
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReaction of Ph2C(X)(CO2H) (X = OH, NH2) with [VO(OR)3] (R = Et, nPr): structure, magnetic susceptibility and ROP capability†
Mollie A.
Glenister
a,
Josef W. A.
Frese
a,
Mark R. J.
Elsegood
 a,
Angelos B.
Canaj
b,
Euan K.
Brechin
b and
Carl
Redshaw
*c
a,
Angelos B.
Canaj
b,
Euan K.
Brechin
b and
Carl
Redshaw
*c
aPlastics Collaboratory, Chemistry, School of Natural Sciences, University of Hull, Cottingham Road, Hull, HU6 7RX, UK. E-mail: c.redshaw@hull.ac.uk
bEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK
cChemistry Department, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK
First published on 27th February 2024
Abstract
Reaction of [VO(OR)3] (R = Et, nPr) with 2,2′-diphenylglycine afforded the alkoxide-bridged dimers {[VO(OR)(μ-OR)][Ph2C(NH2)(CO2)]}2, whereas use of benzilic acid, in the presence of alkali metals, afforded 16-membered metallocycles {V8(O)4M(OR)8[Ph2C(OH)(CO2)]12} (M = <1 Na, K). For the ring systems, magnetic susceptibility data is consistent with mixed-valence vanadium with an average oxidation state of 3.5. The dimer and ring systems are capable of the ring opening polymerisation (ROP) of ε-caprolactone under N2, air, or as melts affording mostly low to medium molecular weight cyclic and linear products.
Current interest in the development of new catalytic species capable of the production of biodegradable polymers via ring opening polymerization (ROP) remains high.1 This is driven by the need to replace materials such as polyethylene, which still finds widespread use despite the difficulties associated with lack of decomposition in landfill sites.2 Metal alkoxides are the preferred catalyst type for ROP and, if necessary, the alkoxide catalyst can be generated in situ by the addition of an external alcohol. The other ligands that complete the coordination environment at the metal can greatly affect both the catalytic activity of the system and the resultant polymer properties. In this respect, we and others have been scoping a variety of ligand types in combination with numerous different metal centres with a view to accessing highly active, controllable ROP systems. As part of our studies, we have recently reported the use of the acids Ph2C(X)(CO2H), where X = OH or NH2, i.e. benzilic acid and 2,2′-diphenylglycine. Our interest in the use of such acids stems from the presence of the motif Ph2C(X), which has been shown to impart high crystallinity on products.3 New ROP catalysts based on titanium, lithium, zinc, aluminium, and rare-earth metals incorporating the motif Ph2C(X) were reported, together with a number of intriguing molecular structures.4 Recently, for the group V metals, we have investigated the use of niobium and tantalum alkoxide precursors, which afforded tetranuclear complexes [M4(OEt)8(L1)4(μ-O)2] in the case of benzilic acid (L1H2), and dinuclear complexes [Nb2(OEt)4(L2H2)4(μ-O)] in the case 2,2′-diphenylglycine (L2H3).5
Herein, as part of an on-going programme investigating the use of earth-abundant metal catalysts, we have extended our studies using this motif to vanadium-based systems, and reacted each of the two Ph2C(X)-containing acids with the vanadyl tris(alkoxides) [VO(OR)3] (R = Et, nPr). For X = NH2, the alkoxide-bridged dimers {[VO(OR)(μ-OR)][Ph2C(NH2)(CO2)]}2 (R = Et, nPr) are formed, whilst for X = OH, 16-membered mixed-valence metallocycles {V8(O)4M(OR)8[Ph2C(OH)(CO2)]12} are formed incorporating eight vanadium centres (Chart 1). In the case of benzilic acid, the presence of small amounts a sodium or potassium salts further aided crystallization. We note that a ferric wheel [Fe(OMe)2(Ph2C(OH)(CO2))]12 derived from benzilic acid was reported by Raptopoulou et al.,6 whilst McInnes et al. have reported the V(III) octanuclear cluster [V8(OEt)8(OH)4(O2CPh)12] via a solvothermal route.7 ROP catalysts based on vanadium have already shown promise when using other ancillary ligand types,8 and there is interest in the use of heterometallic and multinuclear complexes as catalysts for ROP.9
 | ||
| Chart 1 Complexes 1–8 prepared herein (3, R = nPr, M = Na+; 4, R = Et, M = Na+; 5, R = nPr, M = Na+; 6 R = nPr, M = Na+; 7, R = nPr, M = K+; 8 R = nPr, M = K+; L = NCMe). Note there is no L for 1. | ||
Reaction of the vanadyl trisalkoxides [VO(OR)3] (R = Et, nPr) with 2,2′-diphenylglycine, Ph2C(NH2)(CO2H), in refluxing toluene afforded, following work-up (MeCN), the yellow crystalline solids {[VO(OR)(μ-OR)][Ph2C(NH2)(CO2)]}2 (R = Et 1, nPr 2) in moderate to good yield. In the IR spectrum, the peak at 986 cm−1 is assigned to vV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. Both solids can be recrystallized from saturated acetonitrile solutions at 0 °C. The molecular structures of both complexes were determined, and were found to be similar alkoxide-bridged dimers. We will only discuss 2·2MeCN here; for 1 see the ESI (Fig. S1 and S2†). In the case of 2, the formula is {VO(On-Pr)(μ-OnPr)[Ph2C(NH2)(CO2)]}2·2(C2H3N), and there are two half molecules in the asymmetric unit. The molecule containing pseudo octahedral V(1) (Fig. 1) exhibits two intramolecular H-bonds N–H⋯O, plus two intermolecular H-bonds to MeCN molecules. The molecule containing pseudo octahedral V(2) (Fig. S3†) exhibits a different H-bonding pattern. While there is still the N–H⋯O intramolecular H-bond, the second N–H links to O(2) in the molecule containing V(1), not to an MeCN.
O. Both solids can be recrystallized from saturated acetonitrile solutions at 0 °C. The molecular structures of both complexes were determined, and were found to be similar alkoxide-bridged dimers. We will only discuss 2·2MeCN here; for 1 see the ESI (Fig. S1 and S2†). In the case of 2, the formula is {VO(On-Pr)(μ-OnPr)[Ph2C(NH2)(CO2)]}2·2(C2H3N), and there are two half molecules in the asymmetric unit. The molecule containing pseudo octahedral V(1) (Fig. 1) exhibits two intramolecular H-bonds N–H⋯O, plus two intermolecular H-bonds to MeCN molecules. The molecule containing pseudo octahedral V(2) (Fig. S3†) exhibits a different H-bonding pattern. While there is still the N–H⋯O intramolecular H-bond, the second N–H links to O(2) in the molecule containing V(1), not to an MeCN.
 | ||
| Fig. 1 Molecular structure of {VO(OnPr)(μ-OnPr)[(Ph2C(NH2)CO2)]}2·2MeCN (2·2MeCN). Selected bond lengths (Å) and angles (°): V(1)–O(1) 1.9559(10), V(1)–O(3) 1.5859(11), V(1)–O(4) 2.2993(10), V(1)–O(4A) 1.8484(9), V(1)–O(5) 1.7926(11), V(1)–N(1) 2.1845(12); V(1)–O(1)–C(1) 125.06(9), V(1)–N(1)–C(2) 112.73(8), V(1)–O(4)–V(1A) 107.46(4), O(4)–V(1)–O(4A) 72.54(4). | ||
There are two unique MeCNs, i.e. two per V2 complex. One unique MeCN is H-bonded to the molecule containing V(1), the other is not H-bonded. Molecules pack into 1D chains via the intermolecular H-bonds described above (Fig. S4, ESI†).
Reaction of two equivalents of [VO(OnPr)3] with benzilic acid (benzH) in refluxing toluene afforded, following work-up (MeCN), small dark prisms in low yield (<10%). A preliminary crystal structure determination unexpectedly revealed a large macrocyclic structure comprising eight vanadium centres and twelve benzilic acid-derived ligands (Fig. S5, ESI†). At the centre of the metallocycle was electron density suggestive of a sodium ion. This was thought to arise from the drying of the solvent toluene. However, the data associated with this structure, {V8(O)4Na0.75(OnPr)8[Ph2C(OH)(CO2)]12}·8MeCN, 3·8MeCN, was of rather poor quality (see Fig. S5 and Table S1, ESI†).
In order to try and improve both the yield and quality of the crystal data, we conducted the synthesis in the presence of a number of sodium and potassium salts. Indeed, from combinations of [VO(OR)3] (R = Et, nPr), MI or MH (M = Na, K) and benzilic acid, we were able to isolate highly crystalline products in slightly higher yield (ca. 28–40%). The molecular structure of a representative example is shown in Fig. 2, with selected bond lengths and angles given in the caption; a view of the core is given in Fig. S9, ESI.† As for the preliminary structural data mentioned above, the product, namely {V8(O)4Na0.45(OnPr)8[Ph2C(OH)(CO2)]12}·6.38MeCN 5·6.38MeCN, was a 12-membered metallocycle containing eight vanadium centres and twelve benzilic acid derived ligands. Furthermore, there is a central Na+ ion, modelled as partially occupied 0.449(13) with the occupancy refined.
 | ||
| Fig. 2 Molecular structure of {V8(O)4Na0.45(OnPr)8[Ph2C(OH)(CO2)]12}·6.38MeCN (5·6.38MeCN). Selected bond lengths (Å) and angles (°): V(1)–O(1) 2.043(3), V(1)–O(4) 2.033(3), V(1)–O(16) 2.022(3), V(1)–O(19) 1.975(3), V(1)–O(20) 1.954(3), V(1)–O(23) 1.976(3), V(1)⋯V(2) 3.3749(9), Na(1)–O(23) 3.028(2), V(2)–O(2) 2.016(3), V(2)–O(5) 2.031(3), V(2)–O(7) 2.113(3), V(2)–O(21) 1.941(3), V(2)–O(22) 1.962(2), V(2)–O(23) 1.726(3), V(2)⋯V(3) 3.0068(9); V(1)–O(23)–V(2) 131.35(14), V(1)–O(20)–V(4A) 99.81(12), V(2)–O(23)–Na(1) 114.85(11). | ||
For 5·6.38MeCN, the molecule lies on a centre of symmetry so half is unique. There was evidence of non-merohedral twinning, however no twin law could be determined. In all of the similar V8 structures, it is presumed that the charge of the central, partially occupied, alkali metal ion is balanced by the loss of some benzilic acid OH hydrogens, probably disordered. In all cases, the OH atoms needed to be included in a constrained manner with either rotational freedom or aligned to make the most reliable H-bond. In terms of charge, the 28 negative charges (12 × benz1−, 4 × oxo2−, 8 × OnPr1−) suggest mixed-valence V, with a mean oxidation state of 3.5. The benzilic OH hydrogens generally make H-bonds with either adjacent carboxylate oxygens or nearby MeCN of crystallisation. V⋯V distances bridged by two alkoxides and one carboxylate are shorter than those bridged by two carboxylates and an oxo ligand.
This {V8(O)4M(OR)8[Ph2C(OH)(CO2)]12} structural motif appears to be quite general for the various combinations of benzilic acid, MX, and [VO(OR)3], and the molecular structures of 4, 6–8 are provided in the ESI (Fig. S7, S11, S13, S15;† note the quality of 6 is less good but structurally in accord with the others). A notable comparison with the behaviour showcased in the series is that of crown ethers which also possess cavities which bind to alkali metal ions.10 When the cavity diameters are compared with the structures in this series (Fig. S17, ESI†), it can be seen that the V8 rings have a larger diameter and are closer in size to 21-crown-7 (Table S2†). This crown ether configuration favours larger ions such as Rb+ and Cs+, therefore suggesting reasoning for the partial occupancy herein, i.e. the larger cavity of the V8 structures is unfavourable to the smaller Na+ and K+ ions. We note that it has previously been reported that alkali metals can be used to control ring size.11
Dc magnetic susceptibility measurements were performed on a polycrystalline sample of 5 over the temperature range T = 2–275 K, in an applied field (B) of 0.1 T (Fig. 3), where χ = M/B, and M is the magnetisation. At 275 K, the χT value of 4.3 cm3 K mol−1 is below the expected value for spin-only contributions to the susceptibility for a [V8] unit with an average oxidation state of 3.5 (5.5 cm3 mol−1 K, g = 2.0). Upon cooling, the χT product decreases rapidly reaching a value of 0 cm3 K mol−1 at 18 K. The data are therefore suggestive of the presence of very strong antiferromagnetic exchange between neighbouring vanadium ions and a diamagnetic ground state. A quantitative interpretation of the susceptibility data is hampered by the valence-delocalised nature of 5. However, we note that the behaviour of 5 is similar to that of [V8(OEt)8(OH)4(O2CPh)12]8 and to vanadium dimers bridged by [(O)(O2CR)2] and [(OR)2(O2CR)] units, as in 5, which are known to mediate strong antiferromagnetic exchange.12 Similar behaviour is also observed in structurally related [Fe8] wheels with similar bridging units.13
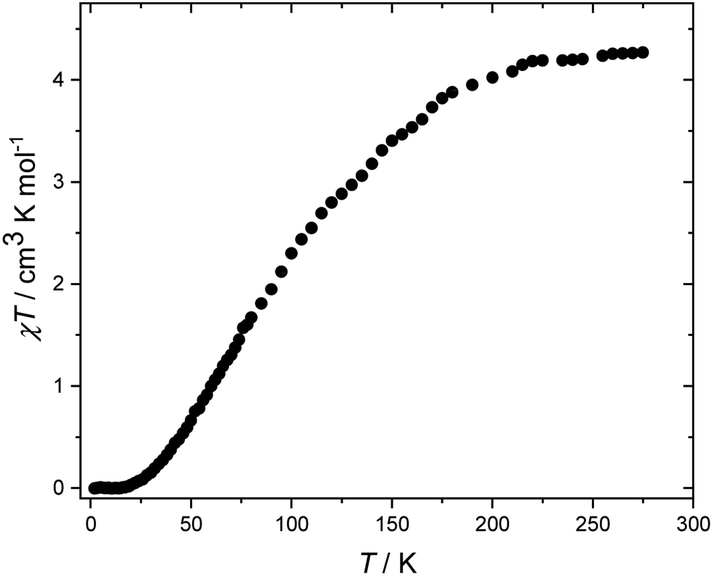 | ||
| Fig. 3 Experimental χT versus T data for 5 measured in the T = 2–275 K temperature range in an applied field of B = 0.1 T. | ||
Complexes 1, 2, 4, and 8 have been screened for their potential to act catalysts for the ROP of ε-caprolactone (Table 1). Using 1, the ratio [CL]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[1], time and temperature were all varied. At ambient temperature, and using a ratio of 500:1 ([CL]:[1]), no activity was observed. Increasing the temperature to 70 °C led to low conversion (ca. 15%), however on further raising the temperature to 130 °C, quantitative conversion was observed. Varying the ([CL]:[1]) proved to be detrimental with lower conversion and less control. Conducting the run under air also led to full conversion, albeit with less control and a lower molecular weight product. When conducted as a melt, a higher molecular weight product was formed with a conversion of ca. 80%. Complex 2 also exhibited full conversion when used either under air in solution or as a melt, affording in the latter case a higher molecular weight product (Mn 8600). For the V8 complex 4, the conversion at 130 °C under N2 (entry 14) was half that observed for 1 (entry 3) and afforded a PCL polymer with half the molecular weight, albeit with slightly better control. A similar run under air (entry 15) led to quantitative conversion, whilst use of 4 as a melt (entry 16) also led to higher conversion (ca. 88%) and an increased molecular weight. However, the use of a melt under air (entry 17) led to a reduction in both the conversion (to ca. 66%) and molecular weight. Use of 8 at 130 °C in toluene under N2 (entry 18) or air (entry 19) led to quantitative conversion, with higher molecular weight but less control for the former. Use of a melt in air (entry 21) proved more effective than a melt under N2 (entry 20).
:[1], time and temperature were all varied. At ambient temperature, and using a ratio of 500:1 ([CL]:[1]), no activity was observed. Increasing the temperature to 70 °C led to low conversion (ca. 15%), however on further raising the temperature to 130 °C, quantitative conversion was observed. Varying the ([CL]:[1]) proved to be detrimental with lower conversion and less control. Conducting the run under air also led to full conversion, albeit with less control and a lower molecular weight product. When conducted as a melt, a higher molecular weight product was formed with a conversion of ca. 80%. Complex 2 also exhibited full conversion when used either under air in solution or as a melt, affording in the latter case a higher molecular weight product (Mn 8600). For the V8 complex 4, the conversion at 130 °C under N2 (entry 14) was half that observed for 1 (entry 3) and afforded a PCL polymer with half the molecular weight, albeit with slightly better control. A similar run under air (entry 15) led to quantitative conversion, whilst use of 4 as a melt (entry 16) also led to higher conversion (ca. 88%) and an increased molecular weight. However, the use of a melt under air (entry 17) led to a reduction in both the conversion (to ca. 66%) and molecular weight. Use of 8 at 130 °C in toluene under N2 (entry 18) or air (entry 19) led to quantitative conversion, with higher molecular weight but less control for the former. Use of a melt in air (entry 21) proved more effective than a melt under N2 (entry 20).
a
| Entry | Cat. | [CL]0:[Cat]0 |
Conv.b (%) |
M
nc (calc.) |
M
nd (obs.) |
Đ |
|---|---|---|---|---|---|---|
| a Conducted at 130 °C under N2 for 24 h unless otherwise stated; [catalyst] = 0.01 mmol. b Determined by 1H NMR spectroscopy. c M n(calc.) = 114.14 × [CL]0/[cat] × %conv + Mend group. d M n(obs) and D obtained by GPC in THF relative to polystyrene standards corrected by the Mark–Houwink correction factor Mn(obs) = MnGPC raw data × 0.56. e Polydispersity index. f Conducted at 20 °C. g Conducted at 70 °C. h Conducted under air. i Conducted for 1 h. j Conducted as a melt (solvent-free conditions). | ||||||
| 1f | 1 | 500:1 |
0 | — | — | — |
| 2g | 1 | 500:1 |
15.4 | — | — | — |
| 3 | 1 | 500:1 |
100 | 57088 |
5890 | 1.58 |
| 4 | 1 | 250:1 |
18.4 | 5268 | 5130 | 1.76 |
| 5 | 1 | 1000:1 |
90.9 | 103771 |
4650 | 2.60 |
| 6h | 1 | 500:1 |
100 | 57088 |
2415 | 1.75 |
| 7i | 1 | 500:1 |
4.1 | — | — | — |
| 8j | 1 | 500:1 |
87 | 49669 |
3560 | 1.92 |
| 9h,j | 1 | 500:1 |
35 | 19993 |
3090 | 2.82 |
| 10 | 2 | 500:1 |
75.2 | 42935 |
3680 | 1.42 |
| 11h | 2 | 500:1 |
100 | 57088 |
3330 | 2.03 |
| 12j | 2 | 500:1 |
81.3 | 46416 |
5370 | 1.84 |
| 13h,j | 2 | 500:1 |
100 | 57088 |
8600 | 3.25 |
| 14 | 4 | 500:1 |
50.5 | 28838 |
2670 | 1.41 |
| 15h | 4 | 500:1 |
100 | 57088 |
3830 | 1.79 |
| 16j | 4 | 500:1 |
88.5 | 50525 |
6320 | 3.70 |
| 17h,j | 4 | 500:1 |
65.8 | 37570 |
2410 | 1.52 |
| 18 | 8 | 500:1 |
100 | 57088 |
3430 | 3.64 |
| 19h | 8 | 500:1 |
100 | 57088 |
2070 | 1.82 |
| 20j | 8 | 500:1 |
70.4 | 40195 |
2550 | 1.81 |
| 21h,j | 8 | 500:1 |
100 | 57088 |
10720 |
1.95 |
In nearly all cases, the molecular weights observed were significantly lower than the theoretical values, suggesting that transesterification had occurred. 1H NMR spectra of the PCL formed indicated the presence of H-PCL-OH end groups (e.g., Fig. S20, ESI†). However, from MALDI-ToF mass spectra (Fig. S21–S26, ESI†), it was evident that several PCL series were present (as sodium adducts). For 1 (entry 4, Table 1, Fig. S21†), these included chain polymers H-PCL-OH and the sodium exchange artefact, and a minor family with no end groups (cyclic) and the sodium exchange artefact. Similarly, for 1 as a melt (entry 9, Table 1, Fig. S22†), chain polymers H-PCL-OEt and the sodium exchange artefact and a minor cyclic family and the sodium exchange artefact. For 2 (entry 15, Table 1, Fig. S23†), the series with end groups H–/–OH was evident, whilst as a melt (entry 17, Table 1, Fig. S24†) at least two families were evident, including those with end groups HO–/–ONa and no end groups (cyclic). For 8 (entry 18, Table 1, Fig. S25†), at least three series of peaks were identified including the sodium exchange artefact for the PrO–/–H end group, the HO–/–H series, together with a number of lower intensity families e.g. MeO–/–H. Use of 8 as a melt under N2 (entry 20, Table 1, Fig. S26†) afforded the PCL series HO–/–OH and the sodium exchange artefact, and PrO–/–H end groups. Use of other V-based complexes for the ROP of ε-CL has led to the isolation of multiple families of products with low to moderate molecular weight.14
Kinetic studies (Fig. S27–S30, ESI†), conducted using 500:1 ([CL]:[Cat]) revealed the rate trend 2 > 8 ≫ 4 > 1, and this order is ascribed to the respective solubilities of the systems.
In conclusion, the reaction of the acids Ph2C(X)CO2H with [VO(OR)3] led to very different products. In the case of X = NH2, alkoxide bridged dimers were formed, whereas for X = OH, large macrocyclic structures of the type {V8(O)4M(OR)8[Ph2C(OH)(CO2)]12} comprising eight vanadium centres, with average oxidation state of 3.5, and twelve benzilic acid derived ligands were isolated. The presence of an alkali metal cation (Na+, K+) favoured the formation of such macrocycles. The systems were capable of the ROP of ε-caprolactone, affording mixtures of low to medium molecular weight products (Mn ≤ 10720) with differing or no end groups, and with varied control (Đ 1.41–3.70).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CR thanks the EPSRC (EP/S025537/1) for financial support. We thank the EPSRC National Crystallography Service, Southampton for data collection. The STFC are thanked for beam time at Daresbury Laboratory SRS. EKB, ABC thank The Leverhulme Trust (grant RPG-2021-176).References
- (a) Y. Sarazin and J.-F. Carpentier, Chem. Rev., 2015, 115, 3564–3614 CrossRef CAS PubMed and references therein; (b) C. Redshaw, Catalysts, 2017, 7, 165–178 CrossRef; (c) J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC; (d) D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 392, 83–145 CrossRef CAS; (e) O. Santoro and C. Redshaw, Catalysts, 2020, 10, 210 CrossRef CAS; (f) O. Santoro, X. Zhang and C. Redshaw, Catalysts, 2020, 10, 165–800 CrossRef.
- L. W. McKeen, Introduction to the use of Plastics in Food Packaging, William Andrew Publishing, 2013, pp. 1–15 Search PubMed; G. Manivasagam, A. Reddy, D. Sen, S. Nayak, M. T. Mathew and A. Rajamanikam, Encyclopaedia of Biomedical Engineering, ed. R. Narayan, Elsevier, Oxford, 2019, pp. 332–347 Search PubMed.
- M. Braun, Angew. Chem., 1996, 108, 565 CrossRef CAS; M. Braun, Angew. Chem. Int. Ed., 1996, 35, 519–522 CrossRef.
- Y. Al-Khafaji, M. R. J. Elsegood, J. W. A. Frese and C. Redshaw, RSC Adv., 2017, 7, 4510–4517 RSC; X. Wang, K.-Q. Zhao, S. Mo, Y. Al-Khafaji, T. J. Prior, M. R. J. Elsegood and C. Redshaw, Eur. J. Inorg. Chem., 2017, 1951–1965 CrossRef CAS; Y. F. Al-Khafaji, T. J. Prior, L. Horsburgh, M. R. J. Elsegood and C. Redshaw, ChemistrySelect, 2017, 2, 759–768 CrossRef; J. Collins, O. Santoro, T. J. Prior, K. Chen and C. Redshaw, J. Mol. Struct., 2021, 1224, 129083 CrossRef; X. Zhang, T. J. Prior, K. Chen, O. Santoro and C. Redshaw, Catalysts, 2022, 12, 935 CrossRef.
- X. Zhang, T. J. Prior and C. Redshaw, New J. Chem., 2022, 46, 14146–14154 RSC.
- C. P. Raptopoulou, V. Tangoulis and E. Devlin, Angew. Chem., Int. Ed., 2002, 41, 2386–3589 CrossRef CAS PubMed.
- R. H. Laye, M. Murrie, S. Ochsenbein, A. R. Bell, S. J. Teat, J. Raftery, H.-U. Güdel and E. J. L. McInnes, Chem. – Eur. J., 2003, 9, 6215–6220 CrossRef CAS PubMed.
- H. Ishikura, R. Neven, T. Lange, A. Galetová, B. Blom and D. Romano, Inorg. Chim. Acta, 2021, 515, 120047 CrossRef CAS.
- (a) W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252 CrossRef CAS PubMed; (b) L.-J. Wo, W. Lee, P. K. Ganta, Y.-L. Chang, Y.-C. Chang and H.-Y. Chen, Coord. Chem. Rev., 2023, 475, 214847 CrossRef.
- (a) R. D. Hancock, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 17, 63–80 CrossRef CAS; (b) J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS.
- R. W. Saalfrank, I. Bernt, E. Uller and F. Hampel, Angew. Chem., Int. Ed. Engl., 1997, 36, 2482–2485 CrossRef CAS.
- K. Kanamori, Coord. Chem. Rev., 2003, 237, 147–165 CrossRef CAS.
- C. Cañada-Vilalta, M. Pink and G. Christou, Chem. Commun., 2003, 1240–1241 RSC.
- See for example: M. R. J. Elsegood, W. Clegg and C. Redshaw, Catalysts, 2023, 13, 998 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details for 1–8; alternative views of 1–8; crystallographic and ROP data. CCDC 2320818–2320821 and 2320775–2320778. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00078a |
| This journal is © The Royal Society of Chemistry 2024 |