO was released from 46 and 47, and in the presence of LCR, from 44–46. 47 does not release CO when esterases are not present. So, the hypothesised enzyme-triggered CO release was clearly shown to exist. Both 45 and 48 showed no toxicity against murine RAW267.4 macrophages up to 100 μM. On the other hand, 44, 46 and 47 had IC20 values in the ranges 11–28 and 14–38 μM using MTT and crystal violet assays, respectively. The impact of ET-CORMs upon NO formation via inducible nitric oxide synthase was assessed. To limit the pseudo positive results due to cytotoxicity, only concentrations lower than the IC20 values were selected. In LPS-induced RAW267.4 cells, the diacetate 47 showed the highest inhibition of NO generation. With concentrations of 15 and 5 μM, 47 suppressed LPS-induced NO production by up to 68 ± 6% and 33 ± 6%, respectively. 5 μM of 46 led to 30 ± 7% suppression of NO production, while 44 (25 μM) reduced NO generation by only 16 ± 10%. For up to 50 μM, 45 and the esterase-insensitive 48 did not notably inhibit NO production.48
Next, the same research group identified the structure–activity relationships of the group of acyloxycyclohexadiene-Fe(CO)3 ET-CORMs, 44–56. PLE and LCR were used to induce the hydrolysis of the various compounds. When compared to the acetate-containing complexes, the rates of CO discharge from palmitate- and pivalate-based complexes were sluggish, correlating to slower hydrolysis rates. Complexes 46, 55 and 56, containing an ester moiety at the diene outer position, showed faster CO release rates than 44, 45 and 54, having the same ester group at the inner position. However, the diacetate complexes showed the fastest CO release rates. This could be brought on by a more rapid ester hydrolysis or by the initial enol complex's reduced stability. Additionally, it cannot be ruled out that the hydrolysis of the second ester function speeds up the CO release. Using murine macrophage cells (RAW264.7), the cytotoxicity and suppression of NO-production were evaluated. In particular, the ET-CORMs 47 and 51 showed promising characteristics. The NO-inhibition was shown to be highly influenced by the enone by-products of the monoester-containing complexes, and not the diester-containing ones.49 The parent compound, whether cyclohexanedione or cyclohexenone, and the ester function location have a significant impact on the influence of ET-CORMs. ET-CORMs with an ester group bonded to the cyclohexenone ring through the outer (46) rather than the inner location (44) possess markedly higher toxicity towards human umbilical vein endothelial cells (HUVEC). This coincided with an increase in CO release from the formerly mentioned ET-CORM. Because the half maximal effective concentration (EC50) values for 46 were much lower than those of FeCl2 or FeCl3 and were unaffected by iron chelation, the toxicity was not found to be mediated by iron. While the 44-derived suppression of the expression of vascular cell adhesion molecule 1 (VCAM-1) in long-term HUVEC cultures decreased over time, the 57-derived inhibition seemed to rise. Both 44 and 57 prevented NFκB without regard to nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor α (IκBα) degeneration. Both 44 and 57 stimulated Nrf-2, which in turn caused HO-1 expression.42
Recently, the acetate-bearing complexes 44 and 46 along with the pivalate-bearing compound 52b were examined ex vivo on small rat mesenteric arteries that had been pre-contracted with methoxamine. Both 44 and 46 caused significant dilation to the pre-contracted mesenteric arteries, while 52b did not produce any effect. Alternatively, 46 did not cause vasodilation in the case of KCl-pre-treated mesenteric arteries. According to these findings, the primary K+-channel by which 46 mediates vasodilation is Kv7.206
Motterlini and co-workers proposed four iron(II)–allyl CORMs, 58, 59, 60 and 61 (Fig. 11) that spontaneously release CO. Complexes 58–60 were soluble in DMSO, while 61 was soluble in water due to its ionic nature. In the concentration range of 10–40 μM, about one mole of CO/mole of each complex was released. Complexes 58–60 were found to be faster CO releasers than the water-soluble 61 (t1/2 = 18 min). Compounds 58–60 caused more significant cytotoxic effects on vascular and inflammatory cells and isolated vessels when compared to 61. Against macrophages, the IC50 values of 58–61 were 9.1, 11.9, 23.6 and 797 μM, respectively. Complex 58 and its iCORM caused substantial vasorelaxation in isolated aortic rings over time and reached maximum after 60 min. These findings imply that the CO-depleted molecule contributes to the observed effect. Treatment with 59 and 60 led to the same results, however their iCORMs had less vasorelaxation effect. Complexes 58–60 caused a total loss in the cell activity of murine smooth muscle cells. For 61, the complex caused fast dose-dependent vasorelaxation that reached maximum after 10 min. In contrast to 58, the iCORM of 61 showed minimum vasorelaxation implying that only the released CO is responsible for such effect. Complex 61 and not its iCORM greatly reduced LPS-induced NO production without any obvious toxicity up to 100 μM.207
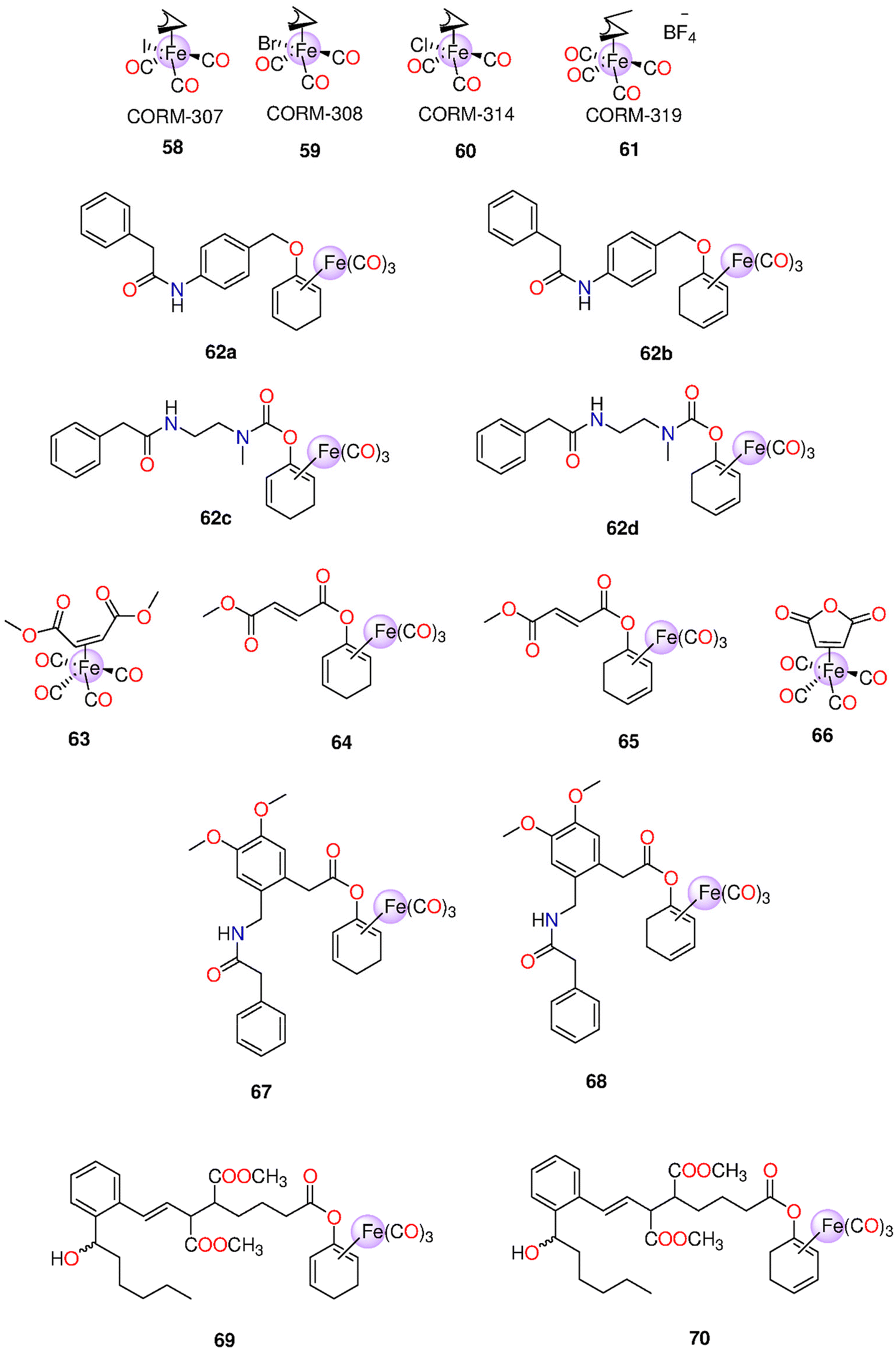 | ||
| Fig. 11 Structures of the Fe(0) CORMs 58–70. | ||
In a concentration range of 1–1000 μM, the cytotoxicity of 44–48, 52b and 53–56 (Fig. 10) was tested on cultures of HUVEC and proximal tubular epithelial cells (PTEC). Complex 48 and three enones were used as controls. Only 46 and 53 showed cytotoxicity at low concentrations. Compound 46 was toxic only against HUVEC, while 53 exhibited cytotoxicity towards PTEC and HUVEC. The hydrolysis products of all complexes did not show any cell toxicity. Changing the substituent from acetate in 46 and 53 to pivalate in 52b and 56 resulted in decreased toxicity. After 24 h of cold preservation, cell damage occurred in both cell lìnes. The cellular damage markedly diminished by treating the cells with 100 μM of 44. Only ET-CORMs containing 2-cyclohexenone were able to reduce the damage caused by cold preservation. The cell protection was dramatically diminished when acetate in 44 was replaced with pivalate in 45. The VCAM-1 expression was significantly suppressed by 44, 52b and 56 and to some extent by 45. For 2-cyclohexenone CORMs, this inhibition could be mediated by 2-cyclohexenone since the ligand itself suppresses VCAM-1 expression.208
Protease-triggered Fe-based CORMs 62a–62d (Fig. 11), containing a PGA (penicillin G amidase)-cleavable site, were prepared.209 Headspace GC and the and the reduction of VCAM-1 expression in a cell-dependent experiment both supported the claim that 62a–62d causes CO release. The phenylacetamide unit is first broken down by an enzyme, followed by linker self-immolation to create the sensitive dienol-Fe(CO)3 intermediate and, finally, oxidation-triggered degeneration of the intermediate to liberate CO. Common biological effects of CO, such as the suppression of the inflammation reaction and the activation of HO-1 expression, were seen only when the CORM and PGA were administered together to human endothelial cells in vitro.
Autoimmune disorders are distinguished with the stimulation of T cells via dendritic cells. Small molecules like dimethyl fumarate or CO are used to treat these serious diseases. Bifunctional iron ET-CORMs 63–66 (Fig. 11), with simultaneous intracellular discharge of CO and methyl fumarate, were developed. The fumarate complexes 63–66 showed enzyme-triggered CO release in vitro using PLE. The substantial suppression of LPS-stimulated pro-inflammatory signalling routes and blocking of (IL)-12 or (IL)-23 generation caused by 63–66 had been shown in bone marrow-derived dendritic cells. The findings also suggest that 63–66 can change dendritic cells into anti-inflammatory phenotypes.210
The effectiveness of iron(0) CORMs that can discharge CO either in response to an enzyme or spontaneously, both extracellularly and intracellularly, was compared. Different CORMs, CORM-2, CORM-3, ET-CORMs (44 and 46) (Fig. 10), and AT-CORMs (62c, 62d, 67, and 68) (Fig. 11), were compared regarding the capacity to release CO, cytotoxicity, the capacity to promote HO-1 expression, and to suppress the production of VCAM-1 in TNF-α-activated HUVEC.50 These impacts are correlated to the quantity of CO discharged by the complex and not to extra- or intracellular CO discharge. As expected, CORM-2 and CORM-3 primarily release CO2 and tiny amount of CO.211,212 However, following the enzymatic hydrolysis, no release of CO2 from the ET- and AT-CORMs was observed. Using certain membrane-linked enzymatic action could allow tissue-targeted CO administration based on the previously mentioned finding that extra- and intracellular CO release generate equivalent anti-inflammatory characteristics.50
To create novel anti-inflammatory drugs, Schmalz and co-workers developed conjugates between lipoxin A4 and an acyloxycyclohexadiene-Fe(CO)3 compound, 69 and 70 (Fig. 11). Both 69 and 70 were toxic to cultured HUVEC in a dose-dependent manner, with 69 being somewhat more toxic. While both Fe-CORMs induced HO-1 sovraexpression in HUVEC, they were unable to decrease VCAM-1 expression induced by TNF-α in the tested cells. The HO-1 expression was greater in M2 polarized macrophages compared to M1 polarized ones. As anticipated, the enhanced HO-1 expression can be attributable to CO release.213
Mitochondria-targeting water-soluble ET-CORMs (Mito-CORMs) with N-methyl-pyridinium triflate motif bonded to the ester moiety (71a–71g) (Fig. 12) were reported.214 When dissolved in PBS/DMSO (5:1) mixture, complexes having an acyloxy motif at 2-location of diene-Fe(CO)3 unit exhibited hydrolysis sensitivity and released CO spontaneously with 1.8 equivalents for 71a (after 2.5 days) and 2 equivalents for 71b and 71c after 2 days. However, this release was significantly suppressed in the presence of PLE (0.6 equivalents from 71b and 71c after 5 days). To determine if the high CO release from 71b and 71c, when PLE is absent, depends on ester moiety activation to hydrolysis, a trans-cyclopropane unit was introduced into 71g to assure electronic and steric isolation of the two functions. Complex 71g exhibited a reduced spontaneous CO release. However, in the presence of PLE, it released just 1.5 equivalents of CO, ruling out any possibility of intramolecular activation of the ester moiety. In contrast, the 1-substituted isomers demonstrated PLE-induced CO release. Three equivalents of CO were released from 71d after 10 h incubation with and without PLE. In the presence of PLE, both 71e and 71f released much more CO (about 3 equivalents) than the spontaneous release without PLE (about one equivalent). Because of the required esterase-triggered CO release, both 71e and 71f were good candidates for biological evaluation. Mito-CORMs 71h and 71i, which were derived from isophorone and not easily oxidized, were developed to rule out the possibility that the phenol produced from cyclohex-2-enone's oxidation contribute to the toxicity of 71e and 71f. Complexes 71e and 71f showed no toxicity against HUVEC up to 500 μM. However, their analogues 71h and 71i, which were supposed to be less toxic, showed pronounced toxicity at concentrations > 50 μM. Neither the hydrolysis by-product isophorone nor the methyl esters were toxic, indicating that the toxicity is probably caused by the released CO. The anti-inflammatory effect of 71h and 71i was shown to be stronger than 71e and 71f. This was noticeable for the suppression of the expression of VCAM-1, but there was not a significant variance in HO-1 inhibition among the two Mito-CORM classes. While both 71h and 71i suppressed mitochondrial respiration in both basal and stressful settings, glycolysis increased. Complexes 71e and 71f elevated both mitochondrial respiration and glycolysis.214
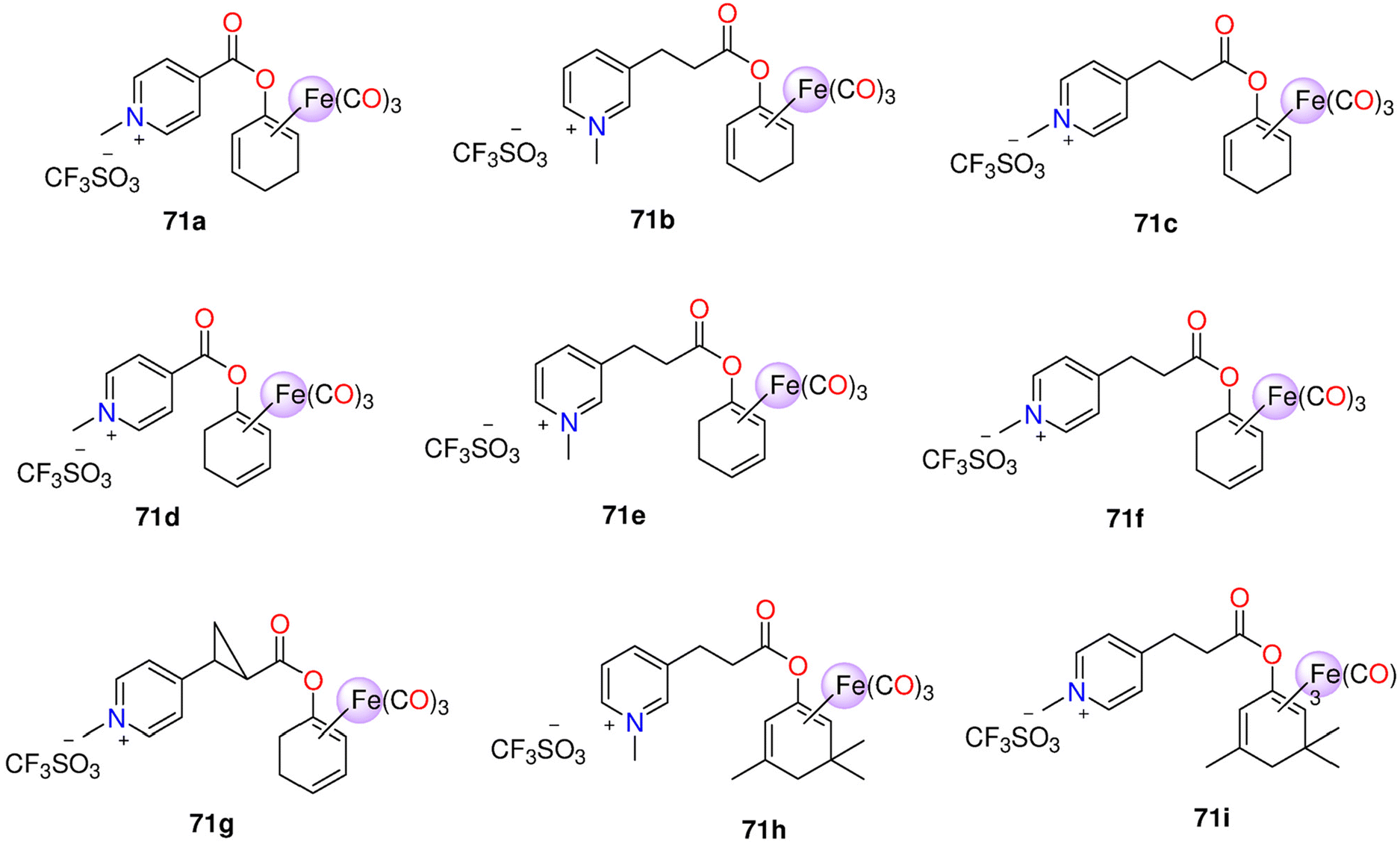 | ||
| Fig. 12 Structures of the Fe(0) CORMs 71a−71i. | ||
6.5 Ruthenium(II) CORMs
Although CORM-2 is widely used to spontaneously release CO when introduced to living tissues, various Ru(II)-based CORMs have been studied as photochemical CO-releasing agents since they do not release CO spontaneously. As a result of the fact that many Ru(II) carbonyls discharge CO after being lighted, such complexes possess the advantageous quality of regulating CO conveying. The CO release from next-generation Ru(II)-based CORMs is induced by a light source or ligand exchange. The solid Ru(II)-based complexes 72a–72i, 73a–73d, 74a and 74b (Fig. 13) are stable even when exposed to light and air. Complexes 72a–72d are water-soluble, while the rest of the compounds exhibit poor solubility in water. The complexes show efficient CO release with t1/2 values ranging from 1.1 to 15.8 min. Complexes with amino acids, 72a–72c, are slightly faster CO releasers than the complexes containing amino-pyridine or Schiff-base ligands (73a and 74a). The complexes show very weak antiproliferative activity against murine L929 fibroblasts (IC50 = 62.69–255.48 mg l−1). Against mice, the complexes have lethal dose LD50 values of 800–1000 mg kg−1 (72a and 72h), 1100–1500 mg kg−1 (72g and 74b) and 150–200 mg kg−1 (73a). When tested on rats in vivo, the complexes have a little impact on liver function, but they do cause physiological harm to liver cells. Additionally, these complexes have a detrimental effect on both functional and physiological processes of the kidney. Metal buildup could be among the causes of adverse effects. The studied complexes do not accumulate in major tissues or organs, and they are unable to pass through the blood–brain barrier. The hepatic P450 enzymes oxidize Ru(II) in CORMs to Ru(III) throughout the metabolism process.215 | ||
| Fig. 13 Structures of the Ru(II) CORMs 72–75. | ||
For Ru(II) complexes 75a–75n (Fig. 13), bearing monodentate ligand, it appeared that the ligand affects the stability of complexes in solution, CO release properties, anti-inflammatory activity and cytotoxicity. The phosphine PTA (1,3,5-triaza-7-phosphaadamantane) was unable to ensure the water solubility of the water-insoluble 75i, despite being employed to enhance the water solubility the complexes. However, the diacetylated form of PTA gave a desirable level of solubility for 75j. The isocyanide-based complexes 75l, 75m, and 75n are slightly soluble in water where they easily degrade. The aqueous solutions of the complexes have shown instability when eluted using H2O/MeOH gradient for 60 or 75 min. In the dark, instead of releasing CO to the headspace, the tricarbonyl complexes release CO2 from aqueous solutions (PBS or water). However, the dicarbonyl (75b and 75c) and the mono-carbonyl 75d do not release CO or CO2 under the same conditions. Complex 75d has a t1/2 of about 50 min and is the most stable complex among the three DMSO-containing complexes. Binding studies of 75f, 75g, and 75h with the model protein HEWL reveal that the Ru(CO)x fragments, i.e. the species missing of one or two COs and of the auxiliary ligands, that are exchanged by H2O molecules, bind with the protein at various metal binding locations. In particular, elucidation of the adducts formed by these CORMs and HEWL demonstrates that Ru(CO)x motifs, formed by the CORMs degradation in solution or due to the presence of the protein, recognize His15, Asp18, Asp52, Asp101 and Asp119 residues with the His15 side chain as the primary binding site. Notably, the X-ray structure of the HEWL conjugated with 75g identified the metallacarboxylate species [His15-RuII(COOH)(CO)(H2O)3], which has been supposed to be an intermediate of the CO release reaction, resulting from the introduction of HO− to the di-cation cis-[Ru(CO)2(H2O)3]2+. The attachment of the CORMs to the cell surface allowed the Ru framework to decompose or internalize into cells. CO could be easily delivered to the cells and its inner target areas in both circumstances. Up to 100 μM, none of the compounds examined were toxic to murine RAW264.7 macrophages. The CORMs reduced NO generation through LPS-activated RAW264.7 cells in a concentration-dependent fashion. The thioether derivative 75k was the most effective at reducing NO generation.216
Bernardes and co-workers synthesized a tricarbonyl Ru(II) CORM with an N-acetyl cysteine ligand, 76 (Fig. 14). In general, CORM-3 generates ROS via a water–gas shift reaction, that dramatically increases its antibacterial activity.103 However, 76 decreases ROS production during the release of CO and has no bactericidal activity. When compared to CORM-3, 76 shows higher stability due to its slower CO release in aqueous media and low toxicity against RAW264.7 cells even at 100 μM. Also, 76 exhibits anti-inflammatory properties through a synergistic effect upon the suppression of the NO production and TNF-α expression. As CORM-3, no CO release was detected from PBS solution of the water-soluble 76 in the dark at ambient conditions using GC-TCD. Both 76 and CORM-3 do not increase the amount of COHb in sheep blood when incubated at 37 °C.217
 | ||
| Fig. 14 Structures of the Ru(II) and Ru(I) CORMs 76–83. | ||
The antiproliferative properties of three Ru(CO)x complexes, 77–79 (Fig. 13), with benzimidazole derivatives, was also examined.218 Complexes 77 and 78 show low solubility in H2O and good solubility in different organic solvents. In PBS, both 77 and 78 show good stability for 72 h at ambient temperature. Both CORMs interact with HEWL and bovine pancreatic ribonuclease (RNase A) to form conjugates having one or two CO ligands, thus losing CO ligands. Similar results219 have been obtained using fac-[RuII(CO)3Cl2(N3-imidazole)] and fac-[RuII(CO)3Cl2(N3-methyl-imidazole)], that are moderately toxic to ovarian and colon cancer cells.220 Both 77 and 78 show toxicity against the CH1/PA-1, A549 and SW480 (colon adenocarcinoma) cells with IC50 values that are nearly the same.218 Complex 77 has no effect on the tumor, while CORM-3 enhance cell growth. With a dose of 2.5 mg, 78 significantly slows tumour growth of CT-26 (murine colorectal carcinoma) cells in Balb/c mice in vivo. Upon illumination at 350 nm, two amide-based Ru(CO)2 bipyridyl complexes, one with an alkyne group (80) and the other with a fluorescent tag (BODIPY) (81) (Fig. 14), showed CO release properties. In particular, the complexes could release CO in a DMSO/H2O (0.8% DMSO) mixture following illumination with 350 nm light. The first CO molecule is rapidly released, while the second one is released more slowly. Both complexes show increased cytotoxicity against A431 (epidermoid carcinoma) cancer cells when illuminated at 350 nm. Complex 81 was promptly taken up by A431 and HEK-293 cell lines and distributed throughout the cytoplasm.221
Ru(II)-based complexes, with 1H-benzimidazol-2-ylmethyl-(N-phenyl) amine derivatives, (R = H (82a), 4-CH3 (82b), 4-Cl (82c), 4-COOCH3 (82d), and 3-COOCH3 (82e)) were developed and photoactivated at 365 nm.222 Except for 82d (IC50 = 45.08 ± 3.5 μM), the studied complexes are non-toxic to MCF-7 in the dark. After being exposed to light, the complexes developed cytotoxicity based on the type of the substituent (82a > 82d > 82b > 82c > 82e). The cytotoxic action of the iCORM of 82a, IC50 = 14.32 ± 1.2 μM, revealed that the gained toxicity might be related to the iCORM and emitted CO.222
To improve the hydrophilic properties and biological applicability of CO, Ru(I)-based sawhorse frame was attached to a water-soluble PEGylated sidearm yielding twelve Ru(I)-based complexes (83a–83l). PhotoCORMs 83a–83j release therapeutic levels of CO under photolysis at 365 nm. By correlating the kinetics of CO release and hydrophilic properties of CORMs, it was revealed that the more hydrophilic the CORM, the faster it releases CO. CORMs 83a–83j are stable in the dark and do not spontaneously deteriorate under physiological conditions. The t1/2 values of 83a–83j at 60 μM were in the range 166–2699 s. Although with a minimum toxicity against murine RAW264.7 macrophages, 83a exhibits anticancer action against HT-29 cancer cells under illumination conditions. With a concentration of 50 μM, 83a and 83h show 12.5% and 6.65% loss in cells activity by illumination.223
6.6 Rhenium(I) and rhenium(II) CORMs
Two trigger methods are utilized to promote the CO release from rhenium carbonyls: changes in pH and exposure to light source. Zobi's group developed CORMs with the formula cis–trans-[ReII(CO)2Br2L2]n84–90 (Fig. 15), and only complexes with monodentate ligands released CO.39 The CO release rate depends on the pH, having t1/2 values ranging from about 6 to 43 min, under the physiological conditions. Selected complexes protected new-born rat ventricular cardiomyocytes from ischemia-reperfusion stress in vitro. The needed time for complete saturation of Mb with the released CO steadily decreased at lower pH levels. The findings seem to suggest that the protection of cardiomyocytes may not depend on the rate of CO released. | ||
| Fig. 15 Structures of the Re(I) and Re(II) CORMs 84–98. | ||
Later, the same group introduced two B12-ReII(CO)2 conjugates, 91a and 91b (Fig. 15), capable of releasing CO in a PBS solution containing Mb, freshly reduced by Na2S2O4. In general, the two conjugates are fully soluble in water, stable in the aqueous media and biocompatible. The rate of CO release from 91a and 91b was shown to be similar, with a t1/2 of around 20 min, while the Re precursor salt, [Et4N]2[ReBr4(CO)2] had a t1/2 of 6 min under the same conditions.39 About one mol of CO per mol of 91a and 91b was released within 2 h.224 The rate of CO discharge could be affected by ligand and pH changes on the basic Re-based moiety. The extracellular discharge of CO, as well as the antioxidant capabilities of both molecules, may contribute to the cytoprotective effects of 91a and 91b. Both complexes are non-toxic against cardiomyocytes, even after CO release, and exhibit cellular protective action from ischemia-reperfusion injury. The iCORM non-toxicity was ascribed to the metal oxidation in water under aerobic conditions and formation of the ReO4− anion, one of the least hazardous fragments among the rare inorganic compounds.225
The highly luminescent tricarbonyl bipyridine Re(I) complex, 92 (Fig. 15), functionalized with tris(hydroxymethyl)phosphine, is stable under aerated aqueous conditions, autoxidation-resistant and releases one CO when exposed to light source at 405 nm.226 Also, the solvated photoproduct is luminescent, a property that enables tracing how 92 transforms into its solvated photoproduct inside cells. Complex 92 displays no activity against the human prostate cancer cell line PPC-1. Based on the luminescence results, it is clear that 92 accumulates throughout the cytoplasm, yet is unable to enter the nuclear membrane of the tested cells.227
Mascharak and co-workers proposed two photoactivatable luminescent tricarbonyl Re(I) complexes, bearing 1,3,5-triaza-7-phospha-adamantane, as well as N,N-bidentate ligand (2-(pyridyl)benzothiazole (93) and 1,10-phenanthroline (94)) (Fig. 15), which discharge CO moderately, via the exposure to low-energy UV light (360 nm). Complexes 93 and 94 are very stable in dark and easily soluble in aerobic/anaerobic aqueous conditions. When exposed to UV-A light, 93 and 94 only release one CO molecule. The luminescence properties of 93 and 94 at 550 nm fade gradually by illumination. The results showed that 93 is rapidly internalized by the MDA-MB-231 cancerous cells.170
The tricarbonyl Re(I) complex functionalized with 2-(pyridyl) benzothiazole 95 (Fig. 15) operates as theranostic two-tone luminescent photoCORM in cell targets. Under low-power UV light, the orange luminescence of 95 at 605 nm is completely replaced by a blue fluorescence at 400 nm as a result of complete CO loss and de-ligation of the benzothiazole ligand. According to Mb assay, the kCO value of 95 is 0.31 min−1. A dose-dependent loss in the survival of MDA-MB-231 cancer cells, caused by the CO-induced apoptosis, was observed when subjected to UV light.228
The dark-stable and promising theranostic Re(I)-based photoCORMs, [ReX(CO)3(Phen)]0/1+ (Phen = 1,10-phenanthroline, X = Cl− (96a), CF3SO3− (96b), MeCN (96c), PPh3 (96d), and methylimidazole (96e)), (Fig. 15) are able to emit CO when subjected to low-power UV light (5 mW cm−2). In acetonitrile, the kCO values of 96a, 96d and 96e are 0.07 ± 0.02, 1.59 ± 0.02 and 0.07 ± 0.02 min−1, respectively. The images obtained using fluorescent confocal microscopy revealed that the luminescent Re(I) compounds, cationic and neutral, with auxiliary ligands with varying lipophilic properties, show significant cellular penetration and are distributed largely throughout the cytoplasm in MDA-MB-231 cells. Complex 96d (containing PPh3) displays moderate nuclear accumulation along with cytosolic distribution.229
Two binuclear metallacyclophanes, (Fig. 15) (97 and 98) have been developed by reaction of [Re(CO)5Cl] with N-(2-hydroxybenzylidene)-benzimidazole. The complexes show good solubility in polar organic solvents and are stable at ambient temperature. The obtained IC50 values indicate that 97 is cytotoxic against HepG2 cells, IC50 = 14.2 ± 4.8 μM in comparison to cisplatin (IC50 = 15.89 ± 5.7 μM). Compound 98 inhibits HeLa malignant cells in a concentration-dependent fashion, having the lowest IC50 value of 12.4 ± 2.9 μM, while cisplatin has an IC50 = 17.56 ± 5.7 μM.171 Cancer cells treated with 98 displayed apoptotic traits in the AO/EB staining, including altered nuclear fragmentation, cell shrinkage and development of apoptotic bodies. Although this has yet to be confirmed, the anticancer effect of 98 could be a result of apoptosis triggered by the CO release.171
7. General remarks, conclusions, and perspectives
It has been established that administering molecules that transport and discharge CO into living systems is a more effective option than using CO as a gas. A method of achieving a controlled, focused CO administration is based on the use of stable compounds that only lose CO upon provoking by an external or internal stimulus. These CO prodrugs are known as carbon monoxide releasing molecules (CORMs). CO, either as a gas or confined within a CORM, could be employed as a useful molecule to treat a broad spectrum of diseases and to preserve organs.230 In this review, we have tried to briefly summarize most of the research done employing CORMs as chemotherapeutic agents focussing our attention on cytotoxic metal carbonyl complexes (MCCs). MCCs were classified according to how they are activated to release CO. Thus, examples of photoinduced CORMs (photoCORMs), solvent-triggered CORMs and enzymatically activated CORMs have been presented. A summary of the results obtained in the last 20 years on the first-generation CORMs is also reported. These molecules are highly reactive. Their chemical reactivities may interfere with the activity of biological macromolecules and/or intercept with CO signalling pathways producing unmanageable problems. These aspects have been critically summarized by Wang and coworkers in a recent review.19To overcome these limitations many other CO-releasing molecules have been prepared. Recently, photoCORMs are attracting more attention than the other CORMs due to their widespread application and medicinal potential.158 Mn(I)- and Ru(II)-based CORMs belong to this class, since they can release CO via illumination. In contrast to the visible-light induced Mn(I) carbonyls complexes, the majority of Ru(II)-based photoCORMs are inactive or lose CO when illuminated by UV light. Because of possible photochemical and biological advantages, tri- and tetra-carbonyl Mn(I)-based complexes have attracted the spotlight.231 Visible light-triggered CORMs, that discharge CO when illuminated by visible light ranging from 468 to 660 nm, could be designed by selecting highly conjugated ligand system. Also, included in the development of photoCORMs, is the utilization of materials such as scaffolds, crystals, matrices or up-conversion particles to either improve photoCORM water-solubility or enable encapsulation of potentially harmful metal-based photo-products.232 Also, the used solvent seems to affect the stability of CORMs and the rate of CO release. Some Re-based CORMs are luminescent and can release CO through illumination. These MCCs can behave as a tracker to identify the site of CO discharge inside the cell.227 Other Re CORMs are pH-dependent39 and others can release CO by oxidation.224 Iron-based CORMs can release CO in response to light, redox state changes, and enzymatic bond breaking at the ligand periphery in enzyme-triggered CORMs. The main mechanism for activating Co(0)-based CORMs is the ligand exchange reaction with the medium. CORMs could be therapeutically active through being anti-inflammatory agents, cytotoxic to cancer cells, preserving transplanted organs, antimicrobial, healing cardiovascular illnesses etc. Overall, these data reveal that the mechanism of MCCs activation is influenced by the type of metals as well as the composition of the auxiliary ligands. The features of the ancillary ligands in CORMs influence the metal core electronic density and the specific ability of a CORM to bind biological (macro)molecules that activate CO release. The properties of the ligands also affect the CORM resistance towards oxidation. In other words, the inner ligand sphere structure is crucial for tailoring a certain CORM stability to tolerate proteins in the plasma, react to a specific stimulus, or generate a particular CO release pattern.36 The benefits that CORMs may provide represent a significant advantage over many typical organic medications, which might focus on just one problem. Instead of requiring a complex mixture of multiple medications, an individual CORM is able to treat a wide range of disorders. Another significant advantage of employing CORMs as medications is the variety of structures that can release CO.230
A key point in designing new CORMs is to take into account their toxicity, appropriate absorption, distribution, metabolism, and excretion (ADME) profile, tissue accumulation and their in vivo stability.233 Unfortunately, little is known on the toxicity profiles of CORMs,234 which could be toxic due to effects produced by both CO release and the formation of metal-containing fragments generated by the original pro-drugs upon reaction with biomolecules, but the few studies carried out on tested complexes suggest severe damages of liver and kidney in both functional and morphological aspects.215 Metal complexes can undergo oxidation or fragmentation prior to CO release. Changes in the metal oxidation state could interfere with electron transfer pathways in cellular components, thus producing harmful effects.34 Furthermore, interaction with biomolecules can also occur prior to CO release. In this frame, further studies are needed to clarify which metal-containing fragments are formed by CORMs, which is their metabolic fate and their toxicity in the cellular environments. New approaches should be developed to understand the different contributions of CO and metal-containing fragments derived from CORMs to the biological properties of these potential drugs. In this respect, it should be also considered that metal-containing fragments generated by CORMs could have a different reactivity with biomolecules when compared to the original prodrugs and thus they could have different targets.
Finally, even after reviewing all the prior achievements in such a large prospective area as metal-based CORMs, several mysteries remain as intriguing questions for scientists. When it comes to physicochemical features of CORMs and their cytotoxic properties, before and after CO induction process, which factor is more important: the CORM itself or the iCORM? As a result, we recommend doing additional extensive studies to fully comprehend such miracles and maximise CORMs medically helpful effect on humanity. These studies should also evaluate the release kinetics, dose control, and safety profile of CORMs and i-CORMs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Mansour thanks United Arab Emirates University, Al-Ain, United Arab Emirates, for financial support. Merlino thanks MIUR PRIN 2022- Cod. 2022JMFC3X, “Protein Metalation by Anticancer Metal-based Drugs” for financial support.References
- S. H. Heinemann, T. Hoshi, M. Westerhausen and A. Schiller, Chem. Commun., 2014, 50, 3644–3660 RSC.
- L. Wu and R. Wang, Pharmacol. Rev., 2005, 57, 585–630 CrossRef CAS PubMed.
- T. Sjöstrand, Nature, 1949, 164, 580–581 CrossRef PubMed.
- R. F. Coburn, W. J. Williams and R. E. Forster, J. Clin. Invest., 1964, 43, 1098–1103 CrossRef CAS PubMed.
- R. Tenhunen, H. S. Marver and R. Schmid, Proc. Natl. Acad. Sci. U. S. A., 1968, 61, 748–755 CrossRef CAS PubMed.
- M. D. Maines, FASEB J., 1988, 2, 2557–2568 CrossRef CAS.
- A. I. Archakov, I. I. Karuzina, N. A. Petushkova, A. V. Lisitsa and V. G. Zgoda, Toxicol. in Vitro, 2002, 16, 1–10 CrossRef CAS PubMed.
- F. P. Guengerich, Biochemistry, 1978, 17, 3633–3639 CrossRef CAS PubMed.
- R. Wang, Can. J. Physiol. Pharmacol., 1998, 76, 1–15 CrossRef CAS PubMed.
- A. Grilli, M. A. De Lutiis, A. Patruno, L. Speranza, F. Gizzi, A. A. Taccardi, P. Di Napoli, R. De Caterina, P. Conti and M. Felaco, Ann. Clin. Lab. Sci., 2003, 33, 208–215 CAS.
- T. M. Brusko, C. H. Wasserfall, A. Agarwal, M. H. Kapturczak and M. A. Atkinson, J. Immun., 2005, 174, 5181–5186 CrossRef CAS PubMed.
- I. C. Alexandreanu and D. M. Lawson, Exp. Biol. Med., 2003, 228, 59–63 CrossRef CAS PubMed.
- S. W. Ryter, H. P. Kim, K. Nakahira, B. S. Zuckerbraun, D. Morse and A. M. K. Choi, Antioxid. Redox Signal., 2007, 9, 2157–2174 CrossRef CAS PubMed.
- S. J. Gibbons and G. Farrugia, J. Physiol., 2004, 556, 325–336 CrossRef CAS PubMed.
- M. Suematsu, N. Goda, T. Sano, S. Kashiwagi, T. Egawa, Y. Shinoda and Y. Ishimura, J. Clin. Invest., 1995, 96, 2431–2437 CrossRef CAS PubMed.
- X. Yang, M. De Caestecker, L. E. Otterbein and B. Wang, Med. Res. Rev., 2020, 40, 1147–1177 CrossRef CAS PubMed.
- A. Halilovic, K. A. Patil, L. Bellner, G. Marrazzo, K. Castellano, G. Cullaro, M. W. Dunn and M. L. Schwartzman, J. Cell. Physiol., 2011, 226, 1732–1740 CrossRef CAS PubMed.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- N. Bauer, Z. Yuan, X. Yang and B. Wang, Biochem. Pharmacol., 2023, 214, 115642 CrossRef CAS PubMed.
- S. S. Mendes, J. Marques, E. Mesterházy, J. Straetener, M. Arts, T. Pissarro, J. Reginold, A. Berscheid, J. Bornikoel, R. M. Kluj, C. Mayer, F. Oesterhelt, S. Friães, B. Royo, T. Schneider, H. Brötz-Oesterhelt, C. C. Romão and L. M. Saraiva, ACS Bio. Med. Chem. Au, 2022, 2, 419–436 CrossRef CAS PubMed.
- L. S. Nobre, H. Jeremias, C. C. Romão and L. M. Saraiva, Dalton Trans., 2016, 45, 1455–1466 RSC.
- S. N. Anderson, J. M. Richards, H. J. Esquer, A. D. Benninghoff, A. M. Arif and L. M. Berreau, ChemistryOpen, 2015, 4, 590–594 CrossRef CAS PubMed.
- L. S. Lazarus, A. D. Benninghoff and L. M. Berreau, Acc. Chem. Res., 2020, 53, 2273–2285 CrossRef CAS PubMed.
- A. M. Mansour, R. M. Khaled, E. Khaled, S. K. Ahmed, O. S. Ismael, A. Zeinhom, H. Magdy, S. S. Ibrahim and M. Abdelfatah, Biochem. Pharmacol., 2022, 199, 114991 CrossRef CAS PubMed.
- M. A. Wright and J. A. Wright, Dalton Trans., 2016, 45, 6801–6811 RSC.
- X. Ji, K. Damera, Y. Zheng, B. Yu, L. E. Otterbein and B. Wang, J. Pharm. Sci., 2016, 105, 406–416 CrossRef CAS PubMed.
- X. Ji and B. Wang, Acc. Chem. Res., 2018, 51, 1377–1385 CrossRef CAS PubMed.
- D. Wang, E. Viennois, K. Ji, K. Damera, A. Draganov, Y. Zheng, C. Dai, D. Merlin and B. Wang, Chem. Commun., 2014, 50, 15890–15893 RSC.
- J. Cheng and J. Hu, ChemMedChem, 2021, 16, 3628–3634 CrossRef CAS PubMed.
- J. S. Ward, R. Morgan, J. M. Lynam, I. J. S. Fairlamb and J. W. B. Moir, MedChemComm, 2017, 8, 346–352 RSC.
- M. Desmard, R. Foresti, D. Morin, M. Dagouassat, A. Berdeaux, E. Denamur, S. H. Crook, B. E. Mann, D. Scapens, P. Montravers, J. Boczkowski and R. Motterlini, Antioxid. Redox Signal., 2012, 16, 153–163 CrossRef CAS PubMed.
- A. F. Tavares, M. R. Parente, M. C. Justino, M. Oleastro, L. S. Nobre and L. M. Saraiva, PLoS One, 2013, 8, e83157 CrossRef PubMed.
- N. Rana, S. McLean, B. E. Mann and R. K. Poole, Microbiology, 2014, 160, 2771–2779 CrossRef CAS PubMed.
- A. Ismailova, D. Kuter, D. S. Bohle and I. S. Butler, Bioinorg. Chem. Appl., 2018, 2018, 1–23 CrossRef PubMed.
- A. C. Kautz, P. C. Kunz and C. Janiak, Dalton Trans., 2016, 45, 18045–18063 RSC.
- C. C. Romão, W. A. Blättler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571 RSC.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, e17–e24 CrossRef CAS PubMed.
- U. Schatzschneider, Br. J. Pharmacol., 2015, 172, 1638–1650 CrossRef CAS PubMed.
- F. Zobi, A. Degonda, M. C. Schaub and A. Yu. Bogdanova, Inorg. Chem., 2010, 49, 7313–7322 CrossRef CAS PubMed.
- B. J. Aucott, J. S. Ward, S. G. Andrew, J. Milani, A. C. Whitwood, J. M. Lynam, A. Parkin and I. J. S. Fairlamb, Inorg. Chem., 2017, 56, 5431–5440 CrossRef CAS PubMed.
- P. C. Kunz, H. Meyer, J. Barthel, S. Sollazzo, A. M. Schmidt and C. Janiak, Chem. Commun., 2013, 49, 4896 RSC.
- E. Stamellou, D. Storz, S. Botov, E. Ntasis, J. Wedel, S. Sollazzo, B. K. Krämer, W. Van Son, M. Seelen, H. G. Schmalz, A. Schmidt, M. Hafner and B. A. Yard, Redox Biol., 2014, 2, 739–748 CrossRef CAS PubMed.
- J. E. Clark, P. Naughton, S. Shurey, C. J. Green, T. R. Johnson, B. E. Mann, R. Foresti and R. Motterlini, Circ. Res., 2003, 9, e2–e8 Search PubMed.
- A. J. Atkin, I. J. S. Fairlamb, J. S. Ward and J. M. Lynam, Organometallic, 2012, 31, 5894–5902 CrossRef CAS.
- R. Motterlini, P. Sawle, S. Bains, J. Hammad, R. Alberto, R. Foresti and C. J. Green, FASEB J., 2005, 19, 1–24 CrossRef PubMed.
- N. Bauer, X. Yang, Z. Yuan and B. Wang, Chem. Sci., 2023, 14, 3215–3228 RSC.
- M. Klein, U. Neugebauer, M. Schmitt and J. Popp, ChemPhysChem, 2015, 17(7), 985–993 CrossRef PubMed.
- S. Romanski, B. Kraus, U. Schatzschneider, J. Neudörfl, S. Amslinger and H. Schmalz, Angew. Chem., Int. Ed., 2011, 50, 2392–2396 CrossRef CAS PubMed.
- S. Romanski, B. Kraus, M. Guttentag, W. Schlundt, H. Rücker, A. Adler, J.-M. Neudörfl, R. Alberto, S. Amslinger and H.-G. Schmalz, Dalton Trans., 2012, 41, 13862 RSC.
- Y. Li, L. Hemmersbach, B. Krause, N. Sitnikov, A. Schlundt Née Göderz, D. O. Pastene Maldonado, H. Schmalz and B. Yard, ChemBioChem, 2022, 23, e202100452 CrossRef CAS PubMed.
- J. D. Seixas, A. Mukhopadhyay, T. Santos-Silva, L. E. Otterbein, D. J. Gallo, S. S. Rodrigues, B. H. Guerreiro, A. M. L. Gonçalves, N. Penacho, A. R. Marques, A. C. Coelho, P. M. Reis, M. J. Romão and C. C. Romão, Dalton Trans., 2013, 42, 5985–5998 RSC.
- Z. Yuan, X. Yang and B. Wang, Chem. Sci., 2021, 12, 13013–13020 RSC.
- R. D. Rimmer, H. Richter and P. C. Ford, Inorg. Chem., 2010, 49, 1180–1185 CrossRef CAS PubMed.
- M. A. Gonzales and P. K. Mascharak, J. Inorg. Biochem., 2014, 133, 127–135 CrossRef CAS PubMed.
- D. Nguyen and C. Boyer, ACS Biomater. Sci. Eng., 2015, 1, 895–913 CrossRef CAS PubMed.
- C. Nagel, S. McLean, R. K. Poole, H. Braunschweig, T. Kramer and U. Schatzschneider, Dalton Trans., 2014, 43, 9986 RSC.
- U. Sachs, G. Schaper, D. Winkler, D. Kratzert and P. Kurz, Dalton Trans., 2016, 45, 17464–17473 RSC.
- H.-M. Berends and P. Kurz, Inorg. Chim. Acta, 2012, 380, 141–147 CrossRef CAS.
- P. Rudolf, F. Kanal, J. Knorr, C. Nagel, J. Niesel, T. Brixner, U. Schatzschneider and P. Nuernberger, J. Phys. Chem. Lett., 2013, 4, 596–602 CrossRef CAS PubMed.
- B. J. Aucott, J. B. Eastwood, L. Anders Hammarback, I. P. Clark, I. V. Sazanovich, M. Towrie, I. J. S. Fairlamb and J. M. Lynam, Dalton Trans., 2019, 48, 16426–16436 RSC.
- R. S. Herrick and T. L. Brown, Inorg. Chem., 1984, 23, 4550–4553 CrossRef CAS.
- S. Rattan, R. A. Haj and M. A. F. De Godoy, Am. J. Physiol.: Gastrointest. Liver Physiol., 2004, 287, G605–G611 CrossRef CAS PubMed.
- E. Fiumana, H. Parfenova, J. H. Jaggar and C. W. Leffler, Am. J. Physiol.: Heart Circ. Physiol., 2003, 284, H1073–H1079 CrossRef CAS PubMed.
- P. Koneru and C. W. Leffler, A Am. J. Physiol.: Heart Circ. Physiol., 2004, 286, H304–H309 CrossRef CAS PubMed.
- Q. Xi, D. Tcheranova, H. Parfenova, B. Horowitz, C. W. Leffler and J. H. Jaggar, Am. J. Physiol.: Heart Circ. Physiol., 2004, 286, H610–H618 CrossRef CAS PubMed.
- B. Arregui, B. López, M. G. Salom, F. Valero, C. Navarro and F. J. Fenoy, Kidney Int., 2004, 65, 564–574 CrossRef CAS PubMed.
- C. Bohlender, S. Gläser, M. Klein, J. Weisser, S. Thein, U. Neugebauer, J. Popp, R. Wyrwa and A. Schiller, J. Mater. Chem. B, 2014, 2, 1454–1463 RSC.
- V. Pathak, K. Roemhild, S. Schipper, N. Groß-Weege, T. Nolte, S. Ruetten, E. M. Buhl, A. El Shafei, M. Weiler, L. Martin, G. Marx, V. Schulz, F. Kiessling, T. Lammers and P. Koczera, Small, 2022, 18, 2200924 CrossRef CAS PubMed.
- J. Zhu, J. Wang, G. Wang, J. Zhang, W. Tao, C. Liu, M. Liu, H. Zhang, R. Xie, F. Ye, Y. Liu, W. Fang, X. Chen and Y. Li, J. Phys. Chem. Lett., 2021, 12, 4658–4665 CrossRef CAS PubMed.
- J. P. Lomont, S. C. Nguyen and C. B. Harris, Organometallics, 2014, 33, 6179–6185 CrossRef CAS.
- K. A. Machovec, D. S. Ushakumari, I. J. Welsby and V. G. Nielsen, Thromb. Res., 2012, 129, 793–796 CrossRef CAS PubMed.
- I. C. Winburn, K. Gunatunga, R. D. McKernan, R. J. Walker, I. A. Sammut and J. C. Harrison, Basic Clin. Pharmacol. Toxicol., 2012, 111, 31–41 CrossRef CAS PubMed.
- P. Sawle, R. Foresti, B. E. Mann, T. R. Johnson, C. J. Green and R. Motterlini, Br. J. Pharmacol., 2005, 145, 800–810 CrossRef CAS PubMed.
- K. Srisook, S.-S. Han, H.-S. Choi, M.-H. Li, H. Ueda, C. Kim and Y.-N. Cha, Biochem. Pharmacol., 2006, 71, 307–318 CrossRef CAS PubMed.
- G. Cepinskas, K. Katada, A. Bihari and R. F. Potter, Am. J. Physiol.: Gastrointest. Liver Physiol., 2008, 294, G184–G191 CrossRef CAS PubMed.
- K. Tsoyi, T. Y. Lee, Y. S. Lee, H. J. Kim, H. G. Seo, J. H. Lee and K. C. Chang, Mol. Pharmacol., 2009, 76, 173–182 CrossRef CAS PubMed.
- Q. Niu, F. Du, X. Yang, X. Yang and X. Wang, Int. Immunopharmacol., 2022, 113, 109441 CrossRef CAS PubMed.
- K.-B. Shiu, S.-J. Yu, Y. Wang and G.-H. Lee, J. Organomet. Chem., 2002, 650, 37–42 CrossRef CAS.
- B.-W. Sun, Q. Jin, Y. Sun, Z.-W. Sun, X. Chen, Z.-Y. Chen and G. Cepinskas, World J. Gastroenterol., 2007, 13, 6183 CrossRef CAS PubMed.
- J. Megías, J. Busserolles and M. J. Alcaraz, Br. J. Pharmacol., 2007, 150, 977–986 CrossRef PubMed.
- H. Soni, P. Patel, A. C. Rath, M. Jain and A. A. Mehta, Vasc. Pharmacol., 2010, 53, 68–76 CrossRef CAS PubMed.
- A. F. N. Tavares, M. Teixeira, C. C. Romão, J. D. Seixas, L. S. Nobre and L. M. Saraiva, J. Biol. Chem., 2011, 286, 26708–26717 CrossRef CAS PubMed.
- C. S. Bang, R. Kruse, I. Demirel, A. Önnberg, B. Söderquist and K. Persson, Microb. Pathog., 2014, 66, 29–35 CrossRef CAS PubMed.
- C. Sahlberg Bang, I. Demirel, R. Kruse and K. Persson, PLoS One, 2017, 12, e0178541 CrossRef PubMed.
- L. S. Nobre, F. Al-Shahrour, J. Dopazo and L. M. Saraiva, Microbiology, 2009, 155, 813–824 CrossRef CAS PubMed.
- T. S. Murray, C. Okegbe, Y. Gao, B. I. Kazmierczak, R. Motterlini, L. E. P. Dietrich and E. M. Bruscia, PLoS One, 2012, 7, e35499 CrossRef CAS PubMed.
- N. A. M. Khir, A. S. M. Noh, I. Long, R. Zakaria and C. A. N. Ismail, Mol. Cell. Biochem., 2023, 479, 539–552 CrossRef PubMed.
- M.-H. Li, Y.-N. Cha and Y.-J. Surh, Biochem. Biophys. Res. Commun., 2006, 342, 984–990 CrossRef CAS PubMed.
- A. Józkowicz, I. Huk, A. Nigisch, G. Weigel, W. Dietrich, R. Motterlini and J. Dulak, Antioxid. Redox Signaling, 2003, 5, 155–162 CrossRef PubMed.
- T. Vera, J. R. Henegar, H. A. Drummond, J. M. Rimoldi and D. E. Stec, J. Am. Soc. Nephrol., 2005, 16, 950–958 CrossRef CAS PubMed.
- M. Allanson and V. E. Reeve, Cancer Immunol. Immunother., 2007, 56, 1807–1815 CrossRef CAS PubMed.
- S. J. Stanford, M. J. Walters, A. A. Hislop, S. G. Haworth, T. W. Evans, B. E. Mann, R. Motterlini and J. A. Mitchell, Eur. J. Pharmacol., 2003, 473, 135–141 CrossRef CAS PubMed.
- L. Shao, C. Liu, S. Wang, J. Liu, L. Wang, L. Lv and Y. Zou, Oncol. Lett., 2018, 16, 3223–3230 Search PubMed.
- M. Juszczak, M. Kluska, D. Wysokiński and K. Woźniak, Sci. Rep., 2020, 10, 12200 CrossRef CAS PubMed.
- K. Fujita, Y. Tanaka, T. Sho, S. Ozeki, S. Abe, T. Hikage, T. Kuchimaru, S. Kizaka-Kondoh and T. Ueno, J. Am. Chem. Soc., 2014, 136, 16902–16908 CrossRef CAS PubMed.
- L. Zhang, L. Laug, W. Münchgesang, E. Pippel, U. Gösele, M. Brandsch and M. Knez, Nano Lett., 2010, 10, 219–223 CrossRef PubMed.
- H. Tabe, K. Fujita, S. Abe, M. Tsujimoto, T. Kuchimaru, S. Kizaka-Kondoh, M. Takano, S. Kitagawa and T. Ueno, Inorg. Chem., 2015, 54, 215–220 CrossRef CAS PubMed.
- P. Kaczara, B. Sitek, K. Przyborowski, A. Kurpinska, K. Kus, M. Stojak and S. Chlopicki, Arterioscler., Thromb., Vasc. Biol., 2020, 40, 2376–2390 CrossRef CAS PubMed.
- M. Ryan, N. Jernigan, H. Drummond, G. Mclemorejr, J. Rimoldi, S. Poreddy, R. Gadepalli and D. Stec, Pharmacol. Res., 2006, 54, 24–29 CrossRef CAS PubMed.
- S. Basuroy, C. W. Leffler and H. Parfenova, Am. J. Physiol.: Cell Physiol., 2013, 304, C1105–C1115 CrossRef CAS PubMed.
- P. Fagone, K. Mangano, S. Mammana, E. Cavalli, R. Di Marco, M. L. Barcellona, L. Salvatorelli, G. Magro and F. Nicoletti, Clin. Immunol., 2015, 157, 198–204 CrossRef CAS PubMed.
- T. R. Johnson, B. E. Mann, I. P. Teasdale, H. Adams, R. Foresti, C. J. Green and R. Motterlini, Dalton Trans., 2007, 1500 RSC.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romão and M. J. Romão, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romao and M. J. Romao, Curr. Med. Chem., 2011, 18, 3361–3366 CrossRef CAS PubMed.
- M. Tinajero-Trejo, K. J. Denby, S. E. Sedelnikova, S. A. Hassoubah, B. E. Mann and R. K. Poole, J. Biol. Chem., 2014, 289, 29471–29482 CrossRef CAS PubMed.
- A. Bagul, S. A. Hosgood, M. Kaushik and M. L. Nicholson, Transplantation, 2008, 85, 576–581 CrossRef CAS PubMed.
- G. Wang, T. Hamid, R. J. Keith, G. Zhou, C. R. Partridge, X. Xiang, J. R. Kingery, R. K. Lewis, Q. Li, D. G. Rokosh, R. Ford, F. G. Spinale, D. W. Riggs, S. Srivastava, A. Bhatnagar, R. Bolli and S. D. Prabhu, Circulation, 2010, 121, 1912–1925 CrossRef CAS PubMed.
- M. D. Musameh, C. J. Green, B. E. Mann, B. J. Fuller and R. Motterlini, J. Heart Lung Transplant., 2007, 26, 1192–1198 CrossRef PubMed.
- R. Foresti, J. Hammad, J. E. Clark, T. R. Johnson, B. E. Mann, A. Friebe, C. J. Green and R. Motterlini, Br. J. Pharmacol., 2004, 142, 453–460 CrossRef CAS PubMed.
- L. Lo Iacono, J. Boczkowski, R. Zini, I. Salouage, A. Berdeaux, R. Motterlini and D. Morin, Free Radicals Biol. Med., 2011, 50, 1556–1564 CrossRef CAS PubMed.
- M. G. Bani-Hani, D. Greenstein, B. E. Mann, C. J. Green and R. Motterlini, Pharmacol. Rep., 2006, 58(Suppl), 132–144 Search PubMed.
- M. L. Ferrandiz, N. Maicas, I. Garcia-Arnandis, M. C. Terencio, R. Motterlini, I. Devesa, L. A. B. Joosten, W. B. Van Den Berg and M. J. Alcaraz, Ann. Rheum. Dis., 2007, 67, 1211–1217 CrossRef PubMed.
- A. Yabluchanskiy, P. Sawle, S. Homer-Vanniasinkam, C. J. Green, R. Foresti and R. Motterlini, Crit. Care Med., 2012, 40, 544–552 CrossRef CAS PubMed.
- M. Desmard, K. S. Davidge, O. Bouvet, D. Morin, D. Roux, R. Foresti, J. D. Ricard, E. Denamur, R. K. Poole, P. Montravers, R. Morterlini and J. Boczkowski, FASEB J., 2009, 23, 1023–1031 CrossRef CAS PubMed.
- S. M. Carvalho, J. Marques, C. C. Romão and L. M. Saraiva, Antimicrob. Agents Chemother., 2019, 63, e00643–e00619 CAS.
- J. L. Wilson, H. E. Jesse, B. Hughes, V. Lund, K. Naylor, K. S. Davidge, G. M. Cook, B. E. Mann and R. K. Poole, Antioxid. Redox Signaling, 2013, 19, 497–509 CrossRef CAS PubMed.
- T. Obara, H. Yamamoto, T. Aokage, T. Igawa, T. Nojima, T. Hirayama, M. Seya, M. Ishikawa-Aoyama, A. Nakao, R. Motterlini and H. Naito, Transplantation, 2022, 106, 1365–1375 CrossRef CAS PubMed.
- J. Wang, D. Zhang, X. Fu, L. Yu, Z. Lu, Y. Gao, X. Liu, J. Man, S. Li, N. Li, X. Chen, M. Hong, Q. Yang and J. Wang, J. Neuroinflamm., 2018, 15, 188 CrossRef PubMed.
- H. Yamamoto-Oka, S. Mizuguchi, M. Toda, Y. Minamiyama, S. Takemura, T. Shibata, G. Cepinskas and N. Nishiyama, Inflammopharmacology, 2018, 26, 435–445 CrossRef CAS PubMed.
- K. Lu, W.-J. Wu, C. Zhang, Y.-L. Zhu, J.-Q. Zhong and J. Li, Curr. Neurovasc. Res., 2020, 17, 464–470 CrossRef CAS PubMed.
- C. Jin, B. Lin, G. Zheng, K. Tan, G. Liu, Z. Yao, J. Xie, W. Chen, L. Chen, T. Xu, C. Huang, Z. Wu and L. Yang, Oxid. Med. Cell. Longevity, 2022, 2022, 1–21 Search PubMed.
- A. Stein, Y. Guo, W. Tan, W. Wu, X. Zhu, Q. Li, C. Luo, B. Dawn, T. Johnson and R. Motterlini, J. Mol. Cell. Cardiol., 2005, 38, 127–134 CrossRef CAS PubMed.
- Y. Guo, A. B. Stein, W.-J. Wu, W. Tan, X. Zhu, Q.-H. Li, B. Dawn, R. Motterlini and R. Bolli, Am. J. Physiol.: Heart Circ. Physiol., 2004, 286, H1649–H1653 CrossRef CAS PubMed.
- Y. Tayem, T. R. Johnson, B. E. Mann, C. J. Green and R. Motterlini, Am. J. Physiol.: Renal Physiol., 2006, 290, F789–F794 CrossRef CAS PubMed.
- L.-M. Zhang, D.-X. Zhang, W.-C. Zheng, J.-S. Hu, L. Fu, Y. Li, Y. Xin and X.-P. Wang, Exp. Neurol., 2021, 341, 113683 CrossRef CAS PubMed.
- Y. Dai, H. Chen, Y. Pan and H. Song, BioMed Res. Int., 2022, 2022, 1–10 Search PubMed.
- M. Seveso, M. Vadori, E. Bosio, F. Fante, F. Besenzon, L. Ravarotto, S. Bedendo, T. Johnson, B. Mann and R. Motterlini, Am. J. Transplant., 2017, 17, 557–564 CrossRef PubMed.
- E. Masini, A. Vannacci, P. Failli, R. Mastroianni, L. Giannini, M. C. Vinci, C. Uliva, R. Motterlini and P. F. Mannaioni, FASEB J., 2008, 22, 3380–3388 CrossRef CAS PubMed.
- M. Vadori, M. Seveso, F. Besenzon, E. Bosio, E. Tognato, F. Fante, M. Boldrin, S. Gavasso, L. Ravarotto, B. E. Mann, P. Simioni, E. Ancona, R. Motterlini and E. Cozzi, Xenotransplantation, 2009, 16, 99–114 CrossRef PubMed.
- R. F. Lyon, H. M. Southam, C. R. Trevitt, C. Liao, S. F. El-Khamisy, R. K. Poole and M. P. Williamson, Biochem. J., 2022, 479, 1429–1439 CrossRef CAS PubMed.
- S. H. Crook, B. E. Mann, A. J. H. M. Meijer, H. Adams, P. Sawle, D. Scapens and R. Motterlini, Dalton Trans., 2011, 40, 4230 RSC.
- S. McLean, B. E. Mann and R. K. Poole, Anal. Biochem., 2012, 427, 36–40 CrossRef CAS PubMed.
- S. V. C. Vummaleti, D. Branduardi, M. Masetti, M. De Vivo, R. Motterlini and A. Cavalli, Chem. – Eur. J., 2012, 18, 9267–9275 CrossRef CAS PubMed.
- R. Motterlini, A. Nikam, S. Manin, A. Ollivier, J. L. Wilson, S. Djouadi, L. Muchova, T. Martens, M. Rivard and R. Foresti, Red. Biol., 2019, 20, 334–348 CrossRef CAS PubMed.
- G. Yang, M. Fan, J. Zhu, C. Ling, L. Wu, X. Zhang, M. Zhang, J. Li, Q. Yao, Z. Gu and X. Cai, Biomaterials, 2020, 255, 120155 CrossRef CAS PubMed.
- J. Chen, D. Chen, J. Chen, T. Shen, T. Jin, B. Zeng, L. Li, C. Yang, Z. Mu, H. Deng and X. Cai, Acta Biomater., 2022, 146, 49–65 CrossRef CAS PubMed.
- W. Ma, X. Chen, L. Fu, J. Zhu, M. Fan, J. Chen, C. Yang, G. Yang, L. Wu, G. Mao, X. Yang, X. Mou, Z. Gu and X. Cai, ACS Appl. Mater. Interfaces, 2020, 12, 22479–22491 CrossRef CAS PubMed.
- J. Liu, R. S. Li, M. He, Z. Xu, L. Q. Xu, Y. Kang and P. Xue, Biomaterials, 2021, 277, 121084 CrossRef CAS PubMed.
- L. K. Wareham, S. McLean, R. Begg, N. Rana, S. Ali, J. J. Kendall, G. Sanguinetti, B. E. Mann and R. K. Poole, Antioxid. Redox Signaling, 2018, 28, 1286–1308 CrossRef CAS PubMed.
- S. Fayad-Kobeissi, J. Ratovonantenaina, H. Dabiré, J. L. Wilson, A. M. Rodriguez, A. Berdeaux, J.-L. Dubois-Randé, B. E. Mann, R. Motterlini and R. Foresti, Biochem. Pharmacol., 2016, 102, 64–77 CrossRef CAS PubMed.
- D. Zhang, Z. Lin, Y. Zheng, J. Song, J. Li, Y. Zeng and X. Liu, ACS Nano, 2020, 14, 8985–8999 CrossRef CAS PubMed.
- M. Stojak, P. Kaczara, R. Motterlini and S. Chlopicki, Pharmacol. Res., 2018, 136, 160–171 CrossRef CAS PubMed.
- R. N. Bhattacharjee, M. Richard-Mohamed, Q. Sun, A. Haig, G. Aboalsamh, P. Barrett, R. Mayer, I. Alhasan, K. Pineda-Solis, L. Jiang, H. Alharbi, M. Saha, E. Patterson, A. Sener, G. Cepinskas, A. M. Jevnikar and P. P. W. Luke, Transplantation, 2018, 102, 1066–1074 CrossRef CAS PubMed.
- D. Babu, G. Leclercq, R. Motterlini and R. A. Lefebvre, Front. Pharmacol., 2017, 8, 31 Search PubMed.
- S. F. Kobeissi, J. L. Wilson, B. Michel, J.-L. Dubois-Randé, R. Motterlini and R. Foresti, Arc. Cardiovasc. Dis. Suppl., 2014, 6, 17 Search PubMed.
- L. Wu, X. Cai, H. Zhu, J. Li, D. Shi, D. Su, D. Yue and Z. Gu, Adv. Funct. Mater., 2018, 28, 1804324 CrossRef.
- F. Yang, W. Yu, Q. Yu, X. Liu, C. Liu, C. Lu, X. Liao, Y. Liu and N. Peng, Small, 2023, 19, 2206124 CrossRef CAS PubMed.
- P. Kaczara, R. Motterlini, G. M. Rosen, B. Augustynek, P. Bednarczyk, A. Szewczyk, R. Foresti and S. Chlopicki, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 1297–1309 CrossRef CAS PubMed.
- L. Braud, M. Pini, L. Muchova, S. Manin, H. Kitagishi, D. Sawaki, G. Czibik, J. Ternacle, G. Derumeaux, R. Foresti and R. Motterlini, JCI Insight, 2018, 3, e123485 CrossRef PubMed.
- U. Hasegawa, A. J. Van Der Vlies, E. Simeoni, C. Wandrey and J. A. Hubbell, J. Am. Chem. Soc., 2010, 132, 18273–18280 CrossRef CAS PubMed.
- Y. Morimoto, W. Durante, D. G. Lancaster, J. Klattenhoff and F. K. Tittel, Am. J. Physiol.: Heart Circ. Physiol., 2001, 280, H483–H488 CrossRef CAS PubMed.
- G. S. Marks, H. J. Vreman, B. E. McLaughlin, J. F. Brien and K. Nakatsu, Antioxid. Redox Signaling, 2002, 4, 271–277 CrossRef CAS PubMed.
- M. Klein, U. Neugebauer, A. Gheisari, A. Malassa, T. M. A. Jazzazi, F. Froehlich, M. Westerhausen, M. Schmitt and J. Popp, J. Phys. Chem. A, 2014, 118, 5381–5390 CrossRef CAS PubMed.
- J. Esteban, J. V. Ros–Lis, R. Martínez-Máñez, M. D. Marcos, M. Moragues, J. Soto and F. Sancenón, Angew. Chem., 2010, 122, 5054–5057 CrossRef.
- A. J. Atkin, J. M. Lynam, B. E. Moulton, P. Sawle, R. Motterlini, N. M. Boyle, M. T. Pryce and I. J. S. Fairlamb, Dalton Trans., 2011, 40, 5755 RSC.
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668–15671 CrossRef CAS PubMed.
- J. Wang, J. Karpus, B. S. Zhao, Z. Luo, P. R. Chen and C. He, Angew. Chem., Int. Ed., 2012, 51, 9652–9656 CrossRef CAS PubMed.
- K. Ling, F. Men, W.-C. Wang, Y.-Q. Zhou, H.-W. Zhang and D.-W. Ye, J. Med. Chem., 2018, 61, 2611–2635 CrossRef CAS PubMed.
- N. C. Burton and T. R. Guilarte, Environ. Health Perspect., 2009, 117, 325–332 CrossRef CAS PubMed.
- M. Liu, Z. J. Lim, Y. Y. Gwee, A. Levina and P. A. Lay, Angew. Chem., Int. Ed., 2010, 49, 1661–1664 CrossRef CAS PubMed.
- B. D. Chatterjee, A. Mitra and G. S. De, Platinum Met. Rev., 2006, 50, 2–12 CrossRef.
- J. Niesel, A. Pinto, H. W. Peindy N'Dongo, K. Merz, I. Ott, R. Gust and U. Schatzschneider, Chem. Commun., 2008, 1798 RSC.
- U. Schatzschneider, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS.
- X. Xie, Y. Yan, N. Zhu and G. Liu, Eur. J. Med. Chem., 2014, 76, 67–78 CrossRef CAS PubMed.
- S. J. Carrington, I. Chakraborty and P. K. Mascharak, Chem. Commun., 2013, 49, 11254 RSC.
- S. J. Carrington, I. Chakraborty, J. M. L. Bernard and P. K. Mascharak, ACS Med. Chem. Lett., 2014, 5, 1324–1328 CrossRef CAS PubMed.
- C. A. Kumar, R. Nagarajaprakash, W. Victoria, V. Veena, N. Sakthivel and B. Manimaran, Inorg. Chem. Commun., 2016, 64, 39–44 CrossRef CAS.
- E. Üstün, A. Özgür, K. A. Coşkun, S. Demir, İ. Özdemir and Y. Tutar, J. Coord. Chem., 2016, 69, 3384–3394 CrossRef.
- E. Üstün, A. Özgür, K. A. Coşkun, S. Demir Düşünceli, İ. Özdemir and Y. Tutar, Transition Met. Chem., 2017, 42, 331–337 CrossRef.
- I. Chakraborty, S. J. Carrington, G. Roseman and P. K. Mascharak, Inorg. Chem., 2017, 56, 1534–1545 CrossRef CAS PubMed.
- C. Ashok Kumar, D. Divya, R. Nagarajaprakash, V. Veena, P. Vidhyapriya, N. Sakthivel and B. Manimaran, J. Organomet. Chem., 2017, 846, 152–160 CrossRef CAS.
- P. Vidhyapriya, D. Divya, M. Bala and N. Sakthivel, J. Photochem. Photobiol., B, 2018, 188, 28–41 CrossRef PubMed.
- J. Jimenez, I. Chakraborty, A. Dominguez, J. Martinez-Gonzalez, W. M. C. Sameera and P. K. Mascharak, Inorg. Chem., 2018, 57, 1766–1773 CrossRef CAS PubMed.
- D. Musib, M. K. Raza, Kh. Martina and M. Roy, Polyhedron, 2019, 172, 125–131 CrossRef CAS.
- H. G. Daniels, O. G. Fast, S. M. Shell and F. A. Beckford, J. Photochem. Photobiol., A, 2019, 374, 84–94 CrossRef CAS.
- M. N. Pinto, I. Chakraborty, J. Jimenez, K. Murphy, J. Wenger and P. K. Mascharak, Inorg. Chem., 2019, 58, 14522–14531 CrossRef CAS PubMed.
- M. Hu, Y. Yan, B. Zhu, F. Chang, S. Yu and G. Alatan, RSC Adv., 2019, 9, 20505–20512 RSC.
- A. M. Mansour and A. Friedrich, Inorg. Chem. Front., 2017, 4, 1517–1524 RSC.
- U. Kumar, S. Roy, R. K. Jha, P. Vidhyapriya, N. Sakthivel and B. Manimaran, ACS Omega, 2019, 4, 1923–1930 CrossRef CAS.
- U. Kumar, S. Jose, D. Divya, P. Vidhyapriya, N. Sakthivel and B. Manimaran, New J. Chem., 2019, 43, 7520–7531 RSC.
- J. Jimenez, M. N. Pinto, J. Martinez-Gonzalez and P. K. Mascharak, Inorg. Chim. Acta, 2019, 485, 112–117 CrossRef CAS.
- C. Zou, H. Zhang, Q. Li, H. Xiao, L. Yu, S. Ke, L. Zhou, W. Liu, W. Wang, H. Huang, N. Ma, Q. Liu, X. Wang, W. Zhao, H. Zhou and X. Gao, Carcinogenesis, 2011, 32, 1840–1848 CrossRef CAS PubMed.
- J. Rossier, J. Delasoie, L. Haeni, D. Hauser, B. Rothen-Rutishauser and F. Zobi, J. Inorg. Biochem., 2020, 209, 111–122 CrossRef PubMed.
- R. M. Khaled, A. Friedrich, M. A. Ragheb, N. T. Abdel-Ghani and A. M. Mansour, Dalton Trans., 2020, 49, 9294–9305 RSC.
- D. Musib, M. K. Raza, M. Pal and M. Roy, Appl. Organomet. Chem., 2021, 35, e6110 CrossRef CAS.
- A. M. Mansour, K. Radacki, R. M. Khaled, M. H. Soliman and N. T. Abdel-Ghani, J. Biol. Inorg. Chem., 2021, 26, 135–147 CrossRef CAS PubMed.
- Y. Zhou, Y. Sun, K. Yi, Z. Wang, Y. Liu and C. He, Inorg. Chem. Front., 2022, 9, 5941–5949 RSC.
- R. M. Khaled, D. A. Habashy, A. Y. Ahmed, O. S. Ismael, S. S. Ibrahim, M. Abdelfatah, K. Radacki and A. M. Mansour, Polyhedron, 2022, 225, 116048 CrossRef CAS.
- D. A. Habashy, R. M. Khaled, A. Y. Ahmed, K. Radacki, S. K. Ahmed, E. K. Tharwat, H. Magdy, A. Zeinhom and A. M. Mansour, Dalton Trans., 2022, 51, 14041–14048 RSC.
- R. M. Khaled, K. Radacki, S. A. Al-Abraq, E. El-Hussieny, G. A. E. Mostafa, E. A. Ali, O. R. Shehab and A. M. Mansour, Polyhedron, 2023, 244, 116574 CrossRef CAS.
- K. Fujita, Y. Tanaka, S. Abe and T. Ueno, Angew. Chem., Int. Ed., 2016, 55, 1056–1060 CrossRef CAS PubMed.
- W. Beck, W. Petri and J. Meder, J. Organomet. Chem., 1980, 191, 73–77 CrossRef CAS.
- M. Marín-García, N. Benseny-Cases, M. Camacho, Y. Perrie, J. Suades and R. Barnadas-Rodríguez, Dalton Trans., 2018, 47, 14293–14303 RSC.
- D. Achatz, M. A. Lang, A. Völkl, W. P. Fehlhammer and W. Beck, Z. Anorg. Allg. Chem., 2005, 631, 2339–2346 CrossRef CAS.
- S. Reiländer, W. Schmehl, K. Popp, K. Nuss, P. Kronen, D. Verdino, C. Wiezorek, M. Gutmann, L. Hahn, C. Däubler, A. Meining, M. Raschig, F. Kaiser, B. Von Rechenberg, O. Scherf-Clavel and L. Meinel, ACS Biomater. Sci. Eng., 2023, 9, 2937–2948 CrossRef PubMed.
- Y. Gong, T. Zhang, H. Liu, Y. Zheng, N. Li, Q. Zhao, Y. Chen and B. Liu, Transition Met. Chem., 2015, 40, 413–426 CrossRef CAS.
- Y. Gong, T. Zhang, M. Li, N. Xi, Y. Zheng, Q. Zhao, Y. Chen and B. Liu, Free Radicals Biol. Med., 2016, 97, 362–374 CrossRef CAS PubMed.
- J. Li, J. Zhang, Q. Zhang, Z. Bai, Q. Zhao, Z. Wang, Y. Chen and B. Liu, J. Organomet. Chem., 2018, 874, 49–62 CrossRef CAS.
- H.-P. Liu, Y. Liao, M.-Z. Ren, Z.-J. Quan and X.-C. Wang, Bioorg. Chem., 2021, 107, 104621 CrossRef CAS PubMed.
- R. Motterlini, B. E. Mann and R. Foresti, Expert Opin. Invest. Drugs, 2005, 14, 1305–1318 CrossRef CAS PubMed.
- P. Sawle, J. Hammad, I. J. S. Fairlamb, B. Moulton, C. T. O'Brien, J. M. Lynam, A. K. Duhme-Klair, R. Foresti and R. Motterlini, J. Pharmacol. Exp. Ther., 2006, 318, 403–410 CrossRef CAS PubMed.
- D. J. Jones and R. J. Mawby, Inorg. Chim. Acta, 1972, 6, 157–160 CrossRef CAS.
- L. Hewison, S. H. Crook, T. R. Johnson, B. E. Mann, H. Adams, S. E. Plant, P. Sawle and R. Motterlini, Dalton Trans., 2010, 39, 8967 RSC.
- R. Kretschmer, G. Gessner, H. Görls, S. H. Heinemann and M. Westerhausen, J. Inorg. Biochem., 2011, 105, 6–9 CrossRef CAS PubMed.
- M.-C. P. Yeh and C.-C. Hwu, J. Organomet. Chem., 1991, 419, 341–355 CrossRef CAS.
- D. Zhang, B. M. Krause, H.-G. Schmalz, P. Wohlfart, B. A. Yard and R. Schubert, Front. Pharmacol., 2021, 12, 702392 CrossRef CAS PubMed.
- R. Motterlini, P. Sawle, J. Hammad, B. E. Mann, T. R. Johnson, C. J. Green and R. Foresti, Pharmacol. Res., 2013, 68, 108–117 CrossRef CAS PubMed.
- S. Romanski, E. Stamellou, J. T. Jaraba, D. Storz, B. K. Krämer, M. Hafner, S. Amslinger, H. G. Schmalz and B. A. Yard, Free Radicals Biol. Med., 2013, 65, 78–88 CrossRef CAS PubMed.
- N. S. Sitnikov, Y. Li, D. Zhang, B. Yard and H. Schmalz, Angew. Chem., Int. Ed., 2015, 54, 12314–12318 CrossRef CAS PubMed.
- B. Bauer, A. Göderz, H. Braumüller, J. M. Neudörfl, M. Röcken, T. Wieder and H. Schmalz, ChemMedChem, 2017, 12, 1927–1930 CrossRef CAS PubMed.
- H. M. Southam, T. W. Smith, R. L. Lyon, C. Liao, C. R. Trevitt, L. A. Middlemiss, F. L. Cox, J. A. Chapman, S. F. El-Khamisy, M. Hippler, M. P. Williamson, P. J. F. Henderson and R. K. Poole, Red. Biol., 2018, 18, 114–123 CrossRef CAS PubMed.
- H. M. Southam, M. P. Williamson, J. A. Chapman, R. L. Lyon, C. R. Trevitt, P. J. F. Henderson and R. K. Poole, Antioxidants, 2021, 10, 915 CrossRef CAS PubMed.
- L. Hemmersbach, R. Adam, C. Plevnali, X. Zhang, B. Yard and H. Schmalz, Eur. J. Org. Chem., 2023, e202201424 CrossRef CAS.
- L. Hemmersbach, Y. Schreiner, X. Zhang, F. Dicke, L. Hünemeyer, J. Neudörfl, T. Fleming, B. Yard and H. Schmalz, Chem. – Eur. J., 2022, 28, e202201670 CrossRef CAS PubMed.
- P. Wang, H. Liu, Q. Zhao, Y. Chen, B. Liu, B. Zhang and Q. Zheng, Eur. J. Med. Chem., 2014, 74, 199–215 CrossRef CAS PubMed.
- J. D. Seixas, M. F. A. Santos, A. Mukhopadhyay, A. C. Coelho, P. M. Reis, L. F. Veiros, A. R. Marques, N. Penacho, A. M. L. Gonçalves, M. J. Romão, G. J. L. Bernardes, T. Santos-Silva and C. C. Romão, Dalton Trans., 2015, 44, 5058–5075 RSC.
- J. D. Seixas, M. Chaves-Ferreira, D. Montes-Grajales, A. M. Gonçalves, A. R. Marques, L. M. Saraiva, J. Olivero-Verbel, C. C. Romão and G. J. L. Bernardes, Chem. – Eur. J., 2015, 21, 14708–14712 CrossRef CAS PubMed.
- G. Tamasi, A. Merlino, F. Scaletti, P. Heffeter, A. A. Legin, M. A. Jakupec, W. Berger, L. Messori, B. K. Keppler and R. Cini, Dalton Trans., 2017, 46, 3025–3040 RSC.
- N. Pontillo, G. Ferraro, L. Messori, G. Tamasi and A. Merlino, Dalton Trans., 2017, 46, 9621–9629 RSC.
- G. Tamasi, A. Carpini, D. Valensin, L. Messori, A. Pratesi, F. Scaletti, M. Jakupec, B. Keppler and R. Cini, Polyhedron, 2014, 81, 227–237 CrossRef CAS.
- S. Geri, T. Krunclova, O. Janouskova, J. Panek, M. Hruby, D. Hernández-Valdés, B. Probst, R. A. Alberto, C. Mamat, M. Kubeil and H. Stephan, Chem. – Eur. J., 2020, 26, 10992–11006 CrossRef CAS PubMed.
- N. M. Ibrahim, R. M. Khaled, M. A. Ragheb, K. Radacki, A. M. Farag and A. M. Mansour, Dalton Trans., 2021, 50, 15389–15399 RSC.
- X. Zhang, N. Guo, S. Yang, H. Khan and W. Zhang, Materials, 2022, 15, 3597 CrossRef CAS PubMed.
- F. Zobi, O. Blacque, R. A. Jacobs, M. C. Schaub and A. Yu. Bogdanova, Dalton Trans., 2012, 41, 370–378 RSC.
- T. J. Haley and F. D. Cartwright, J. Pharm. Sci., 1968, 57, 321–323 CrossRef CAS PubMed.
- K. Koike, N. Okoshi, H. Hori, K. Takeuchi, O. Ishitani, H. Tsubaki, I. P. Clark, M. W. George, F. P. A. Johnson and J. J. Turner, J. Am. Chem. Soc., 2002, 124, 11448–11455 CrossRef CAS PubMed.
- A. E. Pierri, A. Pallaoro, G. Wu and P. C. Ford, J. Am. Chem. Soc., 2012, 134, 18197–18200 CrossRef CAS PubMed.
- S. J. Carrington, I. Chakraborty, J. M. L. Bernard and P. K. Mascharak, Inorg. Chem., 2016, 55, 7852–7858 CrossRef CAS PubMed.
- I. Chakraborty, J. Jimenez, W. M. C. Sameera, M. Kato and P. K. Mascharak, Inorg. Chem., 2017, 56, 2863–2873 CrossRef CAS PubMed.
- J. S. Ward, in Organomet. Chem, ed. I. Fairlamb and J. Lynam, Royal Society of Chemistry, Cambridge, 2015, vol. 40, pp. 140–176 Search PubMed.
- A. M. Mansour, R. M. Khaled, K. Radacki, Z. Younes, M. Gamal, B. Guirguis, G. A. E. Mostafa, E. A. Ali and O. R. Shehab, Dalton Trans., 2023, 52, 10286–10293 RSC.
- E. Kottelat and Z. Fabio, Inorganics, 2017, 5, 24 CrossRef.
- H.-I. Choi, A. Zeb, M.-S. Kim, I. Rana, N. Khan, O. S. Qureshi, C.-W. Lim, J.-S. Park, Z. Gao, H.-J. Maeng and J.-K. Kim, J. Controlled Release, 2022, 350, 652–667 CrossRef CAS PubMed.
- M. I. Khan, M. I. Hossain, M. K. Hossain, M. H. K. Rubel, K. M. Hossain, A. M. U. B. Mahfuz and M. I. Anik, ACS Appl. Bio Mater., 2022, 5, 971–1012 CrossRef CAS PubMed.
Footnote |
| † The authors contributed equally to this review article. |
| This journal is © The Royal Society of Chemistry 2024 |