DOI:
10.1039/D3DT04249A
(Paper)
Dalton Trans., 2024,
53, 4680-4688
Morphology regulation of conductive metal–organic frameworks in situ grown on graphene oxide for high-performance supercapacitors†
Received
19th December 2023
, Accepted 5th February 2024
First published on 15th February 2024
Abstract
In this work, nickel-catecholate (Ni-CAT) nanorods were in situ compounded on graphene oxide (GO) to form a composite Ni-CAT@GO (NCG) with a special “blanket-shape” structure, which was used as an electrode material for supercapacitors. The morphology of Ni-CATs in situ grown on GO was modulated by introducing various contents of GO. With increasing GO, the length of nanorods of Ni-CATs is obviously shortened, and the charge transfer resistance of NCG is significantly reduced as the GO content is relatively low while it increases with further addition of GO, because excessive GO in NCG results in smaller crystal sizes accompanied by smaller stacking pores. Both the over-long Ni-CAT nanorods and the smaller stacking pores can restrict the accessible surface areas for the electrolyte. Optimal nanorod sizes are crucial to achieve good electrochemical performance for electrode materials. Galvanostatic charge–discharge analysis of NCG electrodes shows that their capacity initially increases and then decreases with the addition of more and more GO, and Ni-CAT@GO-0.5 (NCG0.5) with minimal charge transfer resistance exhibits the best electrochemical performance. The results demonstrate that the NCG0.5 electrode with optimal morphology possesses an excellent capacitance of 563.8 F g−1 at 0.5 A g−1 and a good rate performance of 61.9% at 10 A g−1, indicating that Ni-CAT@GO is a new type of promising electrode material for supercapacitors based on conductive metal–organic frameworks.
1. Introduction
Supercapacitors are an emerging class of electrical energy storage devices.1–5 Based on the difference in the mechanism of charge storage, supercapacitors can be divided into two types: electrical double layer capacitors (EDLCs) and pseudo-capacitors.6 EDLCs store charges by accumulating charges on the interfaces between electrodes and electrolytes, which is similar to traditional capacitors.7,8 A large specific surface area can enhance the charge storage ability of EDLCs, and well-controlled hierarchical pores can improve the rate performance by reducing the charge transfer resistance in electrode materials.9 Pseudo-capacitors store charges through a prompt and reversible Faraday reaction,10,11 resulting in better capacitance values than those of EDLCs. Electrode materials for pseudo-capacitors are typical compounds composed of transition metals with variable valence states such as Ni,12 Co,13 Mn,14 and Bi,15,16 and these materials for pseudo-capacitors generally have lower conductivity compared to the electrode materials used in EDLCs, for instance, carbon. Usually, the cycling stability and rate performance of a pseudo-capacitor is lower than those of EDLCs.17 Supercapacitors with high performances require large surface areas, abundant redox sites and good conductivity for various electrode materials.18 Research on a lot of advanced porous materials such as metal–organic frameworks (MOFs)19–23 and covalent organic frameworks (COFs)24,25 for supercapacitor electrodes is rapidly blooming, but their application is still restricted because of their inadequate conductivity. The usage of a conductive substrate and/or conductive materials can enhance the electrochemical performance of electrode materials.
A common way to enhance the electrochemical performance of supercapacitors is to reduce the electrical resistance of the electrode material by compounding it with a conductive substrate, such as carbon nanotubes (CNTs)17,26 or graphene.27 Graphene is a special two-dimensional carbon material with outstanding properties such as high specific surface area and excellent electrical conductivity, hence arousing significant interest within the academic community.28 By Hummers’ method,29 graphite underwent exfoliation and oxidation, resulting in a derivative of graphene known as graphene oxide (GO). This process introduces oxygen-containing functional groups, including hydroxyl and carboxyl groups, onto the surface of graphene. These functional groups make the GO surface more polar and negatively charged in water. As a result, the water dispersibility and hydrophilicity of GO are improved, preventing a reduction in specific surface area due to self-stacking. Furthermore, the positively charged ions are able to be attracted to the negatively charged surface of GO, anchoring the ions to it.30 Nevertheless, there is a reduction in electrical conductivity of GO in comparison with graphene.31
Metal–organic frameworks (MOFs) are composed of metallic ions and organic ligands,32–34 possessing high specific surface areas and ordered pores; such ordered porous structures allow a large number of active metallic sites to be exposed. These properties facilitate their application in various fields, such as catalysis, adsorption, and drug delivery, and as a kind of promising electrode material for supercapacitors. However, MOF materials have seldom been used directly as components of electrode materials due to their poor electrical conductivity.35,36 However, MOFs can function as a precursor of porous electrode materials for EDLCs.19,21,37 Ping et al.38 used ZIF-8 and cellulose as precursors and obtained a type of activated porous carbon with high capacitances of 244 F g−1 and 177 F g−1 at current densities of 1 A g−1 and 20 A g−1, respectively.
Some series of special types of MOFs can exhibit relatively good electrical conductivity.39–42 These materials are expected to have a considerable specific surface area, relatively good electrical conductivity, and numerous exposed active transition-metal sites. Metal catecholates (M-CATs) are a series of conductive MOFs formed by combining metallic ions with 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) as a ligand.43–46 Most M-CATs, such as Ni-CAT and Co-CAT, exhibit two distinct 2D structures within their crystals. One is a 2D structure polymer sheet containing hexagonal holes extending along the plane, as depicted in Fig. 1, where each metal ion coordinates with two water molecules. The other comprises discrete complexes formed by a metal ion coordinating with a deprotonated HHTP molecule and four water molecules. These 2D nanosheets and discrete complexes align alternately and then stack perpendicular to the planes, and rod-shaped crystals are formed, driven by van der Waals forces. In the nanorod crystals, hexagonal holes are allowed to align and 1D pore tunnels are formed parallel to the stacking direction.47 This arrangement enables electrons to migrate in the conjugated π-bond electron cloud within the ligands and also transfer across the hydroxyl group to the metal ion, making these M-CATs electroconductive. Chen et al.48 synthesized Bi(HHTP) nanobelts as negative electrodes for supercapacitors and obtained a remarkable capacitance of 234.0 F g−1 at 1 A g−1, and 72% of the capacitance value was retained after 1000 cycles. In a separate study, Zhao et al.49 grew Ni-CAT crystals on carbon fibers obtained by electrostatic spinning, resulting in an electrode material with a capacitance of 502.95 F g−1 under 0.5 A g−1. On the one hand, nickel is a transition metal element that can act as a Faraday reaction site.30 On the other hand, using GO as a carbon matrix to improve conductivity through Ni-CAT crystals appears to be a viable approach for enhancing capacitance performance.50,51 Therefore, it is rational to expect that the high specific surfaces and good conductivity of both GO and Ni-CAT will enhance the capacitance performance of the composite Ni-CAT@GO.
 |
| | Fig. 1 Chemical structure of HTTP and polymer sheets in the Ni-CAT structure. | |
In this work, a simple one-step hydrothermal strategy was applied to compound GO with Ni-CAT, modulating the content of GO to obtain Ni-CAT@GO (NCG) samples with various compositions and different morphologies. Electrochemical tests were carried out on the supercapacitor electrodes prepared from the NCG samples. Among the tested samples, Ni-CAT@GO-0.5 (NCG0.5) demonstrated great potential with an excellent capacitance of 563.8 F g−1 at 0.5 A g−1. It retained 61.9% capacitance at a high current density of 10 A g−1 and maintained 68.7% of its initial capacitance after 1000 cycles. With an optimal ratio, the morphology of Ni-CAT@GO was tailored by a straightforward strategy of in situ compounding, which significantly increased the accessible surface of the Ni-CAT@GO electrodes, such that the capacitive performance is expected to be significantly enhanced.
2. Materials and methods
2.1 Sample preparation
The GO dispersions were prepared by the conventional Hummers’ method.52 Typically, 3 g of NaNO3, 3 g of graphite, and 100 ml of concentrated sulfuric acid were added to a flask and stirred for 30 minutes under ice bath conditions. Subsequently, 9 g of KMnO4 was slowly added to the flask, and the mixture was stirred thoroughly for 1 hour at 35 °C. Then, 100 ml of distilled water was added while maintaining the temperature of the mixture below 90 °C. After 30 minutes, 80 ml of 30% H2O2 solution was added and maintained for two hours to obtain the mixture. The mixed solution was washed three times by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 r per min with deionized water and then it was centrifuged three times at 7000 r per min and the supernatant was removed, with each centrifugation lasting 5 minutes. Subsequently, deionized water was added to the mixture and vigorously stirred to ensure uniformity, and then it was centrifuged at 3500 r per min for 3 minutes to collect the upper suspension. This process was repeated five times. The collected suspension was subjected to dialysis until the pH value of the dialysate solution reached ∼7, resulting in the GO dispersion. The GO dispersion was stirred and concentrated at 45 °C; then, deionized water was added to form a 10 g l−1 GO dispersion.
000 r per min with deionized water and then it was centrifuged three times at 7000 r per min and the supernatant was removed, with each centrifugation lasting 5 minutes. Subsequently, deionized water was added to the mixture and vigorously stirred to ensure uniformity, and then it was centrifuged at 3500 r per min for 3 minutes to collect the upper suspension. This process was repeated five times. The collected suspension was subjected to dialysis until the pH value of the dialysate solution reached ∼7, resulting in the GO dispersion. The GO dispersion was stirred and concentrated at 45 °C; then, deionized water was added to form a 10 g l−1 GO dispersion.
Fig. 2 illustrates a concise diagram of the sample preparation process.53 Firstly, solution A was prepared by dissolving 0.6 mmol of nickel acetate in 15 ml of deionized water (DI). Subsequently, solution B was prepared by dissolving 0.3 mmol of HHTP in 15 ml of DI. Then, a certain volume of 10 mg ml−1 aqueous dispersion of GO was dispensed in DI to obtain a 15 ml solution, which was denoted as solution C. Solutions A, B, and C were mixed homogeneously and ultrasonicated for 30 min; then, the mixture was settled in an autoclave at 85 °C for 12 hours. The resulting suspensions were then centrifuged 3 times with DI and acetone respectively, followed by overnight drying. Finally, the resultant powder samples were denoted as Ni-CAT@GO-0.25, 0.5, 1, and 2 (NCG0.25, NCG0.5, NCG1, and NCG2), according to the volume (ml) of GO aqueous dispersion in solution C. When 15 ml of DI was directly used as solution C without adding any GO dispersion, the sample was labeled as Ni-CAT (NC).
 |
| | Fig. 2 Preparation process of the samples. | |
2.2 Characterization
A scanning electron microscope (SEM, HITACHI, S-4800) was used to determine the surface morphology and microstructure of the samples. Field-emission transmission electron microscopy (TEM, JEM-F200) was employed to investigate the morphology and lattice pattern of the samples. A nitrogen adsorption and desorption apparatus (JW-BK 122 W, JWGB, China) was applied without any further treatment to estimate the specific surface areas and pore size distributions of the samples at 77 K. An X-ray diffraction analyzer (XRD, D8Advance, Bruker AXS, Germany) was used to analyze the crystal structure and estimate the grain size of the samples. The grain size can be derived using the Debye–Scherrer equation given below:| |  | (1) |
where D (nm) is the grain size, K represents the Scherrer constant, λ (nm) is the wavelength of X-ray (Cu Kα), β denotes the full width at half maximum (FWHM) of the peak, and θ is the Bragg angle of the peak.
2.3 Electrochemical measurement
The electrochemical performance of NC and NCG samples was investigated using an electrochemical workstation (CS 1005, Wuhan Corrtest, China). To prepare the electrode materials, NC and NCG samples were mixed respectively with acetylene black and polyvinylidene fluoride (PVDF), in a mass ratio of 8:1:1. To obtain a uniform slurry, a few drops of N-methylpyrrolidone (NMP) were added to the mixture and ground. The mixture was then applied to a carbon sheet and dried overnight at 60 °C in a vacuum oven to obtain active electrodes. Each electrode contained ∼5 mg of the active material. 3 M KOH was used as the electrolyte.
To evaluate the electrochemical performance of the samples, a series of measurements were performed in a three-electrode system at room temperature, with a 1 × 1 cm platinum plate as the counter electrode and a Hg–HgO electrode as the reference electrode. Cyclic voltammetry (CV), galvanostatic charge/discharge (GCD), and electrochemical impedance spectroscopy (EIS) were performed and analyzed. The CV test was performed with a voltage window of 0 to 0.6 V and a scanning rate selected from 5 mV s−1 to 50 mV s−1. For the GCD test, the potential range was set from 0 to 0.5 V54 and the current density was applied in the range from 0.5 A g−1 to 10 A g−1. The EIS test was performed within a frequency range of 0.01 to 100000 Hz and a voltage amplitude of 10 mV. The precise capacitances of the samples were calculated from the GCD curve, according to the following equation:
| | 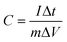 | (2) |
where
I (A) is the electric current, Δ
t (s) represents the duration of the discharging process,
m (g) is the mass of NC or NCGs in the electrode, and Δ
V (V) is the potential range. In addition, the samples underwent 1000 cycles of GCD at a current density of 10 A g
−1 to investigate the cycling stability of the supercapacitor.
To further study the electrochemical properties of the samples, the electrode prepared with NCG0.5 was employed as the positive electrode and paired with a negative electrode prepared using activated carbon (AC); measurements in a two-electrode system were carried out in the same electrolyte as the three-electrode test. The masses of active materials in the positive and negative electrodes were matched using the following equation:
where
m+ (g) and
m− (g) are the masses of NCG0.5 and AC respectively,
C (F g
−1) is the specific capacitance and Δ
V is the potential range. For the CV test, the potential window was set from 0 V to 1.6 V and the scan rate was taken from 5 mV s
−1 to 100 mV s
−1. For the GCD test, the voltage range was also set from 0 V to 1.6 V and the current density was set in a range of 0.5–10 A g
−1. The energy density (W h kg
−1) and power density (W kg
−1) were derived using the following equation:
| | 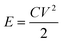 | (4) |
| |  | (5) |
where
C (F g
−1) is the specific capacitance of the system,
V (V) is the voltage window, and
t (s) is the discharging duration.
3. Results and discussion
3.1 Microstructures and morphologies of NC and NCGs
Fig. 3a–e show the microstructure and morphology of NC, NCG0.25, NCG0.5, NCG1, and NCG2 observed by SEM, respectively. The micrographs of NC as shown in Fig. 3a exhibit a typical columnar structure. This is due to the formation of Ni-CAT crystals by stacking 2D sheet layers containing hexagonal holes, which are aligned and stacked along the c-axis to form rod-shaped crystals.55 The morphology of NCG0.25 to NCG2 as shown in Fig. 3b–e displayed a “blanket-like” shape combining 1D Ni-CAT nanorods and 2D GO substrates. Furthermore, the size of nanorods decreases progressively with the incremental addition of GO. This is due to the negatively charged GO surface attracting and adsorbing Ni ions, providing anchor sites for the growth of Ni-CAT rod-shaped crystals.30 As the content of GO increases, the quantity of anchoring sites for the Ni ions also increases. This leads to a depletion of organic ligands before the rod crystals can grow further, which might result in shorter nanorod lengths. The EDS results are presented in Fig. S1.†Fig. 3f presents the TEM image of the NCG0.5 sample. The rod-shaped Ni-CAT crystals exhibit uniform growth on the 2D GO matrix. Furthermore, there is an evident lattice with a spacing of 1.7 nm, corresponding to the (100) crystal plane of the Ni-CAT nanorods.55 The in situ growing process of the NCG samples leads to good contact between Ni-CAT nanorods and GO nanosheets, which contributes to the enhancement of the overall electrical conductivity of the NCG “blanket”.
 |
| | Fig. 3 SEM images of (a) NC, (b) NCG0.25, (c) NCG0.5, (d) NCG1, and (e) NCG2. (f) TEM image of NCG0.5. | |
Fig. 4 presents the X-ray diffraction (XRD) patterns of the samples. The peak located at 13.9° corresponds to the (311) plane of Ni-CAT, and the grain sizes of the five samples are determined based on their FWHM as described in Table 1.44 The main peak of the NC sample exhibits the smallest FWHM, indicating the largest grain size. As the quantity of GO increases, the peaks broaden gradually, indicating a gradual shortening of nanorods. This result is highly consistent with the SEM observation.
 |
| | Fig. 4 XRD patterns of samples. | |
Table 1 Grain sizes of the samples
| Sample |
NC |
NCG0.25 |
NCG0.5 |
NCG1 |
NCG2 |
| FWHM (degree) |
0.374 |
0.412 |
0.429 |
0.438 |
0.489 |
| Grain size (nm) |
21.17 |
19.22 |
18.46 |
18.08 |
16.19 |
Fig. 5a illustrates the nitrogen adsorption/desorption isotherms, and Fig. 5b displays the pore distributions of the samples. The specific surface areas of the samples are 86.28 m2 g−1, 75.93 m2 g−1, 93.40 m2 g−1, 89.38 m2 g−1, and 107.93 m2 g−1, corresponding to NC and NCGs with various GO contents, respectively. Since the samples did not undergo the solvent exchange and vacuum heating process, the majority of the micropores in Ni-CAT are likely to be occupied by solvent molecules, with only a minor portion on the surface being exposed due to the evaporation of solvent molecules. Consequently, the micropores seem to be rare and the isotherms could exhibit a typical IV-type behaviour. Accordingly, all samples contain less micropores and numerous stacking pores formed among the NC nanorods and GO nanosheets. As shown in Fig. 5b, the distribution of mesopores becomes narrower with smaller average pore sizes as the content of GO increases.56 For instance, the size of mesopores in NCG0.25 is primarily distributed between 7 and 15 nm, whereas for NCG2, it dispersed in the range of 4–8 nm. The decrease in the average stacking pore size could be attributed to the smaller size of the Ni-CAT nanorods, which act as spacers between the GO sheets. This narrower pore distribution with a smaller size might influence electrolyte diffusion in the electrode based on NCGs.
 |
| | Fig. 5 (a) Nitrogen adsorption–desorption isotherm curves of samples; (b) pore size distribution of samples. | |
3.2 Electrochemical measurement of NC and NCGs
Fig. 6a–e present the CV curves of samples NC, NCG0.25, NCG0.5, NCG1, and NCG2, respectively. All of these samples exhibit a pair of obvious redox peaks, indicating that the charge storage mechanism is mainly attributed to pseudo-capacitance. The shift in the peak position with increasing scanning rates is due to not only internal resistance, but also the delay in diffusion of ions in the active material.57 By analyzing the correlation between the scanning rate and the peak current in the CV curve, the mechanism that dominates the charge storage in the system can be determined using the following equation:where i (A) is the peak current of the redox peak, and v (mV s−1) is the scan rate. When the b value approaches 1.0, it implies that surface capacitance plays a dominant role in charge storage, and when the value of b is around 0.5, it suggests that ion diffusion is the dominant factor in charge storage. Based on the CV curve, the b values of the samples were calculated with the oxidation peaks (peak 1) and the reduction peaks (peak 2), as shown in the Fig. 6f–j. The data for the calculation are taken with a scanning rate of 5–40 mV s−1. The b values of all samples are approximately 0.5, indicating the dominance of the ion diffusion-controlled mechanism in the energy storage. As shown in Fig. 6f–j, the NC sample exhibits a value of b closer to 0.5, and the b value rises after the GO addition, indicative of an increase in the proportion of surface capacitance mechanism for the NCG composites. To better quantify the contributions of surface-control mechanisms (capacitive) and diffusion-control mechanisms, further analysis of the oxidation peak can be conducted using the following equation:here, k1v and k2v1/2 can be regarded as components corresponding to surface and diffusion controlled contributions, respectively. As shown in Fig. 6k–o, it is evident that the proportion of the capacitive mechanism in the NC sample is exceptionally low, whereas in the NCG samples, the proportion of the surface controlled contribution is significantly higher than that in the NC sample. Particularly, the NCG1 and NCG0.5 samples exhibit a higher proportion of capacitive mechanism compared to the other NCG samples. This result indicates that these two samples might possess superior microstructures, conducive to the accessible surface area for the electrolyte, in comparison with the other samples.
 |
| | Fig. 6 CV curves and the log(i) vs. log(v) plot of (a) NC, (b) NCG0.25, (c) NCG0.5, (d) NCG1 and (e) NCG2; the log(i) vs. log(v) plot of (f) NC, (g) NCG0.25, (h) NCG0.5, (i) NCG1 and (j) NCG2; the contribution ratios of capacitive and diffusion-controlled of (k) NC, (l) NCG0.25, (m) NCG0.5, (n) NCG1 and (o) NCG2 at various scan rates. | |
Fig. 7a–e illustrates the GCD curves of the samples of NC, NCG0.25, NCG0.5, NCG1 and NCG2, respectively. The charging and discharging plateau can be observed, indicating the pseudo-capacitive behavior of the NC and NCG samples, which is consistent with the results of the CV test. The same electrochemical tests were conducted on freeze-dried GO powder samples. As shown in Fig. S2,† GO almost exhibited no capacitive contribution within the potential range of 0–0.5 V; thus, the capacitance of the material was almost entirely contributed by Ni-CAT. Fig. 7f presents the capacitance derived from the GCD data; the capacitance of all samples decreased gradually as the current density increased. The Nyquist plots of all samples shown in Fig. 7g demonstrate comparable small semicircle shapes in the high-frequency range and lines in the low-frequency range. According to the intercept of the Nyquist plot, the equivalent series resistance (Rs) of the samples is obtained, and by the corresponding circuit fitting, the charge transfer resistance (Rct) of the samples is obtained, as shown in Table 2.
 |
| | Fig. 7 GCD curves of (a) NC, (b) NCG0.25, (c) NCG0.5, (d) NCG1 and (e) NCG2 at different scan rates; (f) rate performance of all samples. (g) Nyquist plots and (h) cycling stability at 10 A g−1. | |
Table 2 Resistance of the samples
| Sample |
NC |
NCG0.25 |
NCG0.5 |
NCG1 |
NCG2 |
|
R
s (Ω) |
1.25 |
1.02 |
0.89 |
0.81 |
0.77 |
|
R
ct (Ω) |
13.53 |
9.03 |
5.30 |
7.93 |
9.55 |
As shown in Fig. 7f, it is evident that NC shows the lowest capacitance, possibly due to its oversized crystals and long 1D pores, which significantly restrict the surface areas available for electrolyte ions to penetrate. In cases of slightly rapid charge–discharge, it's believed that Faraday's reaction in the NC sample mainly takes place on the surface of the Ni-CAT crystals. Generally, as the degree of oxidation increases, GO may exhibit conductivity varying from semiconducting to insulating. Since both Ni2+ and HHTP ligands demonstrate considerable reducing properties under the hydrothermal conditions of the synthesis process of NCGs, it is likely that the oxidation degree of GO is decreased, resulting in relatively good conductivity. From Table 2, it's rational that the Rs value decreases gradually with increasing GO content because of the good conductivity of GO. The samples were compressed to tablets at 5 MPa, followed by a four-probe test method to evaluate their conductivity. The results shown in Fig. S3† demonstrate a significant enhancement in conductivity with the increase of GO, agreeing with the results of Rs. Although this conductivity might be influenced by the presence of numerous particle interfaces, the enhancement is likely due to the inherently higher conductivity of the additive GO. The Rct of NC is 13.53 Ω, and it decreases to 5.30 Ω for NCG0.5, while it increases to 9.55 Ω for NCG2 with more GO content. On the one hand, the introduction of GO leads to a reduction in the size of Ni-CAT crystals, allowing more 1D micropores to be exposed and to participate in the Faraday reaction, thereby reducing the Rct of the electrode material, as illustrated in the Nyquist plot shown in Fig. 7g. On the other hand, excessive addition of GO causes the stacks of NCG “blankets” to become denser and the gaps between the GO layers to become too narrow, which increases Rct and reduces the accessible surface area for the electrolyte. Hence, under the total effect of the Ni-CAT nanorods and the GO layer spacing, the Rct of the five samples shows an initial increase and then a decline as the amount of GO added increases.58
According to Fig. 7f, the highest capacitance of 563.8 F g−1 is achieved by NCG0.5 at 0.5 A g−1. The results reveal that elevating the quantity of GO above NCG0.5 leads to a significant reduction in the capacitance at a current density of 0.5 A g−1. The decrease in capacitance is mainly due to the smaller average pore size of the stacking pores of GO within the sample, which leads to a lower accessible surface area. Additionally, a higher proportion of GO may lead to a decrease in specific capacitance since the Ni element dominates the main charge storage. Noteworthily, the rate performance of the samples improved gradually with the addition of GO. For instance, the capacitance of NCG2 only decreased to 71.7% for 0.5 A g−1 at a current density of 10 A g−1, which was significantly better than that for NC (which was 35.3%). The addition of GO effectively improves the capacitance and rate performance of the NCG sample in comparison with pristine Ni-CAT.
Fig. 7h displays the cycling performance of the NC and NCG samples. The cycling stability of the samples improves with increasing GO content. After 1000 cycles, the capacitance of NC and NCGs remains 50.3%, 60.6%, 68.7%, 71.3%, and 82.4% of their maximum values, respectively. The decrease in the size of Ni-CAT crystals may lead to a reduction in deformation during charging and discharging. As we known, the stability of Ni-CAT crystals is very limited in strong alkaline environments.44,59 It is interesting to find that the detachment of the active material is repressed because of its self-assembly on GO surfaces, leading to an improvement in cycling stability.60
The electrochemical performance of the samples was further tested using a two-electrode system. An asymmetric supercapacitor was constructed, with NCG0.5 as the positive electrode material due to its superior performance in the three-electrode system. AC was utilized as the negative electrode material. The electrochemical performance of AC is shown in Fig. S4.†Fig. 8a–c show the electrochemical performance of the NCG0.5//AC supercapacitor. The CV curve of the supercapacitor shows an irregular shape with several redox peaks. The specific capacitance of the supercapacitor obtained from the GCD test can reach 66.8 F g−1 at a current density of 0.5 A g−1, which corresponds to an energy density of 23.76 W h kg−1 at a power density of 399.95 W kg−1. When the current density reaches 10 A g−1, the capacitance decreases to 32.0 F g−1, corresponding to an energy density of 11.38 W h kg−1 at a power density of 7984.09 W kg−1. The Ragone plot of NCG0.5//AC is shown in Fig. S5,† which demonstrates that the supercapacitor based on NCG0.5 possesses a better performance in comparison with those in the literature. Generally, the specific capacitance of an ideal symmetrical two-electrode system should be 1/4 that of a capacitor in three electrode system. Since the negative electrode material was AC, which exhibited a specific capacitance of approximately 150 F g−1, this resulted in the negative electrode material constituting a significant portion of the total mass. Hence, this result is reasonable.
 |
| | Fig. 8 Electrochemical performances of the capacitor: (a) CV curves, (b) GCD curves, (c) rate performance and (d) cycling stability at 5 A g−1. | |
To test the cycling performance of supercapacitors, a GCD test of 1000 cycles was performed at 5 A g−1. A significant decrease in capacitance was observed in the first 100 cycles; however, the specific capacitance increased as cycling continued, which is possibly due to the activation of the carbon negative electrodes by the electrolyte during the charge and discharge processes. After 1000 cycles, the capacitance remained at approximately 92.0% of the initial capacitance, which is a fairly good cycling performance of the supercapacitor. The result suggests that NCG0.5 can be a good reference for similar electrode materials of supercapacitors.
4. Conclusions
In summary, conductive MOF Ni-CAT was successfully in situ grown on GO substrates. SEM observation revealed that NCG samples exhibit a blanket-shaped structure composed of Ni-CAT nanorod crystals and a 2D GO substrate. The grain size of Ni-CAT was observed to decrease gradually with increasing GO content, which was further confirmed by XRD analysis. These shortened Ni-CAT crystals enabled the exposure of 1D pores. The nitrogen adsorption and desorption results indicated that the average size of the mesopores in the NC and NCG samples gradually decreased with increasing addition of GO. This suggests that the reduction in the size of the nanorods reduces the size of the stacking pores between the Ni-CAT nanorods and GO layers. The electrochemical results indicate that the NCG0.5 sample possesses a high capacitance value (563.8 F g−1 at 0.5 A g−1) and a low charge transfer resistance, which can be attributed to the optimal micromorphology of the in situ grown Ni-CAT@GO “blankets”. When the GO content is lower than that in NCG0.5, the elongated 1D pores in the Ni-CAT nanorods prevent the electrolyte ions from adequately penetrating inside. While the GO content is higher than that in NCG0.5, the smaller stacking pores also hinder the diffusion of electrolyte ions. Due to the above synergistic effect, the NCG0.5 sample with optimal micromorphology achieved a highly accessible surface area for the electrolyte. Additionally, as the quantity of GO increases, the equivalent series resistance of the NCG samples gradually decreases, which leads to an improvement in rate performance and cycling stability. NCG0.5 exhibits a slightly lower rate performance and cycling stability (61.9% at 10 A g−1 and 68.7% after 1000 cycles) in comparison with NCG2, but remains in a quite acceptable level. Furthermore, the asymmetric supercapacitor NCG0.5//AC exhibits a high energy density of 23.76 W h kg−1 at a power density of 399.95 W kg−1. The electrochemical properties of the NCGs are considerably enhanced by a straightforward combination strategy and morphology control, which may provide guidance for future research on the application of conductive MOFs in supercapacitor electrode materials.
Author contributions
Haoliang Zhang: writing – original draft, methodology, investigation, visualization, and formal analysis. Lan Yang: visualization and formal analysis. Xu Li: methodology and formal analysis. Yunjie Ping: investigation and formal analysis. Jinzhao Han: methodology and investigation. Si Chen: methodology and investigation. Chunqing He: conceptualization, resources, writing – review & editing, supervision, project administration, and funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 12375288, 12075172 and 12205089) and the National Key R&D Program of China (Grant No. 2019YFA0210003). We thank the Core Facility of Wuhan University for supporting the characterization and analysis of the materials.
References
- P. H. Yang and W. J. Mai, Nano Energy, 2014, 8, 274–290 CrossRef CAS.
- J. Yan, Q. Wang, T. Wei and Z. J. Fan, Adv. Energy Mater., 2014, 4, 43 Search PubMed.
- Y. Huang, M. S. Zhu, Y. Huang, Z. X. Pei, H. F. Li, Z. F. Wang, Q. Xue and C. Y. Zhi, Adv. Mater., 2016, 28, 8344–8364 CrossRef CAS PubMed.
- H. Wang, Y. Yang and L. Guo, Adv. Energy Mater., 2017, 7, 18 Search PubMed.
- W. Raza, F. Z. Ali, N. Raza, Y. W. Luo, K. H. Kim, J. H. Yang, S. Kumar, A. Mehmood and E. E. Kwon, Nano Energy, 2018, 52, 441–473 CrossRef CAS.
- J. Libich, J. Máca, J. Vondrák, O. Cech and M. Sedlaríková, J. Energy Storage, 2018, 17, 224–227 CrossRef.
- J. Z. Han, Y. J. Ping, J. J. Li, Z. Liu, B. Y. Xiong, P. F. Fang and C. Q. He, Diamond Relat. Mater., 2019, 96, 176–181 CrossRef CAS.
- M. Inagaki, H. Konno and O. Tanaike, J. Power Sources, 2010, 195, 7880–7903 CrossRef CAS.
- J. Z. Han, Y. J. Ping, S. J. Yang, Y. M. Zhang, L. B. Qian, J. J. Li, L. Liu, B. Y. Xiong, P. F. Fang and C. Q. He, Diamond Relat. Mater., 2020, 109, 9 CrossRef.
- M. R. Lukatskaya, S. Kota, Z. F. Lin, M. Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P. L. Taberna, M. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 2, 6 Search PubMed.
- T. Nguyen and M. D. Montemor, Adv. Sci., 2019, 6, 41 Search PubMed.
- S. Y. Zhou, S. Wang, S. J. Zhou, H. B. Xu, J. P. Zhao, J. Wang and Y. Li, Nanoscale, 2020, 12, 8934–8941 RSC.
- S. Q. Zhu, J. C. Shu and M. S. Cao, Nanoscale, 2022, 14, 7322–7331 RSC.
- Y. J. Ping, Z. Liu, J. J. Li, J. Z. Han, Y. P. Yang, B. Y. Xiong, P. F. Fang and C. Q. He, J. Alloys Compd., 2019, 805, 822–830 CrossRef CAS.
- S. J. Yang, L. B. Qian, Y. J. Ping, H. L. Zhang, J. J. Li, B. Y. Xiong, P. F. Fang and C. Q. He, Ceram. Int., 2021, 47, 8290–8299 CrossRef CAS.
- S. J. Yang, Y. J. Ping, L. B. Qian, J. Z. Han, B. Y. Xiong, J. J. Li, P. F. Fang and C. Q. He, J. Mater. Sci.: Mater. Electron., 2020, 31, 2221–2230 CrossRef CAS.
- P. Wen, P. W. Gong, J. F. Sun, J. Q. Wang and S. R. Yang, J. Mater. Chem. A, 2015, 3, 13874–13883 RSC.
- D. D. Li, M. Kassymova, X. C. Cai, S. Q. Zang and H. L. Jiang, Coord. Chem. Rev., 2020, 412, 16 CrossRef.
- Y. W. Na, J. Y. Cheon, J. H. Kim, Y. Jung, K. Lee, J. S. Park, J. Y. Park, K. S. Song, S. B. Lee, T. Kim and S. J. Yang, Sci. Adv., 2022, 8, 10 Search PubMed.
- Y. Z. Liu, N. Xu, W. C. Chen, X. L. Wang, C. Y. Sun and Z. M. Su, Dalton Trans., 2018, 47, 13472–13478 RSC.
- L. Wang, H. Z. Liu, J. G. Zhao, X. J. Zhang, C. Z. Zhang, G. H. Zhang, Q. H. Liu and H. G. Duan, Chem. Eng. J., 2020, 382, 10 Search PubMed.
- C. Qu, Y. Jiao, B. T. Zhao, D. C. Chen, R. Q. Zou, K. S. Walton and M. L. Liu, Nano Energy, 2016, 26, 66–73 CrossRef CAS.
- J. Xu, C. Yang, Y. F. Xue, C. Wang, J. Y. Cao and Z. D. Chen, Electrochim. Acta, 2016, 211, 595–602 CrossRef CAS.
- M. A. Khayum, V. Vijayakumar, S. Karak, S. Kandambeth, M. Bhadra, K. Suresh, N. Acharambath, S. Kurungot and R. Banerjee, ACS Appl. Mater. Interfaces, 2018, 10, 28139–28146 CrossRef PubMed.
- L. Li, F. Lu, H. Guo and W. Yang, Microporous Mesoporous Mater., 2021, 312, 10 Search PubMed.
- F. T. Ran, X. Q. Xu, D. Pan, Y. Y. Liu, Y. P. Bai and L. Shao, Nano-Micro Lett., 2020, 12, 13 CrossRef PubMed.
- Q. V. Thi, S. A. Patil, P. K. Katkar, I. Rabani, A. S. Patil, J. Ryu, G. Kolekar, N. T. Tung and D. Sohn, Synth. Met., 2022, 290, 11 CrossRef.
- S. H. Zheng, Z. S. Wu, S. Wang, H. Xiao, F. Zhou, C. L. Sun, X. H. Bao and H. M. Cheng, Energy Storage Mater., 2017, 6, 70–97 CrossRef.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- I. Ibrahim, S. Zheng, C. Y. Foo, N. M. Huang and H. N. Lim, J. Energy Storage, 2021, 43, 11 Search PubMed.
- L. R. Kong and W. Chen, Adv. Mater., 2014, 26, 1025–1043 CrossRef CAS PubMed.
- O. M. Yaghi, G. M. Li and H. L. Li, Nature, 1995, 378, 703–706 CrossRef CAS.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- X. Li, D. Zhang, S. Chen, Y. Geng, Y. Liu, L. Qian, X. Chen, J. Li, P. Fang and C. He, Green Energy Environ., 2023 DOI:10.1016/j.gee.2023.10.007.
- S. S. Xiong, S. Y. Jiang, J. Wang, H. J. Lin, M. X. Lin, S. T. Weng, S. Liu, Y. Jiao, Y. C. Xu and J. R. Chen, Electrochim. Acta, 2020, 340, 10 CrossRef.
- Q. L. Chen, S. J. Lei, P. Q. Deng, X. L. Ou, L. F. Chen, W. Wang, Y. H. Xiao and B. C. Cheng, J. Mater. Chem. A, 2017, 5, 19323–19332 RSC.
- X. G. Han, L. M. Sun, F. Wang and D. Sun, J. Mater. Chem. A, 2018, 6, 18891–18897 RSC.
- Y. J. Ping, S. J. Yang, J. Z. Han, X. Li, H. L. Zhang, B. Y. Xiong, P. F. Fang and C. Q. He, Electrochim. Acta, 2021, 380, 10 CrossRef.
- W. H. Li, W. H. Deng, G. E. Wang and G. Xu, EnergyChem, 2020, 2, 63 Search PubMed.
- D. K. Nguyen, I. M. Schepisi and F. Z. Amir, Chem. Eng. J., 2019, 378, 8 CrossRef.
- T. Deng, X. Y. Shi, W. Zhang, Z. Z. Wang and W. T. Zheng, iScience, 2020, 23, 25 Search PubMed.
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- A. Mähringer, M. Hennemann, T. Clark, T. Bein and D. D. Medina, Angew. Chem., Int. Ed., 2021, 60, 5519–5526 CrossRef PubMed.
- Y. L. Li, J. J. Zhou, M. K. Wu, C. Chen, K. Tao, F. Y. Yi and L. Han, Inorg. Chem., 2018, 57, 6202–6205 CrossRef CAS PubMed.
- M. K. Smith, K. E. Jensen, P. A. Pivak and K. A. Mirica, Chem. Mater., 2016, 28, 5264–5268 CrossRef CAS.
- M. K. Smith and K. A. Mirica, J. Am. Chem. Soc., 2017, 139, 16759–16767 CrossRef CAS PubMed.
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao and F. Zhou, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS.
- S. Chen, H. L. Zhang, X. Li, Y. Liu, M. Y. Zhang, X. Y. Gao, X. Chang, X. J. Pu and C. Q. He, Dalton Trans., 2023, 52, 4826–4834 RSC.
- S. H. Zhao, H. H. Wu, Y. L. Li, Q. Li, J. J. Zhou, X. B. Yu, H. M. Chen, K. Tao and L. Han, Inorg. Chem. Front., 2019, 6, 1824–1830 RSC.
- I. Ibrahim, S. Zheng, C. Foo, N. Huang and H. Lim, J. Energy Storage, 2021, 43, 103304 CrossRef.
- M. S. Rahmanifar, H. Hesari, A. Noori, M. Y. Masoomi, A. Morsali and M. F. Mousavi, Electrochim. Acta, 2018, 275, 76–86 CrossRef CAS.
- M. Muniyalakshmi, K. Sethuraman and D. Silambarasan, Mater. Today: Proc., 2020, 21, 408–410 CAS.
- X. H. Liu, W. L. Hu, W. J. Jiang, Y. W. Yang, S. Niu, B. Sun, J. Wu and J. S. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 28473–28477 CrossRef CAS PubMed.
- Y. Yan, P. Gu, S. S. Zheng, M. B. Zheng, H. Pang and H. G. Xue, J. Mater. Chem. A, 2016, 4, 19078–19085 RSC.
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. F. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS.
- Y. H. Cheng, S. B. Zhou, P. Hu, G. D. Zhao, Y. X. Li, X. H. Zhang and W. B. Han, Sci. Rep., 2017, 7, 11 CrossRef PubMed.
- J. Yan, Z. J. Fan, W. Sun, G. Q. Ning, T. Wei, Q. Zhang, R. F. Zhang, L. J. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- P. C. Du, Y. N. Dong, C. Liu, W. L. Wei, D. Liu and P. Liu, J. Colloid Interface Sci., 2018, 518, 57–68 CrossRef CAS PubMed.
- Y. Li, Y. X. Xu, Y. Liu and H. Pang, Small, 2019, 15, 8 Search PubMed.
- Y. J. Zhou, Z. M. Mao, W. Wang, Z. K. Yang and X. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 28904–28916 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*











