
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterisation and reactivity of a zinc triazenide for potential use in vapour deposition†
Rouzbeh
Samii
 a,
Essi
Barkas
b,
David
Zanders
c,
Anton
Fransson
a,
Manu
Lahtinen
b,
Vadim
Kessler
d,
Heikki M.
Tuononen
b,
Jani O.
Moilanen
b and
Nathan J.
O'Brien
*a
a,
Essi
Barkas
b,
David
Zanders
c,
Anton
Fransson
a,
Manu
Lahtinen
b,
Vadim
Kessler
d,
Heikki M.
Tuononen
b,
Jani O.
Moilanen
b and
Nathan J.
O'Brien
*a
aDepartment of Physics, Chemistry and Biology, Linköping University, Linköping SE-58183, Sweden. E-mail: nathan.o.brien@liu.se
bDepartment of Chemistry, Nanoscience Centre, University of Jyväskylä, P.O. Box 35, FI-40014, Jyväskylä, Finland
cFaculty of Chemistry and Biochemistry, Ruhr University Bochum, Bochum 44801, Germany
dDepartment of Molecular Sciences, Swedish University of Agricultural Sciences, P.O. Box 7015, 75007 Uppsala, Sweden
First published on 28th February 2024
Abstract
In this study, we synthesised and characterised a new zinc(II) triazenide for potential use in vapour deposition of zinc sulphide thin films. The compound is volatile and quantitatively sublimes at 80 °C under vacuum (0.5 mbar). Thermogravimetric analysis showed a one-step volatilisation with an onset temperature at ∼125 °C and 5% residual mass. The compound also reacted with 2 or 4 molar equivalents of triphenylsilanethiol to give dimeric and monomeric zinc thiolates, respectively. The high volatility, thermal stability, and reactivity with sterically hindered thiols makes this compound a potential candidate for use in vapour deposition of zinc containing thin films.
Introduction
Zinc sulphide (ZnS) is a semi-conductor with optical properties suitable for application in a variety of modern-day electronic devices.1 As these devices become smaller with more sophisticated architectures, atomic layer deposition (ALD) has become an important tool for fabrication of future microelectronics. During an ALD process, the metal and non-metal precursors are introduced into the reactor separately. This means the process is highly reliant on the precursors, particularly the metal precursor, to have suitable and favourable surface chemistry. Although there are many publications on ALD of ZnS, there is only a limited number of suitable precursors reported. A majority of the ALD processes use either Me2Zn or Et2Zn with H2S to deposit polycrystalline ZnS.2–6 The popularity of Me2Zn and Et2Zn stems from their high volatility and reactivity, which are desirable properties for ALD precursors. These dialkylzinc precursors are highly pyrophoric, making their transport and handling difficult and dangerous on large scale.7 Safer alternative Zn–O bonded precursors, such as zinc acetate (Zn(OAc)2) and bis(2,2,6,6-tetramethyl-3,5-heptanedionato)zinc (Zn(thd)2), have been investigated for ALD of ZnS.8–11 These films suffer from oxygen and carbon impurities, and non-stoichiometric ratios of Zn and S.Precursors with only M–N bonds are desirable for ALD due to their highly polarised and reactive bonds, which make the formation of sulphides thermodynamically favourable. To date, bis(trimethylsilylamide)zinc (Zn(NSiMe3)2) is the only suitable Zn–N bonded compound, but it has not been used in an ALD process.12 Homoleptic bidentate amidinate and guanidinate ligands have been widely utilised to provide thermally stable and volatile compounds for ALD.13 Although there are many Zn(II) 1,3-dialkylamidinates and guanidinates in the literature, no volatility data has been reported.14–17 Our previous ALD study using amidinates and guanidinates showed that decreasing the size of the carbon substituent on the ligand backbone improved both the physical properties and deposition chemistry of the precursor.18 This makes the triazenide ligand, which differs from amidinates by having a nitrogen atom instead of a substituted carbon, a promising alternative for producing volatile Zn precursors. Only two homoleptic zinc triazenides have been reported to date: bis(1,3-(2,6-diisopropylphenyl)triazenide)zinc(II) (Zn(dippt)2)19 and (1,3-dimethyltriazenide)zinc(II) ((Zn(dmt)2)x).20 No volatility data, however, was stated for either of Zn(dippt)2 or (Zn(dmt)2)x, most likely due to strong intramolecular interactions and polymeric structure, respectively.
Recently, we reported a range of volatile 1,3-dialkyltriazenide compounds for group 11, 13, and 14 metals.21–26 The Ga and In triazenides have been used as ALD precursors to afford excellent quality thin films of GaN, InN, InGaN, and In2O3.21,22,27–29 The group 11 triazenides have great promise as single-source precursors, whilst the group 14 triazenides have desirable thermal and volatility properties.24,26 During our studies, volatile and thermally stable lanthanide dialkyltriazenides have also been reported.30 With the success of the triazenide ligand to produce volatile and thermally stable metal compounds, we decided to investigate its reactivity with divalent Zn. Herein, we report the synthesis, structure, and thermal properties of a Zn(II) triazenide and its reactivity with sterically bulky thiols that mimic a ZnS surface. The high volatility, thermal stability, and reactivity with thiols makes this new precursor extremely interesting for use in vapour deposition of Zn containing thin films.
Results and discussion
Reaction of lithium 1,3-di-tert-butyltriazenide24 with ZnCl2 in THF yielded 1 in good yield after sublimation and recrystallisation (Scheme 1). This synthesis is easy to scale up, which is also the case for the lithium triazenide starting material. The energetic properties of 1 have not been fully characterized and thus care should be taken when handling the compound (see Experimental section) as some metal 1,3-dimethyltriazenides have shown explosive characteristics.20 Compound 1 is stable under nitrogen but immediately degrades when exposed to air. The compound was fully characterised by nuclear magnetic resonance (NMR), elemental analysis, melting point, and single crystal X-ray crystallography.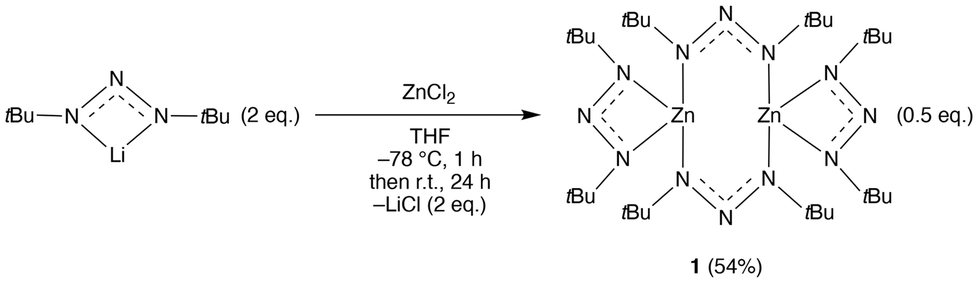 | ||
| Scheme 1 Synthesis of bis(1,3-di-tert-butyltriazenide)zinc(II) 1. | ||
The crystal structure of 1 showed two Zn atoms in distorted tetrahedral geometries each with a terminal chelating ligand, and two ligands bridging the metal centres (Fig. 1). This result is in contrast with the previously reported Zn(dippt)2 analogue,19 which showed a monomeric structure, and suggests that the 1,3-di-tert-butyltriazenide ligand is not sufficiently bulky to inhibit dimerization. The Zn–Zn distance was ∼3.25 Å, which is more than twice the van der Waals radius of Zn (ca. 1.39 Å). This indicates that the metal centres are not engaged in M–M bonding. While the bridging ligands displayed similar Zn–N bond lengths (2.0298(12) and 2.0241(12) Å), each chelating ligand showed a significant variation between the two Zn–N bonds (2.0000(12) and 2.3116(12) Å). This difference is likely caused by steric strain between the tert-butyl groups of the chelating and bridging ligands. Despite the large difference in the Zn–N bond lengths, there is only ~0.01 Å difference between the N–N bond lengths of the chelating ligands (1.3041(18) and 1.2898(18) Å). This suggests a significant degree of electron delocalization over the π-system in the N3 backbone. For the bridging ligands, the Zn atoms are not confined to the plane spanned by their N3 backbone.
 | ||
| Fig. 1 ORTEP drawing of 1 with thermal ellipsoids at the 50% probability level. All hydrogen atoms were omitted for clarity. | ||
Assuming the chelating and bridging ligands exchange slowly in solution state, the 1H NMR spectrum of 1 were expected to give at least two signals: one for the tert-butyl protons on the bridging ligand, and another for those on the chelating ligands. However, the 1H of 1 at 25 °C gave one signal, suggesting a single-proton environment. Variable temperature 1H NMR of 1 showed only minor line broadening between −15 °C and 50 °C. Most likely, 1 adopts a monomeric solution-state structure, in contrast to the dimeric structure found in the solid state. The monomeric solution-state structure is supported by diffusion-ordered spectroscopy (DOSY) in C6D6, where 1 obtained a diffusion coefficient D = 8.47 × 10−10 m2 s−1. Using the Stokes–Einstein equation yielded a diameter of 8.5 Å, which is much smaller than that along the major axis of dimeric 1 obtained from the crystal structure (~13.8 Å). For comparison, DOSY for tris(1,3-di-tert-butyltriazenide)indium(III)25 (In(tBu2N3)3) yielded D = 7.42 × 10−10 m2 s−1, which gave a Stokes–Einstein diameter of 9.7 Å. This diameter matched that from the crystal structure (~10 Å).
The structure of dimeric 1 obtained from density functional theory (DFT) agrees with the crystal structure (see ESI†). Similar to the crystal structure, the DFT gave two similar Zn–N bond lengths for the bridging ligands (2.057 and 2.062 Å) and two significantly different Zn–N bond lengths for each chelating ligand (2.031 and 2.294 Å). The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of 1 are centred on the chelating and bridging ligands, respectively. Natural population analysis gave a charge of +1.47e on each Zn centre with an electron configuration of 3d9.974s0.50. Similar population has previously been reported for EDTA Zn(II) complexes.31 Wiberg bond index (WBI) for Zn–Zn interaction was negligible (∼0.02) for 1, supporting the absence of any M–M bonding.
Thermal analysis and reactivity
Compound 1 sublimed quantitatively between 70 and 80 °C at 0.5 mbar pressure, and the sublimed compound remained unchanged by 1H NMR analysis. Thermogravimetric analysis (TGA) of 1 showed a one-step volatilisation between 125 and 175 °C, with ∼5% residual mass (Fig. 2). Crystals of 1 were sealed in a tube under vacuum and heated to 100 °C for 15 days. The heated solid showed no visible colour change, remained fully soluble in C6D6, and showed only minor changes in the baseline by 1H NMR. Compound 1 is therefore expected to remain intact in a heated CVD or ALD vaporiser. A solution of 1 in toluene-d8 was heated in a flame sealed NMR tube at various temperatures, followed by acquiring 1H NMR spectra (see ESI†). Spectra showed no changes when heating below 140 °C. The compound decomposed slowly at 140 °C, and more rapidly at 150 °C. | ||
| Fig. 2 TGA of 1 (15 mg sample) made under N2 atmosphere with a heating rate of 5 °C min−1. | ||
A previous report on Cd(II) amidinates presented a reactivity study with triphenylsilanethiol, to mimic a thiol-terminated surface in CdS ALD.32 The S–H represents the surface-bound thiol while the bulky triphenylsilane-part represents the substrate surface. Thus, mixing solutions of the potential metal precursor and the thiol mimics the ALD half-cycle where the metal precursor is introduced into an ALD chamber to react with the thiol-terminated substrate. For a successful outcome, the metal precursor exchanges ligands to form the metal thiolate.
The reactivity study was performed for 1 with a successful outcome. That is, 1 was reacted with 2 and 4 equivalents of triphenylsilanethiol to give compounds 2 and 3, respectively (Scheme 2). The new compounds were isolated in good yields after recrystallisation and were fully characterised using the same methods as compound 1.
 | ||
| Scheme 2 Reaction of 1 with 2 and 4 equivalents of triphenylsilanethiol to give 2 and 3, respectively. | ||
The crystal structure of 2 showed a heteroleptic dimeric complex with a chelating triazenide on each metal centre, and two bridging thiolates (Fig. 3a). The Zn atoms were found to adopt distorted tetrahedral geometries with average Zn–N and Zn–S bonds of 2.0482(89) and 2.3526(65) Å, respectively. The Zn–Zn distance of 2 (~3.26 Å) was like that of 1 and implies an absence of M–M bonding. Compound 3 showed a heteroleptic mononuclear structure with the Zn atom in a distorted tetrahedral geometry and bearing two thiolates and a tetramethyl-ethylenediamine ligand (Fig. 3b). The average Zn–N and Zn–S bonds were found to be 2.166(2) and 2.259(5) Å, respectively.
 | ||
| Fig. 3 ORTEP drawings of (a) 2 and (b) 3 with thermal ellipsoids at the 50% probability level. One of two independent molecules in the unit cell of 2 is shown. Hydrogen atoms are omitted for clarity. | ||
Conclusion
We have presented bis(1,3-di-tert-butyltriazenide)zinc(II) (1) as a potential CVD- and ALD precursor. This compound showed a one-step volatilisation by TGA and sublimed quantitatively under reduced pressure (0.5 mbar) at ∼80 °C. Additionally, the compound demonstrated good long-term thermal stability, showing only minor decomposition after being heated to 100 °C for 15 days. Compound 1 was reacted with two and four equivalents of triphenylsilanethiol to successfully displace one or both triazenide ligands, respectively. The volatility, thermal stability and reactivity of 1 makes it highly promising as a new M–N bonded precursor for depositing thin films of ZnS by ALD.Experimental section
General comments
Caution! As catenated nitrogen compounds are known to be associated with explosive hazards, these compounds are possible explosive energetic materials. Although we have not experienced any difficulties or problems in the synthesis, characterization, sublimation and handling of these compounds, their energetic properties have not been investigated and are therefore unknown. We therefore highly recommend all appropriate standard safety precautions for handling explosive materials (safety glasses, face shield, blast shield, leather gloves, polymer apron and ear protection) be always used when working with these compounds. All reactions and manipulations were carried out under a nitrogen atmosphere on a Schlenk line using air-free techniques and in a Glovebox-Systemtechnik dry box. All anhydrous solvents were purchased from Sigma-Aldrich™ and further dried with 4 Å molecular sieves. ZnCl2 (98%) and triphenylsilanethiol (98%) were purchased from Sigma-Aldrich™ and used without further purification. Lithium 1,3-di-tert-butyltriazenide was synthesised according to the literature procedure.24 All NMR spectra were measured with an Oxford Varian and Bruker AvanceNeo 500 MHz spectrometers. Solvent peaks were used as an internal standard for the 1H NMR and 13C{1H} NMR spectra. Melting- and decomposition points were determined in melting point tubes sealed under N2 with a Stuart® SMP10 melting point apparatus and are uncorrected. Elemental analysis was performed by Mikroanalytisches Laboratorium Kolbe, Germany.Synthesis of compounds 1–3
1: Colourless crystals, mp: 128–130 °C. Sublimation: 70–80 °C (0.5 mbar). 1H NMR (500 MHz, C6D6) δ 1.31 (s, 36H, CH3). 13C{1H} NMR (125 MHz, C6D6) δ 30.3 (s, CH3), 56.6 (s, Cq). Anal. calcd for C32H72N12Zn2: C, 50.86%; H, 9.60%; N, 22.24%. Found: C, 51.14%; H, 9.73%; N, 21.97%.
2: Colourless solid, decomp. 118–120 °C. 1H NMR (500 MHz, CD2Cl2) δ 0.88 (s, 18H, CH3), 7.29–7.36 (m, 6H, CH), 7.37–7.43 (m, 3H, CH), 7.53–7.61 (m, 6H, CH). 13C{1H} NMR (125 MHz, CD2Cl2) δ 30.0 (s, CH3), 55.6 (s, Cq), 128.3 (s, CH), 130.4 (s, CH), 135.7 (s, Cq), 135.8 (s, CH). Anal. calcd for C52H66N6S2Si2Zn2: C, 60.86%; H, 6.48%; N, 8.19%. Found: C, 60.41%; H, 6.66%; N, 8.14%.
3: Colourless crystals, decomp. 269–273 °C. 1H NMR (500 MHz, C6D6) δ 1.45 (s, 4H, CH2), 1.66 (s, 12H, CH3), 7.14–7.25 (m, 18H, CH), 8.00–8.07 (m, 12H, CH). 13C{1H} NMR (125 MHz, C6D6) δ 47.5 (s, CH3), 56.7 (s, CH2), 127.8 (s, CH), 129.0 (s, CH), 136.5 (s, CH), 140.1 (s, C). Anal. calcd for C42H46N2S2Si2Zn: C, 65.98%; H, 6.07%; N, 3.66%. Found: C, 66.33%; H, 6.44%; N, 3.58%.
X-ray crystallographic analysis
Single crystals were obtained by recrystallisation from n-hexane at −35 °C for 1, CH2Cl2/n-hexane at −35 °C for 2, and THF/n-hexane at −35 °C for 3. The single crystals were used for X-ray diffraction data collection at 120 or 296 K on a Bruker D8 SMART Apex-II diffractometer, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å), and at 120 K on a Rigaku XtaLAB Synergy-R diffractometer with a HyPix-Arc 100 detector using mirror-monochromated Cu-Kα radiation (λ = 1.54184 Å). All data was collected in hemisphere with over 95% completeness to 2θ < 50.05°. The structures were solved by direct methods. The coordinates of metal atoms were determined from the initial solutions and the N and C atoms by subsequent differential Fourier syntheses. All non-hydrogen atoms were refined first in isotropic and then in anisotropic approximation using Bruker SHELXTL software. Selected crystal data are summarised below.Compound 1. C32H72N12Zn2, M = 755.75, monoclinic, space group P21/c, a = 10.4654(10), b = 11.0754(10), c = 18.2679(2) Å, α = 90°, β = 101.5640(10)°, γ = 90°, V = 2074.42(4) Å3, Z = 2, Dc = 1.21 g cm−3, μ = 1.695 mm−1, T = 120.15 K, 4318 unique reflections measured, 3854 observed [I > 2σ(l)], R1 = 0.0281, wR2 (all data) = 0.0737, GOF = 1.051.
Compound 2. C52H66N6S2Si2Zn2, M = 1026.14, monoclinic, space group P121/c, a = 17.7677(2), b = 19.2739(2), c = 16.2548(2) Å, α = 90°, β = 106.7230(10)°, γ = 90°, V = 5331.08(11) Å3, Z = 4, Dc = 1.279 g cm−3, μ = 2.567 mm−1, T = 120.01(10) K, 10967 unique reflections measured, 9669 observed [I > 2σ(l)], R1 = 0.0308, wR2 (all data) = 0.0803, GOF = 1.030.
Compound 3. C42H46N2S2Si2Zn, M = 764.48, orthorhombic, space group P212121, a = 10.986(5), b = 11.629(6), c = 31.308(15) Å, α = 90°, β = 90°, γ = 90°, V = 4000(3) Å3, Z = 4, Dc = 1.27 g cm−3, μ = 0.81 mm−1, T = 296 K, 7047 unique reflections measured, 6704 observed [I > 2σ(l)], R1 = 0.026, wR2 (all data) = 0.0629, GOF = 1.045.
Diffusion-ordered spectroscopy
Diffusion measurements were performed for 1 and In(tBu2N3)3 using bipolar double-simulated echo pulse sequences with longitudinal eddy-current delay (dstebpgp3s in Topspin).33 Data was acquired using a relaxation delay = 5 s and eddy current delay = 5 ms for both compounds. For acquisition of 1, diffusion time Δ = 46.2 ms, gradient pulse length δ/2 = 1.200 ms. For In(tBu2N3)3 diffusion time Δ = 71.7 ms, gradient pulse length δ/2 = 1.000 ms. Measurements were performed at z-gradient strengths varied linearly between 1 and 47 G cm−1 in 16 increments, with 32 scans at each increment. The diffusion coefficient was extracted using Dynamic Centre 2.8.1.Thermogravimetric analysis
Thermogravimetric analysis (TGA) was performed using a Netzsch STA 409 PC/PG instrument housed in a dry box filled with argon gas. For TGA experiments of 1, a 15 mg sample was heated to 500 °C at a rate of 5 °C min−1. The N2 flow rate (AirLiquide, 99.998%) was adjusted to 90 sccm. The onset of volatilisation was defined as the temperature at which 5% of the precursor mass was lost.Quantum chemical calculations
Gaussian 16 was used for all geometry optimizations and subsequent vibrational frequency calculations.34 Calculations were performed using the hybrid density functional B3LYP,35,36 the def2TZVP37 basis sets, and Grimme's version 3 dispersion correction.38 Minima were confirmed to have no imaginary frequencies. NBO analyses were performed on the optimized geometries using NBO 7.039 interfaced from Gaussian 16.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to Andreas Orthaber for providing access to X-ray single crystal facility at Uppsala University and Lars Ojamäe for access to the Swedish National Supercomputer Centre (NSC). The project was funded by the Swedish Foundation for Strategic Research through the project “Time-resolved low temperature CVD for III-nitrides” (SSF-RMA 15-0018) and by the Knut and Alice Wallenberg foundation through the project “Bridging the THz gap” (No. KAW 2013.0049). E. B. and J. O. M. acknowledge the Research Council of Finland (projects 315829 and 338733) for financial support.References
- X. Fang, T. Zhai, U. K. Gautam, L. Li, L. Wu, Y. Bando and D. Golberg, Prog. Mater. Sci., 2011, 56, 175–287 CrossRef CAS.
- J. Maula, Chin. Opt. Lett., 2010, 8, 53–58 CrossRef.
- Y. S. Kim and S. J. Yun, Appl. Surf. Sci., 2004, 229, 105–111 CrossRef CAS.
- G. Stuyven, P. De Visschere, A. Hikavyy and K. Neyts, J. Cryst. Growth, 2002, 234, 690–698 CrossRef CAS.
- C. Platzer-Björkman and T. Törndahl, J. Appl. Phys., 2006, 100, 044506 CrossRef.
- J. T. Tanskanen, J. R. Bakke, T. A. Pakkanen and S. F. Bent, J. Vac. Sci. Technol., A, 2011, 29, 031507 CrossRef.
- H. Dumont, A. Marbeuf, J.-E. Bourée and O. Gorochov, J. Mater. Chem., 1993, 3, 1075–1079 RSC.
- M. Tammenmaa, T. Koskinen, L. Hiltunen, L. Niinistö and M. Leskelä, Thin Solid Films, 1985, 124, 125–128 CrossRef CAS.
- A. Short, L. Jewell, S. Doshay, C. Church, T. Keiber, F. Bridges, S. Carter and G. Alers, J. Vac. Sci. Technol., A, 2013, 31, 01A138 CrossRef.
- A. Short, L. Jewell, A. Bielecki, T. Keiber, F. Bridges, S. Carter and G. Alers, J. Vac. Sci. Technol., A, 2014, 32, 01A125 CrossRef.
- M. Oikkonen, M. Tammenmaa and M. Asplund, Mater. Res. Bull., 1988, 23, 133–142 CrossRef CAS.
- H. Bürger, W. Sawodny and U. Wannagat, J. Organomet. Chem., 1965, 3, 113–120 CrossRef.
- R. G. Gordon, in Atomic Layer Deposition for Semiconductors, ed. C. S. Hwang and C. Y. Yoo, Springer US, New York, 2014, pp. 15–46 Search PubMed.
- S. Schmidt, S. Schulz, D. Bläser, R. Boese and M. Bolte, Organometallics, 2010, 29, 6097–6103 CrossRef CAS.
- T. Eisenmann, J. Khanderi, S. Schulz and U. Flörke, Z. Anorg. Allg. Chem., 2008, 634, 507–513 CrossRef CAS.
- M. P. Coles and P. B. Hitchcock, Eur. J. Inorg. Chem., 2004, 2662–2672 CrossRef CAS.
- S. Anga, I. Banerjee and T. K. Panda, J. Chem. Sci., 2016, 128, 867–873 CrossRef CAS.
- P. Rouf, N. J. O'Brien, K. Rönnby, R. Samii, I. G. Ivanov, L. Ojamäe and H. Pedersen, J. Phys. Chem. C, 2019, 123, 12691–25700 CrossRef.
- N. Nimitsiriwat, V. C. Gibson, E. L. Marshall, P. Takolpuckdee, A. K. Tomov, A. J. P. White, D. J. Williams, M. R. Elsegood and S. H. Dale, Inorg. Chem., 2007, 46, 9988–9997 CrossRef CAS PubMed.
- F. E. Brinckman, H. S. Haiss and R. A. Robb, Inorg. Chem., 1965, 4, 936–942 CrossRef CAS.
- N. J. O'Brien, P. Rouf, R. Samii, K. Rönnby, S. C. Buttera, C.-W. Hsu, I. G. Ivanov, V. Kessler, L. Ojamäe and H. Pedersen, Chem. Mater., 2020, 32, 4481–4489 CrossRef.
- P. Rouf, R. Samii, K. Rönnby, B. Bakhit, S. C. Buttera, I. Martinovic, L. Ojamäe, C.-W. Hsu, V. Kessler, J. Palisaitis, V. Kessler, H. Pedersen and N. J. O'Brien, Chem. Mater., 2021, 33, 3266–3275 CrossRef CAS.
- R. Samii, D. Zanders, S. C. Buttera, V. Kessler, L. Ojamäe, H. Pedersen and N. J. O'Brien, Inorg. Chem., 2021, 60, 4578–4587 CrossRef CAS PubMed.
- R. Samii, D. Zanders, A. Fransson, G. Bačić, S. T. Barry, L. Ojamäe, V. Kessler, H. Pedersen and N. J. O'Brien, Inorg. Chem., 2021, 60, 12759–12765 CrossRef CAS PubMed.
- R. Samii, S. C. Buttera, V. Kessler and N. J. O'Brien, Eur. J. Inorg. Chem., 2022, e202200161 CrossRef CAS.
- R. Samii, A. Fransson, P. Mpofu, P. Niiranen, L. Ojamäe, V. Kessler and N. J. O'Brien, Inorg. Chem., 2022, 61, 20804–20813 CrossRef CAS PubMed.
- P. Rouf, J. Palisaitis, B. Bakhit, N. J. O'Brien and H. Pedersen, J. Mater. Chem. C, 2021, 9, 13077–13080 RSC.
- P. Mpofu, P. Rouf, N. J. O'Brien, U. Forsberg and H. Pedersen, Dalton Trans., 2022, 51, 4712–4719 RSC.
- C. Lu, N. J. O'Brien, P. Rouf, R. Dronskowski, H. Pedersen and A. Slabon, Green Chem. Lett. Rev., 2022, 15, 658–670 CrossRef CAS.
- C. M. Caroff and G. S. Girolami, Inorg. Chem., 2022, 61, 16740–16749 CrossRef CAS PubMed.
- A. Kovács, D. S. Nemcsok and T. Kocsis, J. Mol. Struct.: THEOCHEM, 2010, 950, 93–97 CrossRef.
- M. J. Foody, M. S. Weimer, H. Bhandari and A. S. Hock, Inorg. Chem., 2021, 60, 6191–6200 CrossRef CAS PubMed.
- A. Jerschow and N. Muller, J. Magn. Reson., 1997, 125, 372–375 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem, 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO7.0, University of Wisconsin, Madison, WI, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation of the compounds and computational calculation details (PDF). CCDC 2324598 for 1, 2324599 for 2 and 2324600 for 3. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00084f |
| This journal is © The Royal Society of Chemistry 2024 |