
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA water-soluble polyphosphorhydrazone Janus dendrimer built by “click” chemistry as support for Ru-complexes in catalysis†
Joel
Cejas-Sánchez
 abc,
Anne-Marie
Caminade
de,
Anna
Kajetanowicz
c,
Karol
Grela
c and
Rosa María
Sebastián
*ab
abc,
Anne-Marie
Caminade
de,
Anna
Kajetanowicz
c,
Karol
Grela
c and
Rosa María
Sebastián
*ab
aDepartment of Chemistry, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, Bellaterra, 08193, Barcelona, Spain. E-mail: rosamaria.sebastian@uab.es
bCentro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Autònoma de Barcelona, Cerdanyola del Vallès, Bellaterra, 08193, Barcelona, Spain
cBiological and Chemical Research Centre, Faculty of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland
dLaboratoire de Chimie de Coordination du CNRS, 205 Route de Narbonne, BP 44099, 31077 Toulouse CEDEX 4, France
eLCC-CNRS, Université de Toulouse, UPS, INPT, Toulouse CEDEX 4, France
First published on 7th May 2024
Abstract
The field of supported catalysis has experienced increased attention with respect to the development of novel architectures for immobilizing catalytic species, aiming to maintain or enhance their activity while facilitating the easy recovery and reuse of the active moiety. Dendrimers have been identified as promising candidates capable of imparting such properties to catalysts through selective functionalization. The present study details the synthesis of two polyphosphorhydrazone (PPH) dendrons, each incorporating azide or acetylene groups at the core for subsequent coupling through “click” triazole chemistry. Employing this methodology, a novel PPH Janus dendrimer was successfully synthesized, featuring ten polyethylene glycol (PEG) chains on one side of the structure and ten Ru(p-cymene) derivatives on the other. This design was intended to confer dual properties, influencing solubility modulation, and allowing the presence of active catalytic moieties. The synthesized dendrimer underwent testing in the isomerization of allyl alcohols in organic solvents and biphasic solvent mixtures. The results demonstrated a positive dendritic effect compared with model monometallic and bimetallic species, providing a proof-of-concept for the first PPH Janus dendrimer with tested applications in catalysis.
Introduction
Dendrimers are highly branched, monodisperse macromolecules with a uniform structure, often taking on a spherical shape.1 These molecules are created through a precise synthetic process that allows for control over their size, shape, and molecular characteristics,2 a capability that enables the production of well-structured polymers with diverse applications in various fields, including sensors,3,4 biology,5,6 biomedicine,1,7 materials science,8,9 and catalysis.10–13 Among these applications, catalysis emerges as a particularly interesting domain, as these macromolecules can serve as efficient catalyst supports.14–17 Anchoring a catalyst onto dendrimers offers several advantages, such as easy catalyst recovery, recycling, and reuse.13,18 This could provide benefits like enhanced catalyst activity, ease of handling and recovery through simple procedures, and the ability to reduce metal contamination in final products. Moreover, dendrimers offer a unique opportunity to combine the advantages of both homogeneous and heterogeneous catalysis while maintaining precise and monodispersed structures.10Due to the inherent uniformity of dendritic structures, dendrimer functionalization has traditionally been limited to one specific area, leading to the development of dendritic structures constrained to a single type of surface functional group. To address this limitation, various methodologies have emerged to expand dendrimer functionalization possibilities. The most common approaches involve attaching two types of functional groups to the surface of dendrimers, either in a stochastic or precise manner,19–21 or the development of new methodologies for dendrimer synthesis. In this context, Janus dendrimers emerge as a promising alternative. Janus dendrimers, named after the Roman God Janus symbolizing doors and transitions, are asymmetric, homogeneous dendritic structures synthesized by combining dendrons (dendrimer wedges) linked to a central core.22–24 Since they were first described by Fréchet in 1991,25 numerous structures have been reported, utilizing various types of dendrimer families and reactions to couple dendrons, with applications in fields as catalysis, drug delivery, nanoparticles, or imaging to cite few examples.23,26 Among various dendrimer families, polyphosphorhydrazone (PPH) dendrimers stand out due to their highly versatile functional groups, ease of surface modification, and convenience of characterization through 31P{1H} NMR.27 Nonetheless, in contrast to other dendrimer families such as PBzE25,28 or PAMAM,29 PPH Janus dendrimers have not been extensively studied, and the work on asymmetric PPH structures has been restricted mostly to AB5 dendrons (dendritic structures formed from cyclotriphosphazene as core, were one P–Cl bond is reacted distinctively to the other five) and their applicability.30,31 In this context, reported examples of PPH Janus dendrimers have primarily focused on core coupling reactions of two dendrons through vinyl/amine reactions32–34 or the use of Staudinger coupling reactions.35,36 Although alternative procedures to synthesize Janus PPH dendrimers have been reported (isolated instances involve coupling through core desulfurization37 or the formation of hydrazone and amide core linkers38), the field remains relatively unexplored, and the applicability of such systems has mostly been related to materials science and biology,39,40 and only showing potential applicability in catalysis.
Nonetheless, a range of examples have been reported in the literature describing dendrimer-supported ruthenium catalysts, such as the ones reported by Cesari and co-workers41 incorporating ruthenium (0) complexes in poly(propyleneimine) dendrimers, and Rodríguez and co-workers42 in carbosilane dendrimers for transfer hydrogenation catalysis; the development of immobilized magnetic PAMAM dendrimers decorated with Ru(II) catalysts for the formation of 1,2,3-triazoles by Wang and co-workers,43 or the isomerization of allyl alcohols with PPH dendrimers by Caminade and co-workers,44 among other examples.45–50
Therefore, having the previous approaches in mind, as well as the established potential of dendrimers in catalysis, with the aim to overcome the issues arising from dendron coupling, we targeted to prepare a novel PPH Janus dendrimer system. Such system incorporates solubility modulation chains on one side of the structure and Ru-complexes on the other, through a “click” reaction to couple two dendrons. The testing of the final Ru@dendrimer was carried out in a model reaction for the isomerization of allyl alcohols, to provide the first example of a PPH Janus dendrimer tested in catalysis.
Results and discussion
Despite the progress made in the development of these Janus systems, ample scope remains for synthesizing such architectures using novel approaches. Integration of “click” triazole chemistry holds tremendous potential as it could enable the generation of complex dendritic structures using straightforward methodologies. In this context, our research has focused on the synthesis of PPH Janus dendrimers by coupling of AB5-type dendrons decorated with azide or acetylene moieties (with similar traits as recently reported by Alami and co-workers51), aiming the formation of a structure coupling a water-soluble modulation PEG-chain on the azide dendron, and an acetylene dendron bearing functional groups to allow the grafting of catalytic moieties.The synthesis of PPH dendrons was accomplished by asymmetrically substituting the hexafunctionalized dendrimer core N3P3Cl6 (1) by means of incorporating substituents based on 4-hydroxybenzaldehyde for the growing to higher generations and including a sixth functionality to incorporate either an azide (2) or acetylene (3) group, to yield the corresponding dendrons with such functionalities at the level of the core which would permit to test the coupling strategy through click chemistry, to afford bifunctional Janus dendrimers. Phenol derivatives 2 and 3 were synthesized according to previously reported methodologies52–54 (see ESI† for experimental details) and characterized by NMR spectroscopy. Of note, the characterization of the 2 also relied on IR spectroscopy, as azide-containing compounds typically exhibit a distinctive sharp N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NN stretching band at ca. 2100 cm−1 (2090 cm−1 for compound 2).
NN stretching band at ca. 2100 cm−1 (2090 cm−1 for compound 2).
The synthesis of dendrons was started following the approach depicted in Scheme 1. For instance, azide derivative 2 was reacted in defect (0.5 equiv.) with N3P3Cl6 under basic Cs2CO3 conditions by dropwise addition at 0 °C, followed by stirring at room temperature for 12 hours. This reaction was designed to maximize the formation of the mono-substituted product 4 while minimizing undesired poly-substitution at the core. The progress of the reaction was closely monitored using 31P{1H} NMR spectroscopy, which stands as the most versatile analytical tool for the characterization of PPH dendrimers.2731P{1H} NMR spectra exhibited the emergence of characteristic signals indicative of a monosubstituted N3P3Cl6 core by means of the formation of a doublet at 22.4 ppm and a triplet at 12.3 ppm. Similarly, the synthesis of the mono-substituted acetylene core 9 was accomplished with slight modification of the previous procedure,55 following previously reported methodologies. Confirmation of the desired compound was achieved through 31P{1H} NMR spectroscopy.
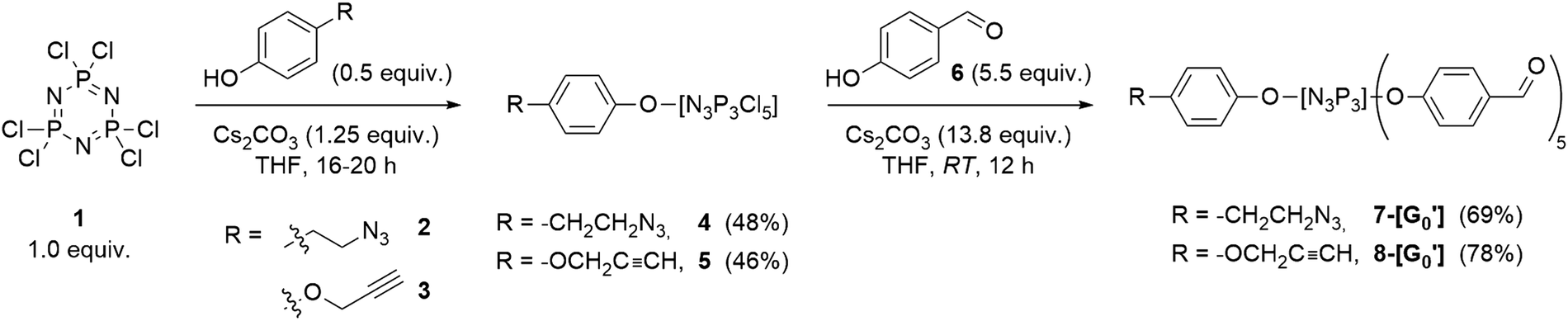 | ||
| Scheme 1 Synthesis of mono-substituted N3P3Cl6 cores incorporating azide (4) and acetylene (5) derivatives, and the corresponding G0′ dendrons 7 and 8. | ||
AB5-type G0′ dendrons were prepared by employing typical reaction conditions for the synthesis of PPH dendrimers and dendrons30 by completing the substitution of compounds 4 and 5 with an excess of 4-hydroxybenzaldehyde under basic conditions. Subsequent filtration of the salts and purification by column chromatography afforded the desired G0′ dendrons in moderate yields (Scheme 1). Verification of the desired products was carried out through 31P{1H} NMR spectroscopy, revealing the formation of a singlet at 7.4 ppm for the azide derivative 7-[G0′] and the emergence of a multiplet (8.7–6.9 ppm) for the acetylene counterpart 8-[G0′].51 The final steps in dendron synthesis were dedicated to the growing of the azide dendron to the first-generation, and the subsequent modification of its surface by incorporating PEG chains to enhance the solubility in water. Firstly, the aldehyde-terminated dendron 7-[G0′] was grown to the first-generation 10-[G1]. Particularly, 7-[G0′] was reacted with an excess of N-methyldichlorothiophosphorhydrazide 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 56 (5.5 equiv.) at 0 °C in a THF/CHCl3 solvent mixture, in the presence of anhydrous Na2SO4 to absorb the water generated during the aldehyde/amine condensation reaction (Scheme 2a). The progress of the obtention of 10-[G1] was closely monitored using 31P{1H} NMR spectroscopy, by the appearance of a new set of signals at 62 ppm, consistent with the formation of terminal –P(S)Cl2 groups. Additionally, 1H NMR revealed the disappearance of aldehyde signals from the 7-[G0′] precursor and the emergence of imine signals in the aromatic region. Of note, AB5-type dendrons such as 10-[G1] have a primary distinction in 31P{1H} NMR when compared to classical PPH dendrimers of the same generation, due to the structural asymmetry of the dendron structures. Notably, while standard first-generation PPH dendrimers would exhibit a singlet at 62.4 ppm for the terminal P(S)Cl2 moieties, 10-[G1] showed the presence of two overlapping singlets in a 3:2 intensity ratio, due to the asymmetry induced in the structure.
56 (5.5 equiv.) at 0 °C in a THF/CHCl3 solvent mixture, in the presence of anhydrous Na2SO4 to absorb the water generated during the aldehyde/amine condensation reaction (Scheme 2a). The progress of the obtention of 10-[G1] was closely monitored using 31P{1H} NMR spectroscopy, by the appearance of a new set of signals at 62 ppm, consistent with the formation of terminal –P(S)Cl2 groups. Additionally, 1H NMR revealed the disappearance of aldehyde signals from the 7-[G0′] precursor and the emergence of imine signals in the aromatic region. Of note, AB5-type dendrons such as 10-[G1] have a primary distinction in 31P{1H} NMR when compared to classical PPH dendrimers of the same generation, due to the structural asymmetry of the dendron structures. Notably, while standard first-generation PPH dendrimers would exhibit a singlet at 62.4 ppm for the terminal P(S)Cl2 moieties, 10-[G1] showed the presence of two overlapping singlets in a 3:2 intensity ratio, due to the asymmetry induced in the structure.
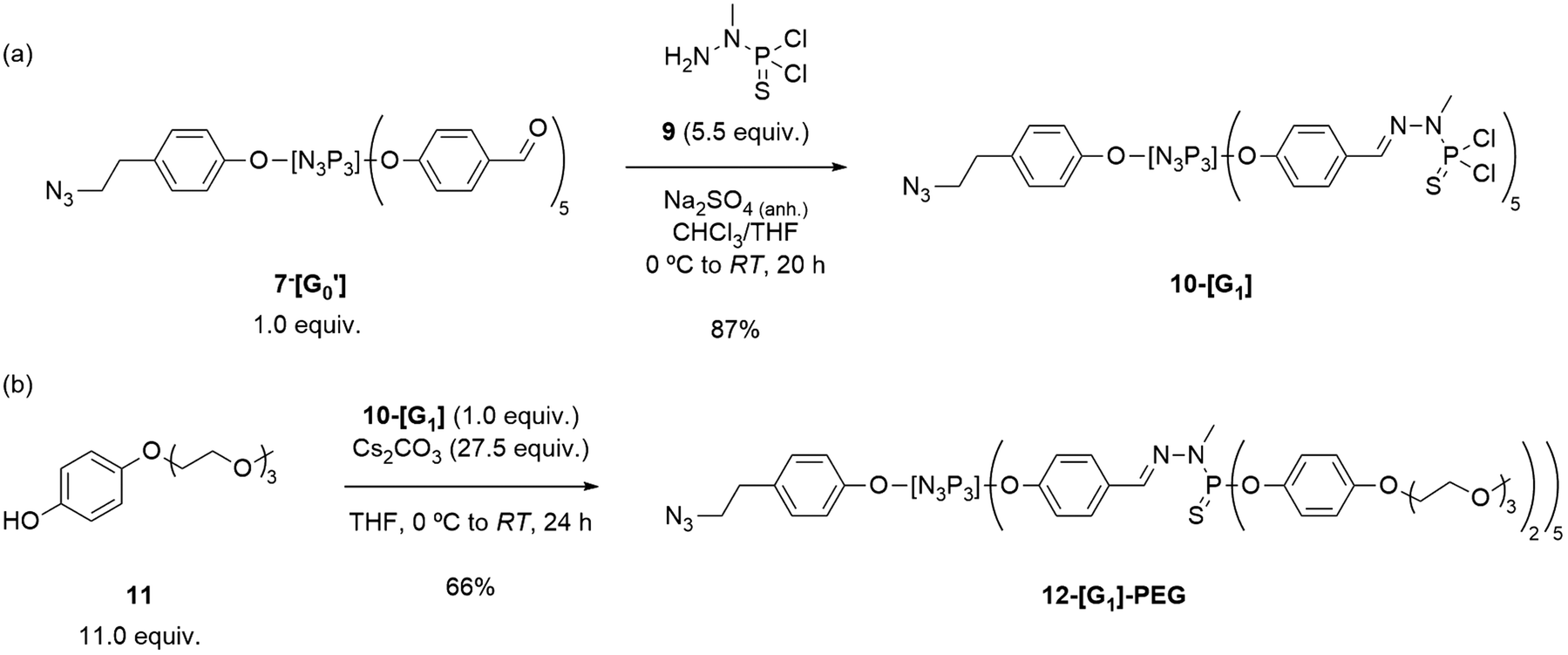 | ||
| Scheme 2 Synthesis of (a) the first-generation azide dendron 10-[G1], and (b) its PEG-surface modified 12-[G1]-PEG dendron. | ||
At this stage, the focus shifted towards developing a dendron that could induce solubility in water, by the introduction of PEG chains. To achieve this objective, the synthesis of the modified phenol 11 was undertaken for the later surface modification of 10-[G1]. PEG-modified phenol 11 was synthesized in two steps from hydroquinone and 2-(2-(2-methoxyethoxy)ethoxy)ethan-1-ol (17) as previously reported6 (see ESI† for details) and later tested for the incorporation onto the dendron structure (Scheme 2b). Of interest, phenol 11 (in slight excess) was dissolved in THF and stirred at 0 °C in presence of Cs2CO3 for 2 h prior to the addition of 10-[G1]. The progress of the reaction was monitored using 31P{1H} NMR spectroscopy. Notably, the substitution reactions of the P–Cl surface bonds with P–OR (where R = 11) induced a downfield shift in the NMR signal from 61.4 to 64.7 ppm. Although the typical reaction of dendrimers to obtain substituted –P(S)(OAr)2 groups results in an upfield shift in the surface phosphorus to 60–62 ppm, Caminade's group has previously reported the formation of similar compounds displaying the same 31P{1H} NMR pattern.6 The desired substituted 12-[G1]-PEG was obtained in a 66% yield after 22 h of reaction, by precipitation from n-pentane and washing with n-pentane/DCM.
Upon synthesis of the desired dendrons, the preparation of a hybrid 13-[G0′][G1]-PEG Janus dendrimer (Scheme 3) was performed by reacting the PEG-modified azide dendron 12-[G1]-PEG and the acetylene dendron 8-[G0′] in stoichiometric amounts by the formation of a 1,2,3-triazole through a copper-catalyzed azide–alkyne cycloaddition at 45 °C in presence of N,N-diisopropylethylamine (DIPEA) and CuI as catalyst. The progress of the reaction was monitored primarily using NMR and IR spectroscopy. Specifically, 1H NMR was analyzed to assess the disappearance of the acetylenic proton signal from the alkyne dendron 9-[G0′] (formerly a triplet at 2.56 ppm), the appearance of a singlet at 5.01 ppm from the methylenic protons in the alkyne moiety of 13-[G0′][G1]-PEG (previously doublet at 4.65 ppm), as well as the emergence of a singlet from the triazole unit at ca. 8 ppm. Furthermore, the progress of the reaction was conveniently monitored through IR spectroscopy, specifically by tracking the disappearance of the NNN stretching band at 2090 cm−1 from the azide dendron 12-[G1]-PEG. The novel hybrid dendrimer was obtained in moderate yields after 30 h of reaction. The final stage in the synthesis of the desired first-generation Janus dendrimer involved extending the aldehyde functionalities of 13-[G0′][G1]-PEG to the full first generation 14-[G1][G1]-PEG (Scheme 3). It entailed the reaction of 13-[G0′][G1]-PEG with N-methyldichlorothiophosphorhydrazide 9 in the presence of anhydrous Na2SO4, as previously described for the growing of dendrons.51 The synthesis of 14-[G1][G1]-PEG was confirmed once more by 31P{1H} NMR by the emergence of a new set of signals corresponding to the terminal –P(S)Cl2 group, manifesting as two singlets at 62.5 ppm and 62.7 ppm.
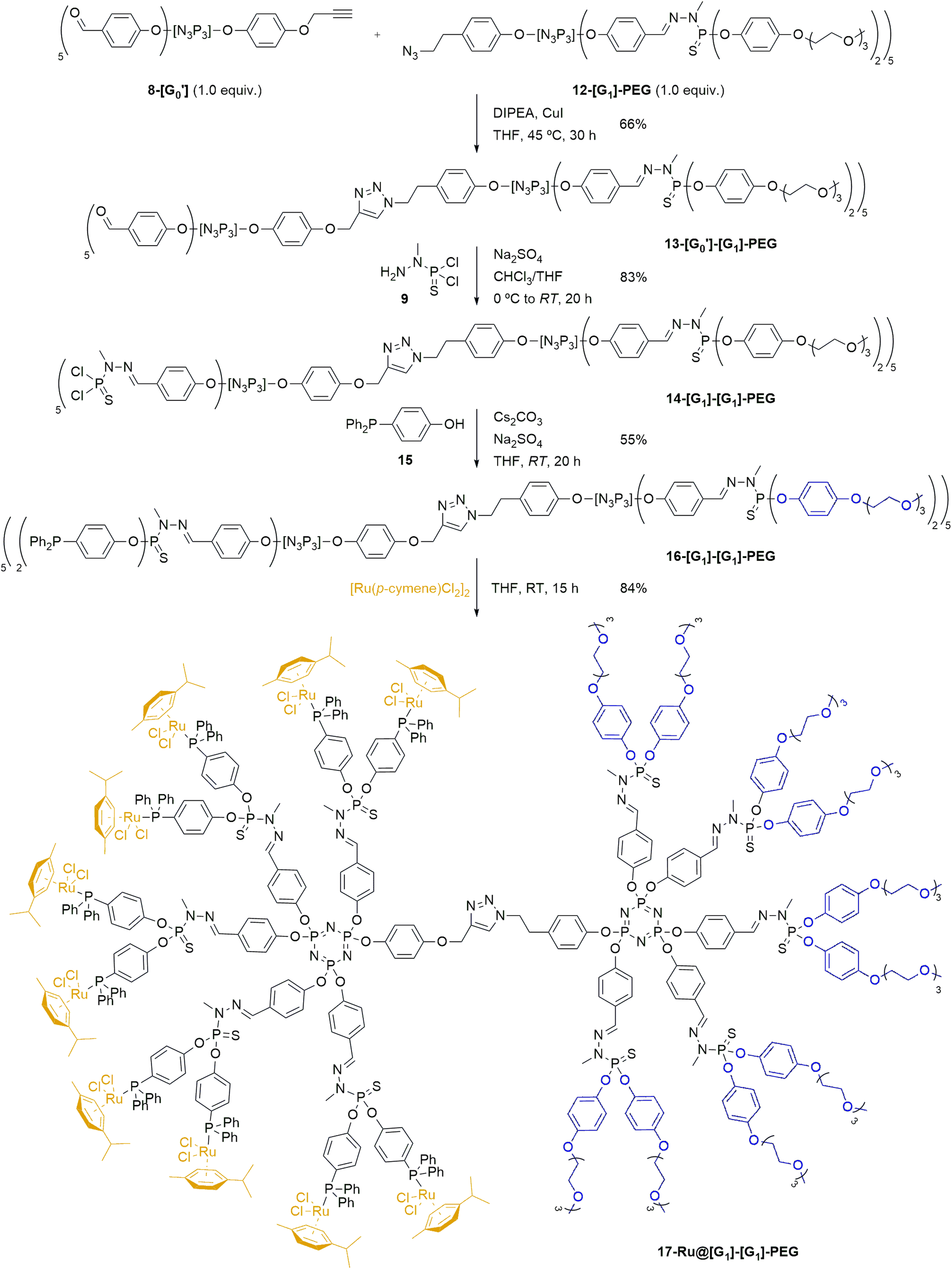 | ||
| Scheme 3 Synthesis of a novel first-generation Ru@PPH Janus dendrimer 17-Ru@[G1][G1]-PEG from the dendrons 8-[G0′] and 12-[G1]-PEG. | ||
Building upon the previous approach, our next objective was to develop a novel catalytic system through the immobilization of metal pre-catalysts on the surface of 14-[G1][G1]-PEG Janus dendrimer. Specifically, we aimed to immobilize [Ru(p-cymene)Cl2]2 complexes on the dendrimer surface, based on the wide availability of such compound. The synthesis of the Ru@Janus dendrimer system commenced with the preparation of a phosphine ligand (4-(diphenylphosphoryl)phenol 15), which would be introduced onto the dendrimer structure before complexation of the pre-catalyst. This compound was chosen due to the presence of both a phenol group –which serves as a grafting point for the reaction with 14-[G1][G1]-PEG– and the phosphine itself, which would be used for the complexation of the ruthenium catalyst. The desired phenol phosphine 15 was synthesized in three steps from 4-bromophenol:57 first, 4-bromophenol was derivatized to introduce a silyl protecting group by reaction with TBDMSCl; later, the conversion into phosphine by a two-step lithiation and phosphination with chlorodiphenyl phosphine; and lastly, the phenolic OH group was regenerated by deprotection with tetrabutylammonium fluoride (TBAF) to reach the desired 15 in 84% yield over three steps (see ESI† for experimental details).
After synthesis of 15, the next step involved introducing it to the first-generation Janus dendrimer 14-[G1][G1]-PEG (Scheme 3). To prepare the desired phosphine-terminated dendrimer, standard reaction conditions for the introduction of phenol derivatives to –P(S)Cl2-terminated dendrimers were employed. Specifically, the Janus dendrimer was dissolved in THF, and phosphine 15 was added under basic Cs2CO3 conditions, with anhydrous Na2SO4 also present in the reaction mixture. The progress of the reaction was monitored using 31P{1H} NMR. In the crude mixture, the appearance of two singlets at 61.0–61.5 ppm was observed, consistent with the substitution of the chlorine atoms from 14-[G1][G1]-PEG by phenoxy derivatives of 15 to give the corresponding –P(S)(OR)2 moieties, which typically appear at ca. 62 ppm. A similar trend confirmed the grafting of 4-(diphenylphosphoryl)phenol onto the dendrimer, as evidenced by a shielding effect on the phosphorus atom of 15, shifting from −7.0 ppm to −6.5 ppm. The desired phosphine-terminated 16-[G1][G1]-PEG was obtained in a moderate 55% yield after work-up.
Lastly, the desired Ru@dendrimer system was obtained by anchoring to the phosphine-terminated dendron 16-[G1][G1]-PEG a commercially available ruthenium complex. To accomplish this, [Ru(p-cymene)Cl2]2 was grafted to the dendrimer through the complexation ability of the terminal phosphines. As illustrated in Scheme 3 (bottom), five equivalents of the ruthenium complex were reacted with one equivalent of dendrimer 16-[G1][G1]-PEG –bearing ten phosphine groups– in THF at room temperature, following a similar procedure as previously reported in the literature.58 The reaction proceeded smoothly, yielding the expected 17-Ru@[G1][G1]-PEG dendrimer as an orange solid in good yield, which was easily purified by precipitation and further washings. The newly formed Ru@dendrimer species was fully characterized, with special mention to the 31P{1H} NMR, revealing a significant deshielding effect of the terminal phosphine upon complexation, shifting from −6.6 ppm to 24.0 ppm, and the formation of a doublet with a coupling constant of 1JP–Ru = 37.2 Hz. Furthermore, inductively coupled plasma (ICP) analysis was used for assessing the ruthenium content in the Janus dendrimer. The analysis demonstrated that the supported 17-Ru@[G1][G1]-PEG contained a Ru content of 9.1%, aligning with the calculated theoretical value.
Dendrimers can have various effects when applied in catalysis, including modifications of catalytic activity due to the high local concentration of catalysts within dendrimers, effects arising from catalyst anchoring, potential electronic modifications, and facilitation of catalyst recyclability and reusability within dendrimeric systems.59,60 To properly assess the performance of the novel immobilized ruthenium complex 17-Ru@[G1][G1]-PEG, it was essential to synthesize analogous ruthenium complexes for comparison purposes. Firstly, we synthesized a monometallic ruthenium complex (20), which involved a two-step process starting from 4-bromoanisole 18 (Scheme 4a). In the initial step, we synthesized 4-(diphenylphosphoryl)anisole 19 through lithiation and phosphination of 18, resulting in a white solid with an 80% yield.61 Subsequently, in the second step, we carried out the reaction with [Ru(p-cymene)Cl2]2, utilizing the coordination ability of phosphines, ultimately yielding the desired complex 20 in excellent yield as an orange powder. The novel ruthenium complex was fully characterized to confirm the formation of the desired structure.
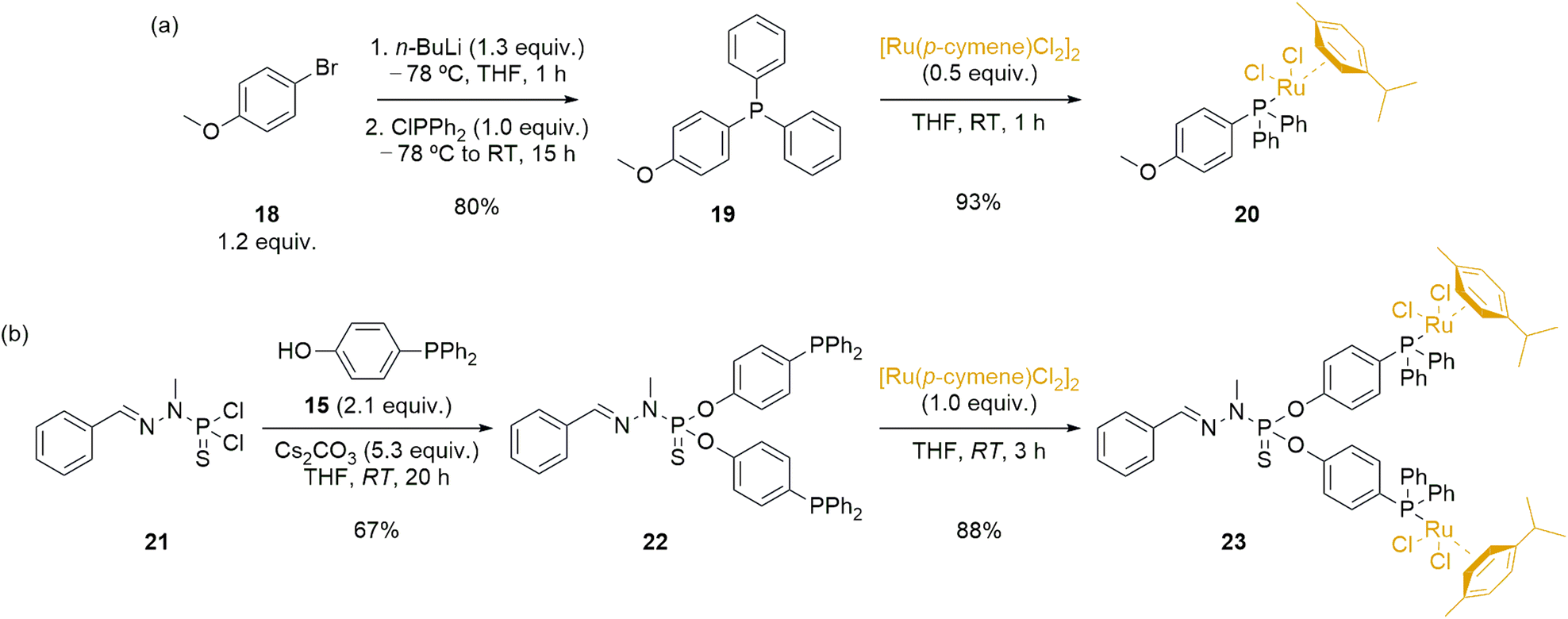 | ||
| Scheme 4 Synthesis of Ru-complexes 20 and 23, monometallic and bimetallic analogous of the Ru@dendrimer 17, respectively. | ||
Simultaneously, we planned the synthesis of a bimetallic species 23 (Scheme 4b), with chemical similarities to the Ru@dendrimer 17. In the first step, we derivatized the model dendrimer 21—synthesized using previously reported methodologies from benzaldehyde and N-methyldichlorothiophosphorhydrazide 9—with a slight excess of 4-(diphenylphosphoryl)phenol 15 under basic Cs2CO3 conditions at room temperature. This reaction resulted in the substitution of the P–Cl bonds in 21 with the phenol phosphine, leading to the formation of the phosphine-modified model dendrimer 22 in a 67% yield. The progress of the reaction was once more monitored using 31P{1H} NMR spectroscopy. Specifically, the phosphorus atom signal from the starting 21 (δ21 = 63.4 ppm) shifted to 61.5 ppm upon substitution with the phenol phosphine 15, confirming the disubstitution pattern. Additionally, the phosphorus atom signal from 22 also experienced a change due to the substitution, showing a slight downfield shift from −7.0 ppm to −6.4 ppm in the final product. The final step to obtain bimetallic species 23 involved reacting with [Ru(p-cymene)Cl2]2, employing the same reaction conditions previously reported for the synthesis of 20. The desired bimetallic species 23 was obtained in good yield 3 hours of reaction, and the product fully characterized to ascertain the formation of the targeted product.
Catalytic tests for the isomerization of allyl alcohols
Having successfully synthesized a PPH Janus dendrimer system containing Ru(p-cymene) complexes within its structure (17), along with analogous monometallic and bimetallic species (20, 23), the next step was to evaluate the catalytic activity of the three prepared complexes. Ru(p-cymene)-type complexes have been well-documented in the literature for their applications as catalysts in various processes, including the isomerization of allyl alcohols and the hydration of acetylenes, among others.44,62 For our evaluation, we selected the isomerization of allyl alcohols as the target reaction to assess the performance of the prepared complexes. This reaction typically involves the conversion of allyl alcohols into ketones or aldehydes through a two-step process that combines oxidation and reduction, but that is carried out in a one-pot manner using organometallic complexes. The initial catalytic tests for the novel Ru-complexes were conducted to evaluate their performance for the isomerization of 1-octen-3-ol to 3-octanone, firstly in conventional organic solvent conditions. In these experiments, a THF solution containing the substrate 24 (1.0 equiv.), 1,3,5-trimethoxybenzene (used as an internal standard), and Cs2CO3 (2 mol%) was prepared (Table 1). The reaction mixture was then heated to 75 °C, and 1 mol% of the ruthenium catalyst was added. It is worth noting that the same reaction conditions were applied for all three Ru-catalysts, and the conversion was assessed by gas chromatography (GC) comparing the crude mixtures with the original samples of substrates and products.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time to full conversiona [h] | Yieldc [%] |
| a Conversion was determined through GC using 1,3,5-trimetoxybenzene as internal standard. b Only traces were detected by GC. c Isolated yield after post-reaction treatment. | ||||
| 1 | 20 – monometallic | THF | 3 | 89 |
| 2 | 23 – bimetallic | 1.5 | 84 | |
| 3 | 17-Ru@[G1][G1]-PEG | 1.5 | 91 | |
| 4 | 20 – monometallic | H2O/n-heptane (1:1) |
n.a.b | — |
| 5 | 23 – bimetallic | n.a.b | — | |
| 6 | 17-Ru@[G1][G1]-PEG | 24 | 98 | |
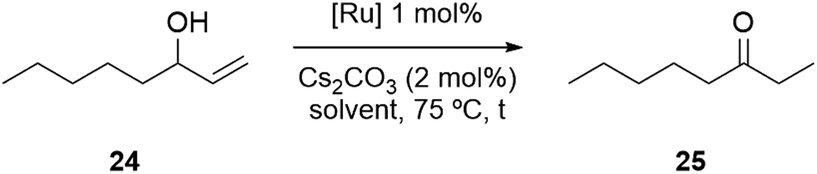
As anticipated, all three Ru-complexes achieved full conversion within the reported time frames in the literature (Table 1, entries 1–3).44,62 Monometallic complex 20 achieved complete conversion after 3 hours of reaction. Remarkably, both the bimetallic complex 23 and the dendrimeric structure 17-Ru@[G1][G1]-PEG yielded 3-octanone in short reaction times (1.5 h). Moreover, the desired ketone derivative was isolated after precipitation of the dendrimeric catalyst from n-pentane, centrifugation, and flash column chromatography affording 3-octanone in yields ranging from 84–91%. These results were highly encouraging, as they indicated that the proximity of ruthenium centers could potentially lead to a cooperative effect, enhancing the efficiency of the reaction.
Following the promising results obtained in organic solvent conditions, our focus shifted to utilizing water as the solvent for the catalytic process. Specifically, we chose an n-heptane/water mixture (1:1) as the solvent system, considering it a valuable approach to reduce the usage of organic solvents by incorporation of a sustainable solvent such as water, and benefiting from the possibility of an easier recovery of the catalyst in biphasic media. Importantly, the presence of an n-heptane/water mixture resulted in the formation of a biphasic system where: (i) the substrate, product, and the internal standard resided in the organic n-heptane phase; and (ii) cesium carbonate and the Ru@dendrimer 17 remained in the water phase making easier its recovering and recycling. Catalytic tests were conducted following the previously established reaction conditions incorporating the use of a biphasic solvent system. Aliquots were sampled at specific intervals, and conversion was determined by GC (Table 1, entries 4–6). The catalytic results revealed that both the monometallic and bimetallic systems yielded no isomerization product after 24 hours of reaction. Intriguingly, when the 17-Ru@[G1][G1]-PEG catalyst was employed, full conversion was achieved after 24 hours of reaction, with an isolated yield of the 98%.
The obtained results were highly positive in terms of the activity of the Ru@dendrimer system. Notably, a positive dendritic effect was observed for the isomerization of 3-octen-1-ol in both organic solvent and biphasic mixtures. Particularly interesting was the use of the biphasic system, where only 17-Ru@[G1][G1]-PEG exhibited catalytic activity for the desired reaction, although activity was reduced, and long reaction times were required.
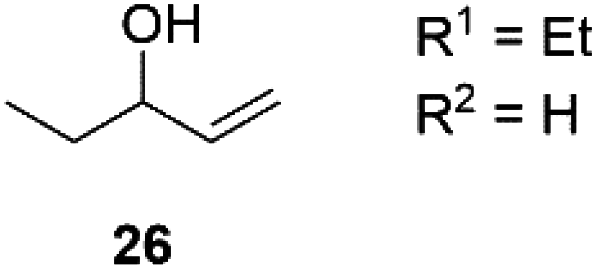
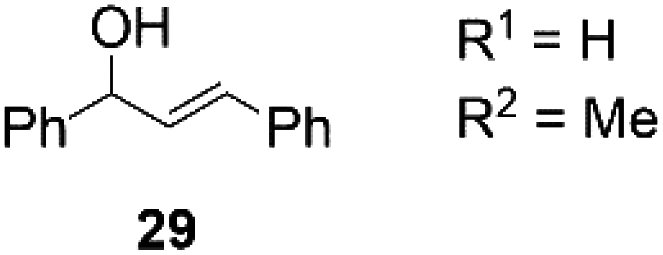
Next, we decided to test the new Ru@dendrimer catalyst 17 on a set of substrates containing different functionalities (26–29) with the standard tested loading of catalyst (1 mol% of “Ru”) and 2 mol% of Cs2CO3 in both solvent conditions (Table 2). Following the same trend as for substrate 24, reactions performed in THF displayed a faster conversion rate, achieving full conversion for all the substrates in times ranging from 0.5–22 hours, showing similar results as reported in the literature.63 In addition to that, the tests in H2O/n-heptane showed also full conversion for almost all the substrates in times ranging from 7 to 24 hours, with the exception of substrate 29 (Table 2, entry 8) which lead to an 86% conversion after one day of reaction, also displaying comparable results to those in the literature.64,65
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Solvent | Conversiona [%] | Time [h] | Yieldb [%] |
| a Conversion was determined through GC using 1,3,5-trimetoxybenzene as internal standard. b Isolated yield after post-reaction treatment. c Not isolated product. | |||||
| 1 |
|
THF | >99 | 0.5 | 96 |
| 2 | H2O/n-heptane | >99 | 7 | 97 | |
| 3 |
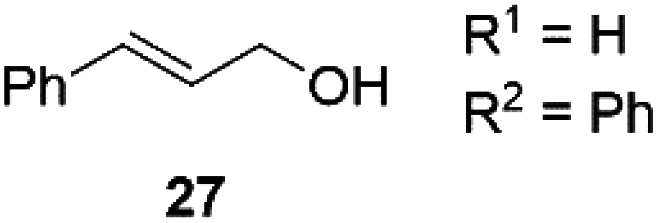
|
THF | >99 | 3 | 98 |
| 4 | H2O/n-heptane | >99 | 22 | 98 | |
| 5 |
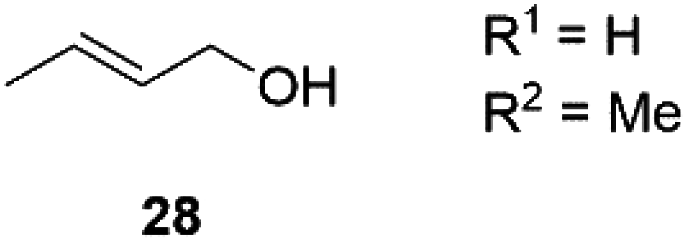
|
THF | >99 | 5 | n.a.c |
| 6 | H2O/n-heptane | >99 | 24 | n.a.c | |
| 7 |
|
THF | >99 | 22 | 95 |
| 8 | H2O/n-heptane | 86 | 24 | 82 | |
The isolation of the isomerization products was achieved for almost all the products with good yields ranging from 82 to 98%, except for butyraldehyde (Table 2, entries 5 and 6) which was not obtained due to the low boiling point of the product and the limited working quantities.
Lastly, we decided to investigate the recyclability of the dendrimeric system, to provide a greener approach to the use of the Ru@dendrimer catalyst. In this context, a catalytic test was conducted in the biphasic solvent conditions, and the reaction was allowed to proceed for 24 hours. After reaction time, the organic phase was separated from the aqueous phase and GC analyses confirmed the full conversion of 24 to 3-octanone in the recovered phase. Subsequently, a mixture of 24 and 1,3,5-trimethoxybenzene in fresh n-heptane was added to the water phase containing the Ru@dendrimer catalyst 17. Repetition of this procedure seven times resulted in the attainment of full conversion to 3-octanone for up to six consecutive runs (Fig. 1), until the drop of the conversion to the 76% in the seventh run. The desired product was isolated after selected cycles affording yields between 94–98%. Moreover, the possibility of metal leaching was also investigated. Analysis of Ru content in the organic phase by ICP revealed no significant leaching, even after full conversion to 3-octenone. ICP results indicated the presence of less than 0.05% Ru contamination in the sample, confirming the excellent stability of the catalyst.
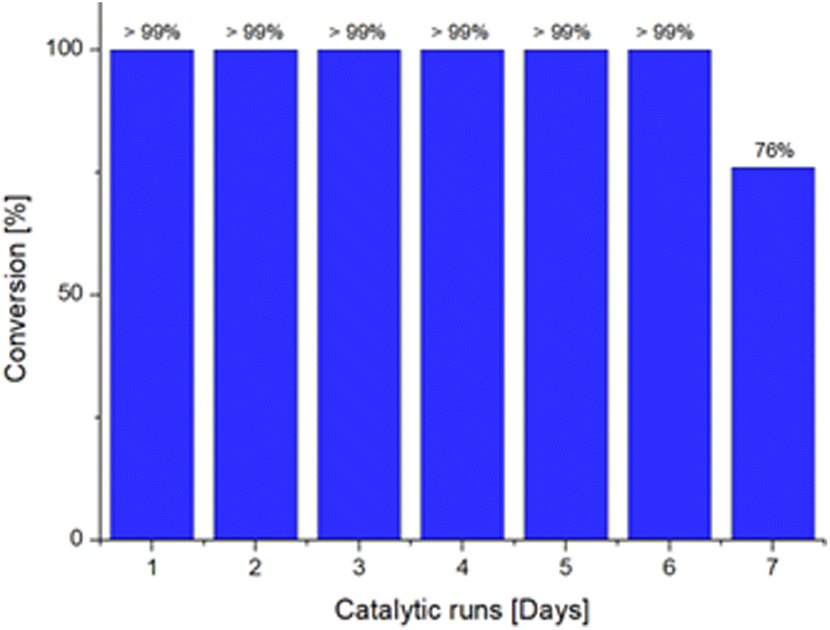 | ||
| Fig. 1 Recyclability experiments for the isomerization of 1-octen-3-ol to 3-octanone catalyzed by Ru@dendrimer 17 for seven consecutive runs. | ||
Conclusions
In summary, we have successfully synthesized a first-first generation PPH Janus dendrimer by connecting two dendrons through the formation of a 1,2,3-triazole linkage, which is a novel approach for phosphorus dendrimers. The Janus dendrimer has been designed to include PEG chains to ensure solubility in water and functional P(S)Cl2 groups. We then tested the dendrimer ability to immobilize ruthenium complexes by incorporating a terminal phosphine on the Janus dendrimer and by reacting it with a commercially available [Ru(p-cymene)Cl2]2 dimer. This resulted in 17-Ru@[G1][G1]-PEG, featuring ten PEG chains on one side and ten Ru-complexes on the other. We then evaluated the catalytic performance of this innovative immobilized Ru-system in the isomerization of 1-octen-3-ol to 3-octanone, assessing its effectiveness compared to two small analogue complexes (20 and 23), designed to mimic the active catalytic moiety found in the dendrimer. Catalytic tests conducted in THF as the solvent produced excellent results for all three Ru systems, particularly for the bimetallic 23 and the dendrimer 17, reaching full substrate conversion in half the time compared to the monometallic analogue 20. Catalysis in biphasic water/n-heptane systems was also explored, where a significant positive dendritic effect was observed. Specifically, only 17-Ru@[G1][G1]-PEG achieved full conversion in the biphasic conditions after 24 hours of reaction, while 20 and 23 did not produce the isomerization product. Furthermore, a range of allyl alcohols was tested employing 17-Ru@[G1][G1]-PEG, showing great results in all the tested cases. Lastly, we tested the recyclability of 17-Ru@[G1][G1]-PEG and found that it could be recycled and reused for up to six consecutive runs without loss of conversion, with ruthenium contamination in the final product amounting to less than 0.05%. Despite the use of a more sustainable solvent system evolved to a reduction of the dendrimeric catalyst activity, the performance of the dendrimer allowed the recyclability of the catalyst without a variation in the activity upon six consecutive runs, being easier its recovering and recycling in the water phase.Through this approach, we have developed an innovative methodology for creating PPH Janus dendrimers by linking azide and acetylene dendrons through click chemistry and reported the first example of PPH Janus dendrimers with applications in catalysis.
Author contributions
Conceptualization, R. M. S.; experimental investigation and formal analysis, J. C.-S.; writing–original draft preparation J. C.-S.; writing–review and editing, J. C.-S., A.-M. C., A. K., K. G. and R. M. S.; supervision R. M. S.; funding acquisition K. G., A.-M. C. and R. M. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 860322 for the ITN-EJD “Coordination Chemistry Inspires Molecular Catalysis” (CCIMC). Thanks are due to Generalitat de Catalunya (2021 SGR 00064 and 2021 SGR 00122) and Ministerio de Ciencia, Innovación y Universidades (PID2019-106171RB-I00 and PID2020-116844RB-C21) for financial support. Authors want to thank the Servei d'Anàlisi Química (Universitat Autònoma de Barcelona) for the EA and ICP analyses, and the Servicio de Espectrometría de Masas (Universidad de Zaragoza) for the HRMS measurements.References
- A.-M. Caminade, C. O. Turrin, R. Laurent, A. Ouali and B. Delavaux-Nicot, Dendrimers: Towards Catalytic, Material and Biomedical Uses, John Wiley & Sons Ltd, 1st edn, 2011 Search PubMed.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117–132 CrossRef CAS.
- S. Thakare, A. Shaikh, D. Bodas and V. Gajbhiye, Colloids Surf., B, 2022, 209, 112174–112186 CrossRef CAS PubMed.
- J. Satija, V. V. R. Sai and S. Mukherji, J. Mater. Chem., 2011, 21, 14367–14386 RSC.
- C. C. Lee, J. A. MacKay, J. M. J. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed.
- A. Sourdon, M. Gary-Bobo, M. Maynadier, M. Garcia, J.-P. Majoral, A.-M. Caminade, O. Mongin and M. Blanchard-Desce, Chem. – Eur. J., 2019, 25, 3637–3649 CrossRef CAS PubMed.
- E. Pedziwiatr-Werbicka, K. Milowska, V. Dzmitruk, M. Ionov, D. Shcharbin and M. Bryszewska, Eur. Polym. J., 2019, 119, 61–73 CrossRef CAS.
- D. Riegert, L. Bareille, R. Laurent, J.-P. Majoral, A.-M. Caminade and A. Chaumonnot, Eur. J. Inorg. Chem., 2016, 2016, 3103–3110 CrossRef CAS.
- B. M. Ali, K. A. Kumar and A. S. Sultan Nasar, ChemistrySelect, 2019, 4, 12983–12991 CrossRef CAS.
- J. N. H. Reek, S. Arévalo, R. van Heerbeek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Adv. Catal., 2006, 49, 71–151 CAS.
- R. van Heerbeek, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Rev., 2002, 102, 3717–3756 CrossRef CAS PubMed.
- P. Neumann, H. Dib, A.-M. Caminade and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2015, 54, 311–314 CrossRef CAS PubMed.
- A.-M. Caminade and R. Laurent, Coord. Chem. Rev., 2019, 389, 59–72 CrossRef CAS.
- Z. Wang, G. Deng, Y. Li, Y. He, W. Tang and Q. Fan, Org. Lett., 2007, 9, 1243–1246 CrossRef CAS PubMed.
- L. Ooe, M. Murata, T. Mizugani, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 1604–1605 CrossRef PubMed.
- A. Miedaner, C. J. Curtis, R. M. Barkley and D. L. DuBois, Inorg. Chem., 1994, 33, 5482–6490 CrossRef CAS.
- X. Liu, D. Gregurec, J. Irigoyen, A. Mantinez, S. Moya, R. Ciganda, P. Hermange, J. Ruiz and D. Astruc, Nat. Commun., 2016, 7, 13152–13160 CrossRef CAS PubMed.
- E. Badetti, A.-M. Caminade, J.-P. Majoral, M. Moreno-Mañas and R. M. Sebastián, Langmuir, 2008, 24, 2090–2101 CrossRef CAS PubMed.
- D. G. Mullen, E. L. Borgmeier, A. M. Desai, M. A. van Dongen, M. Barash, X. Cheng, J. R. Baker and M. M. Banaszak Holl, Chem. – Eur. J., 2010, 16, 10675–10678 CrossRef CAS PubMed.
- T. P. Thomas, B. Haung, S. K. Choi, J. E. Slipe, A. Kotlyar, A. M. Desai, H. Zong, J. Gam, M. Joice and J. R. Baker, Mol. Pharm., 2012, 9, 2669–2676 CrossRef CAS PubMed.
- M. Petriccone, R. Laurent, C.-O. Turrin, R. M. Sebastián and A.-M. Caminade, Organics, 2022, 3, 240–261 CrossRef CAS.
- F. Najafi, M. Salami-Kalajahi and H. Roghani-Mamaqani, J. Mol. Liq., 2022, 347, 118396–118417 CrossRef CAS.
- A.-M. Caminade, R. Laurent, B. Delavaux-Nicot and J.-P. Majoral, New J. Chem., 2012, 36, 217–226 RSC.
- P.-G. de Gennes, Croat. Chem. Acta, 1998, 71, 833–836 CAS.
- K. L. Wooley, C. J. Hawker and J. M. J. Fréchet, J. Chem. Soc., Perkin Trans. 1, 1991,(5), 1059–1076 RSC.
- A. Căta, I. M. C. Ienaşcu, M. N. Ştefănuţ, D. Roşu and O. R. Pop, Pharmaceutics, 2023, 15, 589–613 CrossRef PubMed.
- A.-M. Caminade, R. Laurent, C.-O. Turrin, C. Rebout, B. Delavaux-Nicot, A. Ouali, M. Zablocka and J.-P. Majoral, C. R. Chim., 2010, 13, 1006–1027 CrossRef CAS.
- Z. Bo, J. P. Rabe and A. D. Schlüter, Angew. Chem., Int. Ed., 1999, 38, 2370–2372 CrossRef CAS PubMed.
- C. Ornelas, R. Pennel, L. F. Liebes and M. Weck, Org. Lett., 2011, 13, 976–979 CrossRef CAS PubMed.
- A. Zibarov, A. Oukhrib, J. A. Catot, C.-O. Turrin and A.-M. Caminade, Molecules, 2021, 26, 1–24 CrossRef PubMed.
- J. Cejas-Sánchez, A. Kajetanowicz, K. Grela, A.-M. Caminade and R. M. Sebastián, Molecules, 2023, 28, 5570–5588 CrossRef PubMed.
- V. Maraval, R. Laurent, S. Merino, A.-M. Caminade and J.-P. Majoral, Eur. J. Org. Chem., 2000, 3555–3568 CrossRef CAS.
- V. Maraval, A. Maraval, G. Spataro, A.-M. Caminade, J.-P. Majoral, D. H. Kim and W. Knoll, New J. Chem., 2006, 30, 1731–1736 RSC.
- B. S. Kim, O. V. Lebedeva, D. H. Kim, A.-M. Caminade, J.-P. Majorai, W. Knoll and O. I. Vinogradova, Langmuir, 2005, 21, 7200–7206 CrossRef CAS PubMed.
- L. Brauge, G. Magro, A.-M. Caminade and J.-P. Majoral, J. Am. Chem. Soc., 2001, 123, 6698–6699 CrossRef CAS PubMed.
- S. Gottis, L. I. Rodriguez, R. Laurent, I. Angurell, M. Seco, O. Rossell, J.-P. Majoral and A.-M. Caminade, Tetrahedron Lett., 2013, 54, 6864–6867 CrossRef CAS.
- V. Maraval, R. M. Sebastian, F. Ben, R. Laurent, A.-M. Caminade and J.-P. Majoral, Eur. J. Inorg. Chem., 2001, 7, 1681–1691 CrossRef.
- S. Fuchs, A. Pla-Quintana, S. Mazères, A.-M. Caminade and J.-P. Majoral, Org. Lett., 2008, 10, 4751–4754 CrossRef CAS PubMed.
- M. Blanzat, C.-O. Turrin, A. M. Aubertin, C. Couturier-Vidal, A.-M. Caminade, J.-P. Majoral, I. Rico-Lattes and A. Lattes, ChemBioChem, 2005, 6, 2207–2213 CrossRef CAS PubMed.
- J. Solassol, C. Crozet, V. Perrier, J. Leclaire, F. Béranger, A.-M. Caminade, B. Meunier, D. Dormont, J.-P. Majoral and S. Lehmann, J. Gen. Virol., 2004, 85, 1791–1799 CrossRef CAS PubMed.
- C. Cesari, R. Conti, A. Cingolani, V. Zanotti, M. C. Cassani, L. Rigamonti and R. Mazzoni, Catalysts, 2020, 264–274 CrossRef CAS.
- L. I. Rodríguez, O. Rossell, M. Seco and G. Muller, J. Organomet. Chem., 2007, 692, 851–858 CrossRef.
- D. Wang, C. Deraedt, J. Ruiz and D. Astruc, Acc. Chem. Res., 2015, 48, 1871–1880 CrossRef CAS PubMed.
- P. Servin, R. Laurent, L. Gonsalvi, M. Tristany, M. Peruzzini, J.-P. Majoral and A.-M. Caminade, Dalton Trans., 2009, 4432–4434 RSC.
- A. Daneshvar, M. Moghadam, S. Tangestaninejad, V. Mirkhani, I. Mohammadpoor-Baltork and A. Khalili, Organometallics, 2016, 35, 1747–1755 CrossRef CAS.
- S. Michlewska, M. Ionov, A. Szwed, A. Rogalska, N. S. Del Olmo, P. Ortega, M. Denel, D. Jacenik, D. Shcharbin, F. J. de la Mata and M. Bryszewska, Int. J. Mol. Sci., 2020, 21, 1–13 Search PubMed.
- P. Keshtiara, H. Hadadzadeh, M. Daryanavard, N. Mousavi and M. Dinari, Mater. Chem. Phys., 2020, 249, 122962–122974 CrossRef CAS.
- W. Wang and Q. Wang, Chem. Commun., 2010, 46, 4616–4618 RSC.
- S. Pettirossi, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. – Eur. J., 2009, 15, 5337–5347 CrossRef CAS PubMed.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed.
- O. Alami, R. Laurent, M. Tassé, Y. Coppel, Y. Bignon, S. El Kazzouli, J.-P. Majoral, N. El Brahimi and A.-M. Caminade, Chem. – Eur. J., 2023, 29, e202302198 CrossRef CAS PubMed.
- C. D. Spicer, M. Pujari-Palmer, H. Autefage, G. Insley, P. Procter, H. Engqvist and M. M. Stevens, ACS Cent. Sci., 2020, 6, 226–231 CrossRef CAS PubMed.
- A. Makarem, K. D. Klika, G. Litau, Y. Remde and K. Kopka, J. Org. Chem., 2019, 84, 7501–7508 CrossRef CAS PubMed.
- Y. Q. Jiang, K. Wu, Q. Zhang, K. Q. Li, Y. Y. Li, P. Y. Xin, W. W. Zhang and H. M. Guo, Chem. Commun., 2018, 54, 13821–13824 RSC.
- L. Abbassi, Y. M. Chabre, K. Naresh, A. A. Arnold, S. André, J. Josserand, H.-J. Gabius and R. Roy, Polym. Chem., 2015, 6, 7666–7683 RSC.
- C. Larré, D. Bressolles, C.-O. Turrin, B. Donnadieu, A.-M. Caminade and J.-P. Majoral, J. Am. Chem. Soc., 1998, 120, 13070–13082 CrossRef.
- L. Biancalana, L. K. Batchelor, A. De Palo, S. Zacchini, G. Pampaloni, P. J. Dyson and F. Marchetti, Dalton Trans., 2017, 46, 12001–12004 RSC.
- I. Angurell, C.-O. Turrin, R. Laurent, V. Maraval, P. Servin, O. Rossell, M. Seco, A.-M. Caminade and J.-P. Majoral, J. Organomet. Chem., 2007, 692, 1928–1939 CrossRef CAS.
- L. P. Wu, M. Ficker, J. B. Christensen, P. N. Trohopoulos and S. M. Moghimi, Bioconjugate Chem., 2015, 26, 1198–1211 CrossRef CAS PubMed.
- C.-O. Turrin, V. Maraval, J. Leclaire, E. Dantras, C. Lacabanne, A.-M. Caminade and J.-P. Majoral, Tetrahedron, 2003, 59, 3965–3973 CrossRef CAS.
- D. Prévôte, S. Le Roy-Gourvennec, A.-M. Caminade, S. Masson and J.-P. Majoral, Synthesis, 1997, 1199–1207 CrossRef.
- J.-E. Bäckvall and U. Andreasson, Tetrahedron Lett., 1993, 34, 5459–5462 CrossRef.
- P. Crochet, J. Díez, M. A. Fernández-Zúmel and J. Gimeno, Adv. Synth. Catal., 2006, 348, 93–100 CrossRef CAS.
- V. Cadierno, P. Crochet, J. Francos, S. E. García-Garrido, J. Gimeno and N. Nebra, Green Chem., 2009, 11, 1992–2000 RSC.
- J. García-Álvarez, J. Gimeno and F. J. Suárez, Organometallics, 2011, 30, 2893–2896 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and the data supporting the reported results in this study. See DOI: https://doi.org/10.1039/d3dt04376b |
| This journal is © The Royal Society of Chemistry 2024 |