
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studies on the chemical reduction of polynuclear titanium(IV) nitrido complexes†
Jorge
Caballo
,
Adrián
Calvo-Molina
 ,
Sergio
Claramonte
,
Maider
Greño
,
Adrián
Pérez-Redondo
and
Carlos
Yélamos
*
,
Sergio
Claramonte
,
Maider
Greño
,
Adrián
Pérez-Redondo
and
Carlos
Yélamos
*
Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química “Andrés M. del Río” (IQAR), Universidad de Alcalá, 28805 Alcalá de Henares-Madrid, Spain. E-mail: carlos.yelamos@uah.es
First published on 8th February 2024
Abstract
The redox chemistry of cube-type titanium(IV) nitrido complexes [{Ti4(η5-C5Me5)3(R)}(μ3-N)4] (R = η5-C5Me5 (1), N(SiMe3)2 (2), η5-C5H4SiMe3 (3), and η5-C5H5 (4)) was investigated by electrochemical methods and chemical reactions. Cyclic voltammetry studies indicate that 1–4 undergo a reversible one-electron reduction at ca. −1.8 V vs. ferrocenium/ferrocene. Thus, complex 1 reacts with sodium sand in tetrahydrofuran to produce the highly reactive ionic compound [Na(thf)6][{Ti(η5-C5Me5)}4(μ3-N)4] (5). The treatment of complexes 1–4 in toluene with one equivalent of [K(C5Me5)] in the presence of macrocycles (L) leads to C10Me10 and the formation of more stable derivatives [K(L)][{Ti4(η5-C5Me5)3(R)}(μ3-N)4] (R = η5-C5Me5, L = 18-crown-6 (6), crypt-222 (7); R = N(SiMe3)2, L = 18-crown-6 (8), crypt-222 (9); R = η5-C5H4SiMe3, L = 18-crown-6 (10), crypt-222 (11); R = η5-C5H5, L = crypt-222 (12)). However, the analogous reaction of 4 with [K(C5Me5)] and 18-crown-6 affords [{(18-crown-6)K}2(μ–η5:η5-C5H5)][{Ti4(η5-C5Me5)3(η5-C5H5)}(μ3-N)4] (13) via abstraction of one cyclopentadienide group from a putative intermediate [(18-crown-6)K(μ–η5:η5-C5H5)Ti4(η5-C5Me5)3(μ3-N)4]. In contrast to the cube-type nitrido systems 1–4, the cyclic voltammogram of the trinuclear imido-nitrido titanium(IV) complex [{Ti(η5-C5Me5)(μ-NH)}3(μ3-N)] (14) does not reveal any reversible redox event and 14 readily reacts with [K(C5Me5)] to afford C5Me5H and the diamagnetic derivative [{K(μ4-N)(μ3-NH)2Ti3(η5-C5Me5)3(μ3-N)}2] (15). The treatment of 15 with two equiv. of 18-crown-6 polyethers produces the molecular species [(L)K{(μ3-N)(μ3-NH)2Ti3(η5-C5Me5)3(μ3-N)}] (L = 18-crown-6 (16), dibenzo-18-crown-6 (17)). Complex 17 further reacts with one equiv. of dibenzo-18-crown-6 to yield the ion-separated compound [K(dibenzo-18-crown-6)2][Ti3(η5-C5Me5)3(μ3-N)(μ-N)(μ-NH)2] (18) similar to the ion pair [K(crypt-222)][Ti3(η5-C5Me5)3(μ3-N)(μ-N)(μ-NH)2] (19) obtained in the treatment of 15 with cryptand-222.
Introduction
Polynuclear transition-metal nitrido complexes are an important type of molecule with a wide structural diversity and singular bonding situations.1 While the nitrido ligand usually behaves as a terminal functionality (M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) in the complexes of mid transition metals (groups 6–8) in high oxidation states,2 early transition metal (groups 4 and 5) species have a strong tendency to form dinuclear or polynuclear aggregates with nitrido moieties (μn-N) shared between two or more metal centers. In addition, the bridging nitrido ligand in these polynuclear species is capable of displaying diverse coordination modes in several structural variations.1c Well-defined multimetallic molecular compounds with μn-nitrido groups can shed light on the bridging modes of the chemisorbed nitrogen atoms on the metal surfaces of heterogeneous catalytic systems such as the Haber–Bosch process for the synthesis of ammonia.3
N) in the complexes of mid transition metals (groups 6–8) in high oxidation states,2 early transition metal (groups 4 and 5) species have a strong tendency to form dinuclear or polynuclear aggregates with nitrido moieties (μn-N) shared between two or more metal centers. In addition, the bridging nitrido ligand in these polynuclear species is capable of displaying diverse coordination modes in several structural variations.1c Well-defined multimetallic molecular compounds with μn-nitrido groups can shed light on the bridging modes of the chemisorbed nitrogen atoms on the metal surfaces of heterogeneous catalytic systems such as the Haber–Bosch process for the synthesis of ammonia.3
One of the most common synthetic routes to early transition metal polynuclear nitrido derivatives is the treatment of high-valent organometallic complexes with ammonia.4–8 The resultant multimetallic species usually contain nitrido (μn-N), imido (μn-NH), or amido (μn-NH2) ligands bridging several metal centers. The presence of bulky ancillary ligands (e.g., cyclopentadienyl, alkoxido) strongly bonded to the metals is crucial to isolating discrete and soluble molecular metal nitrides. These compounds contain metal elements in their highest possible oxidation state held together by strong interactions with the bridging ligands. Several theoretical studies on these complexes indicate that the lowest-lying unoccupied orbitals are metal-based and the compounds are able to undergo reduction without sacrificing cluster bonding.8a,9–12 For instance, density functional theory (DFT) calculations on the model titanium(IV) cubane complex [{Ti(η5-C5H5)}4(μ3-N)4] show that the LUMOs are bonding combinations of metal d orbitals and the compound has a high value (2.14 eV) of adiabatic electron affinity.10 Calculations for the isostructural vanadium(IV) model compound [{V(η5-C5H5)}4(μ3-N)4], analogous to [{V(η5-C5Me5)}4(μ3-N)4] isolated by Bottomley and co-workers in the reduction of [{V(η5-C5Me5)Cl}2(μ-N)2] with sodium,13 indicate that the four metal electrons occupy molecular orbitals which show a certain vanadium–vanadium overlap.10
Interestingly, many di- and polynuclear complexes with bridging nitrido ligands have been prepared by dinitrogen fixation via splitting of the triple bond by low-valent species.14 In particular, Hou and co-workers have described the formation of tri- and tetranuclear titanium compounds with μ3-N nitrido and μn-NH imido bridging ligands by cleavage of dinitrogen on multimetallic polyhydride complexes bearing the η5-C5Me4SiMe3 supporting ligand.15 The hydride ligands in these complexes serve as electron and proton sources for N2 splitting and partial hydrogenation of the resultant nitrido units with no need for external reductants or proton sources.16 In a related work, we reported the isolation of a paramagnetic methylidene–methylidyne–nitrido cube-type complex [{Ti(η5-C5Me5)}4(μ3-CH)(μ3-CH2)(μ3-N)2] by the reaction of [Ti(η5-C5Me5)Me3] with forming gas (H2/N2 mixture) under ambient conditions.17 These monocyclopentadienyltitanium nitrido species derived from N2 contain titanium(IV) or titanium(III) centers and their structures resemble those of the trinuclear imido-nitrido [{Ti(η5-C5Me5)(μ-NH)}3(μ3-N)]4 and the tetranuclear nitrido [{Ti(η5-C5Me5)}4(μ3-N)4]5 complexes prepared previously by the reaction of [Ti(η5-C5Me5)R3] (R = Me, NMe2) with ammonia.
Over two decades, we have been investigating the rational synthesis of a family of heterometallic nitrido complexes by incorporation of diverse metal complex fragments into the incomplete cube-type structure of [{Ti(η5-C5Me5)(μ-NH)}3(μ3-N)].1c Remarkably, complexes with cube-type [MTi3N4] cores where M is an electron-rich metal center (e.g., Mo0, IrI) are stabilized by sharing of the electrons of the metal with the Ti3 system.11,18 Within an extensive study of the reactivity of these polynuclear systems, a few examples of reduced heterometallic species have been unexpectedly isolated. For instance, the formation of cube-type species [(RCC)Zn{(μ3-NH)3Ti3(η5-C5Me5)3(μ3-NCCR)}] is accompanied by a two-electron reduction of the Ti3 core according to DFT calculations.19 Later, we have described one-electron reduced species by the reaction of trichloride complexes [Cl3Y{(μ3-NH)3Ti3(η5-C5Me5)3(μ3-N)}] and [Cl3Zr{(μ3-N)(μ3-NH)2Ti3(η5-C5Me5)3(μ3-N)}] with potassium pentamethylcyclopentadienide [K(C5Me5)].20,21 Theoretical calculations on the electronic structure of yttrium–titanium complexes indicated that the additional electron is delocalized among the three titanium atoms.20
Herein we report a systematic study on the electrochemical and chemical reduction of cube-type titanium(IV) nitrido complexes [{Ti4(η5-C5Me5)3(R)}(μ3-N)4] (R = η5-C5Me5 (1), N(SiMe3)2 (2), η5-C5H4SiMe3 (3), and η5-C5H5 (4)). The reduced species [K(18-crown-6)][{Ti(η5-C5Me5)}4(μ3-N)4] (6) derived from 1 was previously isolated as an intermediate in the preparation of the paramagnetic imido–nitrido derivative [{Ti(η5-C5Me5)}4(μ3-N)3(μ3-NH)].22 In addition, we have evaluated the possibility of reducing the trinuclear imido–nitrido titanium(IV) complex [{Ti(η5-C5Me5)(μ-NH)}3(μ3-N)] (14).
Results and discussion
One-electron reduction of cube-type nitrido complexes [{Ti4(η5-C5Me5)3(R)}(μ3-N)4]
The redox chemistry of the cube-type titanium nitrido complexes was initially investigated by electrochemical methods. Cyclic voltammograms of complexes [{Ti4(η5-C5Me5)3(R)}(μ3-N)4] (R = η5-C5Me5 (1), η5-C5H4SiMe3 (3), and η5-C5H5 (4)) were obtained under an argon atmosphere in tetrahydrofuran solutions at 22 °C (see Fig. 1 and S1† for 1, and Fig. S2 and S3† for 3 and 4 in the ESI,† respectively). The cyclic voltammograms revealed a reversible one-electron redox event at E1/2 = −1.84 V for 1, at E1/2 = −1.80 V for 3, and at E1/2 = −1.86 V for 4vs. ferrocenium/ferrocene (Fc+/Fc). No redox waves due to other chemically reversible redox events were observed even at a very low potential of about −3.50 V, where reductive decomposition of tetrahydrofuran began in our setup. For comparison, dinuclear imido titanium(IV) complexes [{Ti(η5-C5H5)X}2(μ-NR)2] (X = Cl, Me; R = tBu, Ph, 3,5-(CF3)2C6H3) exhibited reversible one electron redox waves between −0.72 and −1.42 V vs. Fc+/Fc with no further reduction observable.23 Likewise, the monopentamethylcyclopentadienyltitanium(IV) analogue [{Ti(η5-C5Me5)3Cl}2{μ-NC6H3(CF3)2}2] displayed one reversible redox process at E1/2 = −1.07 V and its reaction with the mild reducing agent [Co(η5-C5H5)2] (E ≈ −1.3 V)24 afforded the mixed-valence Ti(III)–Ti(IV) species [Co(η5-C5H5)2][{Ti(η5-C5Me5)Cl}2{μ-NC6H3(CF3)2}2].23b Similarly, the cyclic voltammogram of the dinuclear tungsten(VI) nitrido complex [{W(CH2tBu)(OAr)2}2(μ-N)2] (Ar = 2,6-iPr2C6H3) revealed a reversible redox event at E1/2 = −1.08 V and chemical reduction of this complex was accomplished with [Co(η5-C5H5)2].25 Furthermore, the trinuclear tantalum(V) nitrido compound [{Ta(η5-C5Me5)Me}3(μ-N)3] showed a reversible reduction wave at E ≈ −2.0 V and was reduced with a Na/K alloy in diethyl ether to yield [K(OEt2)n][{Ta(η5-C5Me5)Me}3(μ-N)3].8a Lastly, the tetranuclear nitrido vanadium(IV) complex [{V(η5-C5Me5)}4(μ3-N)4] structurally analogous to 1 can be reduced electrochemically with a reversible reduction wave at +0.16 V, although no chemical reduction of the complex was reported.13 | ||
| Fig. 1 Cyclic voltammogram of [{Ti(η5-C5Me5)}4(μ3-N)4] (1) in thf/0.1 M [N(nBu)4][PF6] versus Fc+/Fc at 50 mV s−1 scan rate at 22 °C. | ||
In agreement with the electrochemical studies, compounds 1–4 did not react with [Co(η5-C5H5)2] and stronger reductants such as sodium (E ≈ −3.04 V)24 and [K(C5Me5)] were required to obtain one-electron reduced species. The treatment of complex [{Ti(η5-C5Me5)}4(μ3-N)4] (1) with an excess of fine sodium sand (10 equiv.) in tetrahydrofuran at ambient temperature led to a dark purple solution (Scheme 1). Slow diffusion of toluene into this solution gave dark purple crystals of [Na(thf)6][{Ti(η5-C5Me5)}4(μ3-N)4] (5) in 66% yield. Crystals of 5 exhibited good solubility in tetrahydrofuran-d8 but were insoluble in benzene-d6 and pyridine-d5 solvents, in which the release of tetrahydrofuran molecules took place as observed in the 1H NMR spectra. In addition, in the solid state and under vacuum at room temperature, complex 5 lost all tetrahydrofuran molecules to give a dark solid that can be formulated as Na[{Ti(η5-C5Me5)}4(μ3-N)4] according to elemental analyses and the absence of absorptions due to coordinated tetrahydrofuran ligands (1052 and 901 cm−1) in the IR spectrum (KBr). This solid readily dissolves in tetrahydrofuran-d8 to give a dark purple solution, and the 1H NMR spectrum shows a single broad resonance at δ = 9.2 (Δν1/2 = 67 Hz) similar to that observed in a solution of crystals of 5 in the same solvent. These NMR data are consistent with a Td symmetry for the [{Ti(η5-C5Me5)}4(μ3-N)4]− anion in solution. The magnetic moment measurement by the Evans method for solutions of compound 5 in tetrahydrofuran-d8 gave a μeff value of 1.68μB owing to an unpaired electron in the complex.
 | ||
| Scheme 1 Reactions of 1 with Na and [K(C5Me5)]. | ||
The crystal structure of 5 consists of well-separated [Na(thf)6]+ and [{Ti(η5-C5Me5)}4(μ3-N)4]− ions (Fig. S6†). The anionic fragment shows an almost perfect [Ti4N4] cube core with the Ti–N–Ti and N–Ti–N angles very close to 90° (Fig. 2). The average Ti–N (1.952(4) Å) and Ti–Ti (2.793(4) Å) separation distances in 5 are only slightly longer than those found in the molecular structure of complex 1 (1.938(7) and 2.783(2) Å, respectively).5 Most likely, the additional electron is delocalized among the titanium atoms in a fashion similar to those determined by DFT calculations in the electronic structures of heterometallic cube-type complexes [(RCC)Zn{(μ3-NH)3Ti3(η5-C5Me5)3(μ3-NCCR)}]19 and [Cl3Y{(μ3-NH)3Ti3(η5-C5Me5)3(μ3-N)}]−.20
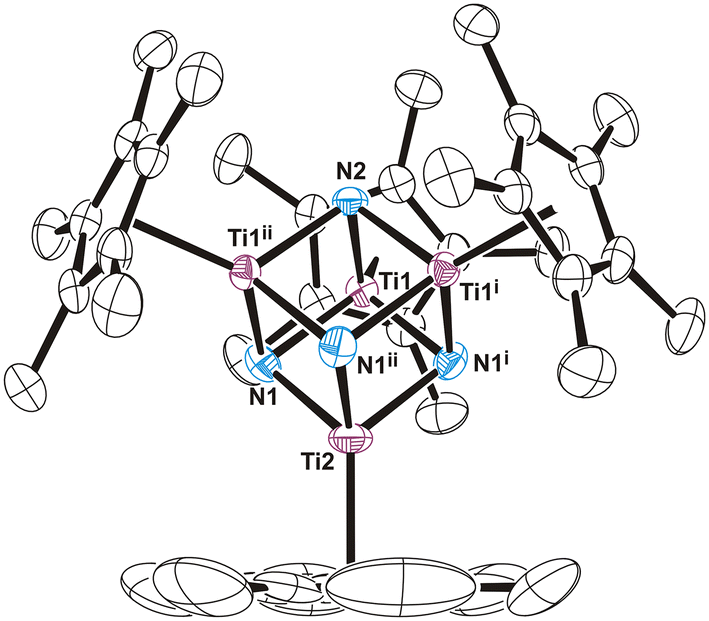 | ||
| Fig. 2 Perspective view of the anion of compound 5 (thermal ellipsoids at the 50% probability level). Hydrogen atoms of the η5-C5Me5 ligands are omitted for clarity. Selected average lengths (Å) and angles (°): Ti–N 1.952(4), Ti–Ti 2.793(4), Ti–N–Ti 91.3(2), N–Ti–N 88.6(2). Symmetry code: (i) z, x, y; (ii) y, z, x. | ||
The nitrido salt 5 was found to be extremely sensitive to H-atom sources such as moisture and organic solvents, precluding the isolation of the compound in its pure form according to elemental analysis. Furthermore, minor resonance signals for other paramagnetic and diamagnetic species in solution were always detected in the 1H NMR spectra of 5. For instance, broad resonances attributable to the paramagnetic imido–nitrido derivative [{Ti(η5-C5Me5)}4(μ3-N)3(μ3-NH)]22 were typically observed in the 1H NMR spectra and a weak νNH vibration at 3333 cm−1 assignable to that compound was identified in the IR spectra. The impurities in the spectra were reduced when the synthesis of 5 was performed in tetrahydrofuran-d8, suggesting that one of the sources of the H atoms is the solvent.
To obtain more stable reduced species from 1, we turned our attention to potassium pentamethylcyclopentadienide which we had previously used as a one-electron reducing reagent.20–22 While [M(C5Me5)] (M = Li, K) compounds are rare reducing agents in organometallic chemistry,24 several examples of metal reduction have been reported in attempts to prepare pentamethylcyclopentadienyl metal complexes.26 The one-electron oxidation of the C5Me5− anion involves the generation of (C5Me5)˙ radicals which are known to dimerize rapidly and irreversibly. The organic (C5Me5)2 byproduct is a liquid at room temperature, and it can be easily separated from the poorly soluble reduced inorganic species in hydrocarbon solvents.
The treatment of 1 with one equivalent of [K(C5Me5)] in toluene in the presence of one equivalent of 18-crown-6 or cryptand-222 led to the precipitation of the ionic derivatives [K(L)][{Ti(η5-C5Me5)}4(μ3-N)4] (L = 18-crown-6 (6),22 crypt-222 (7)) and a solution of C10Me10 (Scheme 1).27 While the reaction was observed at room temperature, the syntheses of complexes 6 and 7 were carried out at 110 °C to ensure complete consumption of 1, which is nearly insoluble in toluene. It is noteworthy that the presence of the macrocyclic ligand for stabilizing K+ is essential because treatments in toluene or tetrahydrofuran attempted without these polyethers showed no reaction and the starting materials were recovered unaltered. This could be related both to the low solubility of potassium pentamethylcyclopentadienide in those solvents and to the presence of aggregates in solution which are hard to oxidize or solvate.28 Most likely, the macrocycles break up these oligomers in solution to give more soluble well-separated C5Me5− ions with higher reduction potential.
In a similar fashion, treatment of cube-type nitrido complexes [{Ti4(η5-C5Me5)3(R)}(μ3-N)4] (R = N(SiMe3)2 (2) and η5-C5H4SiMe3 (3)) with one equivalent of [K(C5Me5)] in the presence of one equivalent of 18-crown-6 or cryptand-222 led to the precipitation of the ionic derivatives [K(L)][{Ti4(η5-C5Me5)3(R)}(μ3-N)4] (R = N(SiMe3)2, L = 18-crown-6 (8), and crypt-222 (9); R = η5-C5H4SiMe3, L = 18-crown-6 (10), and crypt-222 (11)) (Scheme 2). Compounds 8–11 were prepared in toluene at room temperature since the titanium precursors 2 and 3 are soluble in aromatic hydrocarbon solvents.
 | ||
| Scheme 2 Reactions of complexes 2 and 3 with [K(C5Me5)]. | ||
Complexes 6–11 were isolated in 48–83% yields as dark purple or green solids, which are not soluble in aromatic hydrocarbon solvents but exhibit good solubility in pyridine-d5. Compounds 6–11 readily react with chloroform-d1 to give green solutions of the starting materials 1–3 and the corresponding free macrocycles according to 1H NMR spectroscopy. The IR spectra (KBr) of 6–11 show several absorptions in the range 1359–832 cm−1 for the macrocyclic polyethers.29 The 1H NMR spectra of compounds 6–11 in pyridine-d5 reveal one broad signal at δ = 10.7–9.4 (Δν1/2 = 59–89 Hz), attributable to the η5-C5Me5 groups and sharp resonances for one 18-crown-6 or cryptand-222 ligand. In addition, the 1H NMR spectra of complexes 8 and 9 display one broad resonance at δ = 1.22, assignable to the N(SiMe3)2 ligand. Similarly, the spectra of compounds 10 and 11 show a broad signal at δ = 0.77 for the trimethylsilyl group of the C5H4SiMe3 ligand but resonances for the C5H4 protons could not be detected likely because of their broad nature. These NMR data of complexes 8–11 are consistent with a C3v symmetry for the [{Ti4(η5-C5Me5)3(R)}(μ3-N)4]− (R = N(SiMe3)2, η5-C5H4SiMe3) anion in solution. Interestingly, 1H NMR spectroscopy analyses of samples containing 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of pairs of complexes 2/9 and 3/11 in pyridine-d5 at room temperature show one set of signals with averaged chemical shifts. This is consistent with a fast electron exchange between the oxidized and reduced forms of the complexes as those studied in detail for iron heterocubane clusters with [Fe4S4] and [Fe4N4] cores.30 The magnetic moment measurements by the Evans method for solutions of compounds 6–11 in pyridine-d5 gave μeff values of 1.79–1.99μB in good agreement with the presence of an unpaired electron in the complexes. The EPR spectrum of compound 7 in pyridine solution at room temperature was silent. However, the EPR spectrum in a frozen pyridine solution at 93 K (Fig. S10†) shows a broad signal with a g value of 1.996 similar to those previously reported (g = 1.988 and 1.986) for one-electron reduced dinuclear imido-bridged species [Co(η5-C5H5)2][{Ti(η5-C5R5)Cl}2{μ-NAr}2] (R = H, Me; Ar = 3,5-(CF3)2C6H3).23b
:1 mixtures of pairs of complexes 2/9 and 3/11 in pyridine-d5 at room temperature show one set of signals with averaged chemical shifts. This is consistent with a fast electron exchange between the oxidized and reduced forms of the complexes as those studied in detail for iron heterocubane clusters with [Fe4S4] and [Fe4N4] cores.30 The magnetic moment measurements by the Evans method for solutions of compounds 6–11 in pyridine-d5 gave μeff values of 1.79–1.99μB in good agreement with the presence of an unpaired electron in the complexes. The EPR spectrum of compound 7 in pyridine solution at room temperature was silent. However, the EPR spectrum in a frozen pyridine solution at 93 K (Fig. S10†) shows a broad signal with a g value of 1.996 similar to those previously reported (g = 1.988 and 1.986) for one-electron reduced dinuclear imido-bridged species [Co(η5-C5H5)2][{Ti(η5-C5R5)Cl}2{μ-NAr}2] (R = H, Me; Ar = 3,5-(CF3)2C6H3).23b
Dark purple crystals of 11·C6D6 were grown by slow cooling at room temperature of a benzene-d6 solution heated at 80 °C. The X-ray crystal structure shows two independent ion pairs of [K(crypt-222)]+ cations and [{Ti4(η5-C5Me5)3(η5-C5H4SiMe3)}(μ3-N)4]− anions in the asymmetric unit (Fig. S7 and S8†). There are no substantial differences between the ion pairs of 11, and the structure of one of the anions is shown in Fig. 3. The potassium atoms are encapsulated by the cryptand-222 ligands in the cationic fragments with K–O and K–N bond distances of 2.785(5)–2.855(5) Å and 3.004(7)–3.030(6) Å, respectively. The anionic fragments contain an almost perfect [Ti4N4] cube core with Ti–N–Ti (av. 91.3(9)°) and N–Ti–N (av. 88.6(9)°) angles very close to 90°. The average Ti–N (1.93(2) Å) and Ti–Ti (2.77(1) Å) distances in 11 are similar to those found in the anion of compound 5 and in the molecular structures of complexes 1 and 2.5,21 Despite many attempts, we were not able to grow suitable crystals of 18-crown-6 compounds 6, 8 and 10 for X-ray crystal structure determination but most likely the structures contain [K(18-crown-6)]+ cations with the potassium atom oriented toward one of the cyclopentadienyl rings of the [{Ti4(η5-C5Me5)3R}(μ3-N)4]− anions.31 These K⋯cyclopentadienylp interactions in the solid state can be broken upon dissolving the complexes in pyridine according to a disordered crystal structure of [K(18-crown-6)(py)2][{Ti(η5-C5Me5)}4(μ3-N)4] for dark purple crystals of 6 in pyridine.22 However, complexes 6, 8 and 10 in benzene-d6 did not react with an additional equivalent of 18-crown-6 even at high temperatures as determined by 1H NMR spectroscopy.
 | ||
| Fig. 3 Perspective view of one crystallographically independent anion of compound 11 (thermal ellipsoids at the 50% probability level). Hydrogen atoms are omitted for clarity. Selected average lengths (Å) and angles (°): Ti–N 1.94(2), Ti–Ti 2.77(1), Ti–N–Ti 89.8(2)–93.0(2), N–Ti–N 87.2(2)–90.2(2). | ||
In a fashion similar to the preparation of compounds 6–11, the reaction of [{Ti4(η5-C5Me5)3(η5-C5H5)}(μ3-N)4] (4) with [K(C5Me5)] and cryptand-222 at room temperature gave [K(crypt-222)][{Ti4(η5-C5Me5)3(η5-C5H5)}(μ3-N)4] (12) in 76% yield (Scheme 3). However, the treatment of 4 with [K(C5Me5)] and 18-crown-6 afforded a compound with a cyclopentadienyl inverse sandwich countercation [{(18-crown-6)K}2(μ–η5:η5-C5H5)][Ti4(η5-C5Me5)3(η5-C5H5)(μ3-N)4] (13) in 52% yield. Compounds 12 and 13 were isolated as dark purple solids which are only soluble in pyridine and readily react with chloroform to give the starting material 4 and the free macrocycles. The 1H NMR spectra of both compounds in pyridine-d5 showed one broad resonance at δ = 10.6 (Δν1/2 = 82 Hz) attributable to the η5-C5Me5 groups but resonance signals for the η5-C5H5 ligand of the anionic fragment were not observed. While the 1H NMR spectrum of 12 revealed resonances for one cryptand-222 ligand in the countercation, the spectrum of 13 displayed a singlet at δ = 3.43 for two 18-crown-6 ligands and a sharp singlet at δ = 6.50 for the protons of one cyclopentadienide anion. The paramagnetic nature with an unpaired electron of compounds 12 and 13 was confirmed by the determination of their magnetic effective moments (μeff = 1.82 and 1.74μB, respectively) by the Evans method.
 | ||
| Scheme 3 Reactions of 4 with [K(C5Me5)]. | ||
Slow diffusion of a toluene solution of 4 into a toluene solution of [K(C5Me5)] and 18-crown-6 at room temperature afforded dark purple crystals of 13·2C7H8 suitable for X-ray crystal structure determination. The crystal structure of compound 13 consists of well-separated [{(18-crown-6)K}2(μ–η5:η5-C5H5)]+ and [Ti4(η5-C5Me5)3(η5-C5H5)(μ3-N)4]− ions (Fig. S9†). The inverse cyclopentadienyl sandwich cation in 13 contains a C5H5 ring coordinated to two potassium cations which are also bonded to two 18-crown-6 ligands in a fashion similar to other examples in the literature.31d,32 If a centroid (Ct) is considered for the carbon atoms of the cyclopentadienide ring, the K–Ct (2.846 and 2.856 Å) bond lengths are similar to those found in compounds with {K(η5-C5H5)} units such as the polymer [K(η5-C5H5)]n (K–Ct = 2.816 Å).33 The anionic fragment of 13 (Fig. 4) contains an almost perfect [Ti4N4] cube core with Ti–N (av. 1.935(6) Å) and Ti–Ti (av. 2.770(4) Å) distances essentially identical to those found in the crystal structures of the ionic compounds 5 and 11. It appears that the structural parameters in the [Ti4N4] cores are not significantly affected by the addition of an electron which is delocalized among the titanium atoms.
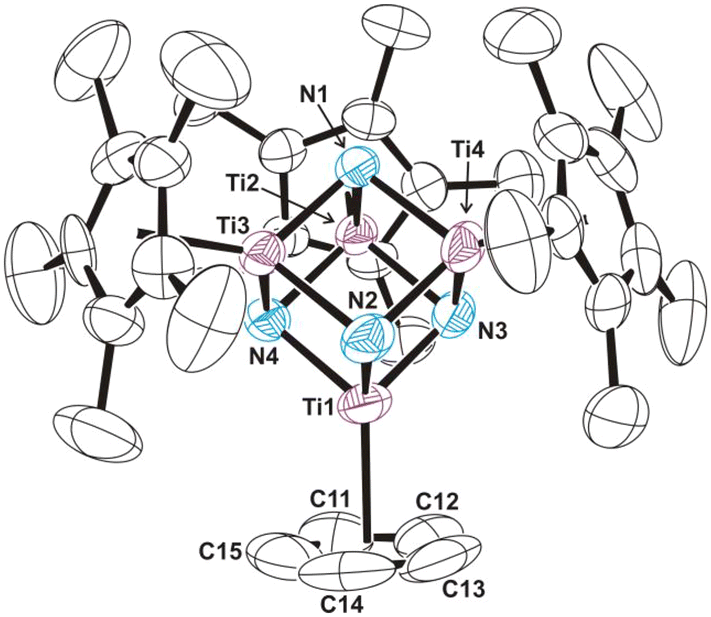 | ||
| Fig. 4 Perspective view of the anion of compound 13 (thermal ellipsoids at the 50% probability level). Hydrogen atoms are omitted for clarity. Selected average lengths (Å) and angles (°): Ti–N 1.935(6), Ti–Ti 2.770(4), Ti–N–Ti 91.4(3), N–Ti–N 88.6(4). | ||
A plausible proposal for the formation of compound 13 is shown in Scheme 4, which is based on reported studies on the reduction of lanthanide complexes [Ln(η5-C5H4SiMe3)3] with potassium in the presence of 18-crown-6 by the Evans group.31c The isolation of 13 with the [{(18-crown-6)K}2(μ–η5:η5-C5H5)]+ countercation could be related to the instability of the anticipated complex [K(18-crown-6)][{Ti4(η5-C5Me5)3(η5-C5H5)}(μ3-N)4] (A) similar to compounds 6–12. In contrast to cryptand-222, the 18-crown-6 macrocycle is not capable of sequestering the K+ cation, and the [K(18-crown-6)]+ unit may interact with the η5-C5H5 ligand bonded to titanium. Such interaction promotes dissociation of the C5H5− group to generate a neutral [K(η5-C5H5)(18-crown-6)] moiety which could react with another equivalent of intermediate A to form the ionic compound 13. Presumably a soluble paramagnetic species [{Ti4(η5-C5Me5)3}(μ3-N)4] (B) was formed but, despite many attempts, we failed to isolate any derivative from the solution resulting in the formation of 13. It is noteworthy that compound 13 was isolated in a similar yield when the reaction of 4 with [K(C5Me5)] was performed in the presence of two equivalents of 18-crown-6 polyether.
 | ||
| Scheme 4 Proposed pathway for the formation of 13. | ||
Attempted reduction of [{Ti(η5-C5Me5)(μ-NH)}3(μ3-N)] (14)
The redox chemistry of the imido–nitrido trititanium complex 14 was also investigated by electrochemical methods. The cyclic voltammogram of 14 in a tetrahydrofuran solution at 22 °C did not reveal any reversible redox event but showed two cathodic reduction peaks at −2.86 and −3.00 V vs. Fc+/Fc (Fig. S4 and S5†). When the scan is reversed, a return oxidation peak is observed at −2.17 V. With these results in hand, the chemical reaction of 14 with one equiv. of [K(C5Me5)] in benzene-d6 at room temperature was monitored by NMR spectroscopy (Scheme 5). Immediately, an abundant yellow solid was deposited at the bottom of the NMR tube. Analysis of the supernatant yellow solution by 1H NMR spectroscopy only showed the formation of C5Me5H while resonances due to C10Me10 were not detected.27 The yellow solid was isolated by filtration, dissolved in pyridine-d5 and characterized by 1H and 13C{1H} NMR spectroscopy as the diamagnetic derivative [{K(μ4-N)(μ3-NH)2Ti3(η5-C5Me5)3(μ3-N)}2] (15). This edge-linked double-cube nitrido complex has been previously synthesized in 57% yield through the reaction of 14 with one equiv. of [K{N(SiMe3)2}].34 | ||
| Scheme 5 Reaction of [{Ti(η5-C5Me5)(μ-NH)}3(μ3-N)] with [K(C5Me5)]. | ||
Treatment of 15 with two equiv. of 18-crown-6 polyethers (L) in toluene at room temperature afforded complexes [(L)K{(μ3-N)(μ3-NH)2Ti3(η5-C5Me5)3(μ3-N)}] (L = 18-crown-6 (16), dibenzo-18-crown-6 (17)) (Scheme 5). Compounds 16 and 17 were isolated in good yields (78 and 87%) as yellow solids which are soluble in toluene or benzene, suggesting a molecular structure. While complex 16 did not react with an additional equivalent of 18-crown-6, the treatment of 17 with dibenzo-18-crown-6 (one equiv.) in toluene at room temperature led to the precipitation of the ion-separated compound [K(dibenzo-18-crown-6)2][Ti3(η5-C5Me5)3(μ3-N)(μ-N)(μ-NH)2] (18) as a yellow solid in 62% isolated yield. An analogous ionic compound [K(crypt-222)][Ti3(η5-C5Me5)3(μ3-N)(μ-N)(μ-NH)2] (19) was obtained in 70% yield through the treatment of 15 with crypt-222 in toluene at 120 °C. Compounds 18 and 19 are not soluble in hydrocarbon aromatic solvents but exhibit good solubility in polar solvents such as pyridine or tetrahydrofuran. This is in good agreement with the ability of two 18-crown-6 units and the cryptand-222 ligand to encapsulate the potassium cation and produce well-separated ion pairs. Compounds 16–19 immediately react with chloroform-d1 to give orange solutions of complex 14 and the corresponding free macrocyclic polyether according to NMR spectroscopy.
Compounds 16–19 were characterized by spectroscopic and analytical methods, as well as by an X-ray crystal structure of 17 on single crystals grown from a benzene-d6 solution at room temperature. The molecular structure of 17 shows a distorted [KTi3N4] cube core (Fig. 5). The potassium atom is bonded to three nitrogen atoms with K(1)–N distances of 2.904(3)–3.034(3) Å and N–K(1)–N angles of average 60.7(8)°. While the K(1) atom is also bound to five oxygen atoms of the 18-crown-6 ligand with K(1)–O bond lengths between 2.862(2) and 3.085(3) Å, the sixth oxygen of the macrocycle is clearly not coordinated to the potassium center (distance K(1)⋯O(43) 3.407(3) Å). Bond distances and angles involving the titanium and nitrogen atoms of the cube do not differ significantly with respect to those found in complex 14.4 Thus, most of the Ti–N distances are in the range 1.909(3)–1.951(3) Å with the exception of those associated with the nitrido ligand N(13), which are slightly shorter (Ti(1)–N(13) 1.886(3) Å and Ti(3)–N(13) 1.882(3) Å).
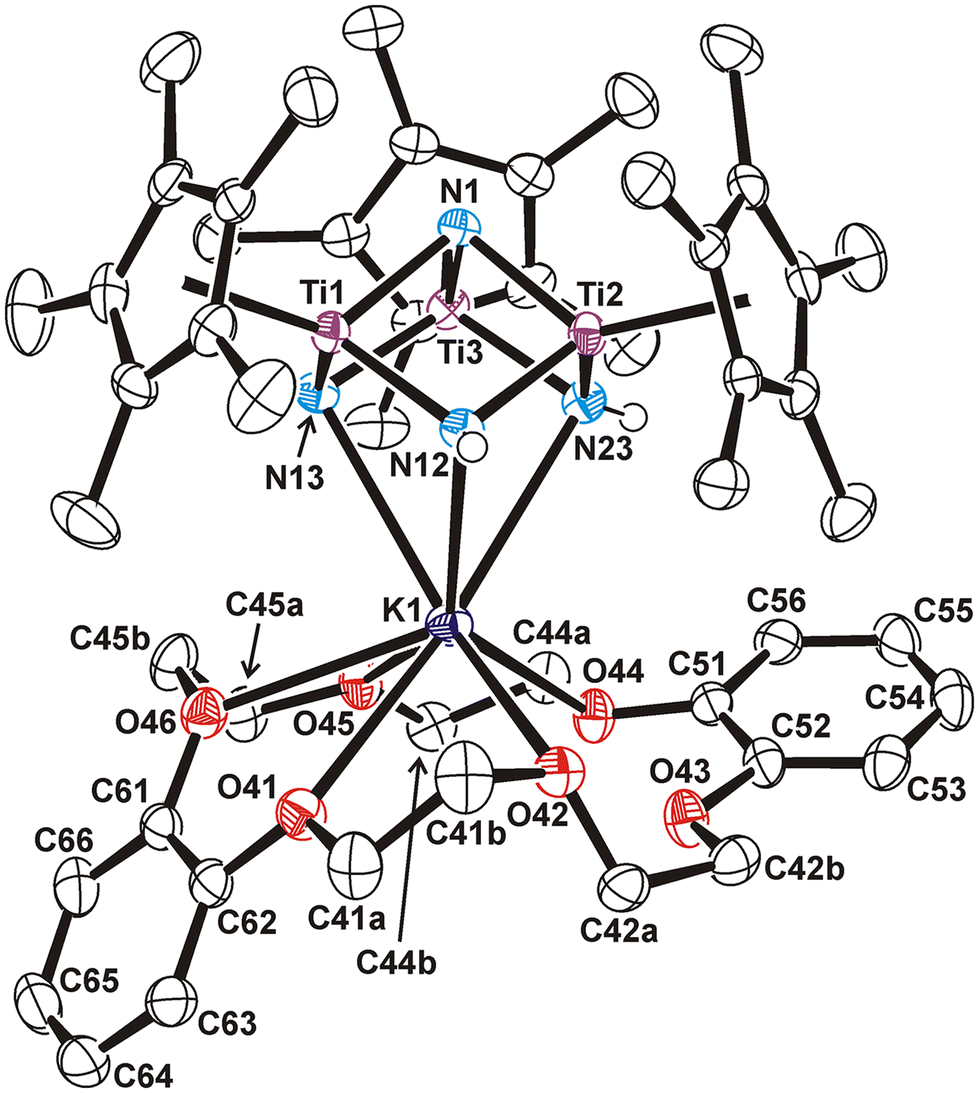 | ||
| Fig. 5 Perspective view of compound 17 (thermal ellipsoids at the 50% probability level). Hydrogen atoms bound to carbon are omitted for clarity. Selected lengths (Å) and angles (°): K(1)–N 2.904(3)–3.034(3), K(1)–O 2.862(2)–3.085(3), K(1)⋯O(43) 3.407(3), Ti–N(1) 1.922(3)–1.951(3), Ti(1)–N(13) 1.886(3), Ti(3)–N(13) 1.882(3), Ti(1)–N(12) 1.918(3), Ti(2)–N(12) 1.909(3), Ti(2)–N(23) 1.934(3), Ti(3)–N(23) 1.941(3), Ti⋯Ti 2.768(1)–2.800(1), K(1)⋯Ti 3.657(1)–3.762(1), N–K(1)–N 59.8(1)–61.2(1), N–Ti–N 102.9(1)–105.0(1), N(1)–Ti–N 86.4(1)–87.4(1), Ti–N–Ti 92.5(1)–94.5(1), Ti–N(1)–Ti 91.1(1)–93.3(1), K(1)–N–Ti 93.1(1)–98.0(1). | ||
The IR spectra (KBr) of 16–19 show one νNH vibration between 3350 and 3327 cm−1 and several absorptions in the range 1360–830 cm−1 for the macrocyclic polyethers.29 The 1H NMR spectra of compounds 16 and 17 in benzene-d6 at room temperature show one broad resonance for the NH imido groups at 13.01 and 12.86 ppm, respectively, and two sharp singlets in a 2:1 ratio for the η5-C5Me5 ligands in accordance with Cs symmetric structures in solution. In addition, the 1H and 13C{1H} NMR spectra of 16 and 17 reveal typical resonances for one 18-crown-6 or dibenzo-18-crown-6 ligand. These resonances are broad indicating a slow exchange between the free and coordinated oxygen atoms of the 18-crown-6 macrocyclic polyethers on the NMR timescale. The 1H NMR spectra of 18 and 19 in pyridine-d5 at room temperature show one resonance for the NH imido groups at 12.96 ppm, two singlets in a 2:1 ratio for the η5-C5Me5 ligands, and one set of resonance signals for two equivalent dibenzo-18-crown-6 or one cryptand-222 ligands. In contrast to those observed for complexes 16 and 17, the resonance signals for the macrocyclic polyethers are sharp in the 1H and 13C{1H} NMR spectra of 18 and 19. However, while the resonances for the η5-C5Me5 ligands in complex 19 are sharp in the NMR spectra, those for complex 18 are broad, suggesting some kind of interaction between the [K(dibenzo-18-crown-6)2]+ and [Ti3(η5-C5Me5)3(μ3-N)(μ-N)(μ-NH)2]− ions in solution. The 13C{1H} NMR spectra of complexes 16–19 show two resonances for the ipso-carbons of the η5-C5Me5 ligands in the narrow ranges of 114.6–114.5 and 113.4–113.1 ppm. These resonances are slightly shifted upfield with respect to that found for 14 (δ = 117.3 ppm) and are similar to those reported for a solution of 15 in pyridine-d5 (δ = 114.9 and 113.5).34b Most likely, the structure of compound 15 in this donor solvent contains a cube-type [KTi3N4] core with solvent molecules coordinated to the potassium atom as a result of the rupture of the edge-linked double-cube structure in the solid state.
Conclusion
The cyclic voltammograms of cube-type titanium(IV) nitrido complexes [{Ti4(η5-C5Me5)3(R)}(μ3-N)4] show a reversible one-electron redox wave at ca. −1.8 V vs. ferrocenium/ferrocene, and chemically reduced titanium complexes are successfully prepared using sodium sand in tetrahydrofuran or [K(C5Me5)] in the presence of macrocycles capable of stabilizing the K+ cation. The anionic fragments [{Ti4(η5-C5Me5)3(R)}(μ3-N)4]− of the resultant ionic species contain one electron delocalized among the titanium atoms and essentially maintain the same structural parameters as the parent neutral cube-type compounds. While cryptand-222 is capable of sequestering the potassium ion in the reduced species to give well-separated ion pairs, the 18-crown-6 polyether appears to allow some interaction between the K+ cation and the cyclopentadienyl ligands of the anion. Such interaction may be responsible for the formation of compound [{(18-crown-6)K}2(μ–η5:η5-C5H5)][{Ti4(η5-C5Me5)3(η5-C5H5)}(μ3-N)4] via abstraction of one cyclopentadienide group of an unstable intermediate [K(18-crown-6)][{Ti4(η5-C5Me5)3(η5-C5H5)}(μ3-N)4].While [K(C5Me5)] is an efficient one-electron reductant to the tetranuclear nitrido complexes, its reaction with the imido–nitrido trinuclear complex [{Ti(η5-C5Me5)(μ-NH)}3(μ3-N)] leads to the deprotonation of one NH imido group and the formation of the double-cube compound [{K(μ4-N)(μ3-NH)2Ti3(η5-C5Me5)3(μ3-N)}2]. The treatment of this potassium complex with 18-crown-6 polyethers or cryptand-222 affords molecular complexes with [KTi3N4] cube cores or well-separated ion pairs with [Ti3(η5-C5Me5)3(μ3-N)(μ-N)(μ-NH)2]− anions.
Experimental section
General considerations
All manipulations were carried out under an argon atmosphere using Schlenk line or glovebox techniques. Toluene and hexane were distilled from the Na/K alloy just before use. Tetrahydrofuran was distilled from purple solutions of sodium benzophenone just prior to use. Pyridine was distilled from calcium hydride just prior to use. NMR solvents were dried with the Na/K alloy (C6D6, C4D8O) or calcium hydride (CDCl3, C5D5N) and distilled before use. Oven-dried glassware was repeatedly evacuated with a pumping system (ca. 1 × 10−3 Torr) and subsequently filled with inert gas. 1,4,7,10,13,16-Hexaoxacyclooctadecane (18-crown-6) and 6,7,9,10,17,18,20,21-octahydrodibenzo[b,k][1,4,7,10,13,16]hexaoxacyclooctadecene (dibenzo-18-crown-6) were purchased from Aldrich and used as received. 4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacoxane (crypt-222) was purchased from Acros and used as received. [{Ti4(η5-C5Me5)3(R)}(μ3-N)4] (R = η5-C5Me5 (1),5 N(SiMe3)2 (2),21 η5-C5H4SiMe3 (3),6 and η5-C5H5 (4)6,21), [{Ti(η5-C5Me5)(μ-NH)}3(μ3-N)] (14),4 [{K(μ4-N)(μ3-NH)2Ti3(η5-C5Me5)3(μ3-N)}2] (15),34 fine sodium sand,35 and [K(C5Me5)]36 were prepared according to published procedures. The synthesis and characterization of [K(18-crown-6)][{Ti(η5-C5Me5)}4(μ3-N)4] (6) have been previously reported.22Samples for infrared spectroscopy were prepared as KBr pellets, and the spectra were obtained using an FT-IR PerkinElmer Spectrum 2000 or FT-IR PerkinElmer Frontier spectrophotometer. 1H and 13C{1H} NMR spectra were recorded on a Varian Unity-300 spectrometer and/or a Mercury-300 spectrometer. Chemical shifts (δ, ppm) in the 1H and 13C{1H} NMR spectra are given relative to residual protons or to the carbon of the solvent: C6D6 (1H: δ = 7.15; 13C: δ = 128.0), C4D8O (1H: δ = 3.58; 13C: δ = 67.2) or C5D5N (1H: δ = 8.71; 13C: δ = 149.9). The effective magnetic moments were determined by the Evans NMR method at 293 K (using a 300 MHz instrument with a field strength of 7.05 Tesla).37 Microanalyses (C, H, N) were performed in a Leco CHNS-932 microanalyzer. CW-EPR spectra were recorded on a Bruker Magnettech ESR5000 spectrometer.
The cyclic voltammograms were recorded using a potentiostat/galvanostat CH Instruments CHI620E. All experiments were carried out using a conventional three-electrode cell. Platinum was used as the working electrode, a platinum wire as the counter electrode, and a silver wire as the pseudo reference electrode. Potentials were internally referenced to the ferrocenium/ferrocene redox couple at 0 mV. Measurements were made under a purified argon atmosphere in a glovebox using tetrahydrofuran solutions of [N(nBu)4][PF6] (0.1 M).
Crystals of compound 5 (0.10 g, 0.080 mmol) were ground with a mortar and pestle to give a fine dark purple powder. This powder was dried under dynamic vacuum for 1 h to afford Na[{Ti(η5-C5Me5)}4(μ3-N)4] (0.063 g, 97%). IR (KBr, cm−1):  2961 (m), 2909 (vs), 2855 (s), 2719 (w), 1494 (w), 1438 (m), 1374 (m), 1021 (m), 789 (m), 650 (m), 627 (m), 531 (s), 427 (s). 1H NMR (300 MHz, C4D8O, 20 °C): δ 9.2 (s br., Δν1/2 = 67 Hz, 60H; C5Me5). Anal. Calcd (%) for C40H60N4NaTi4 (Mw = 811.39): C 59.20, H 7.45, N 6.91. Found: C 59.23, H 7.34, N 6.37.
2961 (m), 2909 (vs), 2855 (s), 2719 (w), 1494 (w), 1438 (m), 1374 (m), 1021 (m), 789 (m), 650 (m), 627 (m), 531 (s), 427 (s). 1H NMR (300 MHz, C4D8O, 20 °C): δ 9.2 (s br., Δν1/2 = 67 Hz, 60H; C5Me5). Anal. Calcd (%) for C40H60N4NaTi4 (Mw = 811.39): C 59.20, H 7.45, N 6.91. Found: C 59.23, H 7.34, N 6.37.
 2962 (s), 2890 (vs), 1477 (m), 1445 (m), 1372 (m), 1354 (s), 1297 (m), 1260 (m), 1134 (s), 1107 (vs), 1078 (s), 951 (s), 933 (m), 832 (w), 792 (m), 752 (m), 623 (m), 543 (s), 425 (s). 1H NMR (300 MHz, C5D5N, 20 °C): δ 9.5 (s br., Δν1/2 = 77 Hz, 60H; C5Me5), 3.40 (s, 12H; OCH2CH2O), 3.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N). Anal. Calcd (%) for C58H96KN6O6Ti4 (Mw = 1204.00): C 57.86, H 8.03, N 6.98. Found: C 57.56, H 7.45, N 6.83. The effective magnetic moment of 7 was determined to be 1.79μB (based on a unit formula of C58H96KN6O6Ti4) in a C5D5N solution.
2962 (s), 2890 (vs), 1477 (m), 1445 (m), 1372 (m), 1354 (s), 1297 (m), 1260 (m), 1134 (s), 1107 (vs), 1078 (s), 951 (s), 933 (m), 832 (w), 792 (m), 752 (m), 623 (m), 543 (s), 425 (s). 1H NMR (300 MHz, C5D5N, 20 °C): δ 9.5 (s br., Δν1/2 = 77 Hz, 60H; C5Me5), 3.40 (s, 12H; OCH2CH2O), 3.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N). Anal. Calcd (%) for C58H96KN6O6Ti4 (Mw = 1204.00): C 57.86, H 8.03, N 6.98. Found: C 57.56, H 7.45, N 6.83. The effective magnetic moment of 7 was determined to be 1.79μB (based on a unit formula of C58H96KN6O6Ti4) in a C5D5N solution.
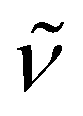 2902 (vs), 1452 (w), 1373 (w), 1352 (m), 1286 (w), 1248 (m), 1108 (vs), 979 (s), 962 (s), 877 (m), 836 (s), 790 (vs), 753 (m), 688 (s), 624 (w), 591 (w), 529 (w), 431 (w). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.6 (s br., Δν1/2 = 59 Hz, 45H; C5Me5), 3.43 (s, 24H; OCH2CH2O), 1.22 (s br., Δν1/2 = 26 Hz, 18H; N(SiMe3)2). Anal. Calcd (%) for C48H87KN5O6Si2Ti4 (Mw = 1116.98): C 51.61, H 7.85, N 6.27. Found: C 51.53, H 7.64, N 6.86. The effective magnetic moment of 8 was determined to be 1.99μB (based on a unit formula of C48H87KN5O6Si2Ti4) in a C5D5N solution.
2902 (vs), 1452 (w), 1373 (w), 1352 (m), 1286 (w), 1248 (m), 1108 (vs), 979 (s), 962 (s), 877 (m), 836 (s), 790 (vs), 753 (m), 688 (s), 624 (w), 591 (w), 529 (w), 431 (w). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.6 (s br., Δν1/2 = 59 Hz, 45H; C5Me5), 3.43 (s, 24H; OCH2CH2O), 1.22 (s br., Δν1/2 = 26 Hz, 18H; N(SiMe3)2). Anal. Calcd (%) for C48H87KN5O6Si2Ti4 (Mw = 1116.98): C 51.61, H 7.85, N 6.27. Found: C 51.53, H 7.64, N 6.86. The effective magnetic moment of 8 was determined to be 1.99μB (based on a unit formula of C48H87KN5O6Si2Ti4) in a C5D5N solution.
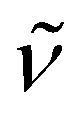 2952 (s), 2892 (vs), 2716 (w), 1477 (m), 1444 (m), 1357 (s), 1298 (m), 1239 (m), 1134 (s), 1107 (vs), 978 (s), 951 (s), 876 (s), 846 (s), 783 (m), 753 (m), 706 (w), 672 (m), 627 (m), 591 (m), 525 (m), 432 (m). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.7 (s br., Δν1/2 = 60 Hz, 45H; C5Me5), 3.40 (s, 12H; OCH2CH2O), 3.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 1.22 (s br., Δν1/2 = 36 Hz, 18H; N(SiMe3)2). Anal. Calcd (%) for C54H99KN7O6Si2Ti4 (Mw = 1229.16): C 52.77, H 8.12, N 7.97. Found: C 52.68, H 7.50, N 7.94. The effective magnetic moment of 9 was determined to be 1.83μB (based on a unit formula of C54H99KN7O6Si2Ti4) in a C5D5N solution.
2952 (s), 2892 (vs), 2716 (w), 1477 (m), 1444 (m), 1357 (s), 1298 (m), 1239 (m), 1134 (s), 1107 (vs), 978 (s), 951 (s), 876 (s), 846 (s), 783 (m), 753 (m), 706 (w), 672 (m), 627 (m), 591 (m), 525 (m), 432 (m). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.7 (s br., Δν1/2 = 60 Hz, 45H; C5Me5), 3.40 (s, 12H; OCH2CH2O), 3.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 1.22 (s br., Δν1/2 = 36 Hz, 18H; N(SiMe3)2). Anal. Calcd (%) for C54H99KN7O6Si2Ti4 (Mw = 1229.16): C 52.77, H 8.12, N 7.97. Found: C 52.68, H 7.50, N 7.94. The effective magnetic moment of 9 was determined to be 1.83μB (based on a unit formula of C54H99KN7O6Si2Ti4) in a C5D5N solution.
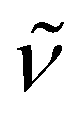 2902 (vs), 1471 (m), 1442 (m), 1373 (m), 1353 (s), 1286 (w), 1248 (s), 1180 (w), 1107 (vs), 1042 (m), 961 (s), 908 (m), 837 (vs), 790 (s), 774 (s), 692 (w), 625 (m), 548 (m), 420 (m). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.3 (s br., Δν1/2 = 89 Hz, 45H; C5Me5), 3.42 (s, 24H; OCH2CH2O), 0.77 (s br., Δν1/2 = 32 Hz, 9H; C5Me4SiMe3). Anal. Calcd (%) for C50H82KN4O6SiTi4 (Mw = 1093.86): C 54.90, H 7.56, N 5.12. Found: C 54.51, H 7.25, N 5.29. The effective magnetic moment of 10 was determined to be 1.92μB (based on a unit formula of C50H82KN4O6SiTi4) in a C5D5N solution.
2902 (vs), 1471 (m), 1442 (m), 1373 (m), 1353 (s), 1286 (w), 1248 (s), 1180 (w), 1107 (vs), 1042 (m), 961 (s), 908 (m), 837 (vs), 790 (s), 774 (s), 692 (w), 625 (m), 548 (m), 420 (m). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.3 (s br., Δν1/2 = 89 Hz, 45H; C5Me5), 3.42 (s, 24H; OCH2CH2O), 0.77 (s br., Δν1/2 = 32 Hz, 9H; C5Me4SiMe3). Anal. Calcd (%) for C50H82KN4O6SiTi4 (Mw = 1093.86): C 54.90, H 7.56, N 5.12. Found: C 54.51, H 7.25, N 5.29. The effective magnetic moment of 10 was determined to be 1.92μB (based on a unit formula of C50H82KN4O6SiTi4) in a C5D5N solution.
 2961 (s), 2889 (vs), 2815 (s), 1477 (m), 1444 (m), 1359 (s), 1355 (s), 1297 (m), 1260 (m), 1242 (m), 1177 (w), 1133 (s), 1106 (vs), 1079 (s), 1040 (w), 951 (s), 933 (m), 907 (w), 833 (s), 796 (vs), 753 (m), 682 (m), 627 (w), 547 (w), 525 (w), 417 (w). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.3 (s br., Δν1/2 = 89 Hz, 45H; C5Me5), 3.40 (s, 12H; OCH2CH2O), 3.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 0.77 (s br., Δν1/2 = 30 Hz, 18H; C5Me4SiMe3). Anal. Calcd (%) for C56H94KN6O6SiTi4 (Mw = 1206.03): C 55.77, H 7.86, N 6.97. Found: C 56.01, H 7.51, N 7.38. The effective magnetic moment of 11 was determined to be 1.92μB (based on a unit formula of C56H94KN6O6SiTi4) in a C5D5N solution.
2961 (s), 2889 (vs), 2815 (s), 1477 (m), 1444 (m), 1359 (s), 1355 (s), 1297 (m), 1260 (m), 1242 (m), 1177 (w), 1133 (s), 1106 (vs), 1079 (s), 1040 (w), 951 (s), 933 (m), 907 (w), 833 (s), 796 (vs), 753 (m), 682 (m), 627 (w), 547 (w), 525 (w), 417 (w). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.3 (s br., Δν1/2 = 89 Hz, 45H; C5Me5), 3.40 (s, 12H; OCH2CH2O), 3.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 0.77 (s br., Δν1/2 = 30 Hz, 18H; C5Me4SiMe3). Anal. Calcd (%) for C56H94KN6O6SiTi4 (Mw = 1206.03): C 55.77, H 7.86, N 6.97. Found: C 56.01, H 7.51, N 7.38. The effective magnetic moment of 11 was determined to be 1.92μB (based on a unit formula of C56H94KN6O6SiTi4) in a C5D5N solution.
 2952 (s), 2891 (vs), 1478 (m), 1446 (m), 1354 (s), 1297 (m), 1261 (m), 1133 (s), 1105 (vs), 1075 (s), 953 (s), 931 (m), 790 (s), 681 (m), 625 (w), 549 (m), 426 (m). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.6 (s br., Δν1/2 = 82 Hz, 45H; C5Me5), 3.39 (s, 12H; OCH2CH2O), 3.34 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.34 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N). Anal. Calcd (%) for C53H86KN6O6Ti4 (Mw = 1133.86): C 56.14, H 7.64, N 7.41. Found: C 56.31, H 7.47, N 7.14. The effective magnetic moment of 12 was determined to be 1.82μB (based on a unit formula of C53H86KN6O6Ti4) in a C5D5N solution.
2952 (s), 2891 (vs), 1478 (m), 1446 (m), 1354 (s), 1297 (m), 1261 (m), 1133 (s), 1105 (vs), 1075 (s), 953 (s), 931 (m), 790 (s), 681 (m), 625 (w), 549 (m), 426 (m). 1H NMR (300 MHz, C5D5N, 20 °C): δ 10.6 (s br., Δν1/2 = 82 Hz, 45H; C5Me5), 3.39 (s, 12H; OCH2CH2O), 3.34 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.34 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N). Anal. Calcd (%) for C53H86KN6O6Ti4 (Mw = 1133.86): C 56.14, H 7.64, N 7.41. Found: C 56.31, H 7.47, N 7.14. The effective magnetic moment of 12 was determined to be 1.82μB (based on a unit formula of C53H86KN6O6Ti4) in a C5D5N solution.
 3057 (w), 2897 (vs), 2855 (s), 1471 (m), 1452 (m), 1434 (m), 1373 (w), 1351 (s), 1284 (m), 1249 (m), 1115 (vs), 1007 (w), 963 (m), 839 (m), 789 (s), 763 (m), 714 (m), 626 (w), 547 (m), 430 (w). 1H NMR (300 MHz, C6D6, 20 °C): δ 10.6 (s br., Δν1/2 = 82 Hz, 45H; C5Me5), 6.50 (s, 5H; C5H5), 3.43 (s, 48H; OCH2CH2O). 13C{1H} NMR (C5D5N, 20 °C): δ 104.8 (μ-C5H5), 70.3 (OCH2CH2O). Anal. Calcd (%) for C64H103K2N4O12Ti4 (Mw = 1390.19): C 55.29, H 7.47, N 4.03. Found: C 54.91, H 7.30, N 3.93. The effective magnetic moment of 13 was determined to be 1.74μB (based on a unit formula of C64H103K2N4O12Ti4) in a C5D5N solution.
3057 (w), 2897 (vs), 2855 (s), 1471 (m), 1452 (m), 1434 (m), 1373 (w), 1351 (s), 1284 (m), 1249 (m), 1115 (vs), 1007 (w), 963 (m), 839 (m), 789 (s), 763 (m), 714 (m), 626 (w), 547 (m), 430 (w). 1H NMR (300 MHz, C6D6, 20 °C): δ 10.6 (s br., Δν1/2 = 82 Hz, 45H; C5Me5), 6.50 (s, 5H; C5H5), 3.43 (s, 48H; OCH2CH2O). 13C{1H} NMR (C5D5N, 20 °C): δ 104.8 (μ-C5H5), 70.3 (OCH2CH2O). Anal. Calcd (%) for C64H103K2N4O12Ti4 (Mw = 1390.19): C 55.29, H 7.47, N 4.03. Found: C 54.91, H 7.30, N 3.93. The effective magnetic moment of 13 was determined to be 1.74μB (based on a unit formula of C64H103K2N4O12Ti4) in a C5D5N solution.
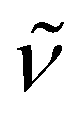 3348 (w) (NH), 2896 (s), 1472 (w), 1452 (w), 1374 (w), 1351 (m), 1286 (w), 1250 (w), 1117 (vs), 964 (m), 842 (w), 769 (m), 735 (s), 697 (s), 633 (m), 530 (w), 467 (w). 1H NMR (300 MHz, C6D6, 20 °C): δ 13.01 (s br., 2H; NH), 3.18 (s br., 24H; OCH2CH2O), 2.29 (s, 30H; C5Me5), 2.16 (s, 15H; C5Me5). 13C{1H} NMR (75 MHz, C6D6, 20 °C): δ 114.6, 113.4 (C5Me5), 69.8 (br., OCH2CH2O), 12.4, 12.3 (C5Me5). Anal. Calcd for C42H71KN4O6Ti3 (Mw = 910.75): C 55.39, H 7.86, N 6.15. Found: C 55.18, H 7.80, N 5.70.
3348 (w) (NH), 2896 (s), 1472 (w), 1452 (w), 1374 (w), 1351 (m), 1286 (w), 1250 (w), 1117 (vs), 964 (m), 842 (w), 769 (m), 735 (s), 697 (s), 633 (m), 530 (w), 467 (w). 1H NMR (300 MHz, C6D6, 20 °C): δ 13.01 (s br., 2H; NH), 3.18 (s br., 24H; OCH2CH2O), 2.29 (s, 30H; C5Me5), 2.16 (s, 15H; C5Me5). 13C{1H} NMR (75 MHz, C6D6, 20 °C): δ 114.6, 113.4 (C5Me5), 69.8 (br., OCH2CH2O), 12.4, 12.3 (C5Me5). Anal. Calcd for C42H71KN4O6Ti3 (Mw = 910.75): C 55.39, H 7.86, N 6.15. Found: C 55.18, H 7.80, N 5.70.
 3329 (w) (NH), 3070 (w), 2898 (m), 2873 (m), 1596 (m), 1505 (s), 1454 (m), 1374 (w), 1327 (w), 1252 (s), 1217 (m), 1128 (s), 1063 (m), 942 (m), 783 (m), 735 (s), 700 (m), 623 (m), 468 (w). 1H NMR (300 MHz, C6D6, 20 °C): δ 12.86 (s br., 2H; NH), 6.85 (m br., 4H; C6H2H2), 6.57 (m br., 4H; C6H2H2), 3.75 (m br., 8H; OCH2CH2O), 3.36 (m br., 8H; OCH2CH2O), 2.24 (s, 30H; C5Me5), 2.08 (s, 15H; C5Me5). 13C{1H} NMR (75 MHz, C6D6, 20 °C): δ 148.4 (br., C6H4), 121.3 (C6H4), 114.6, 113.4 (C5Me5), 112.5 (br., C6H4), 68.6 (br., OCH2CH2O), 12.2 (C5Me5); one OCH2CH2O resonance signal and one C5Me5 resonance signal were not found due to coincidence with those at 68.6 and 12.2 ppm, respectively. Anal. Calcd for C50H71KN4O6Ti3 (Mw = 1006.84): C 59.65, H 7.11, N 5.56. Found: C 59.64, H 6.71, N 4.43.
3329 (w) (NH), 3070 (w), 2898 (m), 2873 (m), 1596 (m), 1505 (s), 1454 (m), 1374 (w), 1327 (w), 1252 (s), 1217 (m), 1128 (s), 1063 (m), 942 (m), 783 (m), 735 (s), 700 (m), 623 (m), 468 (w). 1H NMR (300 MHz, C6D6, 20 °C): δ 12.86 (s br., 2H; NH), 6.85 (m br., 4H; C6H2H2), 6.57 (m br., 4H; C6H2H2), 3.75 (m br., 8H; OCH2CH2O), 3.36 (m br., 8H; OCH2CH2O), 2.24 (s, 30H; C5Me5), 2.08 (s, 15H; C5Me5). 13C{1H} NMR (75 MHz, C6D6, 20 °C): δ 148.4 (br., C6H4), 121.3 (C6H4), 114.6, 113.4 (C5Me5), 112.5 (br., C6H4), 68.6 (br., OCH2CH2O), 12.2 (C5Me5); one OCH2CH2O resonance signal and one C5Me5 resonance signal were not found due to coincidence with those at 68.6 and 12.2 ppm, respectively. Anal. Calcd for C50H71KN4O6Ti3 (Mw = 1006.84): C 59.65, H 7.11, N 5.56. Found: C 59.64, H 6.71, N 4.43.
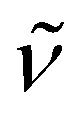 3327 (w) (NH), 3069 (w), 2950 (m), 2896 (s), 2874 (m), 1596 (m), 1505 (s), 1454 (m), 1375 (w), 1329 (m), 1291 (w), 1254 (s), 1232 (m), 1218 (m), 1129 (s), 1063 (m), 1049 (w), 996 (w), 934 (m), 777 (m), 736 (s), 701 (m), 624 (m), 595 (w), 533 (w), 469 (w). 1H NMR (300 MHz, C5D5N, 20 °C): δ 12.96 (s br., 2H; NH), 6.93 (m, 16H; C6H4), 4.12 (m, 16H; OCH2CH2O), 3.75 (m, 16H; OCH2CH2O), 2.24 (s br., 30H; C5Me5), 2.15 (s br., 15H; C5Me5). 13C{1H} NMR (75 MHz, C5D5N, 20 °C): δ 148.9, 121.3 (C6H4), 114.5, 113.2 (C5Me5), 112.7 (C6H4), 69.5 (OCH2CH2O), 68.1 (OCH2CH2O), 12.2 (C5Me5); one C5Me5 resonance signal was not found due to coincidence with that at 12.2 ppm. Anal. Calcd for C70H95KN4O12Ti3 (Mw = 1367.24): C 61.49, H 7.00, N 4.10. Found: C 61.87, H 6.88, N 4.15.
3327 (w) (NH), 3069 (w), 2950 (m), 2896 (s), 2874 (m), 1596 (m), 1505 (s), 1454 (m), 1375 (w), 1329 (m), 1291 (w), 1254 (s), 1232 (m), 1218 (m), 1129 (s), 1063 (m), 1049 (w), 996 (w), 934 (m), 777 (m), 736 (s), 701 (m), 624 (m), 595 (w), 533 (w), 469 (w). 1H NMR (300 MHz, C5D5N, 20 °C): δ 12.96 (s br., 2H; NH), 6.93 (m, 16H; C6H4), 4.12 (m, 16H; OCH2CH2O), 3.75 (m, 16H; OCH2CH2O), 2.24 (s br., 30H; C5Me5), 2.15 (s br., 15H; C5Me5). 13C{1H} NMR (75 MHz, C5D5N, 20 °C): δ 148.9, 121.3 (C6H4), 114.5, 113.2 (C5Me5), 112.7 (C6H4), 69.5 (OCH2CH2O), 68.1 (OCH2CH2O), 12.2 (C5Me5); one C5Me5 resonance signal was not found due to coincidence with that at 12.2 ppm. Anal. Calcd for C70H95KN4O12Ti3 (Mw = 1367.24): C 61.49, H 7.00, N 4.10. Found: C 61.87, H 6.88, N 4.15.
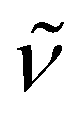 3350 (w) (NH), 2896 (vs), 1477 (m), 1444 (m), 1373 (m), 1354 (s), 1297 (w), 1260 (w), 1133 (s), 1106 (vs), 1079 (s), 1028 (w), 951 (s), 933 (s), 884 (w), 741 (vs), 705 (s), 625 (s), 586 (w), 529 (w), 506 (w), 468 (w), 408 (m). 1H NMR (300 MHz, C5D5N, 20 °C): δ 12.96 (s br., 2H; NH), 3.40 (s, 12H; OCH2CH2O), 3.35 (t, 3J(H,H) = 4.8 Hz, 12H; OCH2CH2N), 2.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.31 (s, 30H; C5Me5), 2.21 (s, 15H; C5Me5). 13C{1H} NMR (75 MHz, C5D5N, 20 °C): δ 114.5, 113.1 (C5Me5), 70.5, 67.7, 53.9 (crypt-222), 12.3, 12.2 (C5Me5). Anal. Calcd for C48H83KN6O6Ti3 (Mw = 1022.93): C 56.36, H 8.18, N 8.22. Found: C 56.71, H 7.78, N 8.08.
3350 (w) (NH), 2896 (vs), 1477 (m), 1444 (m), 1373 (m), 1354 (s), 1297 (w), 1260 (w), 1133 (s), 1106 (vs), 1079 (s), 1028 (w), 951 (s), 933 (s), 884 (w), 741 (vs), 705 (s), 625 (s), 586 (w), 529 (w), 506 (w), 468 (w), 408 (m). 1H NMR (300 MHz, C5D5N, 20 °C): δ 12.96 (s br., 2H; NH), 3.40 (s, 12H; OCH2CH2O), 3.35 (t, 3J(H,H) = 4.8 Hz, 12H; OCH2CH2N), 2.35 (t, 3J(H,H) = 4.5 Hz, 12H; OCH2CH2N), 2.31 (s, 30H; C5Me5), 2.21 (s, 15H; C5Me5). 13C{1H} NMR (75 MHz, C5D5N, 20 °C): δ 114.5, 113.1 (C5Me5), 70.5, 67.7, 53.9 (crypt-222), 12.3, 12.2 (C5Me5). Anal. Calcd for C48H83KN6O6Ti3 (Mw = 1022.93): C 56.36, H 8.18, N 8.22. Found: C 56.71, H 7.78, N 8.08.
X-ray structure determination of 5, 11, 13, and 17
Dark purple crystals of 5, 11·C6D6 and 13·2C7H8, and yellow crystals of 17 were obtained as described in the Results and discussion section. The crystals were removed from the NMR tubes or Schlenk tubes and covered with a layer of viscous perfluoropolyether (FomblinY). A suitable crystal was selected with the aid of a microscope, mounted on a cryoloop, and immediately placed in the low temperature nitrogen stream of a diffractometer. The intensity data sets were collected at 200 K (compound 13) or 150 K (the rest) on a Bruker-Nonius KappaCCD diffractometer equipped with an Oxford Cryostream 700 unit. Crystallographic data for all the complexes are presented in Table S1.†The structures were solved using the WINGX package,38 by intrinsic phasing methods (SHELXT),39 and refined by least-squares against F2 (SHELXL-2014/7).40 Compound 5 crystallized in the cubic space group I213 (Z = 8), and the asymmetric unit is composed of a third of the cation and a third of the anion. The pentamethylcyclopentadienyl ligand linked to Ti(2) is located on a three-fold axis, so it was not possible to define a proper geometry for this C5Me5 group. This disorder was modelled by refining each atom of this fragment with an occupancy of 0.8333, which is the result of the 10/12 quotient. Moreover 5 was treated as a two-component inversion twin using TWIN and BASF instructions. The final value of the BASF parameter was 0.011. All non-hydrogen atoms were anisotropically refined, whereas the hydrogen atoms were positioned geometrically by using a riding model. DELU and SIMU restraints were used for the carbon atoms of the disordered C5Me5 ring (C(21), C(22), C(26) and C(27)) and the atoms of one tetrahydrofuran ligand (O(2), C(5), C(6), C(7) and C(8)).
In the case of 11, several crystalline samples were obtained, and the best possible single crystal was employed for data collection. Compound 11 crystallized with a molecule of benzene in the P21212 space group. The asymmetric unit was constituted by two molecules of benzene, two anions [{Ti4(η5-C5Me5)3(η5-C5H4SiMe3)}(μ3-N)4]−, an entire cation [K(crypt-222)]+, and two halves of two crystallographically independent cations [K(crypt-222)]+. One molecule of benzene and one half of the cation [K(crypt-222)]+ lie on the same crystallographic 2-fold axis, so it was not possible to obtain a sensible chemical model for them. Thus, the PLATON41 squeeze procedure was used to remove their contribution to the structural factors. Crystals of 11 also show disorder for the carbon atoms C(81)–C(90) of the pentamethylcyclopentadienyl group linked to Ti(8). This disorder was treated conventionally by using the PART tool of the SHELXL program and allowing free refinement of the occupancy factor with the FVAR command. The final values of occupancy were 51 and 49%. Simultaneously, the data were refined as a two-component inversion twin using TWIN and BASF instructions, with a final value for the BASF parameter of 0.42. All non-hydrogen atoms were anisotropically refined, except those carbon atoms of the disordered C5Me5 ring (C(81)–C(90) and C(81)′–C(90)′), which were refined isotropically. All the hydrogen atoms were positioned geometrically and refined by using a riding model. DELU and SIMU restraints were applied for the carbon atoms C(61)–C(70) of the pentamethylcyclopentadienyl ligand bound to Ti(6). The carbon atoms C(21)–C(30), C(31)–C(40), C(71)–C(80), C(15)a and C(15)b were also restrained with DELU instructions. Additionally, a DFIX constraint was employed for a carbon–carbon distance in a crypt-222 moiety.
Compound 13 crystallized with two molecules of toluene, which were found in the difference Fourier map, but it was not possible to obtain a chemically sensible model for them, so the PLATON41 squeeze procedure was used to remove their contribution to the structural factors. Crystals of 13 showed disorder for the carbon atoms C(31)–C(40) and C(41)–C(50) of the pentamethylcyclopentadienyl rings linked to Ti(3) and Ti(4) respectively. Moreover the 18-crown-6 connected to the potassium atom K(2) presented disorder as well. These disorders were also treated by using the PART tool. The final values of occupancy were 66 and 34% for the C5Me5 group bound to Ti(3), 66 and 34% for the ring linked to Ti(4), and 78.7 and 21.3% for the macrocyclic ligand. All non-hydrogen atoms were anisotropically refined, except those atoms for the disordered crown ether with minor occupancy (C(71)c, C(71)d, C(72)c, C(72)d, C(73)c, C(73)d, C(74)c, C(74)d, C(75)c, C(75)d, C(76)c, C(76)d and O(71)′–O(76)′), which could be only refined isotropically. The hydrogen atoms were positioned geometrically and refined using a riding model. DELU and SIMU restraints were employed for the disordered pentamethylcyclopentadienyl groups, whereas the disordered macrocyclic ether was treated with SIMU and SADI instructions.
Finally, in the crystallographic study of 17, all non-hydrogen atoms were anisotropically refined. All hydrogen atoms were positioned geometrically and refined by using a riding model, except those linked to nitrogen atoms (H(12) and H(23)), which were located in the difference Fourier map and refined isotropically.
Author contributions
Conceptualization: C. Y.; investigation: J. C., A. C.-M., S. C., M. G. and A. P.-R.; validation: A. C.-M. and C. Y.; supervision: C. Y. and A. P.-R.; visualization: A. C.-M., A. P.-R. and C. Y.; writing – original draft: C. Y.; writing – review & editing: A. C.-M., A. P.-R. and C. Y.; funding acquisition and project administration: C. Y. and A. P.-R. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Spanish MCIU (PGC2018-094007-B-I00) and Universidad de Alcalá (PIUAH22/CC-049 and UAH-GP2022-4) for financial support of this research. J. C. thanks the MEC for a doctoral fellowship. A. C.-M. and M. G. acknowledge the Universidad de Alcalá for fellowships. We thank Dr Alberto Hernán-Gómez for his assistance with EPR data collection for complex 7.References
- (a) K. Dehnicke and J. Strähle, Angew. Chem., Int. Ed. Engl., 1981, 20, 413–426 CrossRef; (b) K. Dehnicke and J. Strähle, Angew. Chem., Int. Ed. Engl., 1992, 31, 955–978 CrossRef; (c) M. Mena, A. Pérez-Redondo and C. Yélamos, Eur. J. Inorg. Chem., 2016, 1762–1778 CrossRef CAS.
- (a) W. A. Nugent and J. M. Mayer, Metal-Ligand Multiple Bonds, Wiley, New York, 1988 Search PubMed; (b) R. A. Eikey and M. M. Abu-Omar, Coord. Chem. Rev., 2003, 243, 83–124 CrossRef CAS; (c) J. F. Berry, Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS; (d) J. M. Smith, Prog. Inorg. Chem., 2014, 58, 417–470 CAS; (e) M. N. Cosio and D. C. Powers, Nat. Rev. Chem., 2023, 7, 424–438 CrossRef CAS PubMed.
- (a) R. Schlögl, Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2nd edn, 2008, vol. 5, pp. 2501–2575 Search PubMed; (b) K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Science, 2005, 307, 555–558 CrossRef CAS PubMed; (c) G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- H. W. Roesky, Y. Bai and M. Noltemeyer, Angew. Chem., Int. Ed. Engl., 1989, 28, 754–755 CrossRef.
- P. Gómez-Sal, A. Martín, M. Mena and C. Yélamos, J. Chem. Soc., Chem. Commun., 1995, 2185–2186 RSC.
- A. Abarca, P. Gómez-Sal, A. Martín, M. Mena, J.-M. Poblet and C. Yélamos, Inorg. Chem., 2000, 39, 642–651 CrossRef CAS PubMed.
- For group 4 polynuclear nitrido complexes prepared with NH3, see: (a) M. M. Banaszak Holl and P. T. Wolczanski, J. Am. Chem. Soc., 1992, 114, 3854–3858 CrossRef CAS; (b) G. Bai, H. W. Roesky, M. Noltemeyer, H. Hao and H.-G. Schmidt, Organometallics, 2000, 19, 2823–2825 CrossRef CAS; (c) G. Bai, P. Müller, H. W. Roesky and I. Usón, Organometallics, 2000, 19, 4675–4677 CrossRef CAS; (d) G. Bai, H. W. Roesky and P. Müller, Bull. Pol. Acad. Sci., Chem., 2002, 50, 1–10 CAS; (e) G. Bai, D. Vidovic, H. W. Roesky and H. Magull, Polyhedron, 2004, 23, 1125–1129 CrossRef CAS; (f) S. Aguado-Ullate, J. J. Carbó, O. González-del Moral, A. Martín, M. Mena, J.-M. Poblet and C. Santamaría, Inorg. Chem., 2011, 50, 6269–6279 CrossRef CAS PubMed.
- For group 5 polynuclear nitrido complexes prepared with NH3, see: (a) M. M. Banaszak Holl, M. Kersting, B. D. Pendley and P. T. Wolczanski, Inorg. Chem., 1990, 29, 1518–1526 CrossRef CAS; (b) M. M. Banaszak Holl, P. T. Wolczanski and G. D. Van Duyne, J. Am. Chem. Soc., 1990, 112, 7989–7994 CrossRef CAS.
- K. A. Lawler, R. Hoffmann, M. M. Banaszak Holl and P. T. Wolczanski, Z. Anorg. Allg. Chem., 1996, 622, 392–400 CrossRef CAS.
- J. P. Sarasa, J.-M. Poblet and M. Bénard, Organometallics, 2000, 19, 2264–2272 CrossRef CAS.
- A. Abarca, M. Galakhov, P. Gómez-Sal, A. Martín, M. Mena, J.-M. Poblet, C. Santamaría and J. P. Sarasa, Angew. Chem., Int. Ed., 2000, 39, 534–537 CrossRef CAS PubMed.
- J. J. Carbó, N. Martínez-Espada, M. Mena, M. E. G. Mosquera, J.-M. Poblet and C. Yélamos, Chem. – Eur. J., 2009, 15, 11619–11631 CrossRef PubMed.
- C. D. Abernethy, F. Bottomley, A. Decken and T. S. Cameron, Organometallics, 1996, 15, 1758–1759 CrossRef CAS.
- For recent reviews on nitrido complexes derived from N2, see: (a) Y. Ishida and H. Kawaguchi, Top. Organomet. Chem., 2017, 60, 45–70 CrossRef; (b) I. Klopsch, E. Y. Yuzik-Klimova and S. Schneider, Top. Organomet. Chem., 2017, 60, 71–112 CrossRef; (c) D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS PubMed; (d) S. J. K. Forrest, B. Schluschaβ, E. Y. Yuzik-Klimona and S. Schneider, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed.
- (a) T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549–1552 CrossRef CAS PubMed; (b) M. M. Guru, T. Shima and Z. Hou, Angew. Chem., Int. Ed., 2016, 55, 12316–12320 CrossRef CAS PubMed; (c) T. Shima, G. Luo, S. Hu, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2019, 141, 2713–2720 CrossRef CAS PubMed; (d) T. Shima and Z. Hou, Eur. J. Inorg. Chem., 2020, 1418–1422 CrossRef CAS.
- (a) T. Shima and Z. Hou, Top. Organomet. Chem., 2017, 60, 23–43 CrossRef; (b) T. Shima, Q. Zhuo and Z. Hou, Coord. Chem. Rev., 2022, 472, 214766 CrossRef CAS.
- M. González-Moreiras, M. Mena, A. Pérez-Redondo and C. Yélamos, Chem. – Eur. J., 2017, 23, 3558–3561 CrossRef PubMed.
- K. Freitag, J. Gracia, A. Martín, M. Mena, J.-M. Poblet, J. P. Sarasa and C. Yélamos, Chem. – Eur. J., 2001, 7, 3645–3651 CrossRef CAS PubMed.
- J. J. Carbó, A. Martín, M. Mena, A. Pérez-Redondo, J.-M. Poblet and C. Yélamos, Angew. Chem., Int. Ed., 2007, 46, 3095–3098 CrossRef PubMed.
- J. Caballo, J. J. Carbó, M. Mena, A. Pérez-Redondo, J.-M. Poblet and C. Yélamos, Inorg. Chem., 2013, 52, 6103–6109 CrossRef CAS PubMed.
- J. Caballo, M. Greño, M. Mena, A. Pérez-Redondo and C. Yélamos, Dalton Trans., 2015, 44, 18145–18157 RSC.
- J. Caballo, M. González-Moreiras, M. Greño, M. Mena, A. Pérez-Redondo and C. Yélamos, Inorg. Chem., 2014, 53, 8851–8853 CrossRef CAS PubMed.
- (a) C. T. Vroegop, J. H. Teuben, F. van Bolhuis and J. G. M. van der Linden, J. Chem. Soc., Chem. Commun., 1983, 550–552 RSC; (b) H. Tsurugi, H. Nagae and K. Mashima, Chem. Commun., 2011, 47, 5620–5622 RSC; (c) C. Lorber, Coord. Chem. Rev., 2016, 308, 76–96 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- Z. J. Tonzetich, R. R. Schrock, K. M. Wampler, B. C. Bailey, C. C. Cummins and P. Müller, Inorg. Chem., 2008, 47, 1560–1567 CrossRef CAS PubMed.
- For examples, see: (a) A. G. Davies and J. Lusztyk, J. Chem. Soc., Perkin Trans. 2, 1981, 692–696 RSC; (b) C. C. Cummins, R. R. Schrock and W. M. Davis, Organometallics, 1991, 10, 3781–3785 CrossRef CAS; (c) W. J. Evans, J. M. Perotti, S. A. Kozimor, T. M. Champagne, B. L. Davis, G. W. Nyce, C. H. Fujimoto, R. D. Clark, M. A. Johnston and J. W. Ziller, Organometallics, 2005, 24, 3916–3931 CrossRef CAS; (d) T. J. Mueller, J. W. Ziller and W. J. Evans, Dalton Trans., 2010, 39, 6767–6773 RSC.
- 1H NMR data for C10Me10 (C6D6, 20 °C, δ): 1.76 (s, 12H), 1.66 (s, 12H), 1.15 (s, 6H). 1H NMR data for C5Me5H (C6D6, 20 °C, δ): 2.49 (m, 1H), 1.81 (s, 6H), 1.76 (s, 6H), 1.00 (d, 3J(H,H) = 7.5 Hz, 3H).
- R. D. Moulton, R. Farid and A. J. Bard, J. Electroanal. Chem., 1988, 256, 309–326 CrossRef CAS.
- (a) S. Al-Rusaese, A. A. Al-Kahtani and A. A. El-Azhary, J. Phys. Chem. A, 2006, 110, 8676–8687 CrossRef CAS PubMed; (b) B. Martinez-Haya, P. Hurtado, A. R. Hortal, S. Hamad, J. D. Steill and J. Oomens, J. Phys. Chem. A, 2010, 114, 7048–7054 CrossRef CAS PubMed.
- (a) J. G. Reynolds, C. L. Coyle and R. H. Holm, J. Am. Chem. Soc., 1980, 102, 4350–4355 CrossRef CAS; (b) C. Lichtenberg, I. Garcia Rubio, L. Viciu, M. Adelhardt, K. Meyer, G. Jeschke and H. Grützmacher, Angew. Chem., Int. Ed., 2015, 54, 13012–13017 CrossRef CAS PubMed.
- For examples, see: (a) M. R. MacDonald, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 15914–15917 CrossRef CAS PubMed; (b) M. R. MacDonald, J. E. Bates, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2012, 134, 8420–8423 CrossRef CAS PubMed; (c) M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857–9868 CrossRef CAS PubMed; (d) R. Zitz, J. Hlina, H. Arp, D. Kinschel, C. Marschner and J. Baumgartner, Inorg. Chem., 2019, 58, 7107–7117 CrossRef CAS PubMed.
- (a) M. Zirngast, U. Flörke, J. Baumgartner and C. Marschner, Chem. Commun., 2009, 5538–5540 RSC; (b) J.-Q. Wang and T. F. Fässler, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 985–988 CrossRef CAS; (c) I. Sänger, T. I. Kückmann, F. Dornhaus, M. Bolte, M. Wagner and H.-W. Lerner, Dalton Trans., 2012, 41, 6671–6676 RSC; (d) T. Han, Y.-S. Ding, J.-D. Leng, Z. Zheng and Y.-Z. Zheng, Inorg. Chem., 2015, 54, 4588–4590 CrossRef CAS PubMed.
- R. E. Dinnebier, U. Behrens and F. Olbrich, Organometallics, 1997, 16, 3855–3858 CrossRef CAS.
- (a) M. García-Castro, A. Martín, A. Pérez-Redondo, M. Mena and C. Yélamos, Chem. – Eur. J., 2001, 7, 647–651 CrossRef; (b) A. Martín, M. Mena, A. Pérez-Redondo and C. Yélamos, Inorg. Chem., 2004, 43, 2491–2498 CrossRef PubMed.
- A. J. Kendall, S. I. Johnson, R. M. Bullock and M. T. Mock, J. Am. Chem. Soc., 2018, 140, 2528–2536 CrossRef CAS PubMed.
- W. A. Herrmann and A. Salzer, Literature, Laboratory Techniques and Common Starting Materials, in Herrmann/Brauer, Synthetic Methods of Organometallic and Inorganic Chemistry, ed. W. A. Herrmann, Georg Thieme Verlag, New York, 1996, vol. 1 Search PubMed.
- (a) D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC; (b) S. K. Sur, J. Magn. Reson., 1989, 169–173 CAS; (c) G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammograms of complexes 1, 3, 4, and 14. Experimental crystallographic data of complexes 5, 11, 13, and 17. Perspective view of the crystal structure of compounds 5, 11, and 13. Tables for selected lengths and angles of the crystal structures of 5, 11, 13, and 17. EPR spectra for complex 7. Selected 1H and 13C{1H} NMR spectra. CCDC 2309190–2309193. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04241c |
| This journal is © The Royal Society of Chemistry 2024 |