
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVolatile lanthanide complexes with fluorinated heptadentate ligands†
Joshua C.
Zgrabik
a,
Balaka
Bhuniya
a,
Thomas
Branstad Phillips
a,
Jorge
Barroso
 b,
Bess
Vlaisavljevich
b and
Scott R.
Daly
*a
b,
Bess
Vlaisavljevich
b and
Scott R.
Daly
*a
aDepartment of Chemistry, The University of Iowa, E331 Chemistry Building, Iowa City, Iowa 52242, USA. E-mail: scott-daly@uiowa.edu
bDepartment of Chemistry, The University of South Dakota, 414 E Clark St, Vermillion, SD 57069, USA
First published on 24th January 2024
Abstract
Understanding factors that influence the volatility of lanthanide complexes remains an important goal for applications such as gas-phase f-metal separations and the synthesis of lanthanide-containing thin films. Lanthanide complexes often exhibit volatility differences that depend on the ability of ligands to saturate the lanthanide coordination sphere and attenuate intermolecular bonding in the solid state. This can make it difficult to assess how electronic factors associated with differing ligand substituents influence volatility. Here we describe the synthesis, structures, and thermal properties of a series of volatile lanthanide complexes (Ln = Nd, Er, and Yb) containing N4O3 ligands decorated with different alkyl and fluoroalkyl substituents (CF3, CF2CF2CF3, Me, and tBu). These ligands completely enveloped the tested lanthanides to form monomeric complexes with 7-coordinate distorted capped octahedral coordination geometries, as determined using single-crystal X-ray diffraction. Thermogravimetric analysis and bulk sublimation studies show how metal encapsulation affords complexes with the same volatility regardless of metal size, even with lanthanide ions with significantly different radii such as Nd3+ and Yb3+. Most notably, the results show that increasing ligand fluorination, a strategy often used to increase the volatility of metal complexes, is not always beneficial and can significantly attenuate the volatility of lanthanide complexes depending on location with respect to other substituents in the ligand framework. A pair-wise model based on density functional theory shows that the net intermolecular interactions in the unit cell can still be stronger when fluorination is present. In other words, even if individual interactions between neighboring molecules are weaker, the total number of interactions in the solid arising from the nature of crystal packing is equally important to consider.
Introduction
Understanding chemical factors that give rise to changes in the volatility of lanthanide complexes is key to their development for applications such as chemical vapor deposition and separations via gas chromatography and thermochromatographic methods.1–4 The ability to sublime or evaporate lanthanide complexes is often dictated first and foremost by coordinative saturation at the metal.5 As compared to transition metals, trivalent lanthanides have relatively large coordination spheres and ionic metal–ligand bonding due to their core-like 4f orbitals. As a result, ligands that have insufficient size to charge ratios often bridge adjacent lanthanides to completely saturate all available metal coordination sites (Fig. 1). These intermolecular bridging modes subsequently attenuate volatility because the lanthanide complexes must then depolymerize to form volatile molecular units that can escape into the gas phase. Even in the absence of formal bridging lanthanide–ligand bonds, accessible voids in the metal coordination sphere can lead to significant intermolecular interactions that suppress volatility. This has led researchers to adopt strategies such as using ancillary, L-type Lewis base donors, in some cases appending them directly to the ligand scaffold,6,7 to fill remaining sites in the lanthanide coordination sphere. Additional anionic ligands can also be added in some cases to afford volatile complex salts, as observed for [Na(tetraglyme)][Ln(hfac)4] (Ln = Y, Gd; hfac = hexafluoroacetylacetonate).8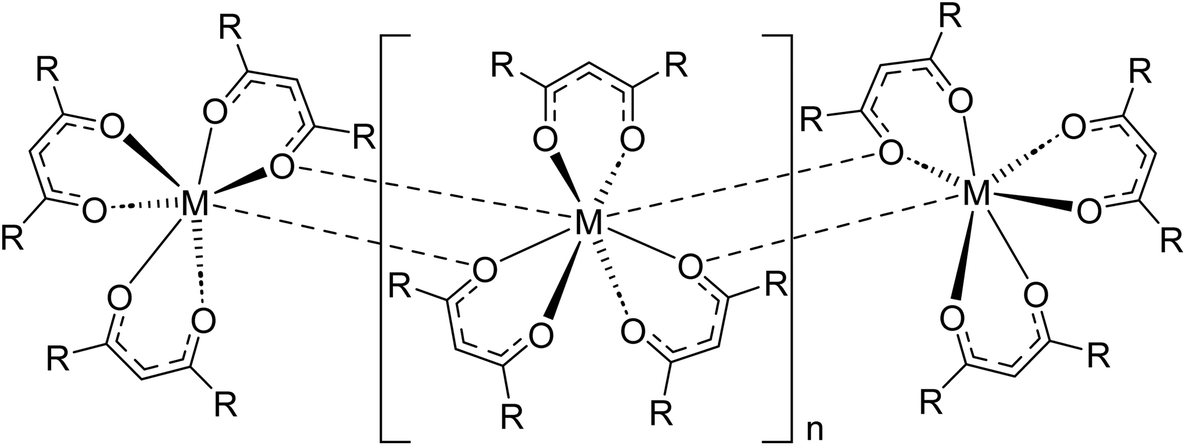 | ||
| Fig. 1 Coordination polymer of sterically undersaturated homoleptic lanthanide complexes, as represented with β-diketonate ligands. Adapted from ref. 1 with permission from Elsevier. | ||
The primary influence of coordinative saturation on the volatility of lanthanide complexes is apparent when looking at complexes that vary only in the size and identity of the lanthanide ion. It has been shown that the sublimation temperatures of homoleptic lanthanide complexes containing identical β-diketonate,5,9–12 amides,13 cyclopentadienyl,14 or aminodiboranate ligands15,16 decrease as the lanthanide series is traversed from large, early lanthanide ions like La3+, Ce3+, and Nd3+ to smaller, late lanthanides like Er3+, Yb3+, and Lu3+. This change occurs primarily because smaller lanthanides are easier to coordinatively saturate, which can make it difficult to evaluate how electronic changes at the metal (particularly with respect to changing Lewis acidity) may also influence volatility.17 Similarly, ligand modifications that alter substituent size and steric profile can also affect electronic properties. This is especially true when decorating complexes with C–F bonds, a common strategy for enhancing the volatility of metal complexes.18 For example, exchange of CH3 groups in the β-diketonate ligand acetylacetonate (acac) for fluorinated CF3 groups in trifluoroacetylacetonate (tfac) and hexafluoroacetylacetonate (hfac) is known to yield a stepwise increase in the volatility of transition metal and aluminum β-diketonate complexes.9 In contrast, it is difficult to assess how such stepwise changes in fluorination influence volatility in the same series of lanthanide β-diketonate complexes because only Ln(hfac)3 are appreciably volatile.19 Homoleptic Ln(acac)3 complexes do not sublime and Ln(tfac)3 complexes typically do so with significant decomposition, which is attributed to significant polymerization in the solid state, as shown in Fig. 1.19
We postulated that one way to better distinguish between steric and electronic substituent contributions to volatility in lanthanide complexes would be to fully encapsulate the metal ion using a single ligand. This would attenuate the possibility of intermolecular interactions so that electronic contributions to volatility could be assessed as a function of metal and ligand variations. The challenge is identifying a single trianionic ligand that not only fully encapsulates the metal to form a charge neutral complex, but also yields some volatility for testing. In general, there are few volatile lanthanide complexes containing ligands with denticities higher than three.20 One of the only known examples that meets these criteria is the heptadentate ligand precursor tris[(4-hydroxypentenylidene-2-imino)ethyl]amine (H3(TRAC), Fig. 2).21 When deprotonated, TRAC is a trianionic N4O3 ligand best described as three β-ketoiminate units tethered together by a tertiary amine linker. Orvig and coworkers showed that TRAC chelates trivalent lanthanide ions to form fully encapsulated metal complexes that can be sublimed, albeit at the relatively high temperature of 180 °C and 10−2 Torr.21 Despite being first reported over 30 years ago, little has been done since then to investigate how ligand substitutions affect the volatility and thermal properties of these complexes.
 | ||
| Fig. 2 Structure of lanthanide TRAC complexes prepared previously, and new fluorinated TRAC derivatives reported here. | ||
Here we report the synthesis of a series of Nd, Er, and Yb complexes with differently-substituted TRAC ligands to investigate the effects of metal encapsulation, metal size, and ligand fluorination on complex volatility. These studies, which include thermogravimetric analysis (TGA) and single-crystal X-ray diffraction (XRD) studies, reveal unexpected trends that help distinguish between steric and electronic ligand contributions to the volatility of lanthanide TRAC complexes. Notably, the results show how fluorination, a common strategy for enhancing the volatility of complexes, can be detrimental to volatility depending on the location of fluorinated substituents within the structure. These results are supported by a set of density functional theory (DFT) calculations of pair-wise interactions that demonstrate how the nature and intensity of intermolecular interactions influence the ease with which these systems can be volatilized. The results show how fully encapsulating different sized lanthanides such as Nd and Yb with the same ligand yields complexes with no appreciable metal-size dependent differences in volatility. Finally, we describe competitive ligand binding studies with Nd and Yb to investigate how differences in metal–ligand reactivity may be used to achieve volatile separation of early and late lanthanides in mixtures.
Results and discussion
Synthesis and structures
Fluorinated H3(TRAC) ligand precursors were synthesized as described for the parent H3(TRAC) by treating tris(2-aminoethyl)amine (TREN) with an excess of the desired β-diketone (Scheme 1a).22 Condensation reactions with hexafluoroacetylacetone, H(hfac), 1,1,1,5,5,6,6,7,7,7-decafluoro-4,6-heptanedione, H(dfhd), 1,1,1,2,2,3,3-heptafluoro-7,7-dimethyl-4,6-octanedione, H(fod), 1,1,1-trifluoroacetylacetone, H(tfac) (Chart 1) yielded the new protonated ligand precursors H3(1) (R, R′ = CF3), H3(2) (R, R′ = CF2CF2CF3, CF3), H3(3) (R, R′ = CF2CF2CF3, tBu), and H3(4) (R, R′ = CF3, Me) respectively. Despite the different substituents, the reactions with unsymmetric β-diketones were quite selective, substituting almost exclusively at the carbon adjacent to the less electron-withdrawing substituent. Even the synthesis of H3(2) with the similar fluoroalkyls R, R′ = CF2CF2CF3, CF3 was remarkably selective. However, we note that the synthesis of the mixed alkyl/fluoroalkyl ligands H3(3) and H3(4) were susceptible to formation of amide-containing side products, especially when performing the reactions in the presence of 3 Å molecular sieves to scavenge water from the reactions. Several monoamide byproducts were isolated and structurally characterized (Fig. S5; ESI†). | ||
| Scheme 1 (a) General synthesis and labeling scheme used for fluorinated TRAC ligands. (b) Protonolysis reactions with Ln[N(SiMe3)2]3 (Ln = Nd, Er, Yb) and labeling scheme used for the prepared complexes. | ||
 | ||
| Chart 1 Substituted β-diketones used to prepare new fluorinated TRAC ligands. | ||
The lanthanide complexes were prepared by mixing one equivalent of the corresponding protonated ligand precursor with one equivalent of Ln[N(SiMe3)2]3 overnight (Scheme 1b). The protonolysis reactions proceeded smoothly in Et2O at room temperature with all four ligands and Yb[N(SiMe3)2]3, as well as for the reaction between Er[N(SiMe3)2]3 and H3(1). Similar reactions with Nd[N(SiMe3)2]3 were not successful under the same conditions in Et2O, but refluxing H3(3) with Nd[N(SiMe3)2] in toluene yielded Nd-3. The six new lanthanide complexes were isolated in good to excellent yields (82–94%) as single crystals by layering concentrated Et2O solutions with pentane followed by cooling to −30 °C. Elemental analysis data collected on the crystals verified their composition and purity.
Single-crystal XRD data show that the lanthanide complexes adopt chiral seven-coordinate distorted capped octahedron coordination geometries. All of the crystallographically-characterized complexes crystallize with both Λ and Δ isomers in the unit cell, as reported previously for the structure of Yb(TRAC) (Fig. 3).21 The twist angle (φ), defined here as the rotation of the O and N triangular faces (Fig. 4), was determined to evaluate the deviation from capped octahedral towards capped trigonal prism as a function of metal and ligand substituents. This value was calculated using the C–N4–Ln–O and C–N4–Ln–N dihedral angles for each β-ketoiminate subunit (C is the carbon atom that tethers the associated β-ketoiminate subunit to the capping atom N4; see Fig. S48 in the ESI† for an example calculation and further details). The twist angles ranged from 51(1)°–56(2)° with the largest twist angles favoring complexes Yb-1, Er-2, and Yb-2 with fully fluorinated CF3 and CF2CF2CF3 substituents (Table 1). The N4–Yb–N and N4–Yb–O angles with the capping amine N define the angles θ2 and θ5, as described by Hoffmann, Muetterties, and coworkers for capped octahedron coordination geometries (Fig. 4).23 The lanthanide TRAC complexes exhibit θ2 and θ5 angles that are slightly more acute than the optimum 75° and 130° angles predicted for d0 and d4 transition metal complexes in the same coordination geometry (Table 1).23
 | ||
| Fig. 3 Molecular structure of Yb-1. Left – Top view looking down the C3 rotation axis defined Yb1–N4 bond. Right – Side-on view. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms were omitted from the figure. Select bond distances and angles: Yb1–O1 = 2.194(3) Å, Yb1–O2 = 2.177(3) Å, Yb1–O3 = 2.178(4) Å; Yb1–N1 = 2.430(6) Å, Yb–N2 = 2.421(4) Å, Yb–N3 = 2.427(5) Å, Yb–N4 = 2.474(5) Å. N1–Yb–O3 = 159.1(1)°, N2–Yb–O1 = 160.5(1)°, N3–Yb–O2 = 157.9(1)°. | ||
 | ||
| Fig. 4 Left – The twist angle (φ) looking down the three-fold rotation axis (Λ isomer). Right – The θ2 and θ5 angles defined previously for 7-coordinate capped octahedral complexes.23 | ||
Yb(TRAC)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 21 |
Yb-1 | Yb-2 | Yb-4 | Er-1 | Nd-3 | |
|---|---|---|---|---|---|---|
| M–O1 | 2.18(1) | 2.194(3) | 2.189(3) | 2.198(4) | 2.211(2) | 2.304(2) |
| M–O2 | 2.161(9) | 2.177(3) | 2.181(2) | 2.195(4) | 2.222(3) | 2.311(2) |
| M–O3 | 2.20(1) | 2.178(4) | 2.187(3) | 2.197(4) | 2.212(3) | 2.301(2) |
| M–N1 | 2.46(1) | 2.430(6) | 2.408(3) | 2.422(5) | 2.443(3) | 2.555(2) |
| M–N2 | 2.45(1) | 2.421(4) | 2.401(3) | 2.457(4) | 2.429(3) | 2.577(2) |
| M–N3 | 2.44(1) | 2.427(5) | 2.408(3) | 2.456(4) | 2.459(2) | 2.579(2) |
| M–N4 | 2.43(1) | 2.474(5) | 2.492(3) | 2.424(4) | 2.558(2) | 2.641(1) |
| N–M–Otrans | 162.8(4) | 160.5(1) | 165.5(1) | 162.0(1) | 167.62(9) | 168.34(6) |
| 161.3(4) | 159.1(1) | 159.7(1) | 161.3(1) | 164.10(9) | 166.90(6) | |
| 159.1(4) | 157.9(1) | 157.9(1) | 160.6(1) | 159.65(9) | 165.26(6) | |
| N4–M–O1 (θ5) | 129.1(4) | 127.5(1) | 125.6(1) | 127.4(1) | 121.69(9) | 122.29(5) |
| N4–M–O2 (θ5) | 126.7(4) | 127.5(1) | 130.3(1) | 128.5(1) | 129.22(9) | 119.31(5) |
| N4–M–O3 (θ5) | 128.8(4) | 129.2(1) | 122.9(1) | 128.9(1) | 118.88(9) | 120.77(5) |
| N4–M–N1 (θ2) | 69.8(4) | 71.6(2) | 71.5(1) | 69.6(1) | 70.74(9) | 68.57(5) |
| N4–M–N2 (θ2) | 68.2(4) | 71.5(2) | 72.6(1) | 68.9(1) | 69.35(9) | 69.39(5) |
| N4–M–N3 (θ2) | 69.0(4) | 72.3(2) | 71.8(1) | 69.0(1) | 70.18(9) | 69.03(5) |
| Twist (φ) | 51(1) | 54(1) | 56(2) | 52(1) | 55(5) | 51(2) |
The Yb–O bond distances are similar in all the complexes and unremarkable when compared to the parent Yb(TRAC) and homoleptic Yb β-ketoiminate complexes reported previously (Table 1).21,24,25 The shortest Yb–O distances in the complexes reported here were found in Yb-1 and range from 2.177(3)–2.194(3) Å, whereas the longest distances found in Yb-4 range from 2.194(4)–2.198(4) Å. In contrast, the Yb–N distances are more sensitive to changes in fluorination and vary by as much as 0.07 Å. The Yb–N bond distances associated with the β-ketoiminate subunit (Yb–N1, Yb–N2, and Yb–N3) increase in the order of Yb-2 < Yb-1 < Yb-4 ∼ Yb(TRAC), whereas the Yb–N4 distances associated with the amine decrease in the same order from 2.492(3) Å in Yb-2 to 2.424(4) Å in Yb-4. The Ln–O and Ln–N bond distances for Er-1 and Nd-3 increase compared to the Yb complexes, as expected due to the increasing size of the lanthanide ions (Yb3+ = 0.868 Å; Er3+ = 0.89; Nd3+ = 0.983; coordination number = 6).26 Like the Yb complexes, the bond distances in Er-1 and Nd-3 are comparable to other lanthanide β-ketoiminate complexes containing these metals.7
NMR studies
1H and 19F NMR spectra collected on the lanthanide complexes in C6D6 revealed paramagnetically shifted and broadened resonances consistent with their formulations and chelation of the ligands (Table 2). The 1H NMR spectra, for example, revealed diastereotopic ethylene resonances diagnostic of ligand chelation, as has been shown for other complexes containing TREN-derived tripodal ligands.27,28 The 19F NMR spectra for Yb-2, Yb-3, and Nd-3 also revealed diastereotopic CF2 resonances from the CF2CF2CF3 chain. No 1H NMR resonances were observed for Er-1, presumably due to paramagnetic broadening, but 19F resonances for the CF3 substituents were observed for Er-1 at δ −68.4 and −78.9 ppm in C7D8.| Complex | 1H (methine) | 1H (ethylene) | 19F |
|---|---|---|---|
| a Tentative assignment. b Collected in C7D8. | |||
| [Yb(TRAC)] | −1.39 | 23.54, 17.51, 6.13, 5.82 | — |
| Yb-1 | −0.82 | 21.27, 10.75, 6.18 | −71.5, −73.6 |
| Yb-2 | −4.18 | 34.10, 32.82, 11.33, 7.66 | −74.8, −78.7, −111.3, −119.5, −122.3 |
| Yb-3 | −12.73 | 60.69, 59.17, 14.73, 10.88 | −70.9, −114.1, −110.2, −120.0 |
| Yb-4 | 1.92a | 6.49, 5.22, 4.73, 2.82 | −76.4 |
| Er-1 | Not observed | Not observed | −68.4, −78.9b |
| Nd-3 | −18.11 | 12.75, 0.53, −3.27, −13.39 | −86.7, −115.8, −131.0 |
The most interesting finding from the NMR studies is that the degree of paramagnetic shifting in the Yb complexes depends significantly on the identity of the ligand substituents (Fig. 5). The 1H resonances for Yb-3 (R = CF2CF2CF3 and R′ = tBu) are spread out over the widest spectral window, ranging from δ 60.7 to −12.7 ppm. In contrast, the resonances for Yb-4 with R = Me and R′ = CF3 fall within the narrow window of δ 6.49 to 1.92 ppm, and the spectral range decreases in the order Yb-3 > Yb-2 > Yb-1 > Yb-4. Given their position with respect to Yb, the paramagnetic NMR shifts for the 1H nuclei are likely governed by the “through-space” pseudocontact shift (as opposed to the “through-bond” contact shift).29 Parker and coworkers recently showed how changing pyridyl substituents in 9-coordinate, tricapped trigonal prismatic Yb complexes yielded similarly dramatic changes in the 1H pseudocontact shifts that were attributed to the influence of ligand dipolar polarizability and solvation on ligand field strength.30 We suspect that a similar phenomenon is operative in the Yb complexes reported here, but further studies will be needed to evaluate this hypothesis. For example, quantifying these sorts of ligand field contributions has been achieved by preparing Eu complexes with the same ligands and measuring their emission spectra.30
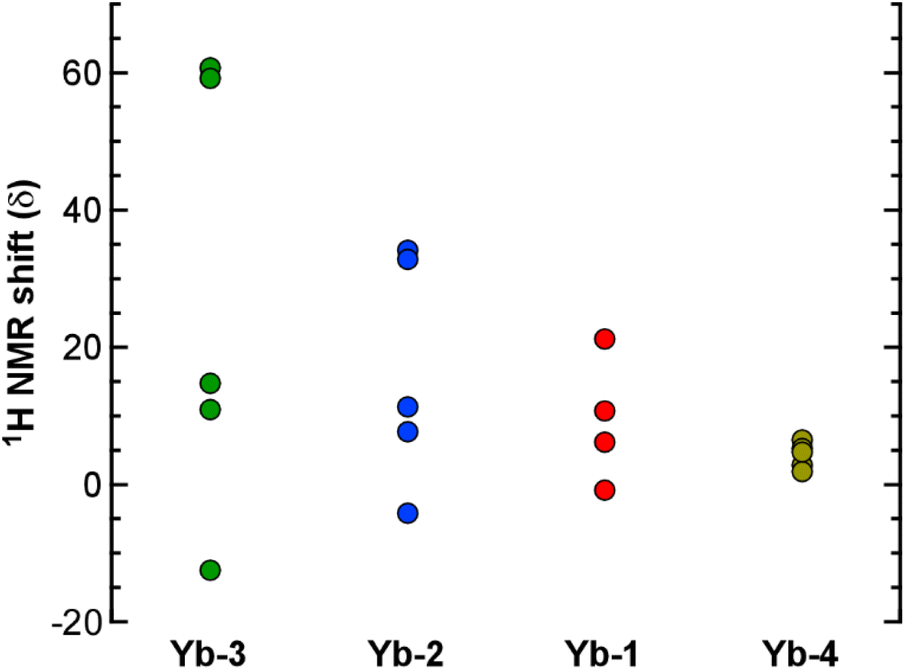 | ||
| Fig. 5 Methine and ethylene 1H NMR shifts for the Yb complexes. | ||
Volatility studies
Thermogravimetric analysis (TGA) and bulk sublimation studies were carried out on all the fluorinated TRAC complexes to determine the influence of substituent modifications and metal identity on complex volatility (Fig. 6). The parent Yb(TRAC) was also prepared and used as a benchmark for comparison. The TGA studies were carried out at atmospheric pressure under a slow purge of N2 gas whereas bulk sublimation studies were carried out under vacuum at 10−2 Torr. Volatility assessments using TGA were determined by taking the 1st derivative of the TGA curves to quantify the temperature of maximum % weight loss (Fig. S38–S44; ESI†).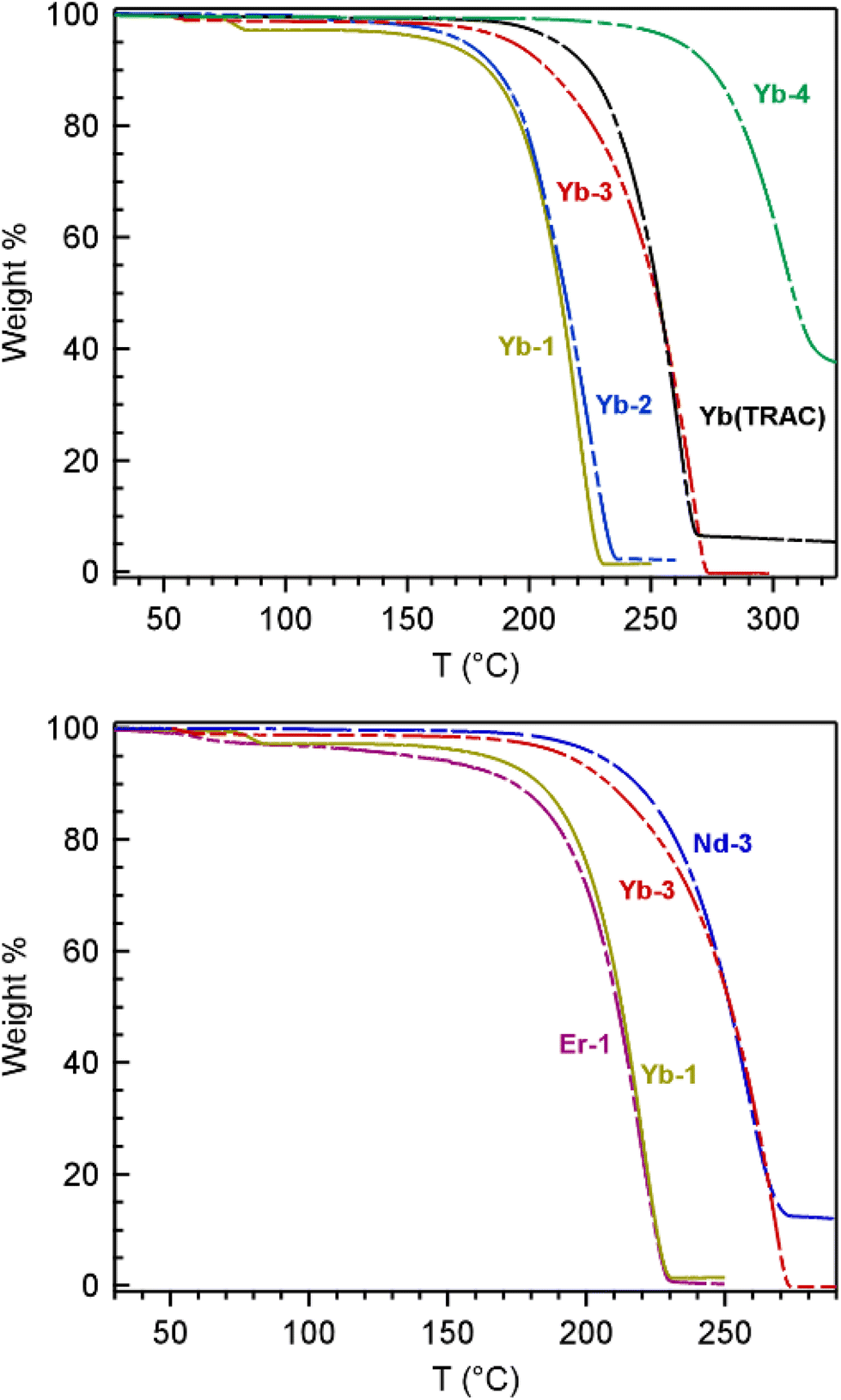 | ||
| Fig. 6 Top –TGA comparison of the Yb complexes: Yb-1 (gold), Yb-2 (blue), Yb-3 (red), Yb-4 (green), and [Yb(TRAC)] (black). Bottom – TGA comparison of lanthanide complexes with different metals: Yb-1 (gold) vs.Er-1 (magenta) and Yb-3 (red) vs.Nd-3 (blue). | ||
The TGA and bulk sublimation studies of the Yb complexes follow the same trend, and show that volatility decreases in the order Yb-1 ∼ Yb-2 > Yb(TRAC) ∼ Yb-3 > Yb-4 (Table 4). Yb-1, Yb-2, and Yb-3 volatilize cleanly with relatively little thermal decomposition, as indicated by >95% weight loss by TGA and during sublimation studies (Fig. 6). Yb-1 and Yb-2 with fully fluorinated substituents are the most volatile, with TGA weight loss maxima of 222 and 225 °C, respectively. Likewise, both complexes are similarly volatile when heated under vacuum, subliming at 120 °C at 10−2 Torr. These results indicate that increasing the length of the fluoroalkyl chains from CF3 in Yb-1 to CF2CF2CF3 in Yb-2 has a negligible impact on volatility despite that higher molecular weight typically portends decreased volatility via increased number of intermolecular forces. For comparison, weight loss maxima and sublimation temperatures for Yb(TRAC) with R, R′ = Me were ca. 40 °C higher than Yb-1 and Yb-2 at 261 and 160 °C, respectively. Notably, the 160 °C sublimation temperature of Yb(TRAC) at 10−2 Torr in our hands occurs even lower than the 180 °C at 10−2 Torr reported previously.21Yb-3 with R = CF2CF2CF3 and R′ = tBu shows similar volatility as the parent Yb(TRAC) complex with a sublimation max of 269 °C by TGA and a bulk sublimation temperature of 160 °C under vacuum.
The most unexpected result was observed with Yb-4 with R = CF3 and R′ = Me. This complex is the least volatile in the series despite increased fluorination with respect to Yb(TRAC) and decreased molecular weight compared to Yb-3 (which also contains alkyl and fluoroalkyl substituents in the same positions). TGA of Yb-4 shows a maximum % weight loss at 306 °C, but this is at least partly attributed to decomposition because the trace levels off after ∼60% weight loss. Yb-4 sublimes more cleanly under vacuum at 190 °C, which is 30 °C higher than Yb-3 and Yb(TRAC) under the same conditions. We tentatively attribute the lower volatility of Yb-4 to the unsymmetric alkyl and fluoroalkyl substituents that enhances intermolecular attractive forces in the solid state via an increased dipole across the complex. It seems likely that a similar dipole increase is present in Yb-3, but the larger tBu and CF2CF2CF3 substituents appear to diffuse the strength of this dipole and/or shield the positively charged metal from significant attractive intermolecular interactions with polarized ligand fragments. The spectral width of the paramagnetic 1H NMR shifts described in the previous section, which are largest for Yb-3 and smallest for Yb-4, indeed suggest a significant difference in ligand dipolar polarizability, which lends support for this hypothesis.
Next, we compare the TGA and sublimation data for Yb-1/Er-1 and Nd-3/Yb-3 complexes containing the same ligand, but different metals. We see no significant differences in volatility when the metal is varied, which is most notable for Nd-3 and Yb-3 given the relatively large differences in metal size and Lewis acidity (Table 3). This finding corroborates previous reports showing that Ln(TRAC) complexes with Ln = Sm, Er, and Yb sublime at identical temperatures.21 Collectively, these results suggest that encapsulation of the metal imparts similar volatility regardless of metal size, which is markedly different compared to most lanthanide complexes, as described in the introduction.
| Complex | Weight loss max (TGA) | Sublimation T (10−2 Torr) |
|---|---|---|
| a Prior studies report bulk sublimation at 180 °C at 10−2 Torr. In our hands, this complex sublimes at temperatures as low as 160 °C at 10−2 Torr. | ||
| Yb-1 | 222 °C | 120 °C |
| Er-1 | 221 °C | 120 °C |
| Yb-2 | 225 °C | 120 °C |
|
Yb(TRAC)a |
261 °C | 160 °C |
| Nd-3 | 263 °C | 160 °C |
| Yb-3 | 269 °C | 160 °C |
| Yb-4 | 306 °C | 190 °C |
Computational analysis of Yb complexes
Density functional theory (DFT) calculations were performed (PBE0-D3/def2-TZVP) to build a lattice model to better understand the observed differences in the volatility of the Yb complexes. Initial assessment of the crystallographic parameters suggested that the differences cannot be attributed solely to differences in crystal packing. For example, the experimental data shows that the most volatile (Yb-1) and the least volatile (Yb-4) have similar atomic packing factor (APF) values, 9.7 and 9.5%, respectively. However, the APF of Yb(TRAC), which is the system with intermediate volatility, is 11.0%. Therefore, to better understand the volatilization tendency between the Yb complexes, we needed to evaluate both the strength and quantity of intermolecular interactions. We carefully analyzed all pair-wise interactions of nearest neighbor complexes in the crystal structures of Yb-1, Yb(TRAC), and Yb-4. Non-covalent interaction energies were measured in a 2 × 2 × 2 supercell for contacts that gave intermolecular Yb⋯Yb distances less than 11 Å, i.e., distances with significant intermolecular interaction energies. This analysis revealed different numbers of intermolecular pairs, or dimers, in the supercell with Yb⋯Yb distances less than 11 Å: six in Yb-1, eleven in Yb(TRAC), and four in Yb-4 (Table 4 and Fig. S45–S47†).| Yb-1 | Yb(TRAC) | Yb-4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| r(Yb⋯Yb) | E inter | n | Total Einter | r(Yb⋯Yb) | E inter | n | Total Einter | r(Yb⋯Yb) | E inter | n | Total Einter |
| 8.73 | −16.3 | 26 | −423.6 | 7.61 | −13.7 | 32 | −439.2 | 7.91 | −17.4 | 80 | −1393.7 |
| 8.79 | −12.0 | 60 | −718.6 | 7.64 | −12.7 | 16 | −203.2 | 9.22 | −24.7 | 38 | −937.4 |
| 10.02 | −2.6 | 44 | −114.7 | 8.13 | −12.0 | 24 | −288.3 | 10.02 | −7.6 | 80 | −610.5 |
| 10.30 | −5.6 | 48 | −268.0 | 8.78 | −8.9 | 24 | −214.4 | 10.81 | −0.9 | 37 | −31.9 |
| 10.85 | −5.0 | 29 | −144.3 | 8.86 | −7.3 | 12 | −87.5 | ||||
| 10.98 | −2.0 | 24 | −47.6 | 8.93 | −7.6 | 4 | −30.5 | ||||
| 9.19 | −2.2 | 10 | −21.8 | ||||||||
| 10.31 | −2.4 | 12 | −28.6 | ||||||||
| 10.36 | −3.1 | 12 | −37.5 | ||||||||
| 10.47 | −0.7 | 24 | −17.8 | ||||||||
| 10.52 | −1.0 | 24 | −23.1 | ||||||||
| −1716.7 (−6.1) | −1391.9 (−7.2) | −2973.6 (−12.7) | |||||||||
As noted above, swapping CH3 groups for CF3 gives rise to different solid-state structures, including the arrangement and packing, and this corresponds to changes in the types of intermolecular interactions in the crystal. In both Yb-1 and Yb(TRAC), the dominant intermolecular interactions take place between the CF3 or CH3 groups of one complex and the O and N atoms in a neighboring complex. Weaker F⋯F and F⋯H contacts are present in Yb-1, and between CH3 and the Schiff arms in the case of Yb(TRAC) (Fig. S45 and S46†). The case of Yb-4 is unique in that the interactions primarily occur between alkyl and CF3 groups except for a single CH3⋯carbonyl interaction (Fig. S47†). This suggests that replacing only half of the CH3 groups in Yb(TRAC) for CF3 results in preferred interactions between these groups in neighboring complexes.
In addition to having differing numbers and type of contacts, the calculations revealed significant differences in their strengths. The non-covalent interaction (NCI) index was computed to identify and quantify the interactions between neighboring moieties in each crystal (Table 4). Noticeably, the strength of interactions between neighboring fragments of Yb-1 and Yb(TRAC) are similar, but the number of instances for the latter is significantly larger. Taking both number and strength yields average interaction energies of −6.1 kcal mol−1 for Yb-1 and −7.2 kcal mol−1 for Yb(TRAC), consistent with the higher volatility of Yb-1. On the other hand, Yb-4 has the strongest and largest number of intermolecular interactions, yielding an average interaction energy of −12.7 kcal mol−1, consistent with it having the lowest volatility in the series. Overall, this simple lattice model supports that the net intermolecular interactions are strongest in Yb-4 and follow the order Yb-4 > Yb(TRAC) > Yb-1 in agreement the observed differences in volatility.
Competitive ligand binding and reactivity studies
The similarities in volatility for Nd-3 and Yb-3 provided an opportunity to investigate if differences in metal–ligand reactivity and ligand binding affinity could be used to achieve volatile separation of early and late lanthanides using 3. As discussed in the introduction, many lanthanide complexes show metal size-dependent differences in volatility that could be used to achieve a separation using methods such as GC or thermochromatography. When there are no significant metal-size dependent differences in complex volatility, as described here, another potential way to achieve volatile separation of lanthanides is to exploit differences in metal–ligand reactivity, as is typically done with selective solvent-based extractants.To investigate this possibility, we examined reactions with one equivalent of Nd[N(SiMe3)2]3, Yb[N(SiMe3)2]3, and H3(3) under different time and temperature conditions (eqn (1)). The resulting mixtures were then evaporated to dryness, the volatile products were sublimed at ∼160 °C at 10−2 Torr until no additional material was observed to deposit on the cold finger from the residue (∼2 hours), and the sublimate was analyzed by 19F NMR spectroscopy to quantify the distributions of products containing 3. We note that the Ln[N(SiMe3)2]3 starting materials used in these studies are also volatile and will certainly sublime to some extent under the tested conditions. This is obviously not desirable from the perspective of lanthanide separations, but the experiment design meets the intended purpose of understanding binding and reactivity preferences of 3 with different lanthanides.
 | (1) |
 | (2) |
The competition reactions of one equivalent of Nd[N(SiMe3)2]3, Yb[N(SiMe3)2]3, and H3(3), as represented by eqn (1), were performed in toluene under three different conditions: stirring at RT for 1 h, stirring at RT for 24 h, and refluxing for 24 h (Table 5 and Fig. S27–S29; ESI†). The reaction performed at 1 h at RT revealed preferential formation of Yb-3 with the smaller Yb3+ ion: the relative distribution of Yb-3 to Nd-3 was 5:1 and 34% of the Yb[N(SiMe3)2]3 was converted to Yb-3 compared to 7% for Nd-3. After 24 h, the distribution remains roughly the same with the conversions of Yb-3 and Nd-3 increasing to 47% and 10%, respectively. By contrast, refluxing the mixture for 24 h results in a marked increase of Nd-3 (48%) relative to Yb-3 (38%), and almost complete consumption of H3(3).
| Reaction conditions | Yb-3 | Nd-3 | Unreacted H3(3) |
|---|---|---|---|
| 1 h/RT | 34% | 7% | 59% |
| 24 h/RT | 47% | 10% | 43% |
| 24 h/reflux | 38% | 48% | 14% |
It was not clear if the preferential conversion to Nd-3 at higher temperatures was a consequence of kinetic or thermodynamic factors, so additional experiments were performed to better evaluate the thermodynamic binding preference of 3 with Nd and Yb relative to their corresponding Ln[N(SiMe3)2]3 complexes. Competition experiments were performed by stirring Nd-3 with Yb[N(SiMe3)2]3 and Yb-3 with Nd[N(SiMe3)2]3 in toluene for 24 h at RT (eqn (2)). No formation of Nd-3 was observed in the mixture containing Yb-3 and Nd[N(SiMe3)2]3 but a small amount Yb-3 (∼6%) corresponding to ligand exchange was observed in the mixture containing Nd-3 and Yb[N(SiMe3)2]3 (Fig. S30 and S31; ESI†). This result suggests that there is a greater thermodynamic preference for Yb-3 and Nd[N(SiMe3)2]3.
Conclusion
In summary, we have described the synthesis, structures, and thermal properties of heptadentate lanthanide complexes containing fluorinated N4O3 ligands derived from TRAC. These encapsulating ligands allowed the influence that fluorinated substituents and metal identity have on the volatility of lanthanide complexes to be quantified without competing intermolecular bonding and steric saturation effects. As reported previously for Ln(TRAC) complexes with Ln = Sm, Er, and Yb, no significant metal-size dependent differences in volatility were observed, which was most notable with Nd-3 and Yb-3 given the relatively large difference in metal ion size. These results suggest that lanthanide identity has little influence on volatility in the absence of solid-state oligomerization.Studies of the differently-substituted Yb complexes reported here show how complexes with fully fluorinated substituents afford significant volatility enhancements when compared to non-fluorinated Yb(TRAC), consistent with the expected result. Substituting the Me substituents in Yb(TRAC) for CF3 and CF2CF2CF3 in Yb-1 and Yb-2 lowered the sublimation temperature of both complexes by 40 °C. However, intermediate fluorination of the TRAC framework caused the opposite effect and was remarkably detrimental to the volatility of Yb-4, increasing its sublimation temperature by 30 °C with respect to Yb(TRAC). A lattice model based on DFT calculations suggests that the reduced propensity to volatilization is due to stronger interactions between CF3 and CH3 groups of neighboring fragments. This observation is in striking contrast with TGA studies reported previously for homoleptic transition metal and aluminum β-diketonates that show stepwise increases in volatility with increasing fluorination in the order of M(acac)3 < M(tfac)3 < M(hfac)3 (M = Al, Cr. Fe, and Rh) with the same substituent combinations as Yb(TRAC) (R, R′ = Me), Yb-4 (R = CF3, R′ = Me), and Yb-1 (R, R′ = CF3).9 Collectively, these studies show how ligand fluorination does not necessarily guarantee an increase in the volatility of lanthanide complexes.
Experimental
General considerations
All reactions were carried out under an atmosphere of N2 or Ar using glovebox or standard Schlenk techniques unless stated otherwise. All glassware was heated at 150 °C for at least two hours and allowed to cool under vacuum before use. Except for those used for chromatography, solvents were dried and deoxygenated using a Pure Process Technologies Solvent Purification System and stored over 3 Å molecular sieves. Deuterated solvents were deoxygenated on the Schlenk line with five freeze–pump–thaw cycles and stored over 3 Å molecular sieves for at least three days before use. H3(TRAC) and Yb(TRAC) were prepared from commercially available starting materials using the method described by Orvig and coworkers.21,22 Nd[N(SiMe3)2]3, Er[N(SiMe3)2]3, and Yb[N(SiMe3)2]3 were prepared by the method described by Bradley and coworkers.31 Reagents and chromatography solvents were purchased from commercial vendors and used as received.1H NMR data were collected on a Bruker AVANCE-300 or DPX-300 operating at 300 MHz, a Bruker AVANCE-400 operating at 400 MHz, or a Bruker AVANCE-500 operating at 500 MHz. The 19F NMR data were collected on a Bruker AVANCE-300 or DPX-300 operating at 282 MHz, a Bruker AVANCE-400 operating at 377 MHz, or a Bruker AVANCE-500 operating at 470 MHz. Chemical shifts are reported in δ units relative to residual NMR solvent peaks (1H) or 10% fluorobenzene in C6D6 (19F; δ −62.9 ppm). Microanalytical data (CHN) were collected using an EAI CE-440 elemental analyzer at the University of Iowa's Shared Instrumentation Facility. IR spectra were acquired with a Thermo Scientific Nicolet iS5 using an ATR accessory with a diamond crystal.
Thermogravimetric analysis data were collected on a TA Instruments Q500 TGA. Samples were transferred from the glovebox in a sealed container and quickly loaded in air. N2 was used as the purge gas. The balance purge flow was set to 10 mL min−1, and the sample purge was set to 90 mL min−1. The ramp rate for all samples was set to 2 °C min−1 with a start point at room temperature and an equilibration point set between 260 and 350 °C. Melting points were determined in capillaries sealed under N2 using a REACH MP device.
H 3 (2). Tris(2-aminoethyl)amine (0.16 mL, 1.1 mmol) and 1,1,1,5,5,6,6,7,7,7-decafluoro-4,6-heptanedione (1.37 mL, 6.49 mmol) were added to toluene (50 mL). The solution became yellow and then turned light pink. The mixture was refluxed with stirring overnight, during which time the mixture turned dark brown. The mixture was allowed to cool to RT, filtered, and evaporated to dryness under vacuum. The residue was purified in air by column chromatography on silica gel (60–200 mesh; 60 Å pore size) using 50% ethyl acetate and 50% hexane as an eluent. The collected fraction was evaporated to dryness, and the oily residue was dissolved in the minimum amount of pentane. Cooling yielded a white solid. Yield: 0.29 g (27%). 1H NMR (300 MHz, CDCl3): δ 10.7 ppm (br s, 3H, OH), 5.86 ppm (s, 3H, CH), 3.63 ppm (q, 6H, CH2), 2.87 ppm (t, 6H, CH2). 19F NMR (282 MHz, CDCl3): δ −65.7 ppm (s, 3F, CF3), −78.9 ppm (t, 3F, CF3), −119.4 ppm (d, 2F, CF2), −125.1 ppm (s, 2F, CF2).
H 3 (3). Tris(2-aminoethyl)amine (0.169 mL, 1.13 mmol) and 1,1,1,2,2,3,3-heptafluoro-7,7-dimethyl-4,6-octanedione (1.57 mL, 6.75 mmol) were added to toluene (10 mL) and the mixture was refluxed with stirring overnight. The mixture was allowed to cool to RT and then evaporated to dryness under vacuum. The mixture was extracted in air with a minimum amount of Et2O, layered with pentane, and cooled to −30 °C to yield white blocks. Yield: 0.694 g (84%). 1H NMR (500 MHz, CDCl3): δ 11.9 ppm (br s, 3H, OH), 5.46 ppm (s, 3H, CH), 3.74 ppm (s, 6H, CH2), 2.83 ppm (s, 6H, CH2), 1.28 ppm (s, 27H, tBu). 19F NMR (377 MHz, CDCl3): δ −86.7 ppm (s, 3F, CF2), −115.9 ppm (br d, 2F, CF2), −131.0 ppm (s, 2F, CF2).
H 3 (1). Prepared as described for H3(3) using tris(2-aminoethyl)amine (5.67 mL, 41.0 mmol) and hexafluoroacetylacetone (1.02 mL, 6.84 mmol). Yield: 1.19 g (25%). 1H NMR (500 MHz, CDCl3): δ 10.6 ppm (br s, 3H, OH), 5.80 ppm (s, 3H, CH), 3.62 ppm (t, 6H, CH2), 2.88 ppm (t, 6H, CH2). 19F NMR (377 MHz, CDCl3): δ −67.4 ppm, −77.5 ppm.
H 3 (4). Tris(2-aminoethyl)amine (0.204 mL, 1.37 mmol) and 1,1,1-trifluoroacetylacetone (1.0 mL, 8.2 mmol) were added to toluene (30 mL). The mixture was refluxed with stirring overnight and then 3 Å molecular sieves were added. Note: we found it important to follow these steps exactly to minimize the formation of difficult to remove side products. The mixture was again refluxed for 2 h and then allowed to cool to RT. The mixture was filtered in air, evaporated to dryness under vacuum, washed with 50 mL of pentane, and again evaporated to dryness to yield a reddish-yellow solid. The solid was extracted with Et2O (60 mL), evaporated again to dryness, and the solid was washed again with pentane (10 mL). The solid was dissolved in Et2O (15 mL) and layered with pentane. Layering yielded a yellow solid that was no longer Et2O soluble. The solid was washed with 80 mL of Et2O and evaporated to dryness. Yield: 0.22 g (29%). 1H NMR (300 MHz, CDCl3): δ 11.19 ppm (br s, 3H, OH), 5.28 ppm (s, 3H, CH), 3.51 ppm (q, 6H, CH2), 2.89 ppm (t, 6H, CH2), 2.06 ppm (s, 9H, CH3). 19F NMR (282 MHz, CDCl3): −77.2 ppm.
Yb-1. Yb[N(SiMe3)2]3 (0.100 g, 0.153 mmol) and H3(1) (0.109 g, 0.153 mmol) were dissolved in Et2O and stirred for 2 h. The solution was evaporated to dryness under vacuum to yield a yellow-orange powder. The powder was redissolved in the minimum amount of Et2O, layered with pentane, and the mixture was stored at −30 °C to yield pale-yellow crystals. Yield: 0.123 g (91%). Mp: 245 °C. Subl. Temp. 120 °C (10−2 Torr). Anal. Calcd for C12H15F18N4O3Yb: C, 28.46; H, 1.71; N, 6.32. Found: C, 28.45; H, 1.95; N, 6.15. 1H NMR (500 MHz, C6D6): δ 21.2 ppm (br s, 6 H, CH2), 10.7 ppm (br s, 3 H, CH2), 6.18 ppm (br s, 3 H, CH2), −0.82 ppm (s, 3 H, CH). 19F NMR (377 MHz, C6D6): δ −71.5 ppm (s), −73.6 ppm (s). IR (cm−1) 3181 (w), 3010 (w), 2950 (w), 2892 (w), 2168 (w), 1625 (s), 1608 (w), 1545 (s), 1517 (m), 1478 (s), 1471 (s), 1316 (s), 1267 (s), 1263 (s), 1192 (s), 1173 (s), 1149 (s), 1125 (s), 1110 (s), 1060 (s), 1031 (s), 966 (w), 947 (m), 917 (m), 867 (m), 848 (m), 792 (s), 737 (s), 663 (s).
Er-1. Prepared as described for Yb-1 with Er[N(SiMe3)2]3 (0.100 g, 0.154 mmol) and H3(1) (0.110 g, 0.154 mmol). Layering yielded purple crystals. Yield: 0.115 g (85%). Mp: 245 °C. Anal. Calcd for C12H15ErF18N4O3: C, 28.64; H, 1.72; N, 6.36. Found: C, 27.99; H, 1.97; N, 6.63. 19F NMR (470 MHz, C7D8): δ −68.4 ppm (s), −78.9 ppm (s). IR (cm−1) 2962 (w), 2875 (w), 2189 (w), 1684 (w), 1626 (m), 1599 (w), 1545 (m), 1535 (w), 1474 (m), 1383 (w), 1316 (m), 1269 (s), 1253 (s), 1175 (s), 1123 (s), 1092 (s), 1059 (s), 1027 (s), 943 (m), 914 (m), 866 (m), 847 (s), 793 (s), 736 (s), 663 (s).
Yb-2. Prepared as described for Yb-1 with Yb[N(SiMe3)2]3 (0.0875 g, 0.134 mmol) and H3(2) (0.136 g, 0.134 mmol). Yield: 0.129 g (90%). Mp: 245 °C. Subl. Temp. 120 °C at 10−2 Torr. Anal. Calcd for C28H24F23N4O3Yb: C, 27.33; H, 1.27; N, 4.72. Found: C, 27.74; H, 1.34; N, 4.79. 1H NMR (500 MHz, C6D6): δ 34.1 ppm (br s, 3 H, CH2), 32.8 ppm (br s, 3 H, CH2), 11.3 ppm (br s, 3 H, CH2), 7.66 ppm (br s, 3 H, CH2), −4.18 ppm (s, 3 H, CH). 19F NMR (377 MHz, C6D6): δ −74.8 ppm (s), −78.7 ppm (s), −111.3 ppm (d), −119.5 ppm (d), −122.3 ppm (s). IR (cm−1) 3175 (w), 3010 (w), 2882 (w), 2168 (w), 2046 (w), 1617 (s), 1534 (s), 1479 (s), 1349 (m), 1309 (w), 1198 (s), 1142 (s), 1114 (s), 1059 (s), 1031 (s), 966 (s), 959 (m), 924 (m), 902 (m), 844 (m), 839 (w), 795 (s), 749 (s), 677 (w).
Yb-3. Prepared as described for Yb-1 with Yb[N(SiMe3)2]3 (0.100 g, 0.153 mmol) and H3(3) (0.150 g, 0.153 mmol). Yield: 0.141 g (82%). Mp: 210 °C. Subl. Temp. 160 °C at 10−2 Torr. Anal. Calcd for C36H42F21N4O3Yb: C, 37.57; H, 3.68; N, 4.87. Found: C, 38.2; H, 3.96; N, 5.14. 1H NMR (500 MHz, C6D6): δ 60.7 ppm (br s, 3 H, CH2), 59.2 ppm (br s, 3 H, CH2), 14.73 ppm (br s, 3 H. CH2), 10.9 ppm (br s, 3 H, CH2), −9.69 ppm (s, 27 H, C(CH3)3), −12.7 ppm (s, 3 H, CH). 19F NMR (470 MHz, C6D6): δ −70.9 ppm (s), −110.2 ppm (d), −114.1 ppm (q), −120.0 ppm (d). IR (cm−1) 3173 (w), 2978 (w), 2933 (w), 2876 (w), 1161 (s), 1512 (s), 1496 (m), 1403 (w), 1371 (w), 1346 (m), 1225 (s), 1191 (s), 1164 (s), 1138 (s), 1135 (s), 1065 (s), 1037 (m), 965 (s), 917 (m), 835 (s), 784 (m), 750 (s), 680 (w), 644 (w).
Nd-3. Nd[N(SiMe3)2]3 (0.100 g, 0.160 mmol) and H3(3) (0.157 g, 0.160 mmol) were dissolved in toluene and refluxed for 2 h. The solution was evaporated to dryness under vacuum to yield a light blue powder. The powder was redissolved in the minimum amount of Et2O, layered with pentane, and the mixture was stored at −30 °C to yield blue crystals. Yield: 0.168 g (94%). Mp: 210 °C. Subl. Temp. 160 °C at 10−2 Torr. Anal. Calcd for C36H42F21N4NdO3: C, 38.54; H, 3.77; N, 4.99. Found: C, 37.22; H, 3.82; N, 5.06. 1H NMR (500 MHz, C6D6): δ 12.8 ppm (s, 3 H, CH), 4.81 ppm (s, 27 H, C(CH3)3), 0.53 ppm (br s, 3 H, CH2), −3.27 ppm (br s, 3 H, CH2), −13.4 ppm (br s, 3 H, CH2), −18.1 ppm (br s, 3 H, CH2). 19F NMR (470 MHz, C6D6): δ −86.7 ppm (s), −115.8 ppm (b), −131.0 ppm (s). IR (cm−1) 3410 (b), 2976 (w), 2911 (w), 1613 (s), 1522 (s), 1454 (m), 1403 (w), 1374 (w), 1344 (m), 1221 (s), 1201 (s), 1174 (s), 1140 (s), 1106 (s), 1086 (m), 1032 (m), 965 (m), 904 (s), 829 (m), 786 (m), 747 (s), 704 (w), 677 (w), 637 (w).
Yb-4. Yb[N(SiMe3)2]3 (0.240 g, 0.160 mmol) and H3(4) (0.200 g, 0.160 mmol) were stirred in THF overnight. The solution was evaporated to dryness under vacuum to yield a pale-yellow powder. The powder was then redissolved in the minimum amount of Et2O, layered with pentane, and the mixture was stored at −30 °C. Yield: 0.227 g (87%). Mp: 250 °C. Subl. Temp. 190 °C at 10−2 Torr. Anal. Calcd for C36H42F21N4NdO3: C, 34.82; H, 3.34; N, 7.73. Found: C, 34.47; H, 3.22; N, 6.98. 1H NMR (500 MHz, C6D6): δ 0.03 ppm (br s, 9 H, CH3), 1.92 ppm (br s, 3 H, CH2), 2.82 ppm (br s, 3 H, CH2), 4.73 (br s, 3 H, CH), 5.22 ppm (br s, 3 H, CH2), 6.49 ppm (br s, 3 H, CH2). 19F NMR (470 MHz, C6D6): δ −76.41 ppm (s). IR (cm−1) 2949 (w), 2892 (w), 1480 (w), 1446 (w), 1426 (w), 1361 (m), 1307 (w), 1285 (m), 1268 (m), 1246 (s), 1223 (m), 1197 (w), 1150 (m), 1134 (s), 1098 (s), 1085 (s), 1050 (s), 1010 (s), 945 (s), 932 (m), 914 (m), 887 (m), 816 (s), 804 (m), 770 (s), 748 (w), 647 (s), 635 (s).
Computational details
While lattice energies can be determined in a more rigorous way using periodic density functional theory (DFT) methods, we hypothesized that a simple lattice model could explain the differences in volatility. Specifically, the number and specific type of contacts (interactions with the nearest neighbor) surrounding a monomer were analyzed by computing the non-covalent interactions (NCI) of the unit cell. The total interaction in the cell can be approximated as a sum of these pair-wise nearest neighbor interaction energies. Therefore, to analyze non-covalent interactions for each crystal, all possible dimers whose Yb–Yb distance is no longer than 11 Å were located. To account for every possible dimer, a supercell of 2 × 2 × 2 was considered. Subsequently, the positions of the hydrogen atoms were optimized at the PBE0-D3/def2-TZVP level of theory.32–35 Heavy atoms were fixed at the experimental positions to ensure the solid-state environment was maintained. The obtained electron density was then used to calculate the NCIs. The identification and strength of the interactions were estimated using the IGMPlot software.36–40Data availability
A data set collection of computational results is available in the ioChem-BD repository41 and can be accessed online using the following link: https://iochem-bd.bsc.es/browse/review-collection/100/314943/93ff121cbebcd98b8a00e40f.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Separation Science program under award DESC0019426. We thank Dale Swenson and Daniel Unruh for collecting the single-crystal XRD data using the instrument supported by NSF CHE-1828117. Some of the NMR data were collected using the instrument supported by NSF CHE-2017828. Computations supporting this project were performed on High Performance Computing systems at the University of South Dakota, funded by NSF Award OAC-1626516. JCZ and SRD thank Tori Forbes for use of the TGA instrument and Daniel Unruh for assistance with modeling C-F disorder in several of the structures. JB and BV thank Kh Mahfuzul Alam for very helpful discussions on intermolecular interaction energies. JB and BV acknowledge that the land their research was performed on is the original homelands of the Dakota, Lakota, and Nakota tribal nations.References
- A. Drozdov and N. Kuzmina, Volatile compounds of lanthanides, Compr. Inorg. Chem. II, 2013, 2, 511–534 CAS.
- A. F. Novgorodov, F. Rösch and N. A. Korolev, Radiochemical Separations by Thermochromatography, in Handbook of Nuclear Chemistry, ed. A. Vértes, S. Nagy, Z. Klencsár, R. G. Lovas and F. Rösch, Springer US, 2011, pp. 2429–2458 Search PubMed.
- I. Zvara, The fundamentals of vacuum thermochromatography, J. Radioanal. Nucl. Chem., 2010, 286, 597–602 CrossRef CAS.
- D. E. Hanson, J. R. Garrison and H. L. Hall, Assessing thermo chromatography as a separation method for nuclear forensics: current capability vis-a-vis forensic requirements, J. Radioanal. Nucl. Chem., 2011, 289, 213–223 CrossRef CAS.
- R. E. Sievers, K. J. Eisentraut, C. S. Springer Jr. and D. W. Meek, Volatile rare earth chelates of β-diketones, Adv. Chem. Ser., 1967, 71, 141–154 CrossRef.
- J. A. Belot, A. Wang, R. J. McNeely, L. Liable-Sands, A. L. Rheingold and T. J. Marks, Highly volatile, low melting, fluorine-free precursors for metal-organic chemical vapor deposition of lanthanide oxide-containing thin films, Chem. Vap. Deposition, 1999, 5, 65–69 CrossRef CAS.
- N. L. Edleman, A. Wang, J. A. Belot, A. W. Metz, J. R. Babcock, A. M. Kawaoka, J. Ni, M. V. Metz, C. J. Flaschenriem, C. L. Stern, L. M. Liable-Sands, A. L. Rheingold, P. R. Markworth, R. P. H. Chang, M. P. Chudzik, C. R. Kannewurf and T. J. Marks, Synthesis and characterization of volatile, fluorine-free β-ketoiminate lanthanide MOCVD precursors and their implementation in low-temperature growth of epitaxial CeO2 buffer layers for superconducting electronics, Inorg. Chem., 2002, 41, 5005–5023 CrossRef CAS PubMed.
- S. Battiato, P. Rossi, P. Paoli and G. Malandrino, Heterobimetallic Sodium Rare-Earth Complexes: “Third-Generation” MOCVD Precursors for the Deposition of NaREF4 (RE = Y, Gd) Films, Inorg. Chem., 2018, 57, 15035–15039 CrossRef CAS PubMed.
- K. J. Eisentraut and R. E. Sievers, Thermogravimetric studies of metal β-diketonates, J. Inorg. Nucl. Chem., 1967, 29, 1931–1936 CrossRef CAS.
- C. S. Springer Jr., D. W. Meek and R. E. Sievers, Rare earth chelates of 1,1,1,2,2,3,3-heptafluoro-7,7-dimethyl-4,6-octanedione, Inorg. Chem., 1967, 6, 1105–1110 CrossRef.
- J. E. Sicre, J. T. Dubois, K. J. Eisentraut and R. E. Sievers, Volatile lanthanide chelates. II. Vapor pressures, heats of vaporization, and heats of sublimation, J. Am. Chem. Soc., 1969, 91, 3476–3481 CrossRef CAS.
- K. Utsunomiya, Gas chromatography of rare-earth chelates of isobutyrylpivalylmethane, Anal. Chim. Acta, 1972, 59, 147–151 CrossRef CAS.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, Low coordination numbers in lanthanide and actinide compounds. I. Preparation and characterization of tris[bis(trimethylsilyl)amido]lanthanides, J. Chem. Soc., Dalton Trans., 1973, 1021–1023 RSC.
- J. M. Birmingham and G. Wilkinson, Cyclopentadienides of scandium, yttrium, and some rare earth elements, J. Am. Chem. Soc., 1956, 78, 42–44 CrossRef CAS.
- S. R. Daly, D. Y. Kim, Y. Yang, J. R. Abelson and G. S. Girolami, Lanthanide, N,N-dimethylaminodiboranates: highly volatile precursors for the deposition of lanthanide-containing thin films, J. Am. Chem. Soc., 2010, 132, 2106–2107 CrossRef CAS PubMed.
- S. R. Daly, D. Y. Kim and G. S. Girolami, Lanthanide, N,N-dimethylaminodiboranates as a new class of highly volatile chemical vapor deposition precursors, Inorg. Chem., 2012, 51, 7050–7065 CrossRef CAS PubMed.
- A. E. Khochenkov, N. V. Belova, A. V. Krasnov, Y. A. Zhabanov, N. I. Giricheva and G. V. Girichev, Sublimation enthalpy of tetrakis(1,1,1,5,5,5-hexafluoro-2,4-pentanedionato)lanthanide(III)-potassium complexes, KLn(C5HF6O2)4: Effect of lanthanide contraction, J. Chem. Thermodyn., 2019, 131, 117–121 CrossRef CAS.
- A. S. Hyre and L. H. Doerrer, A structural and spectroscopic overview of molecular lanthanide complexes with fluorinated O-donor ligands, Coord. Chem. Rev., 2020, 404, 213098 CrossRef CAS.
- E. W. Berg and J. J. Chiang-Acosta, Fractional sublimation of the β-diketone chelates of the lanthanide and related elements, Anal. Chim. Acta, 1968, 40, 101–113 CrossRef CAS.
- J. Schläfer, D. Graf, G. Fornalczyk, A. Mettenbörger and S. Mathur, Fluorinated Cerium(IV) Enaminolates: Alternative Precursors for Chemical Vapor Deposition of CeO2 Thin Films, Inorg. Chem., 2016, 55, 5422–5429 CrossRef PubMed.
- D. J. Berg, S. J. Rettig and C. Orvig, Heptadentate ligands for the lanthanides. The first structurally characterized example of a lanthanide heptadentate ligand complex: [tris(3-aza-4-methyl-6-oxohept-4-en-1-yl)amine]ytterbium(III), J. Am. Chem. Soc., 1991, 113, 2528–2532 CrossRef CAS.
- A. Smith, S. J. Rettig and C. Orvig, Lanthanide complexes of potentially heptadentate ligands including the structure of [tris(3-aza-4-methylhept-4-ene-6-on-1-yl)amine]tris(nitrato)gadolinium(III), Inorg. Chem., 1988, 27, 3929–3934 CrossRef CAS.
- R. Hoffmann, B. F. Beier, E. L. Muetterties and A. R. Rossi, Seven-coordination. A molecular orbital exploration of structure, stereochemistry, and reaction dynamics, Inorg. Chem., 1977, 16, 511–522 CrossRef CAS.
- W. S. Rees Jr., O. Just, S. L. Castro and J. S. Matthews, Synthesis and magnetic and structural characterization of the first homoleptic lanthanide β-ketoiminate, Inorg. Chem., 2000, 39, 3736–3737 CrossRef CAS PubMed.
- Q. Xia, Y. Cui, D. Yuan, Y. Wang and Y. Yao, Synthesis and characterization of lanthanide complexes stabilized by N-aryl substituted β-ketoiminato ligands and their application in the polymerization of rac-lactide, J. Organomet. Chem., 2017, 846, 161–168 CrossRef CAS.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. M. Cohen, M. Meyer and K. N. Raymond, Enterobactin Protonation and Iron Release: Hexadentate Tris-Salicylate Ligands as Models for Triprotonated Ferric Enterobactin, J. Am. Chem. Soc., 1998, 120, 6277–6286 CrossRef CAS.
- S. M. Cohen and K. N. Raymond, Catecholate/Salicylate Heteropodands: Demonstration of a Catecholate to Salicylate Coordination Change1, Inorg. Chem., 2000, 39, 3624–3631 CrossRef CAS PubMed.
- T. Müntener, D. Joss, D. Häussinger and S. Hiller, Pseudocontact Shifts in Biomolecular NMR Spectroscopy, Chem. Rev., 2022, 122, 9422–9467 CrossRef PubMed.
- K. Mason, A. C. Harnden, C. W. Patrick, A. W. J. Poh, A. S. Batsanov, E. A. Suturina, M. Vonci, E. J. L. McInnes, N. F. Chilton and D. Parker, Exquisite sensitivity of the ligand field to solvation and donor polarisability in coordinatively saturated lanthanide complexes, Chem. Commun., 2018, 54, 8486–8489 RSC.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, Three coordination in lanthanide chemistry: tris[bis(trimethylsilyl)amido]lanthanide(III) compounds, J. Chem. Soc., Chem. Commun., 1972, 349–350 RSC.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- C. Lefebvre, G. Rubez, H. Khartabil, J.-C. Boisson, J. Contreras-García and E. Hénon, Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC.
- C. Lefebvre, H. Khartabil, J.-C. Boisson, J. Contreras-García, J.-P. Piquemal and E. Hénon, The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations, ChemPhysChem, 2018, 19, 724–735 CrossRef CAS PubMed.
- J. Klein, H. Khartabil, J.-C. Boisson, J. Contreras-García, J.-P. Piquemal and E. Hénon, New Way for Probing Bond Strength, J. Phys. Chem. A, 2020, 124, 1850–1860 CrossRef CAS PubMed.
- M. Ponce-Vargas, C. Lefebvre, J.-C. Boisson and E. Hénon, Atomic Decomposition Scheme of Noncovalent Interactions Applied to Host–Guest Assemblies, J. Chem. Inf. Model., 2020, 60, 268–278 CrossRef CAS PubMed.
- C. Lefebvre, J. Klein, H. Khartabil, J. C. Boisson and E. Hénon, IGMPlot: A program to identify, characterize, and quantify molecular interactions, J. Comput. Chem., 2023, 44, 1750–1766 CrossRef CAS PubMed.
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, Managing the Computational Chemistry Big Data Problem: The ioChem-BD Platform, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details and molecular structures of Yb-2, Yb-4, Er-1, and Nd-3. NMR and IR spectra, TGA plots. Interacting pairs from DFT calculations. CCDC 2313982–2313988. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04198k |
| This journal is © The Royal Society of Chemistry 2024 |