DOI:
10.1039/D3DT04342H
(Paper)
Dalton Trans., 2024,
53, 4617-4623
Highly selective electrocatalytic reduction of CO2 to HCOOH over an in situ derived Ag-loaded Bi2O2CO3 electrocatalyst†
Received
26th December 2023
, Accepted 2nd February 2024
First published on 5th February 2024
Abstract
The electrochemical reduction of CO2 to HCOOH is considered one of the most appealing routes to alleviate the energy crisis and close the anthropogenic CO2 cycle. However, it remains challenging to develop electrocatalysts with high activity and selectivity towards HCOOH in a wide potential window. In this regard, Ag/Bi2O2CO3 was prepared by an in situ electrochemical transformation from Ag/Bi2O3. The Ag/Bi2O2CO3 catalyst achieves a faradaic efficiency (FE) of over 90% for HCOOH in a wide potential window between −0.8 V and −1.3 V versus the reversible hydrogen electrode (RHE). Moreover, a maximum FE of 95.8% and a current density of 15.3 mA cm−2 were achieved at a low applied potential of −1.1 V. Density functional theory (DFT) calculations prove that the high catalytic activity of Ag/Bi2O2CO3 is ascribed to the fact that Ag can regulate the electronic structure of Bi, thus facilitating the adsorption of *OCHO and hindering the adsorption of *COOH. This work expands the in situ electrochemical derivatization strategy for the preparation of electrocatalysts.
Introduction
The environmental problems caused by CO2 emission are threatening the sustainable development of human society.1 The CO2 reduction reaction (CO2RR) powered by renewable electricity is considered a potential strategy to offset these issues.2,3 Generally, diverse conversion products, such as CO, HCOOH, CH4, C2H4 and C2H5OH, can be obtained from the CO2RR.4–8 Among these products, liquid HCOOH is more favoured due to its high commercial value and convenience for transportation and storage.9
The selectivity of the CO2RR catalyst is believed to be determined by the adsorption energy of *COOH, *OCHO and *H intermediates. Some main group metal elements including Pb, Sn, In, and Sb, which thermodynamically favour the adsorption of *OCHO over *COOH, are efficient electrocatalysts for converting CO2 into HCOOH (Table S1†). However, high toxicity and high cost are two barriers to their large-scale applications. In contrast, as a low-cost and environmentally benign electrocatalyst, Bi has also received much attention for the conversion of CO2 to HCOOH.10–12 Unfortunately, the harsh reaction conditions become a hindrance to its further development. Specifically, Bi-based electrocatalysts are limited by a narrow potential window for high selectivity and poor stability (Table S2†). For example, Bi nanosheets and Bi nanoparticles can only achieve high selectivity (FE > 90%) in a potential window of nearly 300 mV.13,14 On the one hand, the poor stability of *OCHO hinders the formation of HCOOH at low overpotentials. Moreover, H2 and CO ineluctably occupy a larger proportion with increasing cathodic potentials. To this end, its *OCHO adsorption energy remains to be optimized to achieve better selectivity towards HCOOH in a wider working potential. Recent research on Bi-based catalysts demonstrated that bismuth oxide catalysts exhibit better selectivity than metallic Bi because the Bi–O structure is more favourable to stabilize the *OCHO intermediate.15 Among all the Bi-based materials, the valence state of Bi ions in Bi2O2CO3 is higher than that of Bi2O3. While the electron transfer between Bi2O2CO3 and heteroatoms could further modulate the electronic structure of Bi to an optimal level for the adsorption of *OCHO. For example, Ag metal has poor adsorption toward hydrogen, which makes it a good choice to be used in the CO2RR to stabilize *OCHO and suppress the by-reaction HER.16
Therefore, Ag loaded Bi2O2CO3 could be a high activity catalyst for the conversion of CO2 to HCOOH. Here, we prepared Ag/Bi2O3 nanosheets, which could undergo a carbonatization process in KHCO3 electrolyte to generate Ag/Bi2O2CO3. The in situ formed catalysts are believed to possess a lower contact resistance and more active sites. Ag/Bi2O2CO3 demonstrates high selectivity toward HCOOH over a wide potential window. Theoretical calculations suggest that the CO and H2 pathways are repressed due to the optimized adsorption of *OCHO on Ag/Bi2O2CO3. The in situ attenuated total reflection Fourier transform infrared (ATR-FTIR) analysis shows that *CO has a much weaker signal than *OCHO. In addition, durability tests prove that it can maintain an approximate 90% FE towards HCOOH (FEHCOOH) at a current density of −13.0 mA cm−2 in 10 h.
Experimental section
Characterization
The powder X-ray diffraction (XRD) patterns of the samples were collected on a Japan Rigaku D/MAX-γA X-ray diffractometer equipped with Cu Kα radiation (λ = 1.54178 Å) at a scanning rate of 5° min−1 in the 2θ range from 20 to 80°. Scanning electron microscopy (SEM) images were recorded on a Zeiss Supra 40 field-emission scanning microscope operating at 5 kV. Transmission electron microscopy (TEM) images were collected on a Hitachi H-7650 transmission electron microscope using an accelerating voltage of 100 kV and high-resolution transmission electron microscopy (HRTEM) images were recorded on a FEI Talos F200X at an accelerating voltage of 200 kV. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) was conducted with an Optima 7300 DV instrument. XPS was performed on an ESCALAB 250 X-ray photoelectron spectrometer using Al Kα radiation. The C 1s peak at 284.8 eV is used as a reference to correct the binding energy shift caused by the charge effect. The X-ray absorption near edge structures (XANES) of Ag K-edge and Bi L-edge were measured at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF).
Electrochemical measurements
The electrochemical measurements were conducted at 25 °C using an electrochemical workstation (CHI760E) in a three-electrode, two-compartment H-type electrolysis cell. Each compartment of the H-type cell contained 30 mL of 0.5 M KHCO3 solution. Pt foil and a Ag/AgCl (saturated KCl) electrode were employed as the counter and reference electrodes, respectively. Before the investigation of the electrochemical CO2RR, a flow of high-purity CO2 at a flow rate of 30 sccm (standard cubic centimeter per minute) was purged into the 0.5 M KHCO3 solution for 30 min to remove all the oxygen from the electrolyte and achieve CO2 saturation. Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) measurements were carried out in the 0.5 M KHCO3 solution saturated with Ar/CO2. CV was conducted for ten cycles at a scan rate of 100 mV s−1 in the potential range from −1.5 V to −0.3 V. Linear sweep voltammetry (LSV) was performed at a scan rate of 50 mV s−1. Multiple potential step I–t measurements with several selected potentials were performed. All potentials were measured against an Ag/AgCl reference electrode and converted to the RHE scale based on the Nernst equation (ERHE = EAg/AgCl + 0.059 × pH + 0.197). The pH value of the Ar/CO2-saturated 0.5 M KHCO3 electrolyte solution is about 8.8/7.2.
Product analysis
The gas produced during the reaction was carried by high-pure CO2 and analyzed by gas chromatography (Agilent 7890B) equipped with both a thermal conductivity detector (TCD) and a flame ionization detector (FID). The TCD was used to quantify H2 and the FID was used to quantify CO and hydrocarbons. The faradaic efficiency (FE) of the gas product was calculated on the basis of the following equation:
| FE = ix/itot = zx × vgas × cx × F/(itot × Vm) |
where ix is the partial current of product x, itot is the total current, zx represents the number of electrons transferred towards the formation of 1 mol of product x, vgas is the CO2 flow rate (30 sccm), cx represents the concentration of product x detected by gas chromatography (ppm), F is the Faraday constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and Vm is the unit molar volume, which is 24.5 L mol−1 at room temperature (298.15 K). The liquid in the working compartment of the H-cell was collected and analyzed by 1H nuclear magnetic resonance (1H-NMR). 1H-NMR samples were prepared by mixing 1 mL of the collected solution with 50 μL D2O and 10 μL of 2000 ppm DMSO as an internal standard. The FE for formate is calculated as follows:
485 C mol−1), and Vm is the unit molar volume, which is 24.5 L mol−1 at room temperature (298.15 K). The liquid in the working compartment of the H-cell was collected and analyzed by 1H nuclear magnetic resonance (1H-NMR). 1H-NMR samples were prepared by mixing 1 mL of the collected solution with 50 μL D2O and 10 μL of 2000 ppm DMSO as an internal standard. The FE for formate is calculated as follows:
| FE = iformate/itot = z × nDMSO × 6 × F × SDMSO/(itot × Sformate × t) |
where iformate is the partial current of formate, itot is the total current, z represents the number of electrons transferred towards the formation of formate (z = 2), nDMSO is the mole of DMSO, F is the Faraday constant (96485 C mol−1), SDMSO and Sformate are the peak areas of DMSO and formate in 1H-NMR spectra, respectively, and t is the time of each test.
In situ ATR-FTIR test
In situ ATR-FTIR spectroscopy was carried out using a Thermo Fisher NICOLET iS50 FTIR and the measured potential for the CO2RR was in the potential range between 0 V and −1.2 V controlled by an electrochemical workstation (CHI760E). The in situ electrochemical three-electrode cell contained Ag/Bi2O2CO3 as the working electrode, Ag/AgCl as the reference electrode and Pt wire as the counter electrode. The electrolyte used was CO2-saturated 0.5 M KHCO3.
DFT calculations
All calculations were carried out based on density functional theory (DFT) as implemented in the Vienna Ab initio Simulation Package (VASP).17 The projector augmented wave (PAW) pseudopotentials were used to treat the core electrons, while the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) was used for describing the electron interactions.18,19 The cutoff energy was set to 400 eV. The convergence of total energy and forces was set to 1 × 10−5 eV and 0.01 eV Å−1, respectively. A grid of 3 × 3 × 1 Monkhorst–Pack k-points was used for the structural relaxation. A vacuum layer of 15 Å was adopted in the direction perpendicular to the surface to avoid interactions between periodic slabs. The reaction pathway of CO2-to-HCOOH involves the following elementary step, where the symbol ‘*’ represents the active site.| | | CO2 + * + H+ + e− → *OCHO | (1) |
| | | *OCHOH + H+ + e− → *HCOOH | (2) |
And the reaction pathway of CO2-to-CO is as follows:
| | | CO2 + * + H+ + e− → *COOH | (4) |
| | | *COOH + H+ + e− → *CO + H2O | (5) |
The Gibbs free energies were calculated as follows:
where Δ
E is the adsorption or reaction energy based on DFT calculations, ΔZPE is the change in zero-point energy,
T is the temperature (298.15 K), and Δ
S is the change in entropy. The entropies of gas phases CO
2, H
2, CO, HCOOH and H
2O are obtained from the National Institute of Standards and Technology database under standard conditions.
Results and discussion
The synthetic route is depicted in Fig. 1a. Bi2O3 nanosheets were synthesized using a hydrothermal method at 160 °C for 1 h. Ag/Bi2O3 was prepared by a deposition–precipitation method. In short, Bi2O3 aqueous suspension was mixed with AgNO3 solution so that Ag+ could be loaded on the surface of Bi2O3. Then NaOH solution was added to the mixture, followed by centrifugation and heat treatment at 450 °C. The pre-catalyst Ag/Bi2O3 was transformed into Ag/Bi2O2CO3 during the CO2RR.
 |
| | Fig. 1 (a) Scheme of Ag/Bi2O2CO3 preparation. (b) TEM image of Ag/Bi2O3. (c) HRTEM image of Ag/Bi2O2CO3. (d–h) HAADF-STEM images and elemental mapping of Ag/Bi2O2CO3. | |
The as-synthesized Ag/Bi2O3 consists of assembled Bi2O3 nanosheets and Ag nanoparticles (Fig. 1b and S1†). The SEM and TEM images (Fig. 1b and S2†) revealed that the as-prepared Bi2O3, Ag/Bi2O3, and Ag/Bi2O2CO3 exhibited nanosheet morphology. The Brunauer–Emmett–Teller (BET) specific surface areas of the Bi2O3 and Ag/Bi2O3 precursors were found to be 20.77 m2 g−1 and 27.96 m2 g−1, respectively (Fig. S3 and S4†). The XRD patterns of the catalyst (Fig. S5 and S6†) matched with those of Ag and Bi2O2CO3. Interplanar spacings of Bi2O2CO3 (004) and Ag (111) were observed in the HRTEM images (Fig. 1c), indicating that Ag/Bi2O2CO3 was generated after the CO2RR. In Fig. 1d, the HAADF-STEM image proves that Ag/Bi2O2CO3 is also an aggregate of nanosheets.
The elemental composition and chemical states of Ag/Bi2O2CO3 were investigated by X-ray photoelectron spectroscopy (XPS). XPS results revealed that the sample contains Ag, Bi, C and O elements. The binding energy of Bi in Ag/Bi2O2CO3 shows a positive shift compared to Bi2O2CO3, thus Bi is in a higher valence state.11 Moreover, Ag in Ag/Bi2O2CO3 shifted to a lower binding energy compared to Ag nanoparticles (Ag NP), which indicates that Ag is also in a higher valence state.20 The two peaks in the C 1s spectra (Fig. S7a†) were assigned to the carbon species absorbed on the surface of nanoparticles (284.6 eV) and CO32− (288.6 eV), respectively. In Fig. S7b,† three peaks of O 1s were matched to Bi–O bonds (529.2 eV), CO32− (530.4 eV), and absorbed water (532.2 eV).21 The O 1s spectra show that the Bi–O peak and the CO32− peak of Ag/Bi2O2CO3 shifted to a lower binding energy compared to Bi2O2CO3. Therefore, XPS results confirmed that the electrons were transferred from Ag and Bi to O. The electronic state and atomic environment of Bi were further verified by performing X-ray adsorption near-edge structure (XANES) measurements. As shown in Fig. 2c and Fig. S8,† the XANES spectra show that the adsorption edge of Ag/Bi2O2CO3 shifted to a higher energy compared to that of Bi2O3, indicating that the valence of Bi in Ag/Bi2O2CO3 is higher than that of Bi2O3.22 In Fig. 2d, the adsorption edge of Ag/Bi2O2CO3 shows a blue shift compared to Ag foil, which is in agreement with the results of XPS analysis presented in Fig. 2a and b. Furthermore, we analysed k2-weighted Bi L3-edge EXAFS wavelet transform plots of Bi2O3 and Ag/Bi2O2CO3 (Fig. 2e) to determine their coordination environment.23,24 The scattering path signal of Bi–O–C was observed. For k2-weighted Ag K-edge EXAFS wavelet transform plots of Ag foil and Ag/Bi2O2CO3 (Fig. S9†), the existence of the Ag–Ag and Ag–O structures was confirmed, proving that Ag is loaded on the surface of Bi2O2CO3 instead of being doped into the Bi2O2CO3 lattice.
 |
| | Fig. 2 (a) Bi 4f XPS spectra of Bi2O2CO3 and Ag/Bi2O2CO3. (b) Ag 3d XPS spectra of Ag foil and Ag/Bi2O2CO3. (c) Bi L3-edge XANES curves of Bi2O3 and Ag/Bi2O2CO3. (d) Ag K-edge XANES curves of Ag foil and Ag/Bi2O2CO3. (e) Bi L3-edge EXAFS wavelet transform plots of Bi2O3 and Ag/Bi2O2CO3. | |
The electrochemical activities of Bi2O2CO3 and Ag/Bi2O2CO3 were first evaluated by linear sweep voltammetry (LSV) in Ar and CO2 purged 0.5 M KHCO3 (Fig. 3a). Ag/Bi2O2CO3 demonstrates a larger current density than Bi2O2CO3 in CO2 purged 0.5 M KHCO3. Compared to Ag/Bi2O2CO3, Bi2O2CO3 displays a slightly higher current density in Ar purged 0.5 M KHCO3, which means the hydrogen evolution reaction (HER) on Ag/Bi2O2CO3 is hindered. Moreover, constant potential electrolysis at different potentials has been adopted to evaluate their CO2RR performance. Bi2O2CO3 shows nearly 80% FEHCOOH at −1.0 V (Fig. S10†). Further introduction of Ag significantly increases the FEHCOOH. In particular, the FEHCOOH of Ag/Bi2O2CO3 was 95.8% at a potential of −1.1 V with a jHCOOH of −15.3 mA cm−2 (Fig. 3b and c). Moreover, we also investigated the effect of Ag content on CO2RR performance by testing samples with different Ag loadings (denoted as s-1, s-2 and s-3) (Table S3†). Among which, s-2 (0.384 wt% Ag) shows the best performance in terms of FEHCOOH (Fig. 3c and S10†). A high FEHCOOH (>90%) was also obtained for s-2 from −0.8 V to −1.3 V. In addition, there was no apparent change in selectivity and current density during the durability test, showing the outstanding stability of Ag/Bi2O2CO3.
 |
| | Fig. 3 (a) LSV of Bi2O2CO3 and Ag/Bi2O2CO3. (b) Partial current densities for HCOOH (jHCOOH) of Bi2O2CO3 and Ag/Bi2O2CO3. (c) FEHCOOH of Ag/Bi2O2CO3 (s-2). (d) Durability test of Ag/Bi2O2CO3 at −1.0 V for 10 h. (e) In situ ATR-FTIR spectra of Ag/Bi2O2CO3. | |
We carried out in situ ATR-FTIR analysis to identify the key intermediates of the CO2RR (Fig. 3e and S11†). The peaks at 1620 and 1220 cm−1 are assigned to the bending vibration of H2O and Si–O vibration from the silicon prism.25 The peaks at 1430, 1725 and 2940 cm−1 become stronger as the applied potential decreases. These peaks can be assigned to the C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H vibrations of the *OCHO intermediate, respectively. The signal of *CO appears at 1880 cm−1 at high over-potentials, which means HCOOH is the main product during the CO2RR.
O and C–H vibrations of the *OCHO intermediate, respectively. The signal of *CO appears at 1880 cm−1 at high over-potentials, which means HCOOH is the main product during the CO2RR.
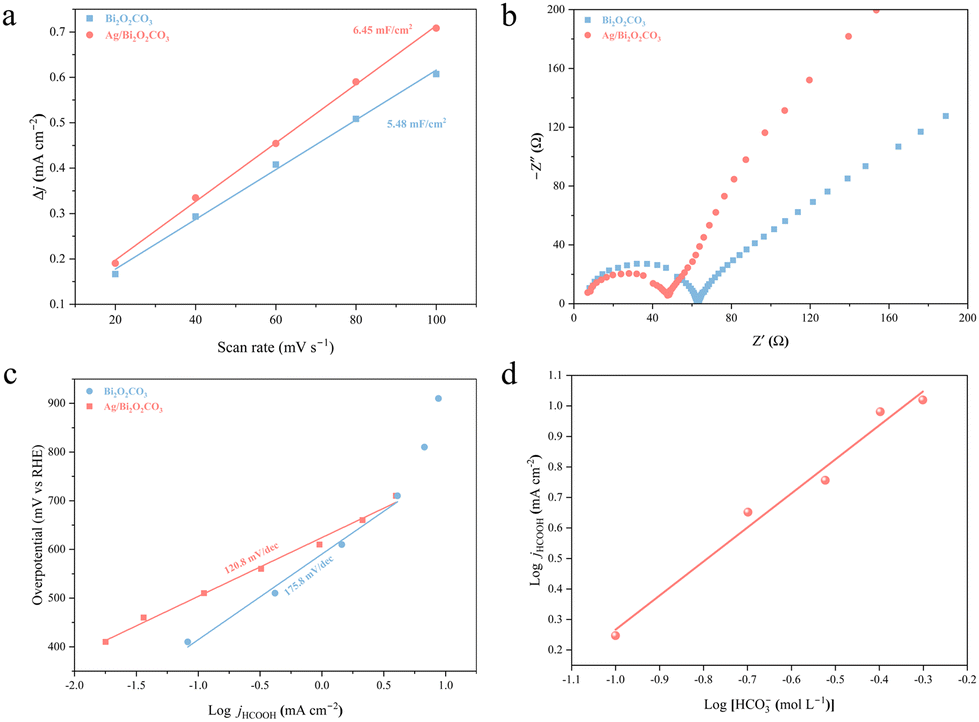
The electrochemically active surface area (ECSA) was assessed by calculating the double-layer capacitance (Cdl). The value of Cdl was determined by cyclic voltammetry (CV) which was measured at different scan rates (Fig. S12†). As shown in Fig. 4a, Ag/Bi2O2CO3 shows a higher Cdl value (6.45 mF cm−2) than Bi2O2CO3 (5.48 mF cm−2). Such results indicate that Ag/Bi2O2CO3 has more exposed active sites than Bi2O2CO3. The charge transfer resistances (Rct) for Bi2O2CO3 and Ag/Bi2O2CO3 were determined by electrochemical impedance spectroscopy (EIS) measurements at −0.6 V (Fig. 4b). The Rct value of Ag/Bi2O2CO3 is smaller than Bi2O2CO3, indicating a faster interface charge transfer. Therefore, it would be easier for CO2 to receive electrons from Ag/Bi2O2CO3 to form the *OCHO intermediate.26 As shown in Fig. 4c, Ag/Bi2O2CO3 exhibited a lower Tafel slope (120.8 mV dec−1) than Bi2O2CO3 (175.8 mV dec−1), revealing that more favourable CO2RR kinetics was attained through the introduction of Ag. Noticeably, this value of 120.8 mV dec−1 for Ag/Bi2O2CO3 was nearing 118 mV dec−1, which means that the first formation of *OCHO was the rate-determining step (RDS).27 Besides, the calculated jHCOOH at −1.0 V delivered a first-order dependence on KHCO3 concentration (Fig. 4d). These results further illustrated that H+ that participated in the formation of the *OCHO intermediate originates from KHCO3 rather than H2O.28
 |
| | Fig. 4 (a) Calculated Cdl values of Bi2O2CO3 and Ag/Bi2O2CO3. (b) Nyquist plots of the impedance spectra of Bi2O2CO3 and Ag/Bi2O2CO3. (c) Tafel slopes of Bi2O2CO3 and Ag/Bi2O2CO3. (d) Logarithm of the partial current density of HCOOH (jHCOOH) versus the logarithm of [KHCO3] concentration at −1.0 V for Ag/Bi2O2CO3. | |
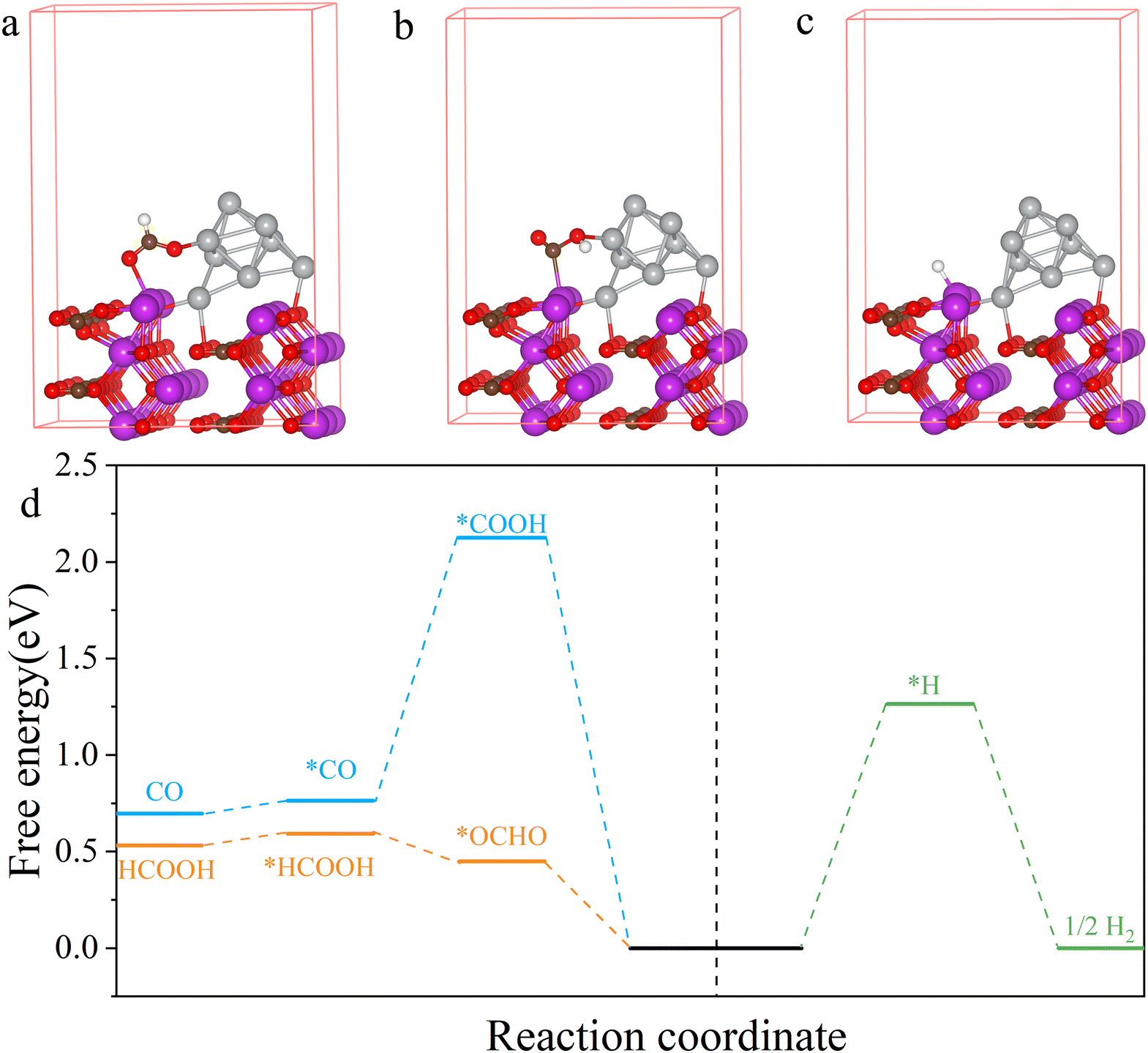
To further explore the reaction mechanism of the CO2RR on the Ag/Bi2O2CO3 catalyst, a theoretical investigation based on density functional theory (DFT) calculations was conducted. The Ag/Bi2O2CO3 catalyst was modelled by Ag clusters supported on the Bi2O2CO3 (100) surface (Fig. S13†). The reaction pathways of the CO2RR to HCOOH and CO as well as the HER on Ag/Bi2O2CO3 and Bi2O2CO3 were considered. The Bi atoms adjacent to Ag clusters are the most active sites for HCOOH production. As shown in Fig. 5a and d, the RDS of the CO2RR to HCOOH on Ag/Bi2O2CO3 is the formation of *OCHO, requiring an energy barrier of 0.45 eV. And the formation of *COOH is the RDS of the CO2RR to CO on Ag/Bi2O2CO3, which needs to overcome an energy barrier of 2.12 eV (Fig. 5b and d). Moreover, the energy barrier of the formation of *H on Ag/Bi2O2CO3 is 1.26 eV (Fig. 5c and d). Therefore, the HCOOH pathway is more energetically-favourable than the CO and H2 pathways, leading to high HCOOH selectivity on Ag/Bi2O2CO3.
 |
| | Fig. 5 The geometrical configuration of (a) *OCHO, (b) *COOH, (c) and *H over Ag/Bi2O2CO3. The white, brown, red, grey, and purple spheres represent hydrogen, carbon, oxygen, silver, and bismuth atoms, respectively. (d) Free energy diagram of formate, carbon dioxide, and hydrogen formation over the Ag/Bi2O2CO3 (100) surface. | |
Similarly, for Bi2O2CO3, HCOOH is more easily generated than CO and H2 (Fig. S14 and S15†). Notably, the energy barrier of *OCHO formation was reduced from 0.86 eV for Bi2O2CO3 to 0.45 eV for Ag/Bi2O2CO3, suggesting that the formation of HCOOH on Ag/Bi2O2CO3 was easier than that on Bi2O2CO3. To investigate the electronic structure of Ag/Bi2O2CO3, charge density difference was also calculated. As shown in Fig. S16,† there is a strong interfacial interaction between Ag and Bi2O2CO3, raising the valence state of Bi and enhancing the adsorption of *OCHO, consequently boosting the reaction activity of CO2 to HCOOH.
Conclusions
In summary, we have successfully prepared Ag/Bi2O2CO3via an in situ transformation from Ag/Bi2O3. The prepared Ag/Bi2O2CO3 exhibits excellent selectivity (FEHCOOH ≥90% over a wide potential window) and satisfactory durability (no activity loss for 10 h) in the CO2RR to HCOOH. XPS results and DFT calculations prove that a higher valence state of the Bi active site possesses optimal binding energy to form the key formate intermediate *OCHO, reducing the energy barrier of the RDS and enhancing the faradaic efficiency of HCOOH. These insights can be further exploited as design principles for other catalysts.
Author contributions
Wei Zheng: conceptualization, methodology, software, investigation, formal analysis, and writing – original draft; Changlai Wang: software, formal analysis, visualization, and writing – original draft; Jing Chen: methodology; Shi Chen: visualization; Zhiyu Lin: methodology; Minxue Huang: software and validation; Hao Huang: data curation and software; Yafei Qu: software, validation, and data curation; Peichen Wang: software, validation, and data curation; Lin Hu: writing – review & editing; Qianwang Chen: conceptualization, funding acquisition, resources, supervision, and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the National Natural Science Foundation (21972145 and 22072140) and the National Key R&D Program of China (Grant No. 2021YFA1600202). The DFT calculations were performed at the Supercomputing Center of the University of Science and Technology of China. We thank the staff of the BL14W1 beamline station for their help with XAFS measurements at the Shanghai Synchrotron Radiation Facility. This work was partially carried out at the Instruments Center for Physical Science, University of Science and Technology of China.
References
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. S. Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- Z. Zhang, M. Chi, G. M. Veith, P. Zhang, D. A. Lutterman, J. Rosenthal, S. H. Overbury, S. Dai and H. Zhu, ACS Catal., 2016, 6, 6255–6264 CrossRef CAS.
- G. Wen, D. U. Lee, B. Ren, F. M. Hassan, G. Jiang, Z. P. Cano, J. Gostick, E. Croiset, Z. Bai, L. Yang and Z. Chen, Adv. Energy Mater., 2018, 8, 1802427 CrossRef.
- M. Huang, S. Gong, C. Wang, Y. Yang, P. Jiang, P. Wang, L. Hu and Q. Chen, Angew. Chem., Int. Ed., 2021, 60, 23002–23009 CrossRef CAS PubMed.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- X. Lv, L. Shang, S. Zhou, S. Li, Y. Wang, Z. Wang, T. Sham, C. Peng and G. Zheng, Adv. Energy Mater., 2020, 10, 2001987 CrossRef CAS.
- Y.-X. Duan, F.-L. Meng, K.-H. Liu, S.-S. Yi, S.-J. Li, J.-M. Yan and Q. Jiang, Adv. Mater., 2018, 30, 1706194 CrossRef PubMed.
- S. Liu, X. F. Lu, J. Xiao, X. Wang and X. W. (David) Lou, Angew. Chem., Int. Ed., 2019, 58, 13828–13833 CrossRef CAS PubMed.
- W. Ma, J. Bu, Z. Liu, C. Yan, Y. Yao, N. Chang, H. Zhang, T. Wang and J. Zhang, Adv. Funct. Mater., 2021, 31, 2006704 CrossRef CAS.
- T. Tran-Phu, R. Daiyan, Z. Fusco, Z. Ma, R. Amal and A. Tricoli, Adv. Funct. Mater., 2020, 30, 1906478 CrossRef CAS.
- L. Yi, J. Chen, P. Shao, J. Huang, X. Peng, J. Li, G. Wang, C. Zhang and Z. Wen, Angew. Chem., Int. Ed., 2020, 59, 20112–20119 CrossRef CAS PubMed.
- D. Wu, X. Wang, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2021, 284, 119723 CrossRef CAS.
- P. Deng, H. Wang, R. Qi, J. Zhu, S. Chen, F. Yang, L. Zhou, K. Qi, H. Liu and B. Y. Xia, ACS Catal., 2020, 10, 743–750 CrossRef CAS.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. M. Ferraria, A. P. Carapeto and A. M. B. do Rego, Vacuum, 2012, 86, 1988–1991 CrossRef CAS.
- Y. Zhang, Y. Chen, R. Liu, X. Wang, H. Liu, Y. Zhu, Q. Qian, Y. Feng, M. Cheng and G. Zhang, InfoMat, 2023, 5, e12375 CrossRef CAS.
- Y. Wang, B. Wang, W. Jiang, Z. Liu, J. Zhang, L. Gao and W. Yao, Nano Res., 2022, 15, 2919–2927 CrossRef CAS.
- M. Muñoz, P. Argoul and F. Farges, Am. Mineral., 2003, 88, 694–700 CrossRef.
- M. Muoz, F. Farges and P. Argoul, Physica Scripta, 2005, 221 CrossRef.
- K. Sun, J. Dong, H. Sun, X. Wang, J. Fang, Z. Zhuang, S. Tian and X. Sun, Nat. Catal., 2023, 6, 1164–1173 CrossRef CAS.
- Z. Chen, K. Mou, X. Wang and L. Liu, Angew. Chem., 2018, 130, 12972–12976 CrossRef.
- J. Zhou, K. Yuan, L. Zhou, Y. Guo, M. Luo, X. Guo, Q. Meng and Y. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14197–14201 CrossRef CAS PubMed.
- K. Fan, Y. Jia, Y. Ji, P. Kuang, B. Zhu, X. Liu and J. Yu, ACS Catal., 2020, 10, 358–364 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Changlai
Wang‡
a,
Jing
Chen
a,
Shi
Chen
a,
Changlai
Wang‡
a,
Jing
Chen
a,
Shi
Chen