
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unusual nucleophilic reactivity of a dithiolene-based N-heterocyclic silane†
Phuong M.
Tran
 ,
Yuzhong
Wang
,
Mitchell E.
Lahm
,
Pingrong
Wei
,
Henry F.
Schaefer
III
and
Gregory H.
Robinson
*
,
Yuzhong
Wang
,
Mitchell E.
Lahm
,
Pingrong
Wei
,
Henry F.
Schaefer
III
and
Gregory H.
Robinson
*
Department of Chemistry, The University of Georgia, Athens, Georgia 30602-2556, USA. E-mail: robinson@uga.edu
First published on 20th March 2024
Abstract
While the dithiolene-based N-heterocyclic silane (4) reacts with two equivalents of BX3 (X = Br, I) to give zwitterionic Lewis adducts 5 and 8, respectively, the parallel reaction of 4 with BCl3 results in 10, a dithiolene-substituted N-heterocyclic silane, via the Si–S bond cleavage. Unlike 5, the labile 8 may be readily converted to 9via BI3-mediated cleavage of the Si–N bond. The formation of 5 and 8 confirms that 4 uniquely possesses dual nucleophilic sites: (a) the terminal sulphur atom of the dithiolene moiety; and (b) the backbone carbon of the N-heterocyclic silane unit.
Silylenes, the silicon analogues of carbenes, have evolved from transient reaction intermediates1,2 to versatile ligands impacting transition metal coordination chemistry, catalysis, small molecule activation, and the stabilization of novel low-oxidation state main group species.3–23 A variety of four-, five- and six-membered N-heterocyclic silylenes (NHSis) have been reported3,8,10,24,25 since the first such molecule was synthesized by the West group three decades ago.26 N-Heterocyclic silylenes have demonstrated considerably different reactivity toward boron halides than their carbon analogues, N-heterocyclic carbenes (NHCs). N-Heterocyclic carbenes usually form stable Lewis adducts with boron halides.27–29 Although stable Lewis adducts have been isolated (Scheme 1a),30,31 reactions between N-heterocyclic silylenes and boron halides often proceed beyond this stage. Braunschweig et al. reported that Xyl-substituted NHSi (A in Scheme 1b, Xyl = 2,6-dimethylphenyl) may react with organoborane halides to give the corresponding oxidative addition products (B), which were subsequently converted to Cvia ring expansion.32 Subsequently, a series of oxidative additions of the B–X (X = halide) bonds of boron halides at the silylene centers and silylene ring expansion reactions have been reported.33–36 When the amidinate-supported four-membered silylene is combined with organoborane halides (Scheme 1c), migration of the amidinate ligand from the silicon atom to the boron atom was reported by Roesky et al.30 In addition, Cui et al. recently reported that reaction of the five-membered NHSi (D) with BBr3 produced the N-heterocyclic boryl-substituted silicon bromide Evia silicon–boron exchange reaction (Scheme 1c).37 Notably, the literature does not reveal any reports of boron halide-mediated backbone activation of N-heterocyclic silylene rings.
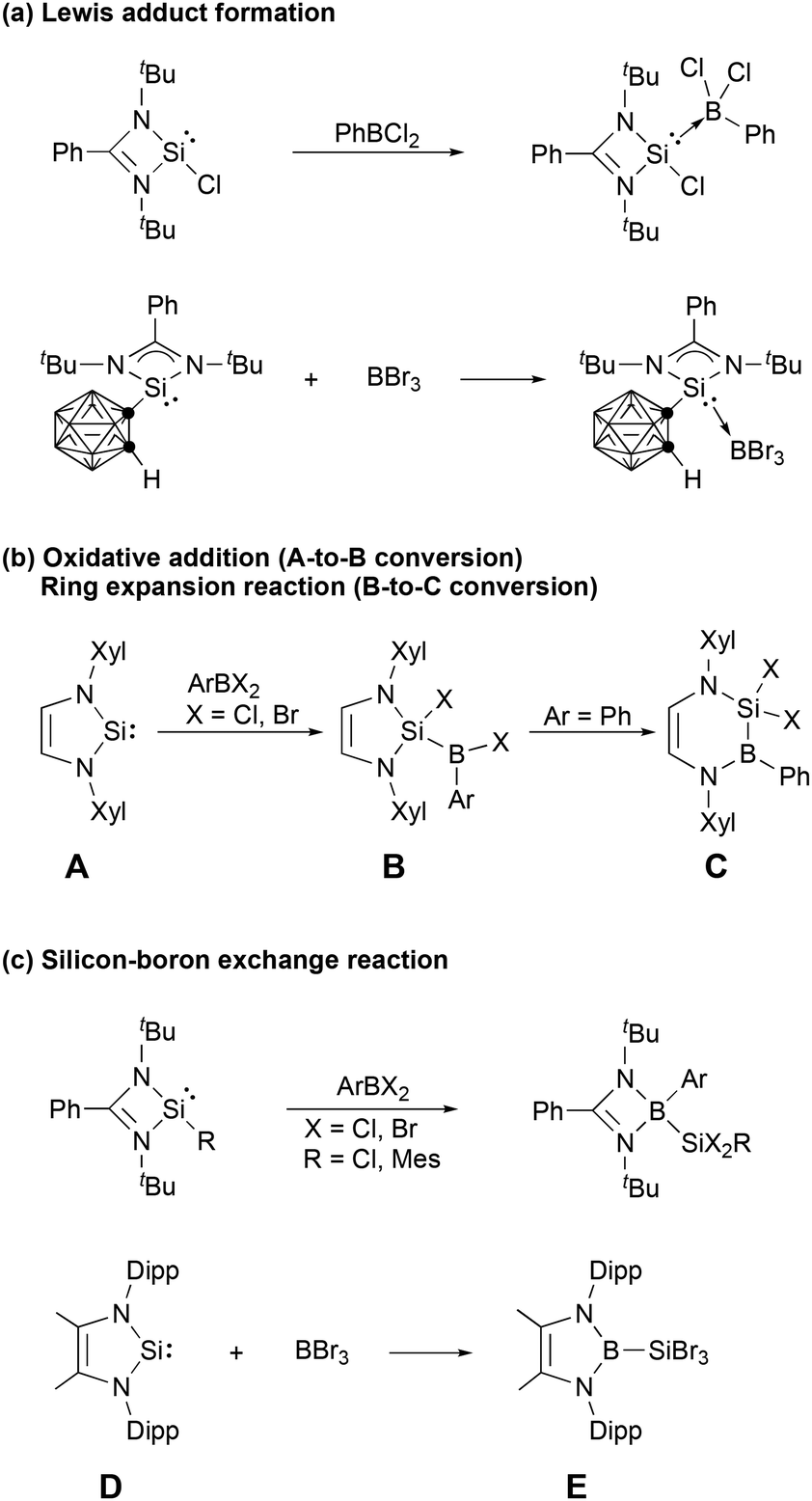 | ||
| Scheme 1 Typical reactions of N-heterocyclic silylenes with boron halides (Xyl = 2,6-dimethylphenyl, Dipp = 2,6-diisopropylphenyl). | ||
Recently this laboratory investigated the silylene38,39 (1, in Scheme 2a)-mediated sulphur–sulphur bond cleavage of an imidazole-based dithione dimer (3),40 affording a dithiolene-based N-heterocyclic silane (4, Scheme 2a).41 Herein, we report the dual nucleophilic reactivity of the carbon backbone of the N-heterocyclic silyl framework and the terminal sulphur atom of the dithiolene unit in 4 with BX3 (X = Br, I)—resulting in the formation of zwitterionic Lewis adducts 5 and 8, respectively. This discovery is a unique example of Lewis acid-induced charge separation of a five-membered N-heterocyclic silyl ring.
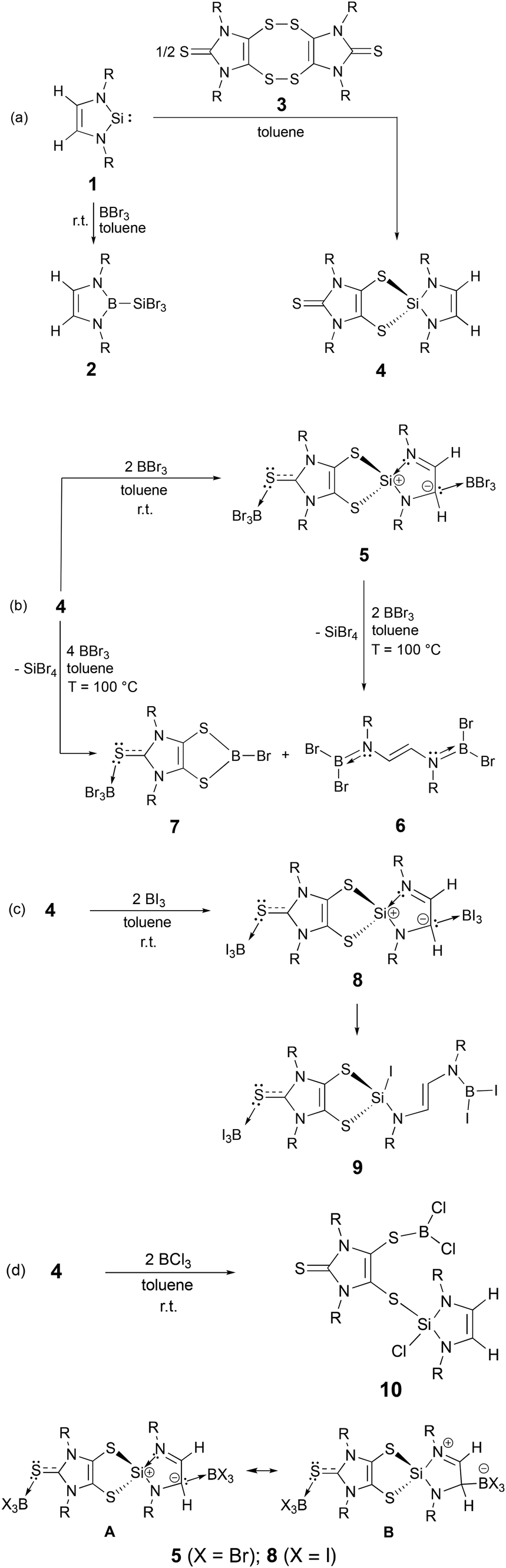 | ||
| Scheme 2 Synthesis of 2, 5–10 (R = 2,6-diisopropylphenyl) and canonical forms of 5 and 8. | ||
Consistent with the D-to-E conversion (Scheme 1c),37 NHSi (1) reacts with BBr3 to give 2 (Scheme 2a). Compound 2 may also be prepared via reaction of the 2-alkoxysilane-1,3,2-diazaborole with BBr3.42 In contrast, room-temperature reaction of 4 with BBr3 (in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) in toluene gave 5 (81% yield) (Scheme 2b).43 X-ray quality yellow crystals of 5 were obtained via recrystallization in toluene. While 4 shows one singlet olefin proton resonance at 5.68 ppm,41 the backbone protons of the N-heterocyclic silyl framework in 5 exhibit two resonances in the 1H NMR spectrum:43 a broad singlet at 5.61 ppm (for HC:BBr3) and a singlet at 6.05 ppm (for N
:2 molar ratio) in toluene gave 5 (81% yield) (Scheme 2b).43 X-ray quality yellow crystals of 5 were obtained via recrystallization in toluene. While 4 shows one singlet olefin proton resonance at 5.68 ppm,41 the backbone protons of the N-heterocyclic silyl framework in 5 exhibit two resonances in the 1H NMR spectrum:43 a broad singlet at 5.61 ppm (for HC:BBr3) and a singlet at 6.05 ppm (for N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH). The singlet (−6.36 ppm) and doublet (−10.97 ppm, 2JBH = 7.8 Hz) 11B NMR resonances of 5 correspond to the BBr3 units bound to the sulphur and carbon atoms, respectively.43 The 4-to-5 conversion results in the downfield shift of the 29Si NMR resonance from −4.67 ppm (for 4, in C6D6)41 to 8.12 ppm (for 5, in toluene-d8).43 Compound 5 may be converted back to 4 in THF. Compound 5, in the presence of BBr3, readily decomposes at room temperature, giving an acyclic doubly borylated (E)-N,N′-diaminoethene (6) and other uncharacterized products. Further reaction of 5 with BBr3 (in a 1:2 ratio) in toluene at an elevated temperature (100 °C) gave a ca. 1:1 mixture of 6 and a dithiolene-based bromoborane complex (7) according to the 1H NMR data (Scheme 2b).43 The mixture of 6 and 7 may also be obtained via the 1:4 reaction of 4 with BBr3 in toluene at 100 °C (Scheme 2b). Due to the similar solubilities, crystals of 6 (square blocks) and 7 (long rods) were manually separated for NMR measurements. The 11B NMR resonance of 6 (28.60 ppm) compares well to that of an aminodichloroborane analogue (13) (32.60 ppm), Cl2BN(Aryl)–CHCH–N(Aryl)BCl2 (Aryl = 2,6-Me2C6H3).44 The 11B NMR spectrum43 of 7 shows a singlet at −6.36 ppm and a broad singlet at 51.15 ppm, which correspond to the four-coordinated boron (in the SBBr3 moiety) and three-coordinated boron (in the five-membered C2S2B ring), respectively. While the mechanistic details of the formation of 6 and 7 from reaction of 5 with BBr3 remain unclear, this transformation may plausibly involve consecutive insertions of the BBr3 species into the Si–N bonds in 5, accompanied by the migration of one bromide from the boron atom to the silicon atom, rendering to 6 and an intermediate 11 (i.e., the dithiolene-based SiBr2 analogue of 7). The BBr3 residing at the backbone carbon in 5 could be released during this process and subsequently react with the intermediate 11 to yield 7 and SiBr4 (as a by-product) via silicon–boron exchange. However, our repeated attempts to isolate intermediate 11 were unsuccessful.
CH). The singlet (−6.36 ppm) and doublet (−10.97 ppm, 2JBH = 7.8 Hz) 11B NMR resonances of 5 correspond to the BBr3 units bound to the sulphur and carbon atoms, respectively.43 The 4-to-5 conversion results in the downfield shift of the 29Si NMR resonance from −4.67 ppm (for 4, in C6D6)41 to 8.12 ppm (for 5, in toluene-d8).43 Compound 5 may be converted back to 4 in THF. Compound 5, in the presence of BBr3, readily decomposes at room temperature, giving an acyclic doubly borylated (E)-N,N′-diaminoethene (6) and other uncharacterized products. Further reaction of 5 with BBr3 (in a 1:2 ratio) in toluene at an elevated temperature (100 °C) gave a ca. 1:1 mixture of 6 and a dithiolene-based bromoborane complex (7) according to the 1H NMR data (Scheme 2b).43 The mixture of 6 and 7 may also be obtained via the 1:4 reaction of 4 with BBr3 in toluene at 100 °C (Scheme 2b). Due to the similar solubilities, crystals of 6 (square blocks) and 7 (long rods) were manually separated for NMR measurements. The 11B NMR resonance of 6 (28.60 ppm) compares well to that of an aminodichloroborane analogue (13) (32.60 ppm), Cl2BN(Aryl)–CHCH–N(Aryl)BCl2 (Aryl = 2,6-Me2C6H3).44 The 11B NMR spectrum43 of 7 shows a singlet at −6.36 ppm and a broad singlet at 51.15 ppm, which correspond to the four-coordinated boron (in the SBBr3 moiety) and three-coordinated boron (in the five-membered C2S2B ring), respectively. While the mechanistic details of the formation of 6 and 7 from reaction of 5 with BBr3 remain unclear, this transformation may plausibly involve consecutive insertions of the BBr3 species into the Si–N bonds in 5, accompanied by the migration of one bromide from the boron atom to the silicon atom, rendering to 6 and an intermediate 11 (i.e., the dithiolene-based SiBr2 analogue of 7). The BBr3 residing at the backbone carbon in 5 could be released during this process and subsequently react with the intermediate 11 to yield 7 and SiBr4 (as a by-product) via silicon–boron exchange. However, our repeated attempts to isolate intermediate 11 were unsuccessful.
As a comparison, we also investigated the parallel reactions of 4 with BX3 (X = Cl, I). The 1:2 reaction of 4 with BI3 in toluene over 2h resulted in the isolation of 8 (the analogue of 5) (Scheme 2c). In contrast to 5, compound 8 may be readily converted to 9via the BI3-mediated silicon–nitrogen bond cleavage (Scheme 2c). While 9 can be isolated as pale-yellow crystalline powder in 72% yield, attempts to obtain pure 8 was unsuccessful due to its high lability. Isolation of 9 further supports our proposed mechanism for the BBr3-mediated decomposition of 5 (Scheme 2b). The formation of 8 has been confirmed by both single crystal X-ray diffraction technique (Fig. 1) and NMR studies. The singlet 1H NMR resonances of 8 [5.58 ppm (HC:BI3) and 6.06 ppm (NCH)] compare well to those for 5 [5.61 ppm (HC:BBr3) and 6.05 ppm (NCH), respectively]. The singlet (−82.90 ppm, CSBI3) and doublet (−70.43 ppm, 2JBH = 7.0 Hz, C(H)BI3) 11B NMR resonances of 8 are shifted highfield compared to those for 5 (−6.36 ppm, CSBBr3 and −10.97 ppm, 2JBH = 7.8 Hz, C(H)BBr3). Due to the high lability of 8 (which was converted to 9 during the 29Si NMR measurement), we only observed the 29Si NMR resonance for 9 (−18.80 ppm). The singlet 11B NMR resonances (at 6.02 ppm and −82.85 ppm) of 9 correspond to the three-coordinate NBI2 and four-coordinate CSBI3 units, respectively.
 | ||
| Fig. 1 Molecular structures of 5–10. Thermal ellipsoids represent 30% probability. All hydrogen atoms (except H(28) and H(29) in 5 and 8) have been omitted for clarity. | ||
Interestingly, the parallel reaction of 4 with BCl3 gave 10 as colourless crystalline powder (in 19% yield) (Scheme 2d) via BCl3-mediated cleavage of the Si–S bond in 4. Formation of the zwitterionic analogue of 5 and 8 was not observed in terms of the 1H NMR tube experiments. 10 exhibits singlet 11B NMR (53.28 ppm) and 29Si NMR (−33.47 ppm) resonances, revealing the presence of three-coordinate boron atom and four-coordinate silicon atom. Compound 10 is labile in solution, which may gradually decompose to give 12, the analogue of 6, in benzene. Compound 12 can be directly synthesized via 1:5 reaction of 4 with BCl3 (in 58% yield). The 11B NMR resonance of 12 (32.27 ppm) compares to that of 6 (28.60 ppm).
The molecular structures of 5–10 were determined by single crystal X-ray diffraction and supported by DFT computations (5-Ph, 7, and 10-Ph models, B3LYP/6-311G** level; 8-Ph and 9-Ph models, mPW1PW91/LANL2DZ level).43 The crystal unit cell contains an enantiomeric pair of 5 (with identical bonding parameters) (Fig. 1). The formation of 5 reveals that 4 can serve as a double donor ligand to bind two equivalents of BBr3 at two nucleophilic sites: the terminal sulphur atom of the dithiolene unit and the backbone carbon of the C2N2Si ring in 4. Each boron atom in 5 is four-coordinate and adopting a distorted tetrahedral geometry. The backbone protons of the C2N2Si ring [i.e., H(28) and H(29)] were located from difference Fourier map.43 With the BBr3 coordination, the C(1)–S(1) bond is elongated from 1.6638(9) Å (as observed in 4)41 to 1.725(2) Å, which compares well to that [1.7256(18) Å] of the zwitterionic boron dithiolene complex with a terminal SR group (R = cyclohexyl) residing at the C2 carbon.45 Accordingly, the Wiberg bond index (WBI) of the C(1)–S(1) bond in 5 (1.17) is somewhat lower than that in 4 (1.49),41 indicating its modest multiple bond character. The S–B bond in 5 [1.932(2) Å] is shorter than that in C4H8S·BBr3 [1.966(13) Å].46 The C–B bond in 5 [1.656(3) Å] is similar to that [1.660(2) Å] in [{Ph2(S)P}(H)(Ph3Si)C(BH3)][Li(THF)3].47 The structural features of the C2N2Si ring in 5 are remarkably different from those of 4.41 While the CC double bond [1.3375(15) Å] in the C2N2Si ring of 4 is elongated to the C(28)–C(29) single bond in 5 [1.462(3) Å], one of the two C–N single bonds in the C2N2Si ring of 4 [1.414 Å, av] is concomitantly shortened to the N(4)–C(29) double bond in 5 [1.293(3) Å]. The Si(1)–N(3) bond in 5 [1.6797(18) Å (experimental value), 1.703 Å (theoretical value)] compares to the covalent Si–N single bonds in 4 [1.713 Å, av].41 The obviously elongated Si(1)–N(4) bond in 5 [1.8197(18) Å (experimental value), 1.829 Å (computed value)] is comparable with the reported dative Si–N single bonds (such as that [1.858(9) Å] in [Me3Si(py)]+[I]−48 and those [1.8290(18) Å and 1.8617(18) Å] in a chlorosilyliumylidene complex).49 Accordingly, the WBI of the Si(1)–N(4) bond in 5 (0.49) is considerably smaller than that (0.70) of the Si(1)–N(3) bond in 5. However, there have no obvious changes for the structural parameters of the C2S2Si rings in both 4 and 5. Compound 5 may be regarded as an intramolecular base-stabilized dithiolene-based silylium species. Natural bond orbital (NBO) analysis of 5-Ph model supports its zwitterionic feature (as shown in Scheme 2)—the silicon atom has a positive charge of +1.57, whereas the carbon atom [i.e., C(28)] bound to the BBr3 unit bears a negative charge of −0.37. The electrostatic potential map of 4 (Fig. S22†) reveals the negative potential resides predominately in the region around the terminal sulphur atom of the dithiolene unit, while the region of the backbone carbon atoms of the N-heterocyclic silyl unit has a very weak negative electrostatic potential. Thus, it is somewhat surprising to observe the nucleophilic behaviour of the backbone carbon atom of the N-heterocyclic silyl ring in 4.
The X-ray structural analysis43 of 6 (Fig. 1) reveals a planar Br2B–N–C–C–N–BBr2 framework, while the two 2,6-diisopropylphenyl substituents are nearly perpendicular to this plane. The structural parameters of the C2N2B2(BBr2)2 core in 6 [dCC = 1.310(9) Å; dNB = 1.387(6) Å] compare well to those for 13 [dCC = 1.333(2) Å; dNB = 1.395(1) Å].44 In the solid state43 (Fig. 1), the terminal S(1) atom of 7, as that in 5, is capped by a boron tribromide species. The C2S2B ring in 7 is nearly planar [bend angle (η) between the BS2 plane and the C2S2 plane = 1.7°]. The three-coordinate boron atom, involved in the five-membered dithiolene ring, adopts a trigonal planar geometry. In 7, the B(1)–S bonds [1.809(4) Å, av; WBI = 1.24, av] are somewhat shorter than the B(2)–S(1) bond [1.935(4) Å, av; WBI = 0.83], which should be due to π-donation of the S-lone pairs into the empty p orbital of the B(1) atom. The C–S bonds (1.737 Å, av) in the C2S2 unit of 7 are longer than those (1.710 Å, av) for the reported four-coordinate boron-based dithiolate complex,45 which may be attributed to the electron donation from the sulphur atoms to the three-coordinate boron in 7.
X-ray structural analysis (Fig. 1) shows that 8 is isostructural with 5. The BI3 bound to the backbone carbon of the N-heterocyclic silyl unit in 8 was released and subsequently cleaved one of the two Si–N bonds to give the NHSi-ring opened product 9. The solid-state structure of 9 (Fig. 1) shows that while a BI2 species is bonded to a nitrogen atom [dNB = 1.393(11) Å], one iodine atom is attached to the central four-coordinate silicon atom. The Si(1)–I(4) bond distance in 9 [2.406(3) Å] is somewhat shorter than the computed value (2.492 Å). The Bsp2–I bonds (2.128 Å, av) in 9 is shorter than those Bsp3–I bonds in 8 and 9 (2.236 Å, av). NBO analysis shows that while the silicon atom in 9-Ph bears a positive charge of +1.10, the silicon atom and the carbon atom (next to BI3) in zwitterionic 8 have a charge of +1.56 and −0.39, respectively. These results, coupled with the elongated Si(1)–N(4) bonds in 5 and 8, suggest that the canonical form A (Scheme 2) represents the predominant formulation of both 5 and 8. The X-ray structure of 10 (Fig. 1) indicates that one Si–S bond in 4 is cleaved by BCl3via the formation of a Si–Cl bond and a B–S bond. The Si–Cl bond distance in 10 [2.0522(11) Å] is marginally shorter than the computed value (2.088 Å). The B–S bond in 10 [1.793(4) Å] compares well to those (involving the three-coordinate boron atom) in 7 [1.809(4) Å, av].
Computations of the simplified 5-Ph model43 (Fig. 2) show that while the LUMO involves both C–B bonding and C–N π-anti-bonding character, the HOMO is dominated by the sulphur- and bromine-based lone pair character of the terminal SBBr3 unit. HOMO−2 and HOMO−3 contain the S–B and C–B σ-bonding character, respectively. According to natural bond orbital (NBO) analysis, the C–B σ bond polarization is 28.2% toward boron and 71.8% toward carbon that has 28.90% s-, 71.08% p-, and 0.02% d-character.
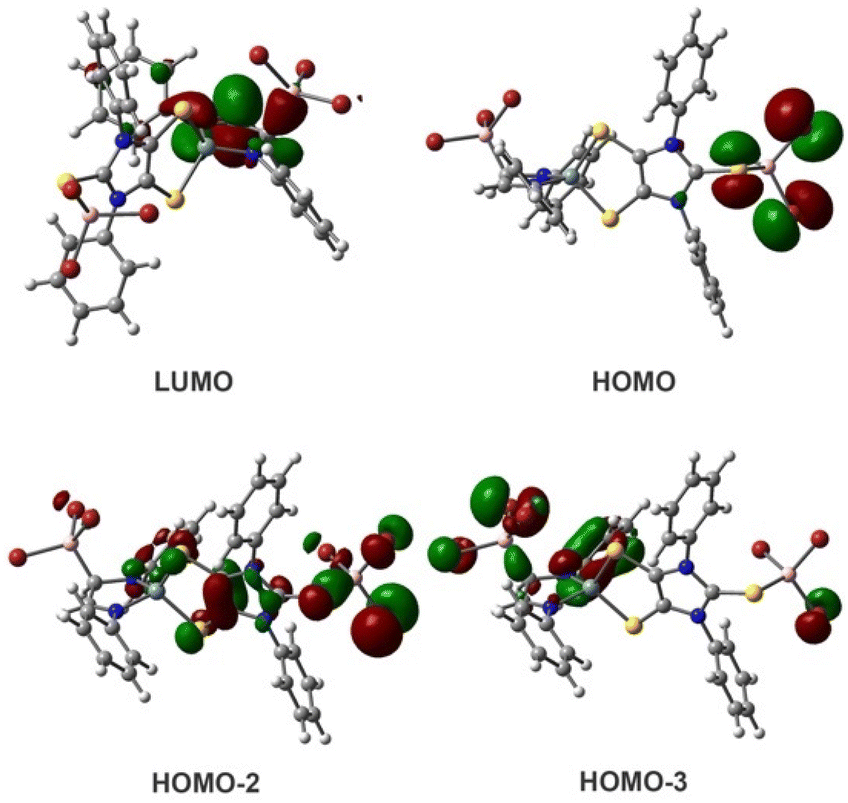 | ||
| Fig. 2 Molecular orbitals of the simplified 5-Ph model. | ||
Conclusions
Dithiolene-based N-heterocyclic silane (4) reacts with two equivalents of BX3 (X = Br, I) to give zwitterionic Lewis adducts 5 and 8, respectively, whereas the parallel reaction of 4 with BCl3 gives 10via the Si–S bond cleavage. Further reaction of 5 with BBr3 (in a 1:2 ratio) in toluene at an elevated temperature (100 °C) resulted in its decomposition, giving a mixture of 6 and 7. In contrast to 5, the labile zwitterion (8) may be readily converted to 9via BI3-mediated Si–N bond cleavage. The 4-to-(5 and 8) conversions reveal that both the terminal sulphur atom of the dithiolene unit and the backbone carbon of the N-heterocyclic silyl moiety in 4 may serve as nucleophilic sites to bind BX3 (X = Br and I) moieties. The potential broad utility of 4 as a species with dual nucleophilic sites is being investigated in this laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Science Foundation (CHE-2153978 to G. H. R. and Y. W.) and the Department of Energy (DOE-SC0015512 to H. F. S.) for support.References
- P. S. Skell and E. J. Goldstein, J. Am. Chem. Soc., 1964, 86, 1442–1443 Search PubMed.
- W. H. Atwell and D. R. Weyenberg, Angew. Chem., Int. Ed. Engl., 1969, 8, 469–477 CrossRef CAS.
- M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed.
- S. S. Sen, S. Khan, P. P. Samuel and H. W. Roesky, Chem. Sci., 2012, 3, 659–682 RSC.
- Y. Xiong, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 4302–4311 CrossRef CAS PubMed.
- R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed.
- B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134–148 RSC.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- B. Blom, M. Stoelzel and M. Driess, Chem. – Eur. J., 2013, 19, 40–62 CrossRef CAS PubMed.
- S. Yao, Y. Xiong and M. Driess, Organometallics, 2011, 30, 1748–1767 CrossRef CAS.
- L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, Coord. Chem. Rev., 2015, 300, 1–28 CrossRef.
- S. Yao, A. Saddington, Y. Xiong and M. Driess, Acc. Chem. Res., 2023, 56, 475–488 CrossRef CAS PubMed.
- W. J. Yang, Y. H. Dong, H. J. Sun and X. Y. Li, Dalton Trans., 2021, 50, 6766–6772 RSC.
- M. J. Krahfuss and U. Radius, Dalton Trans., 2021, 50, 6752–6765 RSC.
- M. Ghosh and S. Khan, Dalton Trans., 2021, 50, 10674–10688 RSC.
- S. Fujimori and S. Inoue, Eur. J. Inorg. Chem., 2020, 2020, 3131–3142 CrossRef CAS PubMed.
- Y. P. Zhou and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3715–3728 CrossRef CAS PubMed.
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- S. Raoufrnoghaddam, Y. P. Zhou, Y. Wang and M. Driess, J. Organomet. Chem., 2017, 829, 2–10 CrossRef.
- R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444–456 CrossRef CAS PubMed.
- M. Driess, Nat. Chem., 2012, 4, 525–526 CrossRef CAS PubMed.
- S. S. Sen, S. Khan, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2012, 45, 578–587 CrossRef CAS PubMed.
- N. Muthukumaran, K. Velappan, K. Gour and G. Prabusankar, Coord. Chem. Rev., 2018, 377, 1–43 CrossRef CAS.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2010, 110, 3850–3850 CrossRef CAS.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- A. J. Arduengo, F. Davidson, R. Krafczyk, W. J. Marshall and R. Schmutzler, Monatsh. Chem., 2000, 131, 251–265 CrossRef CAS.
- Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412–12413 CrossRef CAS PubMed.
- P. Bissinger, H. Braunschweig, K. Kraft and T. Kupfer, Angew. Chem., Int. Ed., 2011, 50, 4704–4707 CrossRef CAS PubMed.
- J. C. Li, Y. S. Liu, S. Kundu, H. Keil, H. P. Zhu, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Inorg. Chem., 2020, 59, 7910–7914 CrossRef CAS PubMed.
- H. Wang and Z. Xie, Organometallics, 2021, 40, 3819–3824 CrossRef CAS.
- A. Gackstatter, H. Braunschweig, T. Kupfer, C. Voigt and N. Arnold, Chem. – Eur. J., 2016, 22, 16415–16419 CrossRef CAS PubMed.
- H. Braunschweig, T. Brückner, A. Deißenberger, R. D. Dewhurst, A. Gackstatter, A. Gärtner, A. Hofmann, T. Kupfer, D. Prieschl, T. Thiess and S. R. Wang, Chem. – Eur. J., 2017, 23, 9491–9494 CrossRef CAS PubMed.
- S. Khoo, Y.-L. Shan, M.-C. Yang, Y. Li, M.-D. Su and C.-W. So, Inorg. Chem., 2018, 57, 5879–5887 CrossRef CAS PubMed.
- L. Zhu, J. Zhang and C. Cui, Inorg. Chem., 2019, 58, 12007–12010 CrossRef CAS PubMed.
- M. K. Bisai, V. S. V. S. N. Swamy, K. V. Raj, K. Vanka and S. S. Sen, Inorg. Chem., 2021, 60, 1654–1663 CrossRef CAS PubMed.
- Y. Ding, Y. Li, J. Zhang and C. Cui, Angew. Chem., Int. Ed., 2022, 61, e202205785 CrossRef CAS PubMed.
- L. Kong, J. Zhang, H. Song and C. Cui, Dalton Trans., 2009, 5444–5446 RSC.
- P. Zark, A. Schäfer, A. Mitra, D. Haase, W. Saak, R. West and T. Müller, J. Organomet. Chem., 2010, 695, 398–408 CrossRef CAS.
- Y. Wang, Y. Xie, P. Wei, H. F. Schaefer III and G. H. Robinson, Dalton Trans., 2019, 48, 3543–3546 RSC.
- Y. Wang, P. M. Tran, Y. Xie, P. Wei, J. N. Glushka, H. F. Schaefer III and G. H. Robinson, Angew. Chem., Int. Ed., 2021, 60, 22706–22710 CrossRef CAS PubMed.
- Z. Liu and C. Cui, J. Organomet. Chem., 2020, 906, 121041 CrossRef CAS.
- See the ESI† for synthetic, computational, and crystallographic details.
- L. Weber, J. Forster, H. G. Stammler and B. Neumann, Eur. J. Inorg. Chem., 2006, 5048–5056 CrossRef CAS.
- Y. Wang, Y. Xie, P. Wei, S. A. Blair, D. Cui, M. K. Johnson, H. F. Schaefer III and G. H. Robinson, Angew. Chem., Int. Ed., 2018, 57, 7865–7868 CrossRef CAS PubMed.
- B. Krebs, G. Schwetlik and M. Wienkenhöver, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 257–261 CrossRef.
- S. Molitor and V. H. Gessner, Chem. – Eur. J., 2013, 19, 11858–11862 CrossRef CAS PubMed.
- K. Hensen, T. Zengerly, P. Pickel and G. Klebe, Angew. Chem., Int. Ed. Engl., 1983, 22, 725–726 CrossRef.
- F. Hanusch, D. Munz, J. Sutter, K. Meyer and S. Inoue, Angew. Chem., Int. Ed., 2021, 60, 23274–23280 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and computational details and structural and spectral characterization. CCDC 2294615–2294617, 2327747–2327749. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03843b |
| This journal is © The Royal Society of Chemistry 2024 |