
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A cross-metathesis approach for polymetallic [FeFe]-hydrogenase mimics†
Sergio
Aguado
 ac,
Pablo
García-Álvarez
bc,
Javier A.
Cabeza
bc,
Luis
Casarrubios
*ac and
Miguel A.
Sierra
*ac
ac,
Pablo
García-Álvarez
bc,
Javier A.
Cabeza
bc,
Luis
Casarrubios
*ac and
Miguel A.
Sierra
*ac
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense, 28040 Madrid, Spain. E-mail: sierraor@ucm.es; lcasarru@ucm.es
bDepartamento de Química Orgánica e Inorgánica. Facultad de Química, Universidad de Oviedo, 33071 Oviedo, Spain
cCentro de Innovación en Química Avanzada, (ORFEO-CINQA), Spain
First published on 24th January 2024
Abstract
A method has been developed for synthesizing [FeFe]-H2ase mimics with diverse structures and properties, employing cross-metathesis of olefins. Vinylmetallocenes (5 and 6) and vinyl half-sandwich complexes (10 and 11) have been used as cross-metathesis partners with [FeFe]-H2ase mimics (4, 8, and 9) bearing a double bond in the moiety attached to the ADT-bridge nitrogen. Electrochemical studies of these complexes, encompassing metallocene-type (7a–b, 12a–b, and 13a–b) as well as half-sandwich derivatives (12c and 13c–d), have demonstrated that the introduction of a redox unit has a marginal impact on the reduction potential of these [FeFe]-H2ase mimics. The application of this cross-metathesis approach has allowed the synthesis of [FeFe]-H2ase mimics featuring an Ir(III) electrochemical antenna (16–18) as well as systems having an electron-donor-photosensitizer structure (ED-PS) (23). The electrocatalytic properties of these complexes have been elucidated through electrochemical studies.
Introduction
Hydrogenases are a class of metalloenzymes capable of catalyzing one of the simplest reactions in chemistry, namely the conversion of protons to molecular hydrogen.1 These enzymes play a fundamental role in the energy metabolism of several microorganisms, as well as in the modulation of the redox potential of the microorganism cell.2 Depending on the metabolic and energetic requisites of the cell, hydrogenase metalloenzymes can promote the evolution or the decomposition of molecular hydrogen. Additionally, these enzymes play a key role in the modulation of transmembrane proton gradients.3 Reversible oxidation of molecular hydrogen occurs in the enzyme active metal-center.4Depending on the nature of the metals present in this active centre, hydrogenases are classified into three different classes: [NiFe]- (A), [FeFe]- (B) and [Fe]-hydrogenases (C) (Fig. 1). The activity in the hydrogen evolution reaction (HER) depends on the enzyme type.2 Thus, [FeFe]-hydrogenases ([FeFe]-H2ases) are highly active in hydrogen production (up to 8000 μmol H2 min−1 mg−1),5 while [NiFe]-hydrogenases efficiently catalyse the oxidation of hydrogen.6 [Fe]-hydrogenases are present in some species of methanogenic archaea, catalysing the reduction of CO2 to CH4.7 In this regard, the similarity of [(μ-S)2Fe2(CO)6] and the active centre of [FeFe]-H2ases opened the possibility of using simplified models (mimics) of these enzymes to effect the HER. The use of these [FeFe]-H2ase mimics has serious drawbacks with respect to the natural enzymes in the HER. Thus, different from the enzymes that can work at neutral pH, their mimics require lower pH values.8 The electrocatalytic reduction of protons by [FeFe]-H2ase mimics occurs at more negative potentials than that in natural enzymes with the subsequent overpotential cost.9 Finally, the systems for H+ and e− transfer that are present in the enzymes are absent in the [FeFe]-H2ase mimics. To solve these problems (knowing that the mimics present a higher tolerance to oxygen than the natural enzymes, which is one of the main shortcomings of these bio-catalysts) hundreds of [FeFe]-H2ases have so far been designed and synthesised.10
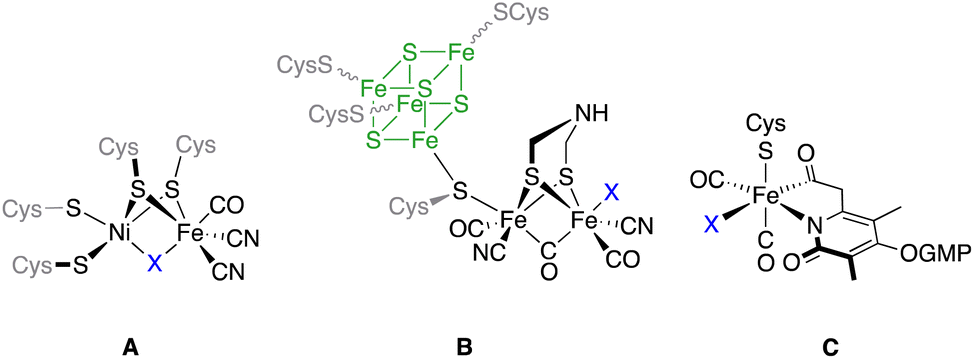 | ||
| Fig. 1 The metal cores present in the three classes of hydrogenases. For [FeFe]-H2ases, the Fe4S4 moiety has been drawn for clarity. | ||
The general approaches for the preparation of [FeFe]-H2ases are limited to the direct reaction of Fe(CO)5 or Fe3(CO)12 with sulphides and disulphides11 to yield complexes 1 and to reactions of highly reduced species [(μ-S)2Fe2(CO)6]2− with formaldehyde imines (either as isolated species or as isolable trimers) or dichlorodimethyl amines, to form compounds 2.12 The [2 + 2]-cycloaddition of [(μ-S)2Fe2(CO)6] to alkenes and alkynes affords complexes 3. This approach is less efficient but it tolerates a large array of functional groups (Scheme 1).13,14 The post-functionalization of simple [FeFe]-H2ase mimics has been restricted to ligand interchange in the inner coordination sphere of the [(μ-S)2Fe2(CO)6] derivative, or in other condensation reactions to attach these complexes to more complex molecular scaffolds.15
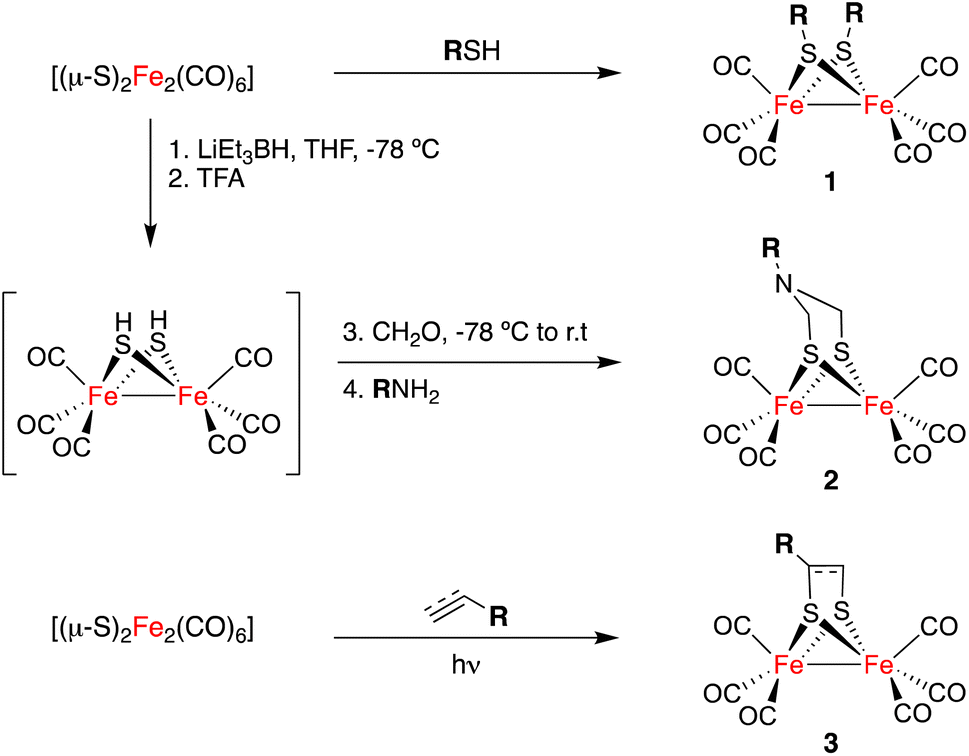 | ||
| Scheme 1 The three main approaches for preparing [FeFe]-H2ase mimics. | ||
Recently, we approached the synthesis of some complex [FeFe]-H2ase mimics by preparing the versatile reagent [(μ-ADT)RFe2(CO)6] (R = p-N3C6H4), and using this azide to introduce the [FeFe]-moiety into several substrates through a Cu-catalysed azide–alkyne cycloaddition.16 In parallel, the use of easily available phosphites having three [FeFe]-moieties allowed the coordination of these polymetallic ligands to different square-planar and piano-stool complexes.17 Moreover, the synthesis of heterometallic [FeFe]-mimics was carried out using tetranuclear complexes, in which the two [FeFe]-moieties were tethered by a bipyridine moiety suitable to complex different metals.18 The electronic and electrocatalytic properties of these compounds were highly influenced by the presence of additional transition metal complexes either enhancing the electronic communication between the [FeFe] centers or inhibiting it, depending on the metal.
Pursuing the idea of developing new methodologies to access heterometallic [FeFe]-H2ase mimics in an easy way and within the concept of precision chemistry, herein we report the preparation of several heteropolymetallic [FeFe]-H2-ase mimics through a simple and efficient cross-metathesis (CM) reaction, also including a new triad having an [ED-PS-F2S2] structure with electron donor (ED) and photosensitizer (PS) moieties, as well as their electrochemical and electrocatalytic properties.
Results and discussion
The initial study was aimed to assess the viability (and optimization of the reaction conditions) of a cross-metathesis reaction (CM) between a vinylmetallocene and an [FeFe]-H2ase mimic having a double bond attached to the nitrogen moiety of the adt-bridge. The reaction was studied using complex 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 and vinylferrocene 5.20 The Grubbs GBI catalyst led to low yields of the desired product 7a, requiring extended reaction times. The Hoveyda–Grubbs catalyst was more efficient but the main reaction products were homocoupling derivatives that were of little interest to fulfil our objectives. Finally, the use of the GBII catalyst led to the desired cross-metathesis product 7a in an acceptable yield (52%, isolated), minimizing the formation of homocoupling reaction products. The optimum solvent for the cross-metathesis process using the GBII catalyst was dichloromethane. These reaction conditions were subsequently applied to the reaction of complex 4 with vinyl ruthenocene 6, forming the cross-metathesis product 7b in 42% yield (Scheme 2).
19 and vinylferrocene 5.20 The Grubbs GBI catalyst led to low yields of the desired product 7a, requiring extended reaction times. The Hoveyda–Grubbs catalyst was more efficient but the main reaction products were homocoupling derivatives that were of little interest to fulfil our objectives. Finally, the use of the GBII catalyst led to the desired cross-metathesis product 7a in an acceptable yield (52%, isolated), minimizing the formation of homocoupling reaction products. The optimum solvent for the cross-metathesis process using the GBII catalyst was dichloromethane. These reaction conditions were subsequently applied to the reaction of complex 4 with vinyl ruthenocene 6, forming the cross-metathesis product 7b in 42% yield (Scheme 2).
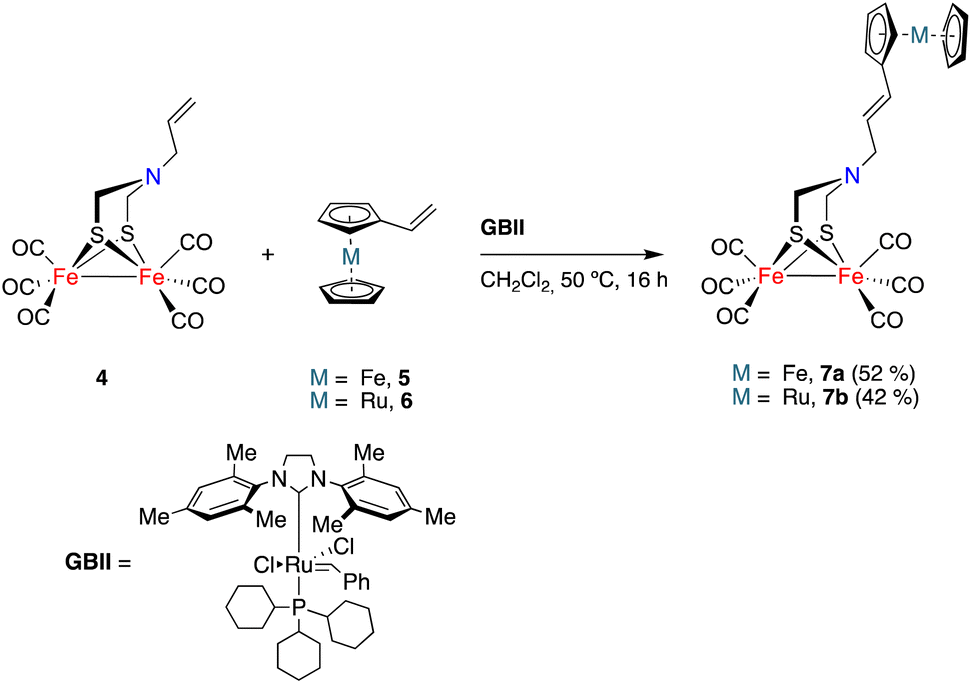 | ||
| Scheme 2 Synthesis of [FeFe]-hydrogenase mimics 7a and 7b. | ||
Complexes 7a and 7b are air-stable and were characterized by spectroscopic and spectrometric techniques. It is remarkable that both [FeFe]-H2ase mimics were formed exclusively as their E-isomers. The signals of the olefinic protons for 7a are clearly identified at 6.19 and 5.57 ppm (J = 15.6 Hz), while those for complex 7b are observed at 6.07 and 5.57 ppm (J = 15.6 Hz). The structure of complex 7a was unambiguously determined by X-ray diffraction (Fig. 2).
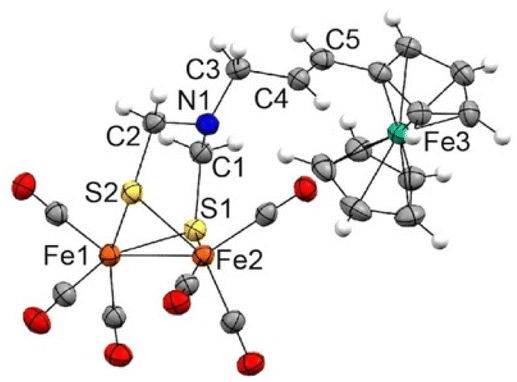 | ||
| Fig. 2 The X-ray structure of polynuclear complex 7a (30% displacement ellipsoids). Selected bond distances (Å): Fe1–Fe2 2.515(1), Fe1–S1 2.250(2), Fe1–S2 2.256(2), Fe2–S1 2.260(2), Fe2–S2 2.248(2), C1–N1 1.44(1), C1–S1 1.829(8), C2–N1 1.433(9), C2–S2 1.845(7), C3–N1 1.485(9), C3–C4 1.492(10), C4–C5 1.32(1). | ||
The generality of the process exemplified in Scheme 2 was proven by affecting the CM reactions of [FeFe]-hydrogenase mimics 8 and 9 with vinylmetallocenes 5 and 6 and half-sandwich complexes 10 and 11 (Scheme 3).21,22 Metallocene derivatives containing the [FeFe]-H2ase mimic moiety 12a–b and 13a–b were obtained in good yields and, consistently, exclusively as their E-isomers. The analogous complexes 12c and 13c–d, having a half-sandwich moiety tethered to the [FeFe]-H2ase mimic, were also obtained in good yields and again as their E-isomers. Yields for heterometallic mimics 13a (87%) and 13b (63%), derived from amides 9, were higher than those of their amine analogues 12a (62%) and 12b (54%).
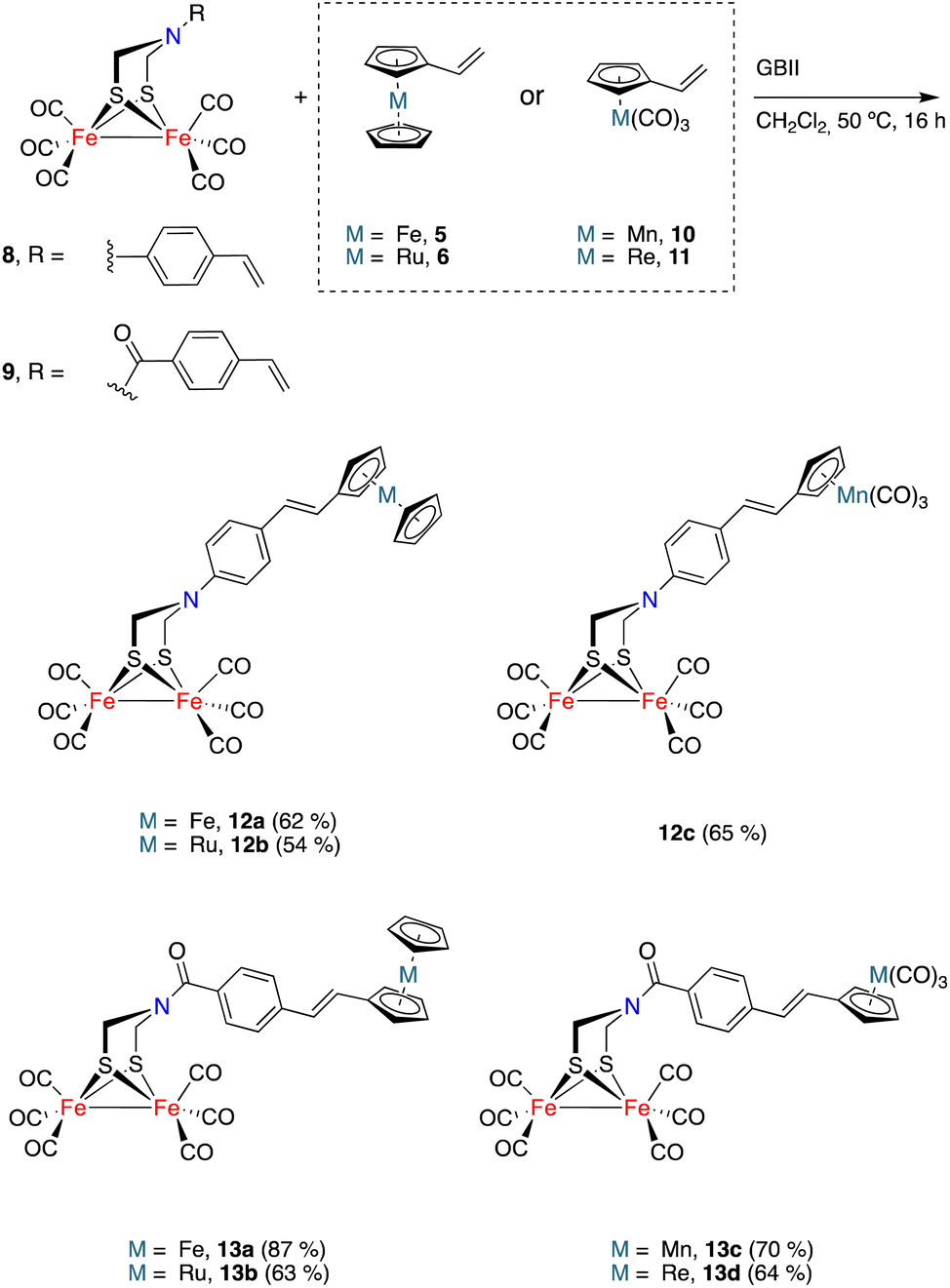 | ||
| Scheme 3 Syntheses of [FeFe]-H2ases bearing sandwich and half-sandwich moieties. | ||
Complexes 12c and 13c–d show two signals in the M-CO region of their 13C{1H} NMR spectra. One, at around 207.0 ppm, is attributable to the six equivalent CO ligands of the [FeFe] moiety and the other is attributable to the M(CO)3 moiety (224.1 ppm for Mn derivatives 12c and 13c, 193.9 ppm for the Re derivative 13d). The X-ray structure of half-sandwich 12c and metallocene 13b confirm the proposed molecular structures and the geometry of the double bond (Fig. 3).
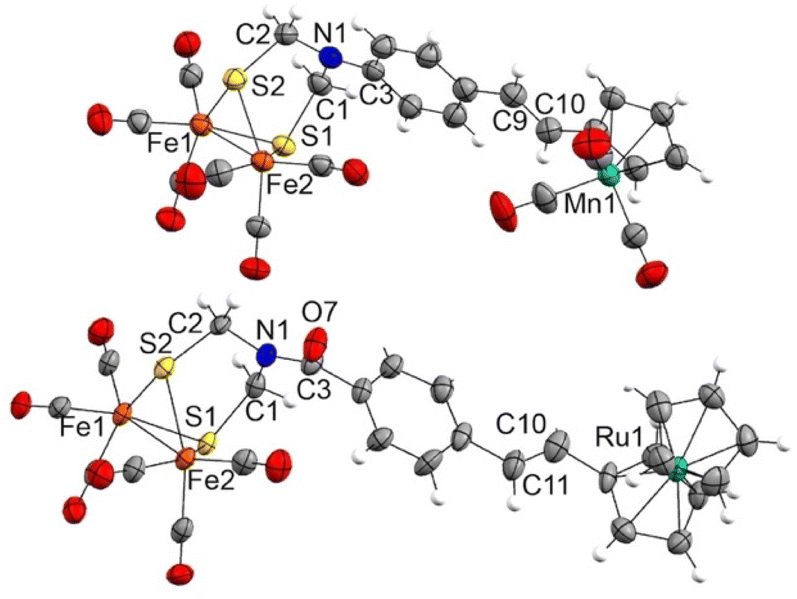 | ||
| Fig. 3 X-ray structures of heterometallic complexes 12c (top) and 13b (bottom) (30% displacement ellipsoids). Selected bond distances (Å): 12c: Fe1–Fe2 2.5070(8), Fe1–S1 2.2620(11), Fe1–S2 2.2580(11), Fe2–S1 2.2723(10), Fe2–S2 2.2533(11), C1–N1 1.428(6), C1–S1 1.870(4), C2–N1 1.420(6), C2–S2 1.871(5), C3–N1 1.397(6), C9–C10 1.311(8). 13b: Fe1–Fe2 2.4944(18), Fe1–S1 2.249(2), Fe1–S2 2.257(3), Fe2–S1 2.271(2), Fe2–S2 2.268(2), C1–N1 1.427(12), C1–S1 1.850(10), C2–N1 1.452(10), C2–S2 1.846(9), C3–N1 1.352(12), C3–O7 1.240(12), C10–C11 1.249(19). | ||
The possibility of applying the CM approach for the synthesis of [FeFe]-H2ase mimics having octahedral Ir(III) moieties was next tested. [FeFe]-H2ase mimics combining an octahedral photoactive moiety tethered to a [(μ-ADT)Fe2(CO)6] fragment have been previously reported,23 since the idea of the octahedral moiety acting as a photocatalyst to induce the electron transfer processes to the [FeFe] moiety is very attractive. Unfortunately, to date, efficient catalytic systems in the HER have not yet been achieved using this approach.24,25
Ir(III) complex 15 acting as a partner in the CM approach was prepared from [Ir(μ-Cl)(C^N)2]214 and 2-(4-vinylphenyl)pyridine26 in the presence of Ag(CF3SO3) in boiling 2-ethoxyethanol. Complex 15 was obtained in 83% yield as an orange solid and as a single isomer, and is a thermodynamically favored25fac complex. Reactions of complex 15 with [FeFe]-mimics 4, 8 and 9, under the conditions reported above, gave heterotrimetallic complexes 16–18 in good yields (Scheme 4). Complexes 16–18 were air-stable and were characterized using spectroscopic techniques. Again, these complexes were obtained as E-isomers (1H NMR JCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH = 15.8 Hz), showing the characteristic 13C{1H} NMR CO signals for the [(μ-ADT)Fe2(CO)6] moiety in the 207–208 ppm range. These spectroscopic data together with the HRMS data support the heterotrimetallic structure proposed for complexes.
CH = 15.8 Hz), showing the characteristic 13C{1H} NMR CO signals for the [(μ-ADT)Fe2(CO)6] moiety in the 207–208 ppm range. These spectroscopic data together with the HRMS data support the heterotrimetallic structure proposed for complexes.
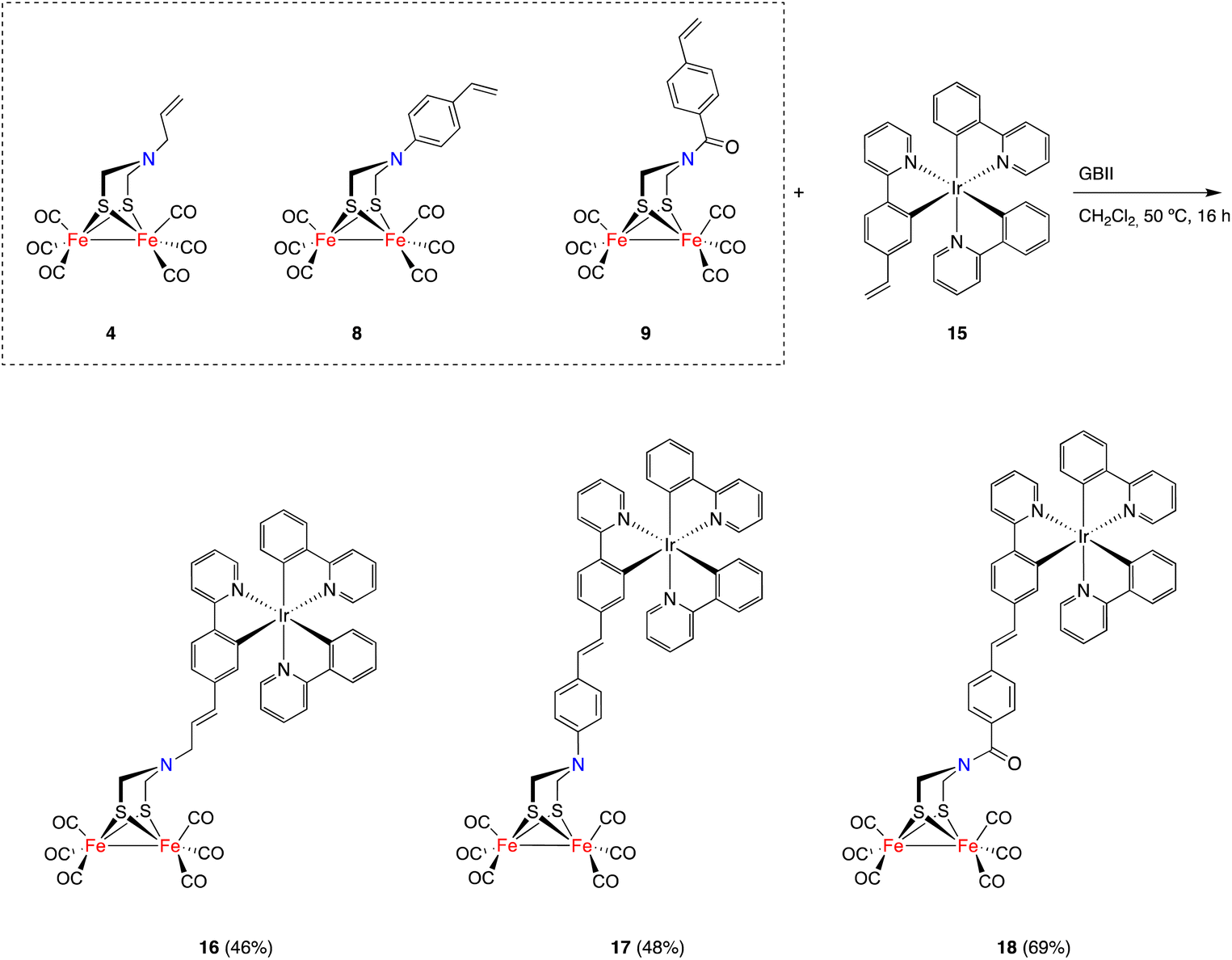 | ||
| Scheme 4 The synthesis of heterometallic [FeFe]-H2ase mimics. | ||
Finally, the CM approach for preparing [FeFe]-H2ase mimics was also used to prepare the [ED-PS-Fe2S2] system 23 (Scheme 5), where ED is an electron donor moiety (ferrocene) and PS is a photoactive moiety (Ir(III) complex). The synthesis began with the preparation of dimeric Ir(III) complex 19 (having formyl groups) by the reaction of 2-(4-formylphenyl)pyridine27 with IrCl3.nH2O in boiling 2-ethoxyethanol. This complex was obtained in 36% yield as a red solid. The dimeric structure of 19 was established using spectroscopic techniques. Its 1H and 13C{1H} NMR spectra show a single set of signals, which pointed to the coexistence of a diastereomeric mixture of compounds in solution. The reaction of dimeric complex 19 with 2-(4-vinylphenyl)pyridine in boiling 2-ethoxyethanol in the presence of silver triflate formed complex 20. The obtained solid was washed with EtOH and purified by silica gel chromatography (using DCM as the eluent) to obtain pure 20 as a red solid (47% yield). The structure of complex 20 was spectroscopically determined. The 1H NMR spectrum shows the characteristic vinyl protons at 5.54 (dd, J1 = 17.6, J2 = 1,3 Hz) and 5.01 (dd, J = 10.8, J2 = 1,3 Hz) ppm. The 13C{1H} NMR spectrum contains two signals, at 194.3 and 194.2 ppm, attributable to inequivalent aldehyde groups. This is consistent with a fac-disposition (thermodynamically favoured isomer).
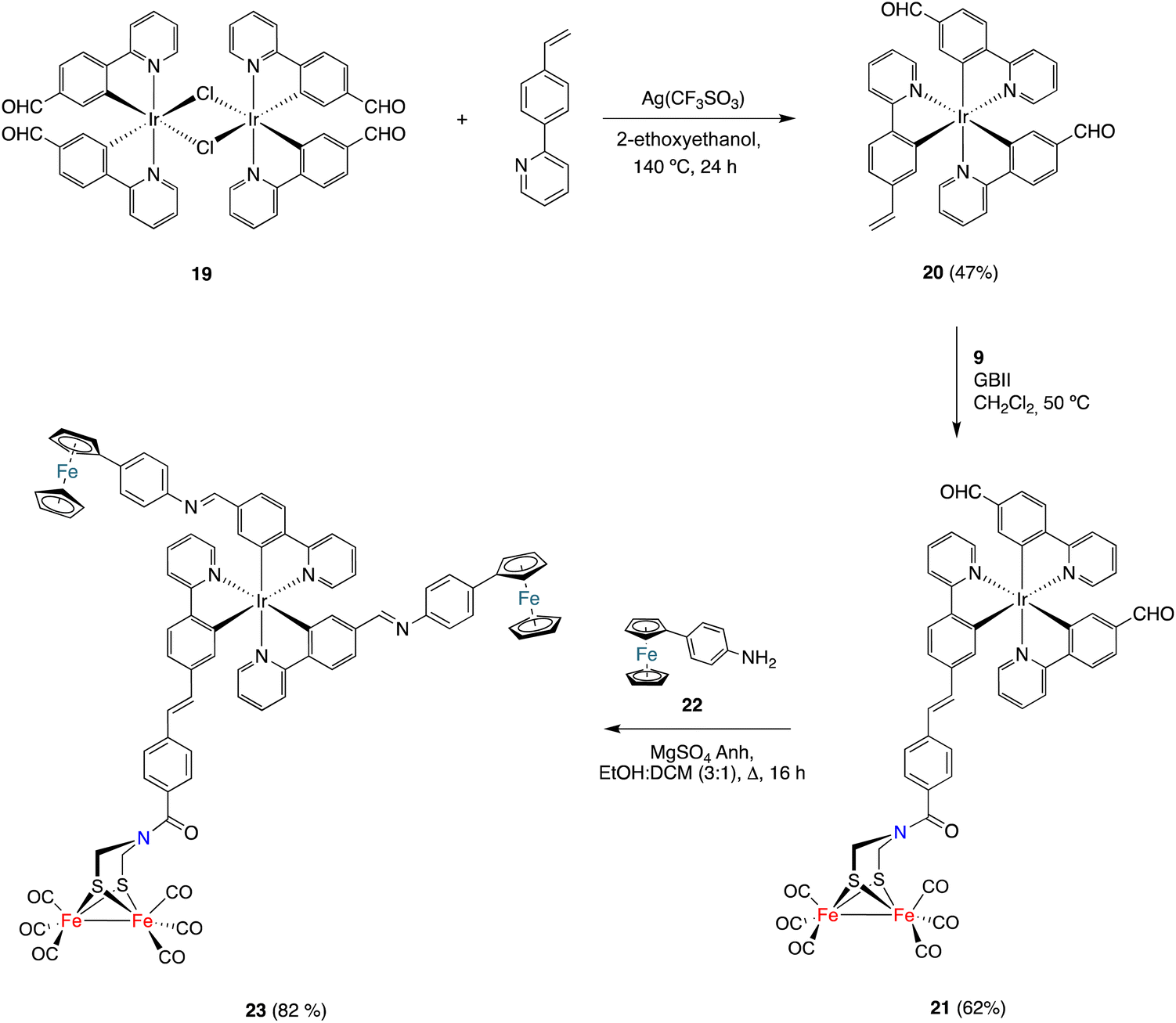 | ||
| Scheme 5 The synthesis of the [ED-PS-Fe2S2] system 23. | ||
Complex 20 was then reacted with alkene 9 under the CM conditions developed in this work. The CM product, namely complex 21, was obtained in 62% yield. The HRMS and spectroscopic data were consistent with the proposed structure. Thus, a signal corresponding to the six CO groups of the [FeFe]-moiety appeared at 207.0 ppm in the 13C{1H} NMR spectrum, while the two signals appearing at 194.5 and 194.3 ppm correspond to the inequivalent formyl groups. Finally, the signal for the amide-CO group appeared at 162.9 ppm. The E-geometry was derived from the coupling of the protons attached to the double bond (J = 15.8 Hz).
To achieve the synthesis of the [ED-PS-Fe2S2] system 23, complex 21 was condensed with p-aminophenyl ferrocene 1928 (2 equiv.) in a boiling solution of 3:1 EtOH/Cl2CH2. The desired complex was isolated as a red solid in 82% yield. Its structure was established using spectroscopic and HRMS techniques. Again, significant signals were observed for the two inequivalent imine protons at 8.29 and 8.23 ppm in the 1H NMR spectrum and a sharp signal at 207.0 ppm was observed in the 13C{1H} NMR spectrum, supporting the integrity of the [(μ-ADT)Fe2(CO)6] moiety in the imine formation reaction.
Electrochemistry
Complexes 7a–b, 12a–c and 13a–d show a quasireversible reduction wave in the range of −1.70 V to −1.60 V (vs. Fc+/0). This wave is attributable to the [FeIFeI] to [FeIFe0] reduction (Fig. S-1† and Table 1).29 Complexes 4, 8 and 9 have been included for comparison. The small displacement in the reduction wave of complex 13a with respect to 7a agrees with the reported scarce influence of the substituent R attached to the nitrogen of the ADT-bridge of [(μ-ADT)NRFe2(CO)6] complexes in their reduction potential.30| Complex | E pc1 | E pa1 | E (1) |
|---|---|---|---|
| 4 | −1.76 | −1.65 (0.11) | −1.70 |
| 7a | −1.74 | −1.64 (0.10) | −1.70 |
| 7b | −1.74 | −1.64 (0.10) | −1.69 |
| 8 | −1.70 | −1.62 (0.08) | −1.66 |
| 12a | −1.69 | −1.62 (0.07) | −1.65 |
| 12b | −1.69 | −1.62 (0.07) | −1.66 |
| 12c | −1.69 | −1.62 (0.07) | −1.65 |
| 9 | −1.64 | −1.57 (0.07) | −1.61 |
| 13a | −1.64 | −1.55 (0.09) | −1.60 |
| 13b | −1.64 | −1.59 (0.05) | −1.62 |
| 13c | −1.64 | −1.58 (0.06) | −1.61 |
| 13d | −1.64 | −1.58 (0.06) | −1.61 |
All complexes show a second irreversible reduction wave at −2.10 V (vs. Fc+/0), attributable to the reduction of [Fe0FeI] to [Fe0Fe0], and a quasireversible oxidation wave at around 0.50 V, attributable to the [FeIFeI] to [FeIFeII] oxidation.29 Additionally, complexes 4a, 12a and 13a show the oxidation waves characteristic of the ferrocene moiety at 0.0 V (vs. Fc+/0), while ruthenocene derivatives 4b, 12b and 13b show the quasireversible oxidation wave at around 0.20 V (vs. Fc+/0), corresponding to the Ru(II) to Ru(III) oxidation.
The electrocatalytic behavior of complexes 7, 12 and 13 in the presence of mild (AcOH, pKaMeCN = 22.331) and strong (CF3COOH, pKaMeCN = 12.632) acids was next studied. Fig. 4 compiles the results of both experiments. The electrochemistry in the presence of AcOH was the standard for related [(μ-ADT)NRFe2(CO)6] complexes.33 The current intensity for the first wave is independent of the amount of AcOH, while a strong increase in the current intensity was observed in the second wave (above 2.0 V). This behaviour is compatible with two successive reduction processes resulting in the HER.
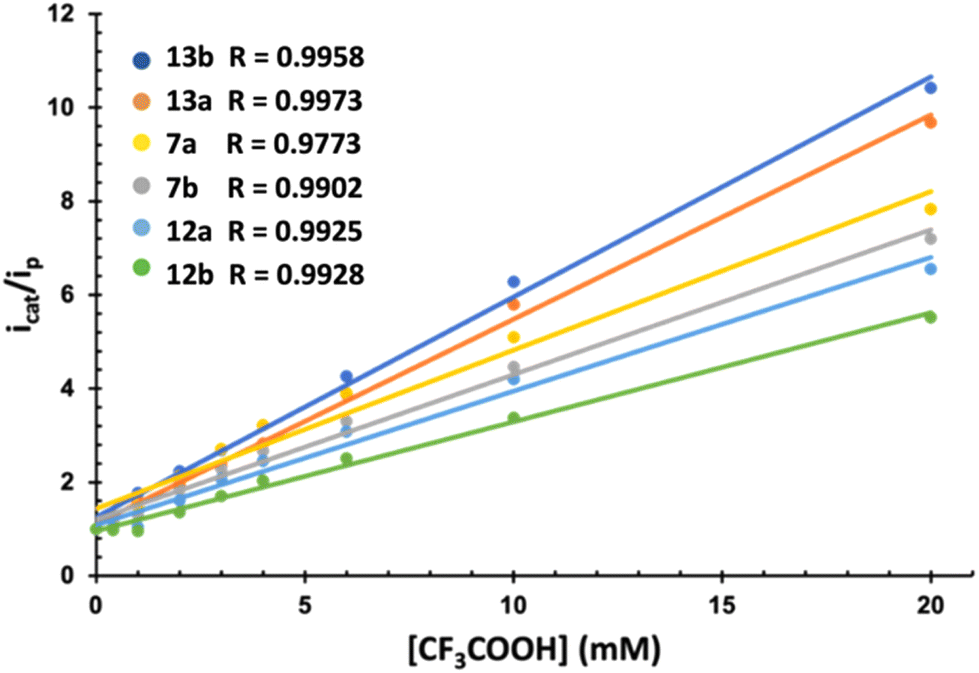 | ||
| Fig. 4 Representation of icat/ip for complexes 7, 12 and 13vs. TFA. | ||
Complexes 7, 12 and 13 behave differently in the presence of TFA. Now the first reduction wave in the range of −1.60 to −1.70 V (vs. Fc+/0) experiences a strong electrocatalytic response (Fig. S-3†). Moreover, this wave splits into two waves upon increasing the concentration of the acid, which is consistent with the reduction of the protonated species.
The different electrocatalytic responses were evaluated through the increase of the current intensity with the acid concentration of the first reduction process (icat) with respect to the basal current intensity of the same process in the absence the acid (ip) (Fig. 4). Similarly, plotting of icat/ipvs. [H+] allowed the calculation of the TOF for the reduction of H+ (Table 2).34 In all cases, the response increases linearly (Fig. 4), showing different sensibilities to the increase of [H+]. Amide derivatives 13b and 13a showed a stronger electrocatalytic response.
| Complex |
E
Catalytica (V) |
ηb (V) |
TOFc (s−1) |
|---|---|---|---|
a Data obtained from Fig. S-3.†
b Overpotential (η) calculated using  .10
c TOF calculated with 20 mM TFA. .10
c TOF calculated with 20 mM TFA.
|
|||
| 7a | −1.74 | 0.85 | 12.1 |
| 7b | −1.67 | 0.78 | 10.2 |
| 12a | −1.65 | 0.76 | 6.0 |
| 12b | −1.64 | 0.75 | 9.9 |
| 12c | −1.69 | 0.80 | 8.9 |
| 13a | −1.64 | 0.75 | 18.2 |
| 13b | −1.65 | 0.76 | 21.4 |
| 13c | −1.66 | 0.77 | 16.6 |
| 13d | −1.64 | 0.75 | 20.8 |
Table 2 compiles the overpotentials (η) and the TOFs for the corresponding processes of all complexes. Amide derivatives 13d and 13b show higher TOFs. These values are 200% higher than that of 12a, which clearly demonstrates the strong influence that the substituent in the ADT-bridge of [(μ-ADT)NRFe2(CO)6] exerts on the kinetics (TOF) of these processes. These results may be explained by the fact that protonating the amide nitrogen of 13d and 13b is more difficult than protonating the amine moiety of complexes 7a–b and 12a–c. This difficulty may direct the protonation directly to the Fe nucleus or may trigger the proton transfer from the more acidic protonated amide to the Fe-nucleus.
The electrochemistry of [FeFe]-H2ase mimics having octahedral Ir(III) moieties was next studied. Firstly, we registered the electrochemistry of complex 15. Fig. 5 shows a reversible oxidation wave at 0.33 V (IrIII to IrIV) (vs. Fc+/0) and two reduction waves at −2.69 and −2.92 V (vs. Fc+/0), attributable to both types of ppy ligands (ppy = 2-phenylpyridine).
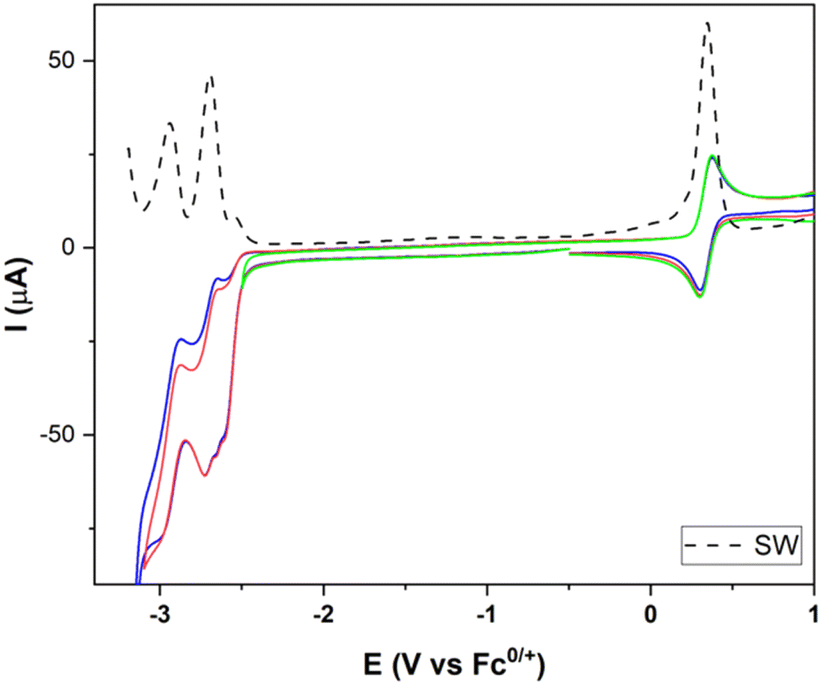 | ||
| Fig. 5 CVs and SW of complex 15 in 10−3 M MeCN solutions containing [nBu4N]PF6 10−1 M as the supporting electrolyte (25 °C), Pt as the counter electrode, vitreum carbon as the working electrode; potentials in V vs. Fc+/Fc; scan rate: 100 mV s−1. | ||
Complexes 16–18 show a quasireversible reduction wave between −1.48 and −1.57 V (vs. Fc+/0) (Fig. 6 and Table 3) that are attributable to the [FeIFeI] to [FeIFe0] reduction. Comparing with the reference voltammogram shown in Fig. 5, the additional reduction processes at around −1.98 and −2.09 V (vs. Fc+/0) are attributable to the [FeIFeI] to [Fe0Fe0] reduction. Reduction of the Ir(III) moiety was not seen in the window used. The oxidation waves of these compounds appeared in the range expected for the oxidation of Ir(III) to Ir(IV) moieties (0.33 V vs. Fc0/+).35 The main difference between complexes 15 and 16–18 in their oxidation waves is the irreversibility of the Ir(III) to Ir(IV) process observed for complex 16, while complex 18 maintains the reversibility of the oxidation wave. Obviously, this behaviour should be related to the linker that connects the Ir and the FeFe centers, which is efficient in the case of the amide linker (probably due to its iminol tautomer), but it fails in the case of the enamine linker.
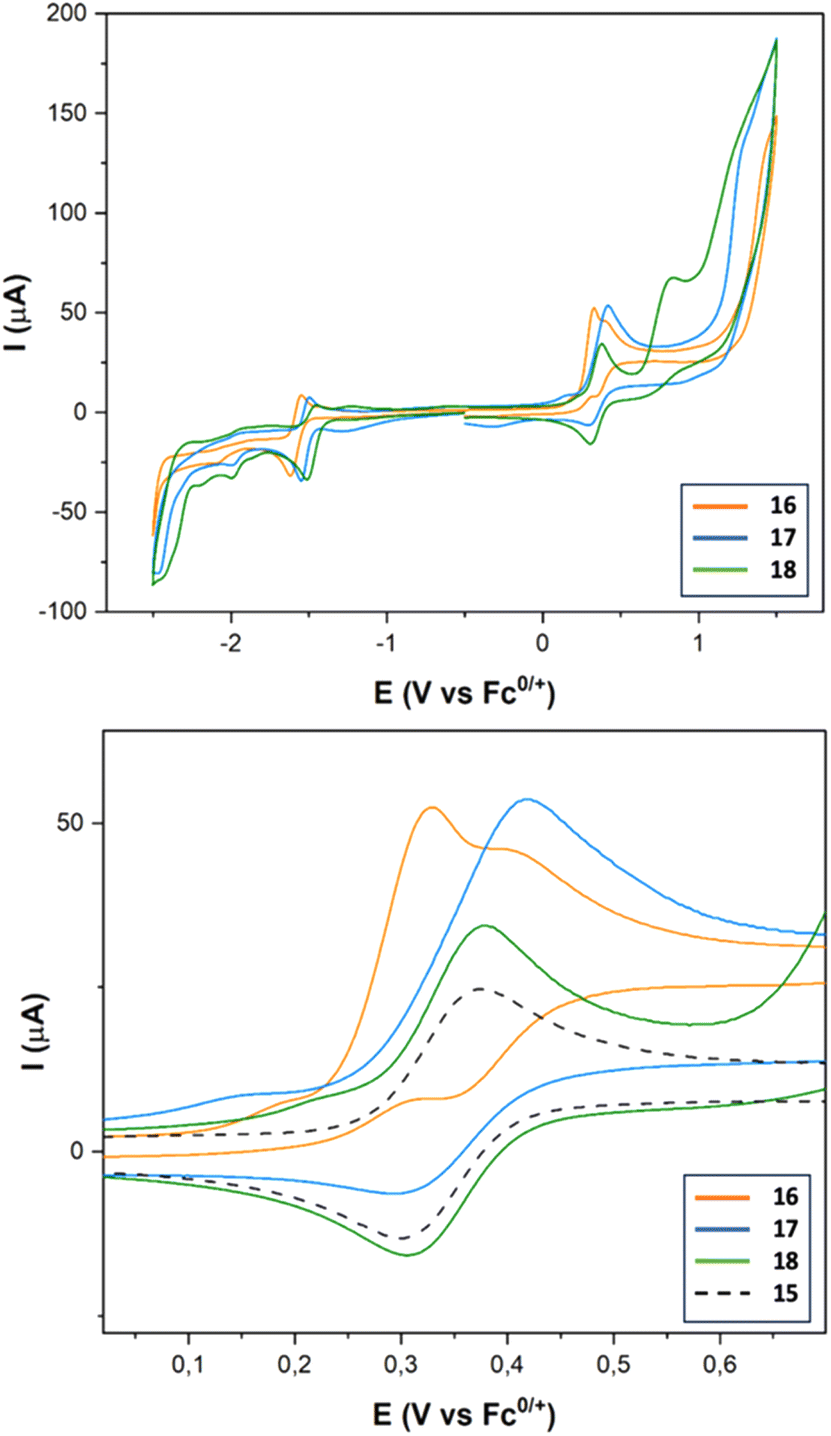 | ||
| Fig. 6 CVs of complexes 16–18 (top) and expansion of the IrIII to IrIV oxidation wave for 16–18vs.15 (bottom), obtained from 10−3 M solutions in MeCN containing [nBu4N]PF6 10−1 M as the supporting electrolyte, recorded at 25 °C. Counter-electrode: Pt; working electrode: vitreous carbon; potentials given in V vs. Fc+/Fc; scan rate: 100 mV s−1. | ||

To assess the electrocatalytic properties, an electrochemical study was conducted in the presence of a strong acid (CF3COOH, pKaMeCN = 12.632). Complexes 16–18 exhibit a strong electrocatalytic response (see Fig. S-4†). TOF values were calculated using the same H+ source. Complex 18 shows the highest value (200-fold with respect to that of 16), like the series of complexes 7, 12 and 13. Evidently, the nature of the linker between the new metal and the FeFe centres determine the electrocatalytic activity of these complexes.
To complete the study, the electrochemical response of the [ED-PS-Fe2S2] system 23 was measured. The [FeIFeI] to [FeIFe0] reduction appears at −1.43 V (vs. Fc+/0) and the [FeIFe0] to [Fe0Fe0] reduction process appears at −1.95 V (vs. Fc+/0). The successive oxidation processes at 0.08 V (reversible) and 0.53 V (vs. Fc+/0) correspond to the ferrocene and iridium moieties (Fig. 7).
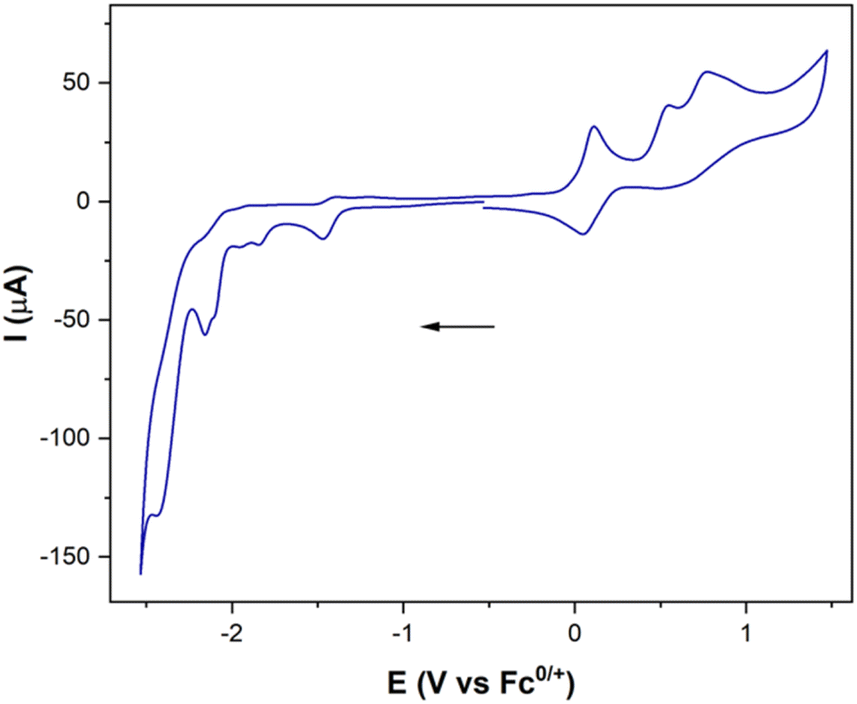 | ||
| Fig. 7 CV of complex 23, obtained from 10−3 M solutions in MeCN containing [nBu4N]PF6 10−1 M as the supporting electrolyte, recorded at 25 °C. Counter-electrode: Pt; working electrode: vitreous carbon; potentials given in V vs. Fc+/Fc; scan rate: 100 mV s−1. | ||
Conclusions
The cross-metathesis coupling of [FeFe]-H2ase mimics having double bond-containing moieties attached to the nitrogen of the adt bridge with vinyl metallocenes and half-sandwich and octahedral Ir(III) complexes having a vinyl functionality enables the syntheses of diverse heteropolymetallic [FeFe]-H2ase mimics in an easy and efficient way. The new complexes have an E-stereochemistry across the formed double bond.Electrochemical studies on both metallocenes (7a–b, 12a–b, and 13a–b) and half-sandwich derivatives (12c and 13c–d) have revealed that the placement of a redox unit has only a minimum impact on the reduction potentials of these [FeFe]-H2ase mimics.
The application of this cross-metathesis approach also allowed the synthesis of complexes having an electron-donor-photosensitizer structure (ED-PS) (23) bonded to the nitrogen of the adt-bridge. The electrocatalytic properties of these complexes have been elucidated. We observed a clear increase in the electrocatalytic response (a 200-fold increase in the TOF values for the HER) upon replacing an amine group with an amide group. This significant enhancement is attributed to the potential of the iminol tautomer to act as an efficient proton transfer group. In summary, an easy, efficient and versatile synthetic approach for [FeFe]-H2ase mimics has been developed.
Author contributions
M. A. S. and L. C. designed and supervised the experimental work. S. A. performed the synthetic and characterisation experiments. J. A. C. and P. G.-A. determined the X-ray structures. All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this work under grants PID2019-108429RB-I00 (to M. A. S), PID2019-104652GB-I00 (to J. A. C and P. G-A) and RED2022-134287-T (ORFEO-CINQA network), from the MCIN (Spain), is gratefully acknowledged. Technical support provided by Servicios Científico-Técnicos de la Universidad de Oviedo is also acknowledged.References
- (a) P. M. Vignais and B. Billoud, Occurrence, Classification, and Biological Function of Hydrogenases: An Overview, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed; (b) B. L. Greene, Progress and Opportunities in Photochemical Enzymology of Oxidoreductases, ACS Catal., 2021, 11, 14635–14650 CrossRef CAS.
- (a) J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and A. Volbeda, Structure-function relationships of anaerobic gas-processing metalloenzymes, Nature, 2009, 460, 814–822 CrossRef CAS PubMed; (b) S. T. Stripp, B. R. Duffus, V. Fourmond, C. Léger, S. Leimkühler, S. Hirota, Y. Hu, A. Jasniewski, H. Ogata and M. W. Ribbe, Second and Outer Coordination Sphere Effects in Nitrogenase, Hydrogenase, Formate Dehydrogenase, and CO Dehydrogenase, Chem. Rev., 2022, 122, 11900–11973 CrossRef CAS PubMed.
- P. M. Vignais, B. Billoud and J. Meyer, Classification and phylogeny of hydrogenases1, FEMS Microbiol. Rev., 2001, 25, 455–501 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- S. T. Stripp and T. Happe, How algae produce hydrogen—news from the photosynthetic hydrogenase, Dalton Trans., 2009, 9960–9969 RSC.
- H. Ogata, K. Nishikawa and W. Lubitz, Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase, Nature, 2015, 520, 571–574 CrossRef CAS PubMed.
- B. Schwörer, V. M. Fernandez, C. Zirngibl and R. K. Thauer, H2-forming N5,N10-methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum. Studies of the catalytic mechanism of H2 formation using hydrogen isotopes, Eur. J. Biochem., 1993, 212, 255–261 CrossRef PubMed.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, [FeFe]-Hydrogenases: maturation and reactivity of enzymatic systems and overview of biomimetic models, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- G. A. N. Felton, C. A. Mebi, B. J. Petro, A. K. Vannucci, D. H. Evans, R. S. Glass and D. L. Lichtenberger, Review of electrochemical studies of complexes containing the Fe2S2 core characteristic of [FeFe]-hydrogenases including catalysis by these complexes of the reduction of acids to form dihydrogen, J. Organomet. Chem., 2009, 694, 2681–2699 CrossRef CAS.
- (a) H. Reihlen, A. Gruhl and G. V. Hessling, Über den photochemischen und oxydativen Abbau von Carbonylen, Justus Liebigs Ann. Chem., 1929, 472, 268–287 CrossRef CAS; (b) W. Hieber and P. Spacu, Metal carbonyls. XXVI. Effect of organic sulfur compounds on the carbonyls of iron and cobalt, Z. Anorg. Allg. Chem., 1937, 233, 353–364 CrossRef CAS; (c) J. A. Cabeza, M. A. Martínez-García, V. Riera, D. Ardura and S. García-Granda, Binuclear Iron(I), Ruthenium(I), and Osmium(I) Hexacarbonyl Complexes Containing a Bridging Benzene-1,2-dithiolate Ligand. Synthesis, X-ray Structures, Protonation Reactions, and EHMO Calculations, Organometallics, 1998, 17, 1471–1477 CrossRef CAS; (d) U.-P. Apfel, M. Rudolph, C. Apfel, C. Robl, D. Langenegger, D. Hoyer, B. Jaun, M.-O. Ebert, T. Alpermann, D. Seebach and W. Weigand, Reaction of Fe3(CO)12 with octreotide—chemical, electrochemical and biological investigations, Dalton Trans., 2010, 39, 3065–3071 RSC.
- (a) J. L. Stanley, T. B. Rauchfuss and S. R. Wilson, Studies on the Condensation Pathway to and Properties of Diiron Azadithiolate Carbonyls, Organometallics, 2007, 26, 1907–1911 CrossRef CAS PubMed; (b) Y. Li and T. B. Rauchfuss, Synthesis of diiron(I) dithiolato carbonyl complexes, Chem. Rev., 2016, 116, 7043–7077 CrossRef CAS PubMed; (c) L.-C. Song, Investigations on butterfly Fe/S cluster S-centered anions (μ-S)2Fe2(CO)6, (μ-S)(μ-RS)Fe2(CO)6, and related species, Acc. Chem. Res., 2005, 38, 21–28 CrossRef CAS PubMed; (d) L.-C. Song, F.-X. Luo, H. Tan, X.-J. Sun, Z.-J. Xie and H.-B. Song, Synthesis, structures, and properties of diiron azadithiolate complexes containing a subphthalocyanine moiety as biomimetic models for [FeFe]-hydrogenases, Eur. J. Inorg. Chem., 2013, 2549–2557 CrossRef CAS.
- (a) I. Silaghi-Dumitrescu, T. E. Bitterwolf and R. B. King, Butterfly Diradical Intermediates in Photochemical Reactions of Fe2(CO)6(μ-S2), J. Am. Chem. Soc., 2006, 128, 5342–5343 CrossRef CAS PubMed; (b) A. Kramer and I. P. Lorenz, Photochemically induced [2 + 2] cycloadditions of alkenes and dienes with the sulfur-sulfur bond of the nido-clusters [(O(C)3Fe S]2, J. Organomet. Chem., 1990, 388, 187–193 CrossRef CAS; (c) A. Kramer, R. Lingnau, I. P. Lorenz and H. L. Mayer, Complex-chemical synthesis of cyclic 1,2-dithiolato ligands with the nido cluster [(CO)3FeS]2. Molecular structure and partial S-oxidation of hexacarbonyl(μ2-cis-1,2-cyclohexanedithiolato-S,S)diiron, Chem. Ber., 1990, 123, 1821–1826 CrossRef CAS; (d) R. D. Adams and S. Miao, Metal Carbonyl Derivatives of Sulfur-Containing Quinones and Hydroquinones: Synthesis, Structures, and Electrochemical Properties, Inorg. Chem., 2004, 43, 8414–8426 CrossRef CAS PubMed; (e) M. D. Westmeyer, T. B. Rauchfuss and A. K. Verma, Iron sulfido derivatives of the fullerenes C60 and C70, Inorg. Chem., 1996, 35, 7140–7147 CrossRef CAS PubMed.
- S. Aguado, L. Casarrubios, C. Ramírez de Arellano and M. A. Sierra, Revisiting the photochemical synthesis of [FeFe]-hydrogenase mimics: reaction optimization, mechanistic study and electrochemical behaviour, RSC Adv., 2020, 10, 29855–29867 RSC.
- (a) M. Lian, J. He, X.-Y. Yu, C. Mu, X.-F. Liu, Y.-L. Li and Z.-Q. Jiang, Diiron ethanedithiolate complexes with acetate ester: Synthesis, characterization and electrochemical properties, J. Organomet. Chem., 2018, 870, 90–96 CrossRef CAS; (b) S. Ghosh, S. Basak-Modi, M. G. Richmond, E. Nordlander and G. Hogarth, Electrocatalytic proton reduction by thiolate-capped triiron clusters [Fe3(CO)9(μ3-SR)(μ-H)] (R = iPr, tBu), Inorg. Chim. Acta, 2018, 480, 47–53 CrossRef CAS; (c) P.-H. Zhao, Z.-Y. Ma, M.-Y. Hu, J. He, Y.-Z. Wang, X.-B. Jing, H.-Y. Chen, Z. Wang and Y.-L. Li, PNP-Chelated and -Bridged Diiron Dithiolate Complexes Fe2(μ-pdt)(CO)4{(Ph2P)2NR } Together with Related Monophosphine Complexes for the [2Fe]H Subsite of [FeFe]-Hydrogenases: Preparation, Structure, and Electrocatalysis, Organometallics, 2018, 37, 1280–1290 CrossRef CAS.
- A. D. Merinero, A. Collado, L. Casarrubios, M. Gómez-Gallego, C. Ramírez de Arellano, A. Caballero, F. Zapata and M. A. Sierra, Triazole-Containing [FeFe] Hydrogenase Mimics: Synthesis and Electrocatalytic Behavior, Inorg. Chem., 2019, 58, 16267–16278 CrossRef CAS PubMed.
- A. Torres, D. J. Vicent, A. Collado, M. Gómez-Gallego, C. R. de Arellano and M. A. Sierra, Phosphite Bearing [(μ-ADT)RFe2(CO)6] (ADT = Azadithiolate) Moieties: A Tool for the Building of Multimetallic [FeFe]-Hydrogenase Mimics, Organometallics, 2023, 42, 316–326 CrossRef CAS.
- A. Torres, A. Collado, M. Gómez-Gallego, C. R. de Arellano and M. A. Sierra, Heteropolymetallic [FeFe]-Hydrogenase Mimics: Synthesis and Electrochemical Properties, Inorg. Chem., 2023, 62, 3409–3419 CrossRef CAS PubMed.
- J. D. Lawrence, H. Li and T. B. Rauchfuss, Beyond Fe-only hydrogenases: N-functionalized 2-aza-1,3-dithiolates Fe2[(SCH2)2NR](CO)x (x = 5, 6), Chem. Commun., 2001, 1482–1483 RSC.
- X. Wang, G. Kehr, C. G. Daniliuc and G. Erker, Internal Adduct Formation of Active Intramolecular C4-bridged Frustrated Phosphane/Borane Lewis Pairs, J. Am. Chem. Soc., 2014, 136, 3293–3303 CrossRef CAS PubMed.
- J.-M. Heldt, N. Fischer-Durand, M. Salmain, A. Vessières and G. Jaouen, Preparation and characterization of poly(amidoamine) dendrimers functionalized with a rhenium carbonyl complex and PEG as new IR probes for carbonyl metallo immunoassay, J. Organomet. Chem., 2004, 689, 4775–4782 CrossRef CAS.
- M. L. Lage, I. Fernández, M. J. Mancheño, M. Gómez-Gallego and M. A. Sierra, The electronic structure and photochemistry of group 6 bimetallic (Fischer) carbene complexes: beyond the photocarbonylation reaction, Chem. – Eur. J., 2010, 16, 6616–6624 CrossRef CAS PubMed.
- (a) S. Ott, M. Kritikos, B. Åkermark and L. Sun, Synthesis and Structure of a Biomimetic Model of the Iron Hydrogenase Active Site Covalently Linked to a Ruthenium Photosensitizer, Angew. Chem., Int. Ed., 2003, 42, 3285–3288 CrossRef CAS PubMed; (b) H. Wolpher, M. Borgström, L. Hammarström, J. Bergquist, V. Sundström, S. Styring, L. Sun and B. Åkermark, Synthesis and properties of an iron hydrogenase active site model linked to a ruthenium tris-bipyridine photosensitizer, Inorg. Chem. Commun., 2003, 6, 989–991 CrossRef CAS; (c) S. Ott, M. Borgström, M. Kritikos, R. Lomoth, J. Bergquist, B. Åkermark, L. Hammarström and L. Sun, Model of the iron hydrogenase active site covalently linked to a ruthenium photosensitizer: synthesis and photophysical properties, Inorg. Chem., 2004, 43, 4683–4692 CrossRef CAS PubMed; (d) J. Ekström, M. Abrahamsson, C. Olson, J. Bergquist, F. B. Kaynak, L. Eriksson, L. Sun, H.-C. Becker, B. Åkermark, L. Hammarström and S. Ott, Bio-inspired, side-on attachment of a ruthenium photosensitizer to an iron hydrogenase active site model, Dalton Trans., 2006, 4599–4606 RSC; (e) W. Gao, J. Liu, W. Jiang, M. Wang, L. Weng, B. Åkermark and L. Sun, An azadithiolate bridged Fe2S2 complex as active site model of FeFe-hydrogenase covalently linked to a Re(CO)3(bpy)(py) photosensitizer aiming for light-driven hydrogen production, C. R. Chim., 2008, 11, 915–921 CrossRef CAS.
- R. Lomoth and S. Ott, Introducing a dark reaction to photochemistry: photocatalytic hydrogen from [FeFe] hydrogenase active site model complexes, Dalton Trans., 2009, 45, 9952–9959 RSC.
- P. Poddutoori, D. T. Co, A. P. S. Samuel, C. H. Kim, M. T. Vagnini and M. R. Wasielewski, Photoinitiated multistep charge separation in ferrocene–zinc porphyrin–diiron hydrogenase model complex triads, Energy Environ. Sci., 2011, 4, 2441–2450 RSC.
- H. Mizuno, J. Takaya and N. Iwasawa, Rhodium(I)-Catalyzed Direct Carboxylation of Arenes with CO2 via Chelation-Assisted C−H Bond Activation, J. Am. Chem. Soc., 2011, 133, 1251–1253 CrossRef CAS PubMed.
- J. Hu, M. Gao, Y. Zhang, Y. Wang, Z. Qiao, W. Zhang, Q. Wang, L. Yan and H. Qian, Novel piperazine urea derivatives as highly potent transient receptor potential vanilloid 1 (TRPV1) antagonists, Bioorg. Chem., 2021, 115, 105229 CrossRef CAS PubMed.
- T. Mochida, H. Shimizu, S. Suzuki and T. Akasaka, Synthesis and properties of azole-substituted ferrocenes, J. Organomet. Chem., 2006, 691, 4882–4889 CrossRef CAS.
- (a) F. Wang, M. Wang, X. Liu, K. Jin, W. Dong and L. Sun, Protonation, electrochemical properties and molecular structures of halogen-functionalized diiron azadithiolate complexes related to the active site of iron-only hydrogenases, Dalton Trans., 2007, 34, 3812–3819 RSC; (b) G. Eilers, L. Schwartz, M. Stein, G. Zampella, L. de Gioia, S. Ott and R. Lomoth, Ligand versus Metal Protonation of an Iron Hydrogenase Active Site Mimic, Chem. – Eur. J., 2007, 13, 7075–7084 CrossRef CAS PubMed; (c) L. Schwartz, G. Eilers, L. Eriksson, A. Gogoll, R. Lomoth and S. Ott, Iron hydrogenase active site mimic holding a proton and a hydride, Chem. Commun., 2006, 5, 520–522 RSC.
- M. Watanabe, Y. Honda, H. Hagiwara and T. Ishihara, [FeFe]-Hydrogenase and its organic molecule mimics—Artificial and bioengineering application for hydrogenproduction, J. Photochem. Photobiol., C, 2017, 33, 1–26 CrossRef CAS.
- W.-G. Wang, H.-Y. Wang, G. Si, C.-H. Tung and L.-Z. Wu, Fluorophenyl-substituted Fe-only hydrogenases active site ADT models: different electrocatalytic process for proton reduction in HOAc and HBF4/Et2O, Dalton Trans., 2009, 15, 2712–2720 RSC.
- G. A. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Iron-only hydrogenase mimics. Thermodynamic aspects of the use of electrochemistry to evaluate catalytic efficiency for hydrogen generation, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS PubMed.
- S. Gao, Q. Liang, Q. Duan, D. Jiang and J. Zhao, Electrochemical proton reductions in varying acidic media by a simple synthetic hydrogenase mimic, Int. J. Hydrogen Energy, 2018, 43, 7245–7256 CrossRef CAS.
- M. P. Stewart, M.-H. Ho, S. Wiese, M. L. Lindstrom, C. E. Thogerson, S. Raugei, R. M. Bullock and M. L. Helm, High Catalytic Rates for Hydrogen Production Using Nickel Electrocatalysts with Seven-Membered Cyclic Diphosphine Ligands Containing One Pendant Amine, J. Am. Chem. Soc., 2013, 135, 6033–6046 CrossRef CAS PubMed.
- H.-M. Cui, M.-Q. Hu, H.-M. Wen, G.-L. Chai, C.-B. Ma, H. Chen and C.-N. Chen, Efficient [FeFe] hydrogenase mimic dyads covalently linking to iridium photosensitizer for photocatalytic hydrogen evolution, Dalton Trans., 2012, 41, 13899–13907 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, analytical data, and complementary XRD data. CCDC 2308698–2308700. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt04197b |
| This journal is © The Royal Society of Chemistry 2024 |