
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of porphyrin ligands on the catalytic CH4 oxidation activity of monocationic μ-nitrido-bridged iron porphyrinoid dimers by using H2O2 as an oxidant†
Yasuyuki
Yamada
 *ab,
Yusuke
Miwa
a,
Yuka
Toyoda
b,
Yoshiki
Uno
a,
Quan Manh
Phung
ac and
Kentaro
Tanaka
*a
*ab,
Yusuke
Miwa
a,
Yuka
Toyoda
b,
Yoshiki
Uno
a,
Quan Manh
Phung
ac and
Kentaro
Tanaka
*a
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan. E-mail: Yamada.yasuyuki.i6@f.mail.nagoya-u.ac.jp; kentaro@chem.nagoya-u.ac.jp
bResearch Center for Materials Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan
cInstitute of Transformaytive Bio-Molecules (ITBM), Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan
First published on 19th March 2024
Abstract
The μ-nitrido-bridged iron phthalocyanine homodimer is a potent molecule-based CH4 oxidation catalyst that can effectively oxidize chemically stable CH4 under mild reaction conditions in an acidic aqueous solution including an oxidant such as H2O2. The reactive intermediate is a high-valent iron-oxo species generated upon reaction with H2O2. However, a detailed comparison of the CH4 oxidation activity of the μ-nitrido-bridged iron phthalocyanine dimer with those of μ-nitrido-bridged iron porphyrinoid dimers containing one or two porphyrin ring(s) has not been yet reported, although porphyrins are the most important class of porphyrinoids. Herein, we compare the catalytic CH4 and CH3CH3 oxidation activities of a monocationic μ-nitrido-bridged iron porphyrin homodimer and a monocationic μ-nitrido-bridged heterodimer of an iron porphyrin and an iron phthalocyanine with those of a monocationic μ-nitrido-bridged iron phthalocyanine homodimer in an acidic aqueous solution containing H2O2 as an oxidant. It was demonstrated that the CH4 oxidation activities of monocationic μ-nitrido-bridged iron porphyrinoid dimers containing porphyrin ring(s) were much lower than that of a monocationic μ-nitrido-bridged iron phthalocyanine homodimer. These findings suggested that the difference in the electronic structure of the porphyrinoid rings of monocationic μ-nitrido-bridged iron porphyrinoid dimers strongly affected their catalytic light alkane oxidation activities.
Introduction
Methane (CH4) is a naturally abundant molecule commonly found in natural gas or as methane hydrate. Moreover, the Sabatier process enables the efficient conversion of CO2 into CH4 through a reaction with H2 in the presence of a Ni catalyst. Therefore, CH4 has long been regarded as a next-generation carbon resource for the production of industrially valuable chemical raw materials. However, CH4 is well-recognized as a chemically stable compound with a particularly high C–H bond dissociation energy of 104.9 kcal mol−1. This feature implies that achieving efficient low-temperature conversion into industrially useful organic molecules, such as MeOH, is highly challenging.1,2 Therefore, significant efforts have been devoted to developing novel catalysts for the efficient low-temperature conversion of CH4.In nature, soluble methane monooxygenase (sMMO) is known to efficiently convert CH4 into MeOH in an aqueous solution under ambient reaction conditions by utilizing a high-valent iron-oxo species as its catalytic intermediate.3–5 This observation has stimulated the interest of chemists to investigate the mechanism at the sMMO reaction center and inspire the production of a large variety of biomimetic high-valent iron-oxo species. These studies successfully elucidated the reaction mechanism of sMMOs, revealing that the proton-coupled electron transfer (PCET) pathway facilitates the low-temperature and efficient oxidation of CH4 by high-valent iron-oxo species.6–10
However, there is still a limited number of artificial molecular iron-oxo-based biomimetic catalysts that effectively activate the C–H bonds of CH4 at temperatures below 100 °C.5,11,12 This is most likely due to the high stability of CH4. μ-Nitrido-bridged iron porphyrinoid dimers, such as μ-nitrido-bridged iron phthalocyanine homodimers 1 or 1+ (Fig. 1a), are among few iron-oxo-based molecular catalysts capable of actually activating the C–H bonds of CH4 in acidic aqueous solutions containing H2O2 at a temperature lower than 100 °C.11,13–17 The reactive intermediate of the CH4 oxidation catalyzed by either 1 or 1+ is considered to be the high-valent iron-oxo species 1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O generated in situ upon reaction with H2O2.11,18 A DFT calculation study indicated the importance of the core μ-nitrido-bridged dinuclear iron structure (Fe–NFe) for the high CH4 oxidation activity of 1O.19 It has also been demonstrated that catalysts based on μ-nitrido-bridged iron porphyrinoid dimers are applicable to the oxidation of other stable organic chemicals such as benzene and ethane in aqueous media.20–23
O generated in situ upon reaction with H2O2.11,18 A DFT calculation study indicated the importance of the core μ-nitrido-bridged dinuclear iron structure (Fe–NFe) for the high CH4 oxidation activity of 1O.19 It has also been demonstrated that catalysts based on μ-nitrido-bridged iron porphyrinoid dimers are applicable to the oxidation of other stable organic chemicals such as benzene and ethane in aqueous media.20–23
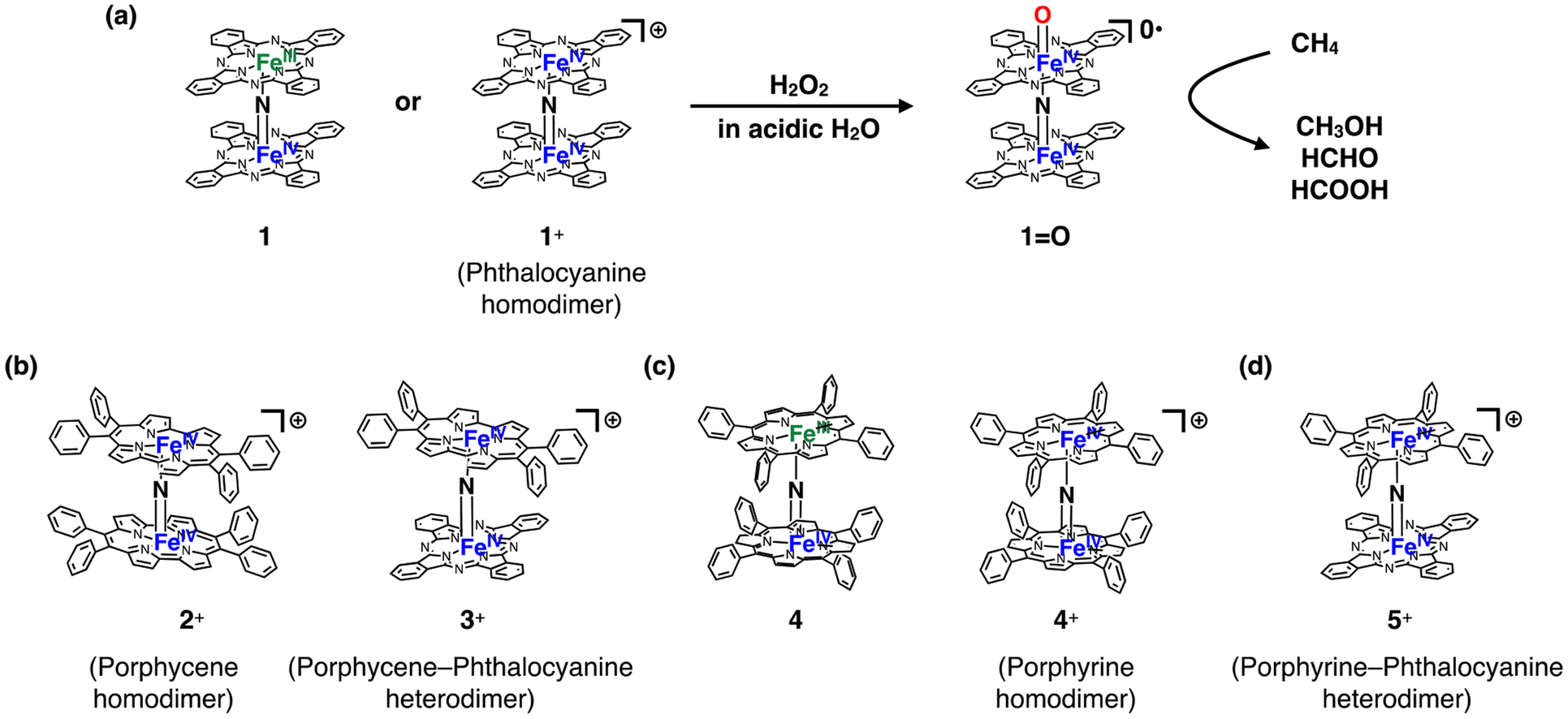 | ||
| Fig. 1 (a) Formation of high-valent iron oxo species 1O with a potent CH4 oxidation ability from a neutral (1) or monocationic (1+) μ-nitrido-bridged iron phthalocyanine homodimer upon reaction with H2O2. (b) Structures of a monocationic μ-nitrido-bridged iron porphycene homodimer (2+) and monocationic μ-nitrido-bridged heterodimer of an iron porphycene and an iron phthalocyanine (3+). (c) Structures of a neutral μ-nitrido-bridged iron porphyrin homodimer (4) and monocationic μ-nitrido-bridged iron porphyrin homodimer (4+). (d) Structure of a μ-nitrido-bridged heterodimer of an iron porphyrin and an iron phthalocyanine (5+). | ||
On the other hand, we recently demonstrated that the introduction of porphycene ring(s), a well-known class of porphyrinoids, into a monocationic μ-nitrido-bridged iron porphyrinoid dimer [2+ (μ-nitrido-bridged iron porphycene homodimer) or 3+ (μ-nitrido-bridged heterodimer of an iron phthalocyanine and an iron porphycene), Fig. 1b] dramatically changed the reactivity and decreased the stability of the corresponding high-valent iron-oxo species generated through the reaction with H2O2.24,25 This suggests that the porphyrinoid structure is also an important factor in determining the reactivity of μ-nitrido-bridged iron porphyrinoid dimers.
Inspired by these previous studies, we became interested in the reactivity of porphyrin-containing μ-nitrido-bridged iron porphyrinoid dimers. Obviously, porphyrin is the representative pigment among the porphyrinoids. The synthesis of neutral μ-nitrido-bridged iron porphyrin homodimer 4 (Fig. 1c) was reported by Cohen et al. in 1976,26 which is 8 years earlier than the first report on the synthesis of neutral phthalocyanine homodimer 1 by Ercolani et al.27 Sorokin et al. demonstrated that a high-valent iron-oxo species with a spin state of S = 1/2 was generated through the reaction of a neutral μ-nitrido-bridged iron porphyrin homodimer with m-chloroperbenzoic acid (mCPBA).18 Although it was confirmed that a neutral form of μ-nitrido-bridged iron porphyrin homodimer 4 exhibited catalytic CH4 oxidation activity in the presence of mCPBA,18 no report exists directly comparing the reactivities of μ-nitrido-bridged iron porphyrin homodimers (4 or 4+, Fig. 1c) and μ-nitrido-bridged heterodimers of an iron phthalocyanine and an iron porphyrin (5 or 5+, Fig. 1d) with that of μ-nitrido-bridged iron phthalocyanine homodimers (1 or 1+) under the same reaction conditions, despite the similar structure of porphyrin and phthalocyanine. Especially, the catalytic oxidation activities of μ-nitrido-bridged heterodimers of an iron phthalocyanine and an iron porphyrin (5 or 5+) have not been described yet. Herein, we present a comparison of the catalytic CH4 and CH3CH3 oxidation activities of a monocationic μ-nitrido-bridged iron porphyrin homodimer (4+) and a monocationic μ-nitrido-bridged heterodimer of an iron porphyrin and an iron phthalocyanine (5+) with those of a monocationic μ-nitrido-bridged iron phthalocyanine dimer (1+) in an acidic aqueous solution containing H2O2 as oxidant.
Results and discussion
Synthesis of monocationic μ-nitrido-bridged iron porphyrinoid dimers containing porphyrin rings (4+ and 5+)
The monocationic iron porphyrin dimer 4+ was synthesized by modifying the methods reported by the Suslick as well as our groups (Fig. 2a).28,29 Iron tetraphenyl porphyrin chloride 6 was heated with an excess of NaN3 in 1-chloronaphthalene at 280 °C for 2 h to afford neutral μ-nitrido bridged iron porphyrin dimer 4, which underwent subsequent oxidation by I2 to give the desired monocationic species 4+·I3−. A monocationic μ-nitrido-bridged heterodimer of an iron porphyrin and an iron phthalocyanine was synthesized in a similar manner by heating a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of iron porphyrin 6 and iron phthalocyanine 7 in 1-chloronaphthalene in the presence of an excess of NaN3 at 280 °C for 2 h, followed by oxidation with I2 to afford 5+·I3− in a moderate yield (71%). Elemental analysis suggested that the counter anion of the monocationic heterodimer was triiodide, which was further confirmed by the results of single-crystal X-ray structural analysis (vide infra).
:1 mixture of iron porphyrin 6 and iron phthalocyanine 7 in 1-chloronaphthalene in the presence of an excess of NaN3 at 280 °C for 2 h, followed by oxidation with I2 to afford 5+·I3− in a moderate yield (71%). Elemental analysis suggested that the counter anion of the monocationic heterodimer was triiodide, which was further confirmed by the results of single-crystal X-ray structural analysis (vide infra).
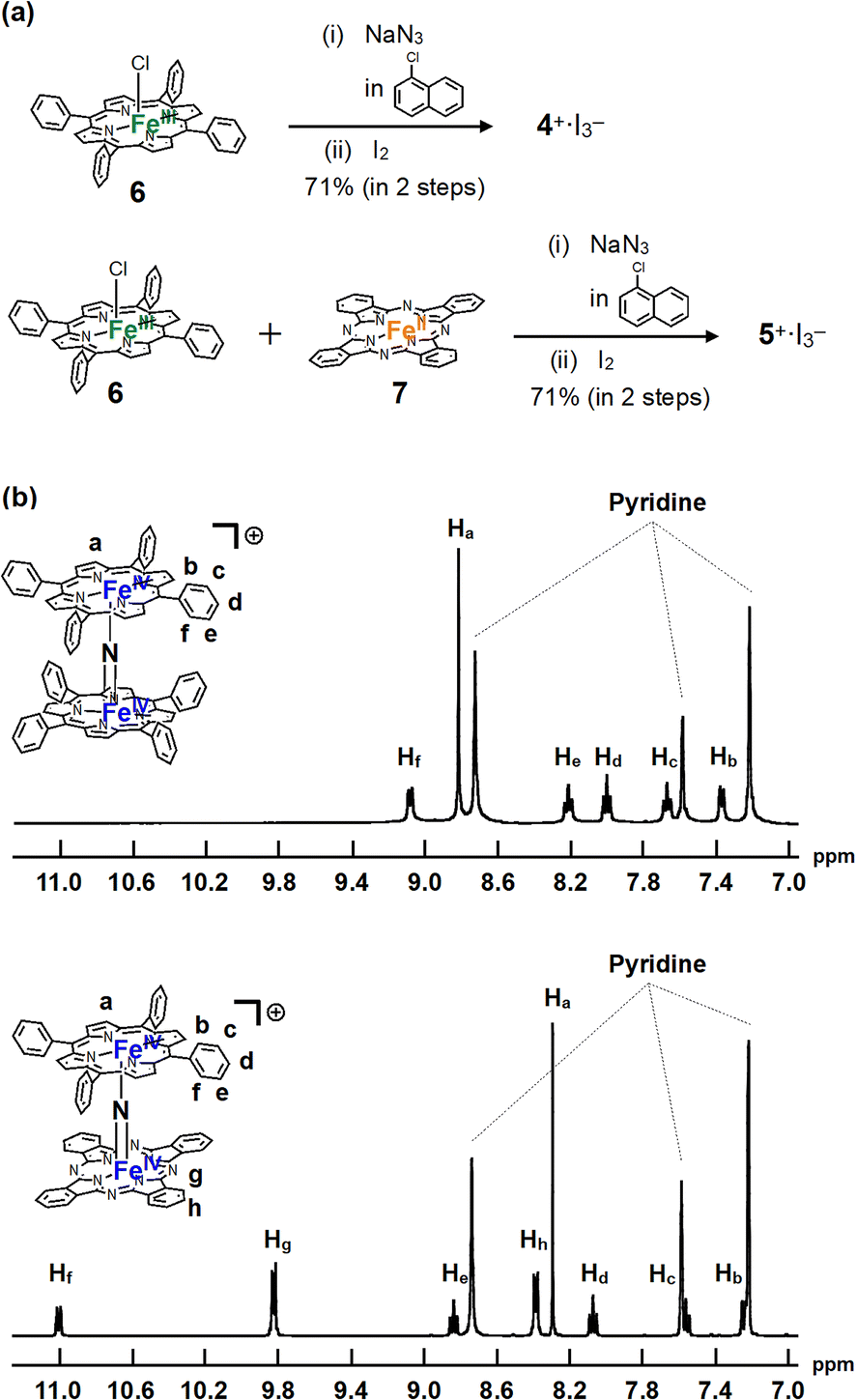 | ||
| Fig. 2 (a) Synthesis of monocationic homodimer 4+·I3− and heterodimer 5+·I3−. (b) 1H-NMR spectra of 4+·I3− and 5+·I3− in pyridine-d5. | ||
1H-NMR and MALDI-TOF MS analyses were performed to characterize 4+·I3− and 5+·I3−. In the MALDI-TOF MS spectra shown in Fig. S1 and S2 in the ESI,† signals corresponding to 4+ and 5+ were observed at around m/z = 1350.3 and m/z = 1250.4, respectively, which corresponded to the theoretical isotope distribution pattern of 4+ and 5+. As shown in Fig. 2b, all the 1H-NMR peaks of both 4+·I3− and 5+·I3− were observed as sharp signals within the 7–11 ppm range, indicating that 4+·I3− and 5+·I3− include two Fe(IV) centers interacting with each other in an antiferromagnetic fashion as in the case of other monocationic μ-nitrido-bridged iron porphyrinoid dimers (1+, 2+, or 3+).30,31 Furthermore, two sets of signals attributable to the o- and m-protons of the peripheral phenyl groups of tetraphenyl porphyrins (TPPs) were observed, suggesting that the rotation of the peripheral phenyl groups of TPPs was restricted due to the steric repulsion between the phenyl groups and porphyrin rings in both 4+·I3− and 5+·I3−. On the other hand, the fact that the porphyrin and phthalocyanine rings of 4+·I3− and 5+·I3− were observed as C2 symmetrical structures implies that the rotation of the TPP or phthalocyanine rings along the Fe–NFe axis was sufficiently fast compared to the NMR timescale for both 4+·I3− and 5+·I3−.
A comparison of the UV-Vis spectra of 4+·PF6− and 5+·PF6− with that of a monocationic μ-nitrido-bridged iron phthalocyanine homodimer 1+·I− in pyridine is shown in Fig. 3. Under these conditions, pyridine is expected to coordinate to the iron centers of 4+·PF6−, 5+·PF6−, and 1+·I−. The bands at 425 nm of 5+·PF6− were assignable to the Soret bands of porphyrin, which were slightly shifted to longer wavelengths compared to that of 4+·PF6− (423 nm). More conspicuously, the Q-band of the phthalocyanine of 5+·PF6− (740 nm) was shifted to a much longer wavelength compared to that of 1+·I− (636 nm). Single crystal X-ray structural analysis of 1+·I− demonstrated that the iodide anion (I−) in 1+·I− was not coordinated to the iron center in a pyridine solution.32 Considering that PF6− would not coordinate to the iron center of 5+ in pyridine, which can coordinate more strongly than PF6−, it could be concluded that the electronic structure of both the iron porphyrin and iron phthalocyanine in 5+·PF6− differed significantly from those in 4+·PF6− and 1+·I− due to the pronounced electronic interaction between these two moieties.
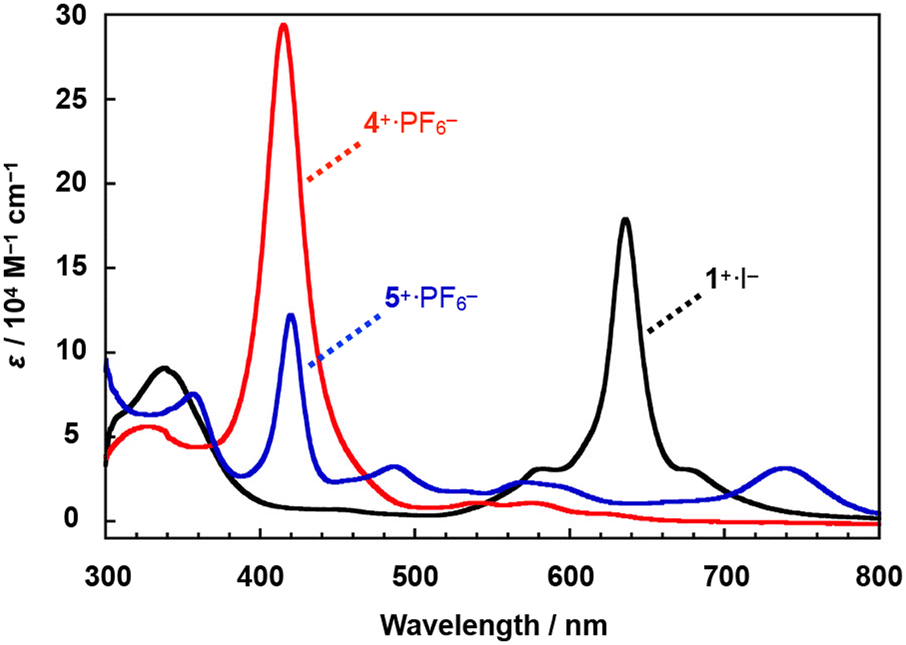 | ||
| Fig. 3 (a) Comparison of the UV-Vis spectra of heterodimer 5+·PF6− and homodimers 1+·I− and 4+·PF6− in pyridine at 20 °C. | ||
Single crystal X-ray structural analysis
Dimers 4+·I3− and 5+·I3− were successfully crystallized in the triclinic space group (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) by slow evaporation of solvent from a mixture of CHCl3 and pyridine. The molecular structures of 4+·I3− and 5+·I3− are shown in Fig. 4a–d. A comparison of some structural parameters of 4+·I3− and 5+·I3− with the previously reported crystal structure of 1+·I− is summarized in the table in Fig. 4e.
) by slow evaporation of solvent from a mixture of CHCl3 and pyridine. The molecular structures of 4+·I3− and 5+·I3− are shown in Fig. 4a–d. A comparison of some structural parameters of 4+·I3− and 5+·I3− with the previously reported crystal structure of 1+·I− is summarized in the table in Fig. 4e.
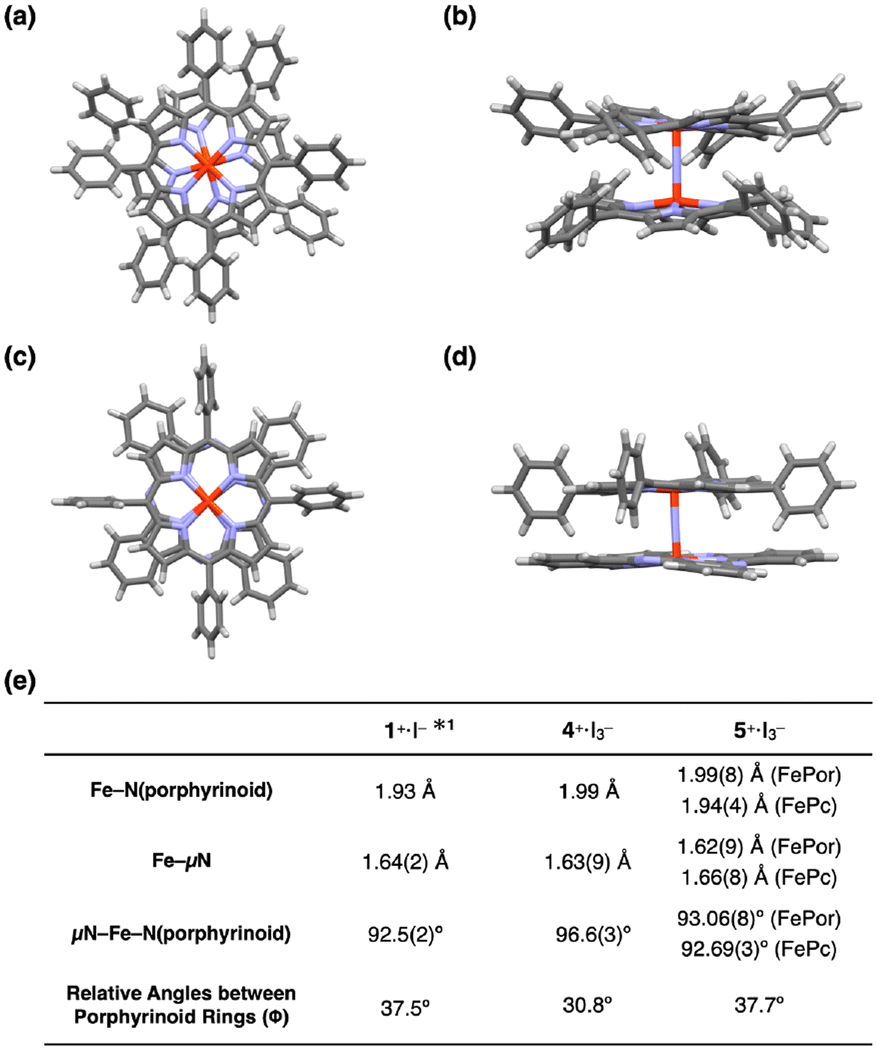 | ||
| Fig. 4 (a) Top and (b) side views of the single crystal X-ray structure of 4+·I3−. (c) Top and (d) side views of the single crystal X-ray structure of 5+·I3−. I3−, coordinating pyridines, and crystalline solvents were omitted for clarity. (e) Comparison of some mean bond lengths and angles of 1+·I−, 4+·I3−, and 5+·I3−. *1: ref. 32. | ||
The porphyrin rings in the porphyrin homodimer 4+·I3− were significantly distorted from the flat π-plane (Fig. 4b), whereas no significant distortion was observed in the porphyrin rings of heterodimer 5+·I3−. The observed porphyrin ring distortion was likely due to the steric repulsion of the peripheral phenyl rings in 4+·I3−. In contrast, the peripheral phenyl rings of the iron tetraphenyl porphyrin of 5+·I3− located between the benzene rings of the iron phthalocyanine prevented steric repulsion. The mean bond distances between the four porphyrin nitrogen atoms and the coordinated Fe(IV) ion in 4+·I3− was 1.99 Å, which is almost identical to that between the four porphyrin nitrogens and the coordinated Fe(IV) ion in 5+·I3− [1.99(8) Å]. Similarly, the mean bond distances between the four phthalocyanine nitrogens and the coordinated Fe(IV) ion in 5+·I3− were almost identical to that in phthalocyanine homodimer 1+·I− (1.93 Å). These differences in mean distances between the porphyrinoid nitrogens (Fe–N(porphyrinoid)) and their coordinated Fe(IV) ions reflected the fact that phthalocyanines generally possess narrower central cavities than porphyrins.
The average distance between the μ-nitrogen and iron ions of 4+·I3− was 1.64 Å, which is almost identical to that of 1+·I−. In both complexes, the μ-nitrogen is located almost in the center of the Fe–μN–Fe structures, suggesting the conjugation of a single Fe–μN bond with a double FeμN bond in 4+·I3− and 1+·I−. The Fe–μN bond can be, therefore, assigned as 1.5 (vide infra, see DFT calculation section). In the case of 5+·I3−, the distance between the μ-nitrogen and iron ion of the iron porphyrin (Fe(Por)–μN) was 1.62(9) Å, whereas the distance between the μ-nitrogen and iron ion of the iron phthalocyanine (Fe(Pc)–μN) was 1.66(8) Å. Considering the relatively large standard deviations of these values, it seems to be difficult to claim the difference from 1.64 Å. However, the fact that the Fe(Por)–μN was larger than the Fe(Pc)–μN might imply that, in 5+·I3−, the Fe(Por)–μN has a higher double-bond (FeN) character, while the Fe(Pc)–μN has a higher single-bond (Fe–N) character. It should also be noted that a similar difference in the distance between μ-N and Fe ion was observed in a monocationic μ-nitrogen-bridged heterodimer of an iron(IV) phthalocyanine and iron(IV) porphycene.25
As for the average bond angles of the μ-nitrogen–Fe–nitrogens(porphyrinoid) (μN–Fe–N(porphyrinoid)), the value of 4+·I3− was slightly larger [96.6(3)°] than those in 1+·I− and 5+·I3−. This could be related to the structural distortion of the porphyrin ring in 4+·I3−. The reason why the relative angle between the porphyrinoid rings in 4+·I3− (30.8°) was slightly smaller than those of 1+·I− and 5+·I3− can be attributable to the steric repulsion of the peripheral phenyl rings and porphyrin core of 4+·I3−.
Among a variety of μ-heteroatom-bridged iron porphyrinoid dimers, the structures of μ-oxo- or μ-hydroxo-bridged iron porphyrin dimers, where iron ions are generally in the 3+ states and the Fe–μO bondings have single-bond characters with slightly bent Fe–μO–Fe angles, are well-examined by single crystal X-ray structural analyses, allowing us to compare the structures with those of 4+·I3− and 5+·I3−.33 The Fe–N(porphyrin) distances of μ-oxo- or μ-hydroxo-bridged iron porphyrin dimers are approximately 2.08 Å, which are apparently longer than that in 4+·I3− and 5+·I3−. These differences might be because of the differences in the oxidation states of the iron centers. The μ-oxo- or μ-hydroxo-bridged iron porphyrin dimers have the Fe–μO distances of 1.76–1.90 Å, which are apparently longer than those of the Fe–μN distances in 4+·I3− and 5+·I3−. These results clearly indicate the double-bond character of the Fe–μN bondings in 4+·I3− and 5+·I3−.
Cyclic voltammetry
The cyclic voltammograms of 1+·I−, 4+·PF6−, and 5+·PF6− in pyridine solutions containing 100 mM nBu4N+·PF6− were compared, as shown in Fig. 5. The cyclic voltammograms of a μ-nitrido-bridged iron phthalocyanine dimer and μ-nitrido-bridged iron porphyrin dimer have already been studied by the groups of Bottomley and Ercolani, and Kadish and Goff, respectively.30,34 According to these previous reports, the four reversible 1e− reductions of 1+·I− at −0.58, −1.52, −1.74, and −2.00 V vs. Fc/Fc+ were assignable to the reduction of the iron centers, whereas the one irreversible oxidation wave at −0.19 V was due to that of the phthalocyanine ring. Similarly, the two reversible 1e− reductions of 4+·PF6− at −0.82 and −1.69 V vs. Fc/Fc+ were attributable to the reduction of the iron centers, whereas the irreversible oxidation wave at 0.55 V was assignable to that of the porphyrin ring.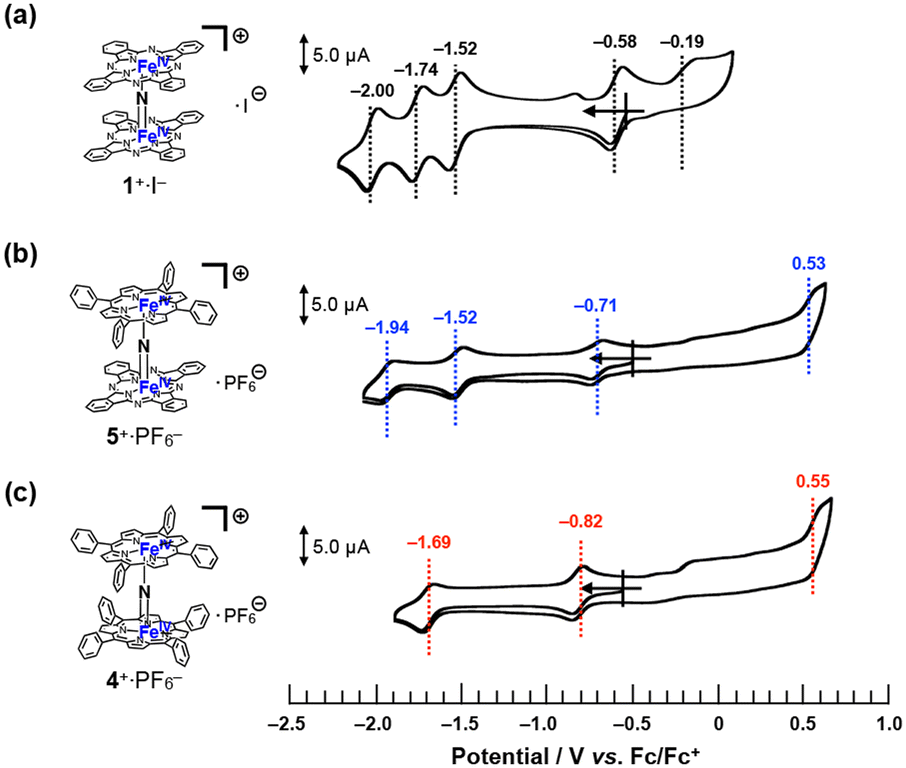 | ||
| Fig. 5 Comparison of cyclic voltammograms of (a) 1+·I−, (b) 5+·PF6−, and (c) 4+·PF6− in a pyridine solution containing 100 mM of nBu4N+·PF6− at room temperature. [Substrate] = 200 μM, [scan rate] = 100 mV s−1. | ||
Based on these results, the three reversible 1e− reductions of 5+·PF6− at −0.71, −1.52, and −1.94 V vs. Fc/Fc+ were attributed to the reduction of the iron centers. As for the oxidation of 5+·PF6− at 0.53 V, theoretical calculations suggested that the phthalocyanine ring was more easily oxidized than the porphyrin ring (vide infra, see “DFT calculations” section).
Overall, it could be concluded that the reduction potential assignable to Fe(IV)Fe(IV)/Fe(III)Fe(IV) of these monocationic μ-nitrido-bridged iron porphyrinoid dimers showed negative shift as the number of tetraphenylporphyrins increased (−0.58 V for 1+·I−, −0.71 V for 5+·PF6−, and −0.82 V for 4+·PF6−), whereas the oxidation potential of the porphyrinoid ring was shifted to a higher potential (−0.19 V for 1+·I−, 0.53 V for 5+·PF6−, and 0.55 V for 4+·PF6−). Thus, it was confirmed that the difference in the porphyrinoid structure significantly affected the electronic structure of the μ-nitrido-bridged iron porphyrinoid dimer.
ESI-TOF MS analysis of high-valent iron-oxo species
To confirm the generation of high-valent iron-oxo species, i.e., the reactive intermediates generated in the oxidation reaction upon treatment of either 4+·I3− or 5+·I3− with H2O2 in an aqueous solution, ESI-TOF MS measurements were performed on an acetonitrile solution of either 4+·I3− or 5+·I3− after addition of aqueous H2O2.As shown in Fig. 6, the signals assignable to the high-valent iron-oxo species ([4 + O]+ (see Fig. 6a) and [5 + O]+ (see Fig. 6b)) were observed after addition of aqueous H2O2 at room temperature, and their isotopic distribution patterns corresponded to those calculated. In the case of 5+·I3−, the signals due to the hydroperoxo species [5 + OOH + H]+ were clearly observed at around m/z = 1284. These results demonstrate that high-valent iron-oxo species were generated by treating either 4+ or 5+ with H2O2via the corresponding hydroperoxo species, as in the case of other μ-nitrido-bridged iron phthalocyanine dimers. However, in the case of the heterodimer of an iron porphyrin and iron phthalocyanine 5+, it was difficult to determine the accurate position of the FeO moiety, i.e., whether it was located on the iron porphyrin or iron phthalocyanine group.25
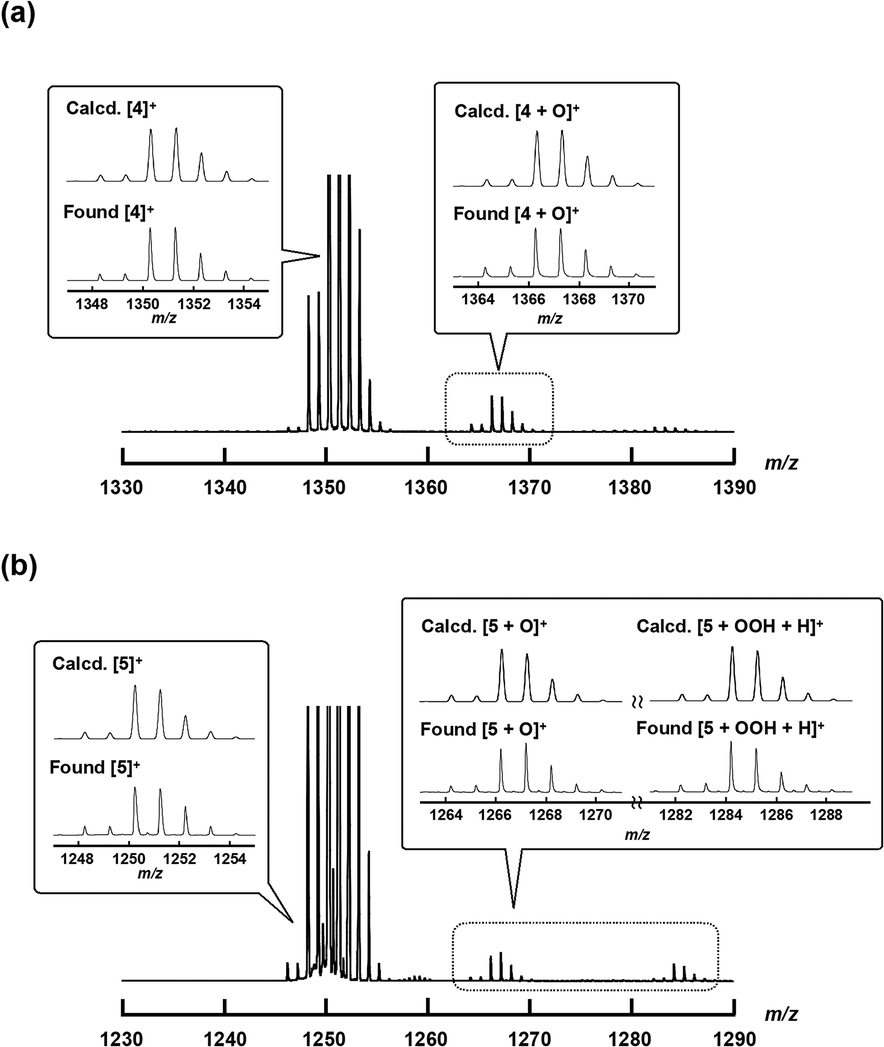 | ||
| Fig. 6 ESI-TOF MS spectra of 4+·I3− or 5+·I3− in an aqueous CH3CN solution containing an excess amount of H2O2 at room temperature (the detailed experimental procedure is shown in the Experimental section). Insets: Comparison of calculated (top) and experimental (bottom) isotopic distribution patterns of the molecular peaks assignable to [4]+, [4O]+, [5]+, [5O]+, and [5 + OOH + H]+. | ||
DFT calculations
In order to better understand and explain the experimental data, we conducted density functional theory (DFT) calculations on 4, 5, and 1. The computational details can be found in the Experimental section. Our primary focus was directed towards analyzing the redox processes involving these species (for example 4+, 42+, and 4). The electronic configurations of the monocationic complexes have been previously elucidated by Ghosh et al.35 and Sorokin et al.,11 and can be succinctly described as (dxy)2 (dxy)2 (dxz − dxz)2 (dyz − dyz)2. In agreement with the experimental data, the calculations revealed the symmetry of complexes 4+ and 1+. Specifically, we found that in 1+, the Fe–N(Pc) and Fe–μN bond lengths were 1.953 and 1.590 Å, respectively. In 4+, the corresponding bond distances were slightly longer, i.e., 1.986 and 1.601 Å. The Fe–N results confirmed the notion that the cavity of phthalocyanine is smaller than that of porphyrin. In all cases, Fe–N bond distances of less than 2.0 Å are compatible with the low-spin nature of the Fe centers. In both 4+ and 1+, the Wiberg Fe–μN bond order is 1.61, with a contribution of around 0.8 from Fe(dxz)–N(px) and Fe(dyz)–N(py) π interactions (Table S12†). In 5+, the two macrocycles caused a subtle distortion of the Fe–N–Fe axis. The Fe–μN bond distances were 1.587 and 1.600 Å. Although the DFT analysis suggested shorter bond distances than the experimental data (probably due to crystal packing), it correctly predicts a shorter bond distance between the Fe of iron porphyrin and μ-N, with a Wiberg bond order of 1.66. This bond exhibits a π character of around 0.90 from Fe(dxz)–N(px) and Fe(dyz)–N(py) interactions. The observed π character is slightly larger than that found in the bond between Fe of iron phthalocyanine and μN (0.76). Similar to what was observed in 4+ and 1+, the Fe–N(P) average bond distance (2.001 Å) was around 0.05 Å longer than the Fe–N(Pc) average bond distance (1.954 Å).In analogy to our previous work,25 the Fe–μN bonds became weaker upon reduction of 4+, 1+, and 5+ due to the occupation of an antibonding orbital Fe(dz2) − 2s + Fe(dz2). The Fe–μN bond distances in the reduced complexes were systematically longer by 0.02 to 0.04 Å. Spin populations as well as spin density (Fig. 7) revealed that only the Fe–N–Fe axis was involved in the reduction. We found a major spin population on the Fe atoms, whereas the spin population of the macrocycles was small (less than 0.04). The two Fe centers were very similar, indicating that the oxidation state of iron in all reduced complexes could be formally assigned to 3.5. The reduction free energies, defined as the difference between the Gibbs free energies of the monocationic and neutral complexes ΔGred = GX − GX+, were calculated. The reduction free energies of 1+, 4+, and 5+ were computed as −5.1, −4.4, and −4.8 eV, respectively. The results were consistent with the cyclic voltammetry data, where the first reduction potentials (vs. Fc/Fc+) for 1+, 4+, and 5+ were recorded at −0.58 V, −0.82 V, and −0.71 V, respectively.
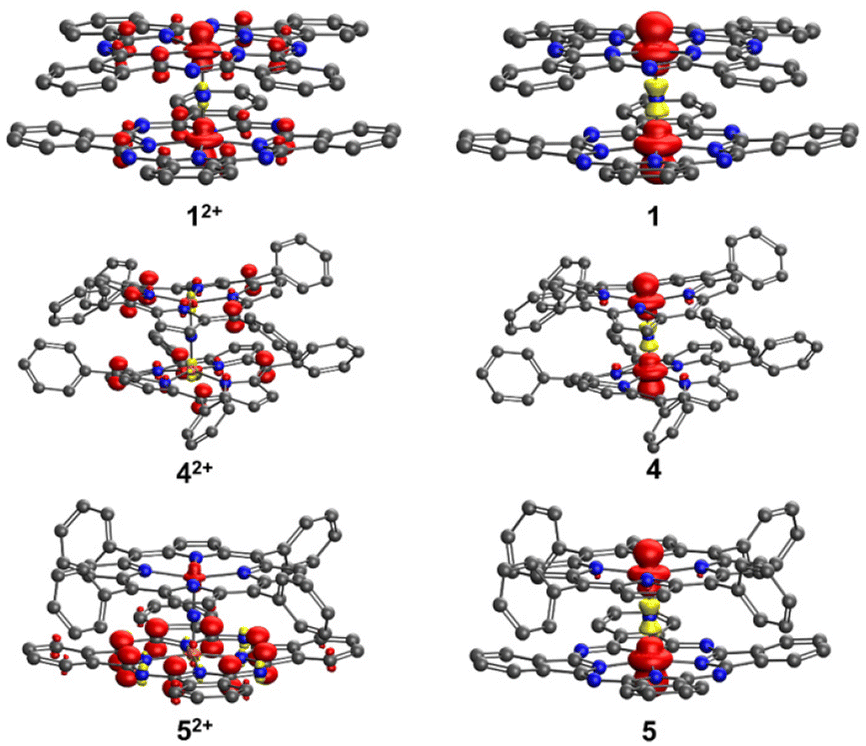 | ||
| Fig. 7 Spin density in 1, 4, 5, 12+, 42+, and 52+. In 1+, 4+, and 5+, the spin density is zero everywhere. Hydrogen atoms are neglected for clarity. | ||
The 1e−-oxidation of 4+, 1+, and 5+ induced negligible changes in the iron–ligand bond distances, suggesting that the oxidation mainly occurred within the macrocycles. Indeed, the spin density of μN in 42+, 12+, and 52+ (Fig. 7) was negligible. For 12+ and 42+, we observed that both macrocycles exhibited very similar densities, indicating that both complexes were partly oxidized. Conversely, in the case of 52+, the spin population of P was only 0.122, whereas the spin population of Pc was up to 0.8. This unambiguously pointed towards the phthalocyanine ring being the primary site of oxidation. The relatively higher tendency of phthalocyanine to be oxidized could be attributed to its longer conjugation length compared to that of porphyrin. The oxidation ΔGox = GX2+ − GX+ was also calculated. The oxidation energy of 1+ was 5.6 eV, whereas for 4+ and 5+ very similar oxidation energies were observed (around 5.8 eV). These findings are consistent with the cyclic voltammetry data, according to which the first oxidation potentials (vs. Fc/Fc+) for 1+, 4+, and 5+ were found at −0.19 V, 0.55 V, and 0.53 V, respectively.
Further calculations were conducted to determine the reduction energy of the high-valent iron oxo species [1O], [4O], and [5O] in water. As a result, we found the reduction free energies of [1O], [4O], and [5O] to be very similar (around 4.6 eV). Moreover, it was found that catalytic CH4 and CH3CH3 oxidation reactions via [1O] in an acidic aqueous solution could not be quenched by addition of an excess Na2SO3, reducing reagent, and radical scavenger (entry 14 in Table S2,† (vide infra)),36 whereas oxidation reactions via ˙OH could be significantly quenched under these conditions. It can be assumed that the oxidation reactions by [4O] and [5O] are less likely to be quenched by addition of Na2SO3.
CH4 and CH3CH3 oxidation activity
In order to investigate the catalytic CH4 and CH3CH3 oxidation activities of 1+·I−, 4+·I3−, and 5+·I3− in an aqueous solution, we prepared silica gel supported catalysts (1+·I−/SiO2, 4+·I3−/SiO2, and 5+·I3−/SiO2) since these complexes are not soluble in aqueous solutions. The CH4 and CH3CH3 oxidation reactions were performed in an acidic aqueous solution containing 57 μM of the catalyst molecule, 189 mM H2O2, and 51 mM trifluoroacetic acid (TFA) at 60 °C for 24 h under a CH4 (or CH3CH3) atmosphere of 1.0 MPa.15–17,22,24,25 After completion of the oxidation reaction, the oxidized products (MeOH, HCHO, and HCOOH for the CH4 oxidation, and EtOH, CH3CHO, AcOH, and HCOOH for the CH3CH3 oxidation) were quantified by GC-MS analysis, as summarized in Tables S1 and S2.†To appropriately evaluate the catalytic CH4 oxidation activity, the effective total turnover number (TTNeff) was defined according to eqn (i) and (ii) for the CH4 oxidation, and eqn (iii) and (iv) for the CH3CH3 oxidation, based on the idea that both CH4 and CH3CH3 can be oxidized in a stepwise manner and summarized in Tables S1, S2† and Fig. 8.15–17,22,24,25 We assumed that the small amount of oxidized products observed in the absence of CH4 or CH3CH3 (under a N2 atmosphere) could mainly originate from the adsorption of organic solvents on the silica-gel surface of the catalysts.
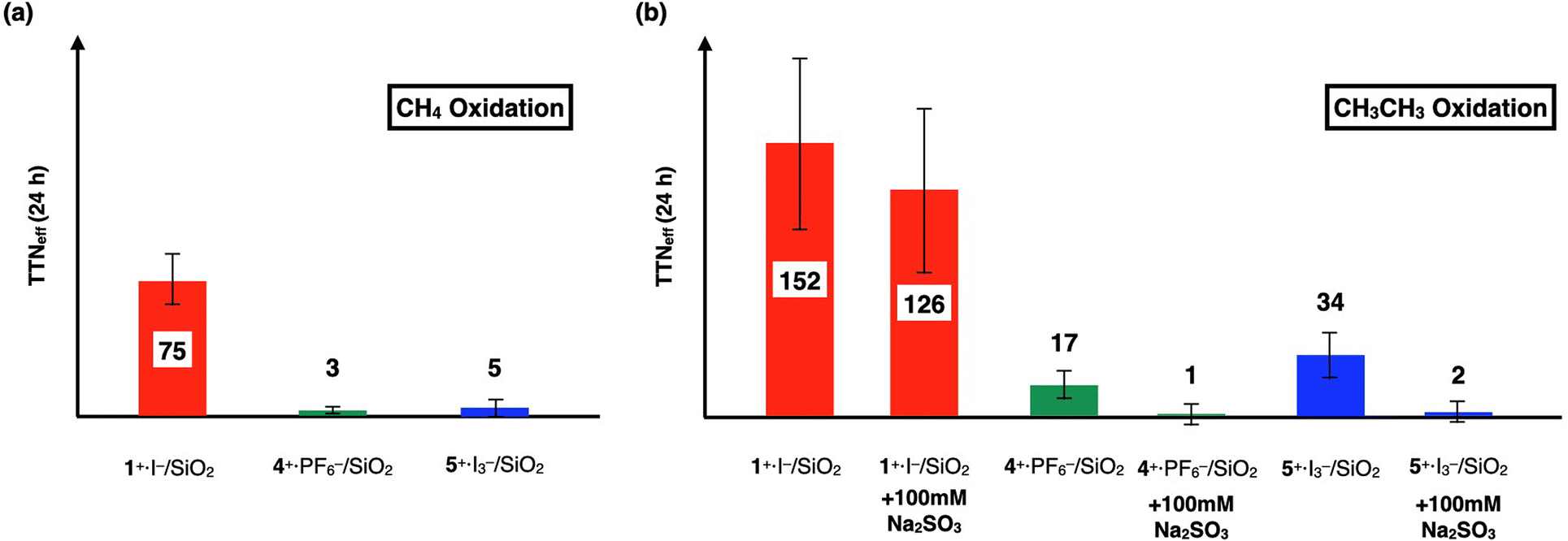 | ||
| Fig. 8 (a) Comparison of TTNeff for CH4 oxidation for 24 h by 1+·I−, 4+·I3−, and 5+·I3−. (b) Comparison of TTNeff for CH3CH3 oxidation for 24 h by 1+·I−, 4+·I3−, and 5+·I3−. Reactions were performed by using a silica gel-supported catalyst (30 mg, containing 57 μM of the catalyst molecule) in an aqueous solution (3.0 mL) containing H2O2 (189 mM) and TFA (51 mM) at 60 °C for 24 h under a CH4, CH3CH3, or N2 atmosphere (1.0 MPa). The amounts of each oxidized product in each oxidation reactions are summarized in Tables S1 and S2.† | ||
(CH4 oxidation)
| TTNeff(CH4) = TTN(CH4) − TTN(N2) | (i) |
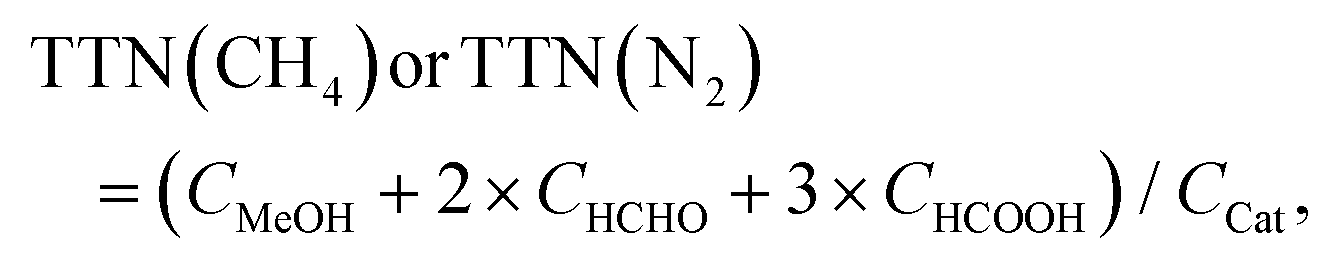 | (ii) |
(CH3CH3 oxidation)
| TTNeff(CH3CH3) = TTN(CH3CH3) − TTN(N2) | (iii) |
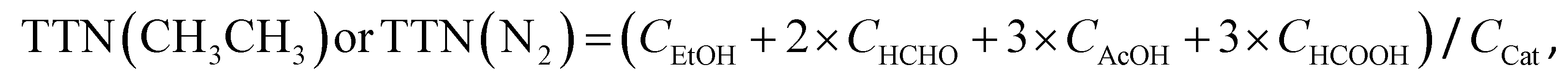 | (iv) |
We previously demonstrated that monocationic μ-nitrido-bridged iron phthalocyanine dimer 1+·I− acts as a CH4 oxidation catalyst under similar reaction conditions.16 Specifically, it was confirmed that both CH4 and CH3CH3 were oxidized by 1+·I−/SiO2, as shown in Tables S1, S2,† and Fig. 8. Since the CH3CH3 oxidation reaction by 1+·I−/SiO2 was not significantly quenched in the presence of an excess amount of Na2SO3, a radical scavenger that could quench ˙OH (entry 14 in Table S2,† and Fig. 8b),36 and the color of 1+·I−/SiO2 was not apparently bleached after 24 h oxidation (Fig. S6†), the oxidation reactions promoted by 1+·I−/SiO2 were assumed to proceed through the formation of a high-valent iron-oxo species (1O) rather than a Fenton-type reaction via ˙OH. Time dependencies of the TTNeffs of CH3CH3 oxidations by the three catalysts are summarized in Table S2 and Fig. S5.† TTNeffs seemed to be increased especially after 24 h for all of the catalysts, implying that decomposed catalysts showed Fenton-type oxidation, which can also be confirmed by the fact that the amounts of HCOOH were apparently increased at 48 h even in the absence of CH3CH3. We found it difficult to reuse these silica gel-supported catalysts for the catalytic oxidation reactions because the silica gel supports became pulverized and some of the catalysts were detached and aggregated after long time stirring.
In contrast, both 4+·I3−/SiO2 and 5+·I3−/SiO2 did not show apparent CH4 oxidation activities under these reaction conditions (entry 3 and 5 in Table S1,† and Fig. 8a). Although 4+·I3−/SiO2 and 5+·I3−/SiO2 showed CH3CH3 oxidation activities, as shown in entries 16 and 19 in Table S2,† and Fig. 8b, their catalytic CH3CH3 oxidation activities were significantly lower than that of 1+·I−/SiO2. Moreover, it was confirmed that the catalytic CH3CH3 oxidation activities of 4+·I3−/SiO2 and 5+·I3−/SiO2 could be apparently quenched in the presence of an excess amount of Na2SO3 (entries 17 and 20 in Table S2,† and Fig. 8b).
It should also be noted that both 4+·I3−/SiO2 and 5+·I3−/SiO2 did not show apparent color bleaching even after 24 h oxidation under these reaction conditions (Fig. S6†), indicating that the continuous occurrence of Fenton-type reactions is less probable. This is in clear contrast with the observation that monocationic μ-nitrido-bridged iron porphycene dimer 2+·I3− showed complete bleaching under the same reaction conditions, which was elucidated by the instability of the high-valent iron-oxo species, as reported in our previous study.24
Considering that the results of the ESI-TOF MS experiments for 4+·I3− and 5+·I3− in the presence of H2O2 indicated the generation of high-valent iron-oxo species (4O and 5O), it seemed that the high-valent iron-oxo species of 4+ and 5+ were actually generated as in the case of 1 and 1+. In addition, the majority of the CH3CH3 (or CH4) oxidation reactions using 4+·I3−/SiO2 and 5+·I3−/SiO2 might be attributed to ˙OH, which could be catalytically generated from the small amount of the decomposed products of 4+ and 5+, since it was significantly quenched upon addition of Na2SO3 although the DFT calculations suggested that 4O and 5O could have similar reduction potential as 1O.
Therefore, the results of the CH4 and CH3CH3 oxidation reactions in the presence of 4+·I3−/SiO2 and 5+·I3−/SiO2 at 24 h (Fig. 8) implied that the catalytic alkane oxidation ability of 4O and 5O generated under these reaction conditions appeared to be much lower than that of 1O. Considering the differences in the crystal structures of the Fe–NFe moieties in 1+, 4+, and 5+ were not significant, the difference in catalytic activities could originate from that in the electronic structures of porphyrin and phthalocyanine, as indicated by the cyclic voltammetric studies of 1+, 4+, and 5+. Notably, the 1e−-oxidation of 1+ corresponding to the 1e−-oxidation of the porphyrinoid ring occurred at a much lower potential (−0.19 V) than those of 4+ (0.55 V) and 5+ (0.53 V) could imply that the phthalocyanine moiety of 1+ possessed a more electron-rich character than those of the phthalocyanine moiety of 4+ and the porphyrin moiety of 5+.
The proposed reaction mechanism and possible elucidation of the contribution of the electron-rich character of the phthalocyanine moiety to the high CH4 oxidation activity of 1+·I− are summarized in Fig. 9. The reactive intermediate is considered to be the in situ generated 1O species.11 In general, the CH4 oxidation by high-valent iron oxo species proceeds via a proton-coupled electron transfer (PCET) mechanism, in which the proton and electron abstraction from CH4 occur in a concerted manner.10 It can be expected that the electron-rich character of the phthalocyanine ring could increase the basicity of the oxo species. Another possibility is that the heterolytic cleavage of the O–O bonding of hydroperoxo species to give the high-valent iron-oxo species (1O) is facilitated by the electron-rich phthalocyanine moiety, as in the case with a push effect.16,37,38 Exact determination of rate constants for the reactions by 4+ and 5+ were difficult because they apparently include Fenton-type reaction. On the other hand, although the reaction by 1+·I−/SiO2 is considered to be proceeded mainly via high-valent iron-oxo species, determination of the production rate of 1O was also difficult due to both of the catalase reaction, which competed with the alkane oxidation by converting H2O2 into O2, and the silica-gel support, which readily adsorbed the organic dyes added in the solution in order to determine the production rate of the oxo species.
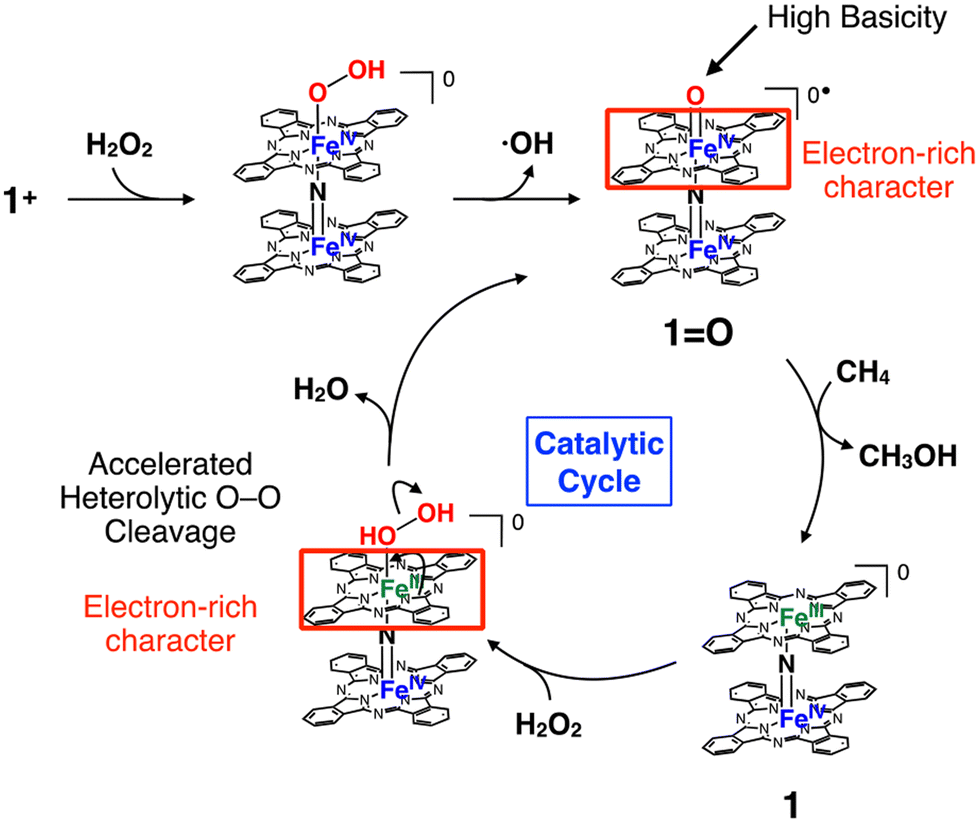 | ||
| Fig. 9 Proposed reaction mechanism and possible elucidation for the contribution of electron-donation by phthalocyanine ring to the high methane oxidation activity of 1+. | ||
Sorokin et al. reported that 4/SiO2 exhibited an apparent catalytic CH4 oxidation activity to give HCOOH as a major product in an aqueous solution by using mCPBA as an oxidant instead of H2O2.18 Considering that both 4 and 4+ can generate the same high-valent iron-oxo species 4O upon reacting with H2O2, it is unlikely that the difference in the oxidation state (4 and 4+) significantly affected the catalytic activity. Actually, in the case of a μ-nitrido-bridged iron phthalocyanine dimer, 1 and 1+ showed a similar catalytic activity in an acidic aqueous solution containing H2O2.17 Therefore, we concluded that the reason why 4+ did not show apparent CH4 oxidation ability could mainly be due to the different oxidant (mCPBA and H2O2). It is probable that the coordination of the m-chlorobenzoic acid anion onto the other side of the iron center of the high-valent iron-oxo species, as suggested by the DFT calculations conducted by de Visser et al., could have changed the electronic character and reactivity of the high-valent iron-oxo species of 4+.19
Conclusions
In this study, we synthesized a monocationic μ-nitrido-bridged porphyrin heterodimer (4+) along with a monocationic μ-nitrido-bridged heterodimer of iron phthalocyanine and iron porphyrin (5+) and compared their electrochemical and spectroscopic properties with those of a monocationic μ-nitrido-bridged iron phthalocyanine homodimer (1+), which showed potent catalytic CH4 oxidation activity in an acidic aqueous solution in the presence of H2O2. Single-crystal X-ray structural analysis demonstrated that the Fe–NFe core structures of 4+·I3− and 5+·I3− were similar to that of 1+·I. We also confirmed that the high-valent iron-oxo species of 4+ and 5+ were actually observed by ESI-TOF MS spectroscopy in the presence of H2O2. It was demonstrated that the catalytic CH4 and CH3CH3 oxidation activities of 4+·I3− and 5+·I3− were significantly lower than that of 1+·I− in an acidic aqueous solution at 60 °C in the presence of an excess of H2O2. These results clearly indicated that the difference in the porphyrinoid ring of the monocationic μ-nitrido-bridged iron porphyrinoid dimer strongly affected the electronic structure and reactivity of the high-valent iron-oxo species generated from monocationic μ-nitrido-bridged iron porphyrinoid dimers.
Experimental
General
All reagents and solvents were purchased at the highest commercial quality available and used without further purification, unless otherwise stated. 1H NMR spectra were recorded on a JEOL JNM-ECS400 (400 MHz for 1H) spectrometer at a constant temperature of 298 K. Elemental analysis was performed on a Yanaco MT-6 analyzer. The absorption spectrum was recorded with a Hitachi U-4100 spectrophotometer in pyridine solutions at 20 ± 0.1 °C in 1.0 cm quartz cells. MALDI-TOF MS was performed on Bruker Daltonics ultrafleXtreme using α-CHCA as a matrix. MALDI-TOF MS was performed on Bruker Daltonics compact.Synthesis of 4+·I3−
A mixture of 6 (201 mg, 0.29 mmol), NaN3 (212 mg, 3.3 mmol) and 1-chloronaphthalene (5.0 mL) was heated under air at 280 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with CH2Cl2 (200 mL). The solution was washed with H2O (100 mL × 2) and brine (100 mL × 2) then evaporated to give a reddish-brown crude solid. The crude product was dissolved in CH2Cl2 (150 mL). After iodine (552 mg, 4.3 mmol as I) was added, the resulting solution was stirred for 2 h at room temperature, followed by evaporation of the volatile compounds. The residue was washed with Et2O (300 mL) to remove the remaining iodine, and then, dried under reduced pressure to give reddish brown solid. The crude was purified by silica gel column chromatography (∅ 4.5 cm × 16.0 cm, CH2Cl2, then CH2Cl2/MeOH = 20/1) and recrystallization of CH2Cl2/Et2O to yield 4+·I3− as a reddish brown solid (176 mg, 71%). 1H-NMR (400 MHz, pyridine-d5/TMS): δ = 9.08 (d, J = 7.6 Hz, 8H), 8.81 (s, 16H), 8.21 (t, J = 7.4 Hz, 8H), 8.00 (t, J = 7.6 Hz, 8H), 7.67 (t, J = 7.6 Hz, 8H), 7.37 (d, J = 7.6 Hz, 8H). MALDI-TOF MS: m/z = 1350.34: calcd for C88H56Fe2N9 ([M]+) found: 1350.35. Anal. calcd for C88H56Fe2I3N9O5 (4+·I3−): C; 61.03, H; 3.26, N; 7.28, found: C; 61.15, H; 3.31, N; 6.95 (0.33% error).Synthesis of 4+·PF6−
4 +·I3− (50 mg, 29 μmol) was dissolved in 10 mL of 1:1 (v/v) mixture of pyridine and CH3CN. After KPF6 (541 mg, 2.9 mmol) in 1:1 (v/v) mixture of pyridine and CH3CN (10 mL) was added to the solution, H2O (80 mL) was added. The resulting precipitate was collected by centrifugation, followed by decantation to yield reddish brown solid (49.7 mg). The crude product was further purified by recrystallization from CH2Cl2/Et2O to give the desired product 4+·PF6− as a reddish brown solid (41 mg, 27 μmol, 94%). 1H-NMR (400 MHz, pyridine-d5/TMS): δ = 9.08 (d, J = 8.0 Hz, 8H), 8.82 (s, 16H), 8.21 (t, J = 7.6 Hz, 8H), 8.00 (t, J = 7.4 Hz, 8H), 7.66 (t, J = 8.0 Hz, 8H), 7.36 (d, J = 8.4 Hz, 8H). MALDI-TOF MS: m/z = 1350.34: calcd for C88H56Fe2N9 ([M]+) found: 1350.35. Anal. calcd for C76H44F6Fe2N13P (4+·PF6−·2Py·2CH2Cl2): C; 65.84, H; 3.87, N; 8.45, found: C; 66.18, H; 3.55, N; 8.49 (0.34% error).
Synthesis of 5+·I3−
A mixture of 6 (105 mg, 0.15 mmol), 7 (85 mg, 0.15 mmol), NaN3 (201 mg, 3.1 mmol) and 1-chloronaphtalene (5.0 mL) was heated under air at 280 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with hexane (400 mL), the precipitate was collected by suction filtration, washed with H2O (300 mL), dried under vacuum to give blackish green crude solid (234 mg). The crude product was dissolved in pyridine (200 mL). After iodine (215 mg, 0.85 mmol as I) was added, the resulting solution was stirred for 2 h at room temperature, followed by evaporation of the volatile compounds. The residue was washed with Et2O (300 mL) to remove the remaining iodine, and then, dried under reduced pressure to give dark green solid. The crude was purified by silica gel column chromatography (∅ 4.5 cm × 16.0 cm, CH2Cl2/AcOEt = 1/1, then CH2Cl2/AcOEt/MeOH = 10/10/1) and recrystallization of CH2Cl2/Et2O to yield 5+·I3− as a dark green solid (174 mg, 71%). 1H-NMR (400 MHz, pyridine-d5/TMS): δ = 11.00 (d, J = 8.0 Hz, 4H), 9.82 (dd, J = 5.6, 3.2 Hz, 8H), 8.83 (t, J = 7.8 Hz, 4H), 8.38 (dd, J = 5.6, 2.8 Hz, 8H), 8.29 (s, 8H), 8.07 (t, J = 8.0 Hz, 4H), 7.53–7.58 (m, 4H), 7.21–7.25 (m, 4H). MALDI-TOF MS: m/z = 1250.25: calcd for C76H44Fe2N13 ([M]+) found: 1250.39. Anal. calcd for C90H64Fe2I3N15O (5+·I3−·2Py·Et2O): C; 57.99, H; 3.46, N; 11.27, found: C; 57.83, H; 3.11, N; 11.15 (0.35% error).Synthesis of 5+·PF6−
5 +·I3− (60 mg, 37 μmol) was dissolved in 10 mL of 1:1 (v/v) mixture of pyridine and CH3CN. After KPF6 (704 mg, 3.8 mmol) in 1:1 (v/v) mixture of pyridine and CH3CN (10 mL) was added to the solution, H2O (80 mL) was added. The resulting precipitate was collected by centrifugation, followed by decantation to yield dark green solid (80 mg). The crude product was further purified by recrystallization from CH2Cl2/Et2O to give the desired product 5+·PF6− as a dark green solid (48 mg, 34 μmol, 93%). 1H-NMR (400 MHz, pyridine-d5/TMS): δ = 11.00 (d, J = 8.4 Hz, 4H), 9.82 (dd, J = 5.6, 3.0 Hz, 8H), 8.83 (t, J = 8.0 Hz, 4H), 8.38 (dd, J = 5.4, 2.2 Hz, 8H), 8.29 (s, 8H), 8.07 (t, J = 7.8 Hz, 4H), 7.5–7.58 (m, 4H), 7.21–7.28 (m, 4H). MALDI-TOF MS: m/z = 1250.25: calcd for C76H44Fe2N13 ([M]+) found: 1250.39. Anal. calcd for C77H46Cl2F6Fe2N13P (5+·PF6−·CH2Cl2): C; 62.45, H; 3.13, N; 12.30, found: C; 62.24, H; 3.02, N; 12.50 (0.21% error).
Single-crystal X-ray structural analyses of 4+·I3− and 5+·I3−
Crystal suitable for single-crystal X-ray structural analysis was obtained by slow evaporation of solvent from a 4+·I3− (or 5+·I3−) solution in a 1:50 (v/v) mixture of pyridine and CHCl3. Single-crystal X-ray diffraction measurements were performed using a Rigaku X-ray diffractometer equipped with a molybdenium MicroMax-007 and Saturn 70 CCD detector. The measurement was performed at 123 K. The structure was solved via the direct method (SHELXT) and refined via full-matrix least-squares on F2 (SHELX-2018) using Olex2-1.3 program All non-hydrogen atoms were refined anisotropically. Geometrical restraints were applied: DFIX, SADI, DELU, ISOR, and OMIT. The crystal datas are as follows:
4
+·I3−: Formula C360H232Cl48Fe8I12N60, FW = 9069.594, crystal size 0.04 × 0.16 × 0.18 mm3, triclinic, space group P, a = 15.1304(17) Å, b = 20.032(2) Å, c = 29.169(3) Å, α = 90°, β = 92.653(7)°, γ = 90°, V = 8831.5(18) Å3, Z = 1, R1 = 0.0941(35119) (I > 2(I)), wR2 = 0.2427(40518) (all), GOF = 1.031. CCDC identification code 2242976.† All the checkCIF Level-B alerts are due to the low crystal quality.
5
+·I3−: Formula C388H260Cl48Fe8I12N40, FW = 9153.994, crystal size 0.10 × 0.10 × 0.10 mm3, triclinic, space group P, a = 20.2412(6) Å, b = 15.9489(5) Å, c = 27.7869(8) Å, α = 90°, β = 90°, γ = 90°, V = 8970.3(5) Å3, Z = 1, R1 = 0.0382(22741) (I > 2(I)), wR2 = 0.1515(25785) (all), GOF = 1.300. CCDC identification code 2242975.† All the checkCIF Level-B alerts are due to the low crystal quality.
Cyclic voltammogram of 4+·PF6−, 5+·PF6−
Cyclic voltammograms were measured with a BAS Electrochemical Analyzer Model 750Ds at room temperature in pyridine solutions containing 100 mM TBAPF6 in a standard one-component cell under an N2 atmosphere equipped with a 3 mm-O.D. glassy carbon disk working electrode, platinum wire counter electrode, and Ag/AgCl reference electrode. All solutions were deoxygenated by N2 bubbling for at least 20 min. Obtained E°′ vs. Ag/AgCl were converted to those vs. Fc/Fc+ based on measured redox potential of ferrocene. Tetrabutylammonium hexafluorophosphate (TBA+PF6−) was recrystallized from 95% EtOH and dried under vacuum overnight at 100 °C.ESI-TOF MS measurement of 5+·I3− in CH3CN in the presence of H2O2
To a solution of 5+·I3− in CH3CN (2.0 mL, 1.0 μM) was added 35% aqueous H2O2 (0.1 mL, 1.16 mmol). After addition, the resulting mixture was subjected to ESI-TOF MS measurement immediately.DFT calculation
DFT calculations of 1, 4, and 5 were performed with the BP86-D3(BJ)/def2-TZVP level of theory implemented in the Turbomole software package.39–42 This level was chosen based on our previous study as well as the works of Sorokin et al.11 To ensure the validity of the optimized structures and to acquire Gibbs free energies,43 frequency calculations were carried out. On top of the optimized structures, single-point calculations combined with the COSMO model were done to account for the solvent effect (pyridine with a dielectric constant of 12.5).44 Wiberg bond order indices in the natural atomic orbital (NAO) basis were obtained at the same level of theory, using the Gaussian 16 and Multiwfn packages.45,46The structures of the oxo complexes were obtained at the B3LYP-D3(BJ)/def2-TZVP level of theory. For the neutral species, only the doublet state FeIVFeIVO(P˙+) was considered. For the anion species, the open-shell singlet state FeIVFeIVO(P) was calculated. To account for the water solvent, CPCM solvation model was employed.47 The calculations were done with the Orca 5.0 package.48
CH4 or CH3CH3 oxidation reactions
Heterogeneous CH4 or CH3CH3 oxidation reaction was performed in a stainless-steel autoclave with a glass tube. A mixture containing the catalyst on SiO2 (30 mg, 55 μM as 4+·I3−, or 5+·I3−, or 1+·I−), 35% aqueous H2O2 (50 μL, 189 mM), and TFA (12 μL, 51 mM) in H2O (3.0 mL) was heated at 60 °C under 1.0 MPa of CH4 (or CH3CH3, or N2) atmosphere for 24 h with continuous stirring (900 rpm). After the autoclave was opened, the reaction mixture was filtrated through a disposable membrane filter (DISMIC-13CP, ADVANTEC). The filtrate was analyzed by GC-MS (system: Agilent 7890A equipped with JEOL JMS-T100GCV, detection: EI, column: Agilent DB-WAX UI, external standard: isovaleric acid (5 mM), temperature conditions: initial: 70 °C to 220 °C (10 °C min−1) − hold (5 min)). The yields of CH3OH, EtOH, CH3CHO, AcOH and formic acid were determined based on the results of GC-MS. The yield of formaldehyde was examined using the method reported in our previous paper.15–17,22,24,25Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by a JSPS KAKENHI Grant-in-Aid for Scientific Research (B) (22H02094) awarded to KT, a JSPS KAKENHI Grant-in-Aid for Challenging Exploratory Research (Number 22K19045), a Grant-in-Aid for Scientific Research (B) (Number 22H02156) awarded to YY. YY thanks to the financial supports by Tatematsu Foundation, Iwatani Naoji Foundation, and Toyoaki Scholarship Foundation. The single-crystal synchrotron radiation X-ray diffraction measurements were carried out at the BL02B1/Spring-8 in Japan (Proposal No. 2020A1077, 2021A1105, and 2022A1134). The computations were performed using Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C987).References
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44(4), 280–288 CrossRef CAS PubMed.
- S. Sirajuddin and A. C. Rosenzweig, Biochemistry, 2015, 54, 2283–2294 CrossRef CAS PubMed.
- V. C.-C. Wang, S. Maji, P. P.-Y. Chen, H. K. Lee, S. S.-F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- A. R. McDonald and L. Que Jr., Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que Jr., S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef CAS PubMed.
- Y. J. Jeong, Y. Kang, A.-R. Han, Y.-M. Lee, H. Kotani, S. Fukuzumi and W. Nam, Angew. Chem., 2008, 120, 7431–7434 CrossRef.
- J. E. M. N. Klein and G. Knizia, Angew. Chem., Int. Ed., 2018, 57, 11913–11917 CrossRef CAS PubMed.
- P. Afanasiev and A. B. Sorokin, Acc. Chem. Res., 2016, 49, 583–593 CrossRef CAS PubMed.
- H. Fujisaki and T. Kojima, Catal. Sci. Technol., 2023, 13, 4270–4284 RSC.
- A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC.
- Ü. İşci, A. S. Faponle, P. Afanasiev, F. Albrieux, V. Briois, V. Ahsen, F. Dumoulin, A. B. Sorokin and S. P. de Visser, Chem. Sci., 2015, 6, 5063–5075 RSC.
- Y. Yamada, K. Morita, N. Mihara, K. Igawa, K. Tomooka and K. Tanaka, New J. Chem., 2019, 43, 11477–11482 RSC.
- Y. Yamada, J. Kura, Y. Toyoda and K. Tanaka, Dalton Trans., 2021, 50, 6718–6724 RSC.
- Y. Yamada, K. Morita, T. Sugiura, Y. Toyoda, N. Mihara, M. Nagasaka, H. Takaya, K. Tanaka, T. Koitaya, N. Nakatani, H. Ariga-Miwa, S. Takakusagi, Y. Hitomi, T. Kudo, Y. Tsuji, K. Yoshizawa and K. Tanaka, JACS Au, 2023, 3, 823–833 CrossRef CAS PubMed.
- E. V. Kudrik, P. Afanasiev, L. X. Alvarez, P. Dubourdeaux, M. Clémancey, J.-M. Latour, G. Blondin, D. Bouchu, F. Albrieux, S. E. Nefedov and A. B. Sorokin, Nat. Chem., 2012, 4, 1024–1029 CrossRef CAS PubMed.
- M. G. Quesne, D. Senthilnathan, D. Singh, D. Kumar, P. Maldivi, A. B. Sorokin and S. P. de Visser, ACS Catal., 2016, 6, 2230–2243 CrossRef CAS.
- E. V. Kudrik and A. B. Sorokin, Chem. – Eur. J., 2008, 14, 7123–7126 CrossRef CAS PubMed.
- L. X. Alvarez and A. B. Sorokin, J. Organomet. Chem., 2015, 793, 139–144 CrossRef CAS.
- N. Mihara, Y. Yamada, H. Takaya, Y. Kitagawa, K. Igawa, K. Tomooka, H. Fujii and K. Tanaka, Chem. – Eur. J., 2019, 25, 3369–3375 CrossRef CAS PubMed.
- Y. Yamada, C.-M. Teoh, Y. Toyoda and K. Tanaka, New J. Chem., 2022, 46, 955–958 RSC.
- Y. Yamada, Y. Miwa, Y. Toyoda, T. Yamaguchi, S. Akine and K. Tanaka, Dalton Trans., 2021, 50, 16775–16781 RSC.
- Y. Yamada, Y. Miwa, Y. Toyada, Q. M. Phung, K. Oyama and K. Tanaka, Catal. Sci. Technol., 2023, 13, 1725–1734 RSC.
- D. A. Summerville and I. A. Cohen, J. Am. Chem. Soc., 1976, 98, 1747–1752 CrossRef CAS PubMed.
- C. Ercolani, M. Gardini, G. Pennesi, G. Rossi and U. Russo, Inorg. Chem., 1988, 27, 422–424 CrossRef CAS.
- D. R. English, D. N. Hendrickson and K. S. Suslick, Inorg. Chem., 1985, 24, 122–127 CrossRef.
- Y. Yamada, T. Sugiura, K. Morita, H. Ariga-Miwa and K. Tanaka, Inorg. Chim. Acta, 2019, 489, 160–163 CrossRef CAS.
- K. M. Kadish, R. K. Rhodes, L. A. Bottomley and H. M. Goff, Inorg. Chem., 1981, 20, 3195–3200 CrossRef CAS.
- L. A. Bottomley and B. B. Garrett, Inorg. Chem., 1982, 21, 1260–1263 CrossRef CAS.
- T. Shimizu, K. Wakamatsu, Y. Yamada, Y. Toyoda, S. Akine, K. Yoza and H. Yoshikawa, ACS Appl. Mater. Interfaces, 2021, 13, 40612–40617 CrossRef CAS PubMed.
- T. Guchhait, S. Sasmal, F. S. T. Khan and S. P. Rath, Coord. Chem. Rev., 2017, 337, 112–144 CrossRef CAS.
- L. A. Bottomley, J.-N. Gorce, V. L. Goedken and C. Ercolani, Inorg. Chem., 1985, 24, 3733–3737 CrossRef CAS.
- A. Ghosh, E. Tangen, E. Gonzalez and L. Que Jr., Angew. Chem., 2004, 116, 852–856 CrossRef.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- K. Yamaguchi, Y. Watanabe and I. Morishima, Inorg. Chem., 1992, 31, 156–157 CrossRef CAS.
- Y. Watanabe, High-Valent Intermediates, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Elsevier, 2003, vol. 4, ch. 30 Search PubMed.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2242975 and 2242976. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04313d |
| This journal is © The Royal Society of Chemistry 2024 |