
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubtle adjustments for constructing multi-nuclear luminescent lanthanide organic polyhedra with triazole-based chelates†
Xiao-Qing
Guo
a,
Li-Peng
Zhou
 a,
Shao-Jun
Hu
a and
Qing-Fu
Sun
*ab
a,
Shao-Jun
Hu
a and
Qing-Fu
Sun
*ab
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China. E-mail: qfsun@fjirsm.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
First published on 6th February 2024
Abstract
Controlled self-assembly of predetermined multi-nuclear lanthanide organic polyhedra (LOPs) still presents a challenge, primarily due to the unpredictable coordination numbers and labile coordination geometries of lanthanide ions. In this study, through introducing triazole-based chelates to increase the chelating angle of C2-symmetric linear ligands and stabilize the coordination geometry of Eu(III) centers, M4L6-type (M = EuIII, L = ligand) tetrahedra were efficiently synthesized, especially a biphenyl-bridged ligand which is well known to form M2L3-type helicates. A series of LOPs were formed and characterized by high-resolution electrospray ionization time-of-flight mass spectroscopy (ESI-TOF-MS) and X-ray crystallography. Moreover, the europium complexes exhibit bright emission (luminescence quantum yield up to 42.4%) and circularly polarized luminescence properties (|glum| up to 4.5 × 10−2). This study provides a feasible strategy for constructing multi-nuclear luminescent LOPs towards potential applications.
Introduction
Coordination-directed self-assembly is a compelling and versatile approach in the realm of supramolecular chemistry, holding significant promise for addressing the evolving needs of the scientific community.1–5 Within this context, 3D metallosupramolecular architectures have emerged as intriguing candidates with diverse applications.6–16 However, while the potential of coordination-directed self-assembly is evident, the utilization of lanthanide ions in constructing such assemblies, especially high-nuclear assemblies, has faced unique challenges.17 Lanthanides exhibit remarkable spectroscopic and electromagnetic properties that make them attractive building blocks;18–21 however, their application has been constrained by factors such as variable coordination numbers and low steric requirement. Inspired by the pioneering research conducted by Piguet,22,23 Bünzli,24 Gunnlaugsson,25 Park26 and others,27–40 primarily including mononuclear bundles and dinuclear helicates, the field of lanthanide assembly chemistry has witnessed significant advancements. These achievements serve as valuable benchmarks, offering valuable insights and guidance for the future design and synthesis of more intricate and functionalized LOPs.17,19,41–46Following the symmetry principles elucidated by Raymond's group,47 our group published a structural evolution of Ln2nL3n (Ln, lanthanides; L, ligands) edifices from helicates and tetrahedra to cubes by systematically increasing the offset between two chelating arms on C2-symmetric bis(tridentate) ligands.48 Notably, when compared to malposed C2-symmetric ligands, linear ligands demonstrate greater ascendancy in maximizing the inner cavity volumes and window sizes of LOPs. However, the resulting structures assembled by linear amide–pyridine–amide ligands (APA-L, e.g., chelating arms bridged by phenyl or biphenyl unit) and LnIII are almost Ln2L3 helicates (Scheme 1).49–52 In sharp contrast to LnIII ions, the self-assembly of transition metal ions (M) with similar linear ligands typically results in the formation of M4L6 tetrahedra.53 The underlying reason for this difference lies in the variable coordination configurations of LnIII ions, a consequence of their unique electrostatic interactions, which result in a wide range of twist angles. Therefore, to construct Ln4L6 tetrahedra or higher-nuclear LOPs based on linear ligands, a rational strategy needs to be considered in the design of ligands.
 | ||
| Scheme 1 Ligand modification for constructing multi-nuclear LOPs (Linker: phenyl/biphenyl/terphenyl; Con.: concentration). | ||
According to Albrecht-Gary's report,54,55 the self-assembly mechanism of triple-stranded helicates proceeds though a pre-organized “side-by-side” intermediate species, Ln2L2. Hence, the key to preventing the formation of low-nuclear Ln2L3 helicates lies in inhibiting the occurrence of the Ln2L2 intermediate, which can be achieved by enhancing the chelating angle and the ligand rigidity. To efficiently avoid the formation of Ln2L3, we have implemented two modifications to the ligands: (i) increasing the chelating angle and rigidity of C2-symmetric linear ligands by introducing an amide–pyridine–triazole (APT) chelating arm and (ii) stabilizing the coordination geometry of the metal center by modifying the electron-rich naphthylethyl group on the periphery of the ligands (Scheme 1).
Herein, a series of LOPs were constructed by self-assembling APT-based C2-symmetric ligands with EuIII ions (Fig. 1). In contrast to our earlier findings and those of others,48,50 we observed that linear ligands based on biphenyl spacers primarily formed Ln4L6 tetrahedra instead of the previously observed Ln2L3 helicates. This shift in preference can be attributed to the larger chelating angle, effectively inhibiting the formation of triple-stranded helicates. As a control experiment, isopropyl-modified L2AC exhibited a tendency to form di-nuclear helicates relative to naphthylethyl-modified L2RR/SS, suggesting the feasibility of stabilizing the coordination geometry of the metal center through peripheral auxiliary groups. Furthermore, a significant |glum| magnitude (4.5 × 10−2) for Eu4(L2SS)6 was observed in CPL spectroscopy, attributed to the enhanced metal coordination environment through inter-ligand synergistic effects.
 | ||
| Fig. 1 The ligand length/concentration-dependent self-assembly behavior of linear C2-symmetric bis(tridentate) ligands with Eu(OTf)3. | ||
Results and discussion
The APT-based bis(tridentate) ligands were synthesized in a three-step process: firstly, the chiral naphthylethyl group was attached to 6-bromopicolinic acid by amide coupling, followed by a Sonogashira-coupling reaction to introduce the alkyne group; then three spacers with different lengths bridged the chelating arms via a high-yield Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction.56 All ligands were fully characterized using NMR and high-resolution ESI-TOF-MS spectra (see the Experimental section in the ESI† for details).By simply increasing the linear length of the ligand, the assembly behavior of LnRR/SS (n = 1–3) shows significant variations under the same conditions. When 3 eq. of LnRR/SS (n = 1–3) were treated with 2 eq. of Eu(OTf)3 in a CD3CN/CD3OD (v/v 4/1) mixture at 50 °C for several minutes, the initially turbid suspensions quickly turned into clear solutions. However, the architectural features of the resulting assemblies exhibited substantial diversity due to the inherent dissimilarities in the respective ligands.
In the case of L1SS or L1AC, a singular set of 1H NMR signals was observed, signifying the formation of a single species with high symmetry (Fig. 2A and B, S22 and S42†). Compared with free L1SS, most signals originating from the assembly experienced shifts attributed to the effects of lanthanide-induced shifts and relaxation rate enhancements.57 The 1H DOSY measurement of the solution showed that all signature signals had the same diffusion coefficient D1 = 5.68 × 10−10 m2 s−1, indicating the formation of a single species with a hydrodynamic diameter of 2.3 nm, estimated by the Stokes–Einstein equation (Fig. 2E). The composition of Eu4(L1SS)6(OTf)12 in solution was further confirmed by ESI-TOF-MS analysis (Fig. S81†), which revealed a series of prominent peaks observed at m/z = 721.1652, 845.4669, 1011.2031, 1243.2347, and 1591.2808, corresponding well to [Eu4(L1SS)6(OTf)n]12−n+ (n = 4–8). It is worth noting that the assembly of APA-L, bridged by a biphenyl linker with a similar length of 16.5 Å (distance between pyridine nitrogen atoms, 16.0 Å for L1SS or L1AC, Fig. S66†), only resulted in the triple helicate,50 verifying the feasibility of chelating angle regulation for high-nuclear LOPs.
 | ||
Fig. 2
1H NMR spectra (400 MHz, 298 K) of (A) L1SS (CDCl3), (B) Eu4(L1SS)6 ( ), the assemblies of Eu(OTf)3 with L2SS under different concentrations (C) 7.2 × 10−3 M, (D) 1.4 × 10−2 M (CD3CN/CD3OD, v/v 4/1, Eu2(L2SS)3 ( ), the assemblies of Eu(OTf)3 with L2SS under different concentrations (C) 7.2 × 10−3 M, (D) 1.4 × 10−2 M (CD3CN/CD3OD, v/v 4/1, Eu2(L2SS)3 ( ), Eu4(L2SS)6 ( ), Eu4(L2SS)6 ( ). 1H DOSY of E) Eu4(L1SS)6, and (F) the mixture of Eu2(L2SS)3 and Eu4(L2SS)6. ). 1H DOSY of E) Eu4(L1SS)6, and (F) the mixture of Eu2(L2SS)3 and Eu4(L2SS)6. | ||
However, two sets of signals were observed during the assembly of L2SS (with an N⋯N distance of 19.5 Å) with Eu(OTf)3, implying the possible presence of a mixture comprising Eu4(L2SS)6 and Eu2(L2SS)3 (Fig. 2C). This observation is consistent with the distinct diffusion coefficients observed in 1H DOSY, where D2 = 4.85 × 10−10 m2 s−1 and D3 = 7.53 × 10−10 m2 s−1 (Fig. 2F). The composition of the mixture was also confirmed by ESI-TOF-MS (Fig. S82†). By integrating the 1H NMR signals associated with Eu2(L2SS)3 and Eu4(L2SS)6 at a concentration of [L2SS] = 4.8 mM, the apparent equilibrium constant (K) for 2Eu2(L2SS)3 ⇌ Eu4(L2SS)6 was determined to be 2.2 × 103 M−1 (ΔG = −19.1 kJ mol−1, Fig. S55†). When the concentration of [L2SS] was increased from 4.8 to 14.4 mM, the complete transformation of Eu4(L2SS)6 was observed (Fig. 2D, S52†). In addition, once formed, the tetrahedral cage can maintain long-term stability under dilution conditions (3 days, conversion rate <25%, Fig. S53†). Furthermore, in comparison with L2AC, it was observed that the assembly of L2SS tended to generate a higher proportion of tetrahedra under the same ligand concentration conditions (Fig. S54†). The corresponding apparent equilibrium constant (K) for 2Eu2(L2AC)3 ⇌ Eu4(L2AC)6 was calculated as 39.4 M−1 (ΔG = −8.9 kJ mol−1, Fig. S56†). Therefore, it can be inferred that the peripheral auxiliary group played a pivotal role in regulating the coordination geometry around the EuIII center.
VT-NMR experiments were carried out to determine the thermodynamic parameters of the equilibria 2Eu2(L2AC)3 ⇌ Eu4(L2AC)6 (1) and 2Eu2(L2SS)3 ⇌ Eu4(L2SS)6 (2) using the van't Hoff equation (Figs. S57–S62†). The negative ΔG° values for the dimerization equilibria suggest that the transformation from helicates to tetrahedra is a spontaneous process at room temperature. To analyze the internal driving forces that regulate the helicate/tetrahedron equilibrium, the statistical factors (ωEu,L2ACdim = 3/2 and ωEu,L2SSdim = 3) and the corresponding free energy contributions (ΔGEu,L2ACdim = −1.0 kJ mol−1 and ΔGEu,L2SSdim = −2.7 kJ mol−1) to equilibria (1) and (2) were calculated based on the literature23 (see the detailed calculations in the ESI†). The negative free energies ΔGEu,Ldim indicate that the statistical factors favor the formation of tetrahedral cages due to the relaxation of the rotational degeneracy. Moreover, from a purely statistical point of view, ΔGEu,L2SSdim < ΔGEu,L2ACdim demonstrates that the trend of dimerization in equilibrium (2) is stronger than that in equilibrium (1).
To quantitatively analyze the contribution of enthalpic/entropic driving forces to the dimerization process, the relationship between the dimerization constants (βEu,Ldim) and the effective molarities (EMEu,L2,3) was deduced as βEu,L2ACdim = 3/2(0.71)3/EMEu,L2AC2,3 = 0.54/EMEu,L2AC2,3 and βEu,L2SSdim = 3(0.75)3/EMEu,L2SS2,3 = 1.27/EMEu,L2SS2,3, respectively.23 Firstly, the term 3/2 or 3 from the statistical factors implies an entropic driving preference for the formation of tetrahedral cages, coinciding with positive ΔS° (0.016 kJ mol−1 K−1 and 0.030 kJ mol−1 K−1) obtained by the VT-NMR experiments. Secondly, the transformation from triple-stranded helicates (30-membered metallomacrocycle, Fig. S65†) to tetrahedra (45-membered metallomacrocycle), accompanied by a reduction in enthalpic ring strain, further supports the favorability of the dimerization process. Experimental EM values at different temperatures were calculated to compare the relative trends for equilibria (1) and (2) (Table S1†). With the increase of temperature, the EM values showed a positive correlation, indicating that the temperature rise overcomes the gain of enthalpic ring strain and promotes the formation of helicates. Notably, the significantly lower EMEu,L2SS2,3 compared to EMEu,L2AC2,3 (about two orders of magnitude) indicates a stronger inclination of [Eu2(L2SS)3]6+ to form tetrahedral cages. This preference may be attributed to serve ring strains or poor preorganization, which is consistent with the lower ΔH° = −9.923 kJ mol−1 of equilibrium (2) compared to ΔH° = −4.288 kJ mol−1 of equilibrium (1).
Given the high charge state of the complexes and the use of polar solvents, both the alterations in entropy and enthalpy largely depend on the change of solvent energies that can be evaluated by the Born equation.58 The contributions of solvation energies (ΔsolvGEu,L4,6 − 2ΔsolvGEu,L2,3) for equilibria (1) and (2) were determined to be −976 kJ mol−1 and −2777 kJ mol−1, respectively. Therefore, considerable changes in solvation energies suggest a pivotal role in influencing the alterations of entropy and enthalpy during the dimerization process. Furthermore, from the perspective of solvation energies, the trend to form tetrahedra in equilibrium (2) emerges as notably more pronounced than that in equilibrium (1).
As anticipated, in the case of the extended-version ligand L3SS (with an N⋯N distance of 23.8 Å), it exclusively forms the Eu2(L3SS)3 helicate, which was confirmed by 1H DOSY, NMR spectroscopy and ESI-TOF-MS (Figs. S38, S41 and S83†). These findings suggest that the strategy of regulating the chelating angle and coordination geometry configuration becomes unwieldy when using more flexible linear ligands.
Fortunately, colorless crystals suitable for X-ray diffraction analysis were obtained by the slow diffusion of dichloromethane vapour into the complex solution. In the case of Eu4(L1RR)6, a tetrahedral arrangement with approximate T symmetry was revealed, with four facially coordinated Eu3+ centers being bridged by six bis(tridentate) linear ligands (Fig. 3A). All Eu3+ centers adopt the same Λ configuration induced by the R,R-ligand point chirality. The average distance between two adjacent EuIII centers was found to be 14.2 Å, defining a theoretically tetrahedral cavity of ca. 342 Å3. Notably, intriguing triple π–π stacking interactions were observed between the electron-rich naphthylethyl groups at the periphery and the electron-deficient pyridine groups from adjacent L1RR ligands. These interactions, resembling a mechanical contraction, appeared to enlarge the distance between the long arms of the complex, effectively preventing the formation of the helicate.
 | ||
| Fig. 3 Crystal structures of (A) Eu4(L1RR)6 and (B) Eu4(L2AC)6 (atom colours: red, Eu(III); blackish green, C; white, H; azarin, O; blue, N). | ||
Regrettably, repeated attempts to crystallize Eu4(L2SS)6 were unsuccessful, possibly due to the low symmetry and the large cavity of the complex. As an alternative, a high-quality crystal structure for Eu4(L2AC)6 was obtained to unambiguously verify the tetrahedral architecture (Fig. 3B). In comparison with Eu4(L1SS)6, it is evident that the central cavity of the tetrahedron was significantly increased to 736 Å3 (Fig. S71†) by extending the distance between the central Eu3+ ions to 18.5 Å. This expansion suggests its potential application in host–guest chemistry.
The UV-Vis absorption of LnSS (n = 1–3) and their corresponding assemblies was measured at room temperature (Fig. S72†). With the extension of the spacer, the aromaticity of the ligands was gradually increased, as evidenced by a noticeable bathochromic-shift in absorption (Fig. 4A). Circular dichroism (CD) spectroscopy was employed to verify the enantiomeric nature of the assemblies in solution. In the case of Eu4(L2SS/RR)6, the enantiomers exhibited a mirror image with a strong Cotton effect, manifesting as split peaks located at 221, 281 and 320 nm, corresponding to the UV-Vis region and arising from π → π* transitions (Fig. 4B). Similarly, the enantiomeric nature and optical activity of Eu4(L1SS/RR)6 or Eu2(L3SS/RR)3 were also confirmed by their CD spectra. The emission spectra of EuIII assemblies were obtained in an acetonitrile solution. For Eu4(L2SS/RR)6, characteristic emission peaks were observed at 580, 594, 615, 649, 693 and 750 nm, corresponding to the 5D0 → 7FJ (J = 0, 1, 2, 3, 4, 5) transitions (Fig. 4C). The excitation window for the Eu4(L1SS)6 cage is limited to <340 nm, while the Eu2(L3SS)3 helicate can be excited at 370 nm. Notably, except for L1SS, highly efficient sensitization for EuIII ions was demonstrated by the other two ligands. The quantum yields (Φ) of these supramolecular assemblies were determined to be 1.7% for Eu4(L1SS)6, 37.2% for Eu4(L2SS)6 and 42.4% for Eu2(L3SS)3 (Table S2†). Compared to analogous bis(tridentate) ligands featuring amide groups,48,50,51 the rigid ligands with triazole-based chelates demonstrate enhanced efficacy in sensitizing the emission of europium ions. Typically, the quantum yields of the europium complexes with biphenyl-bridged bis(tridentate) ligands significantly increase from Φ < 1% to 37.2%. We speculate that the triazole group effectively suppresses the dissipation of energy transfer from the sensitizing group (biphenyl) to the EuIII centers. Therefore, in addition to its effect on the assembly behavior, the triazole-based chelate group manifests a universally potent sensitizing performance for chiral europium complexes.
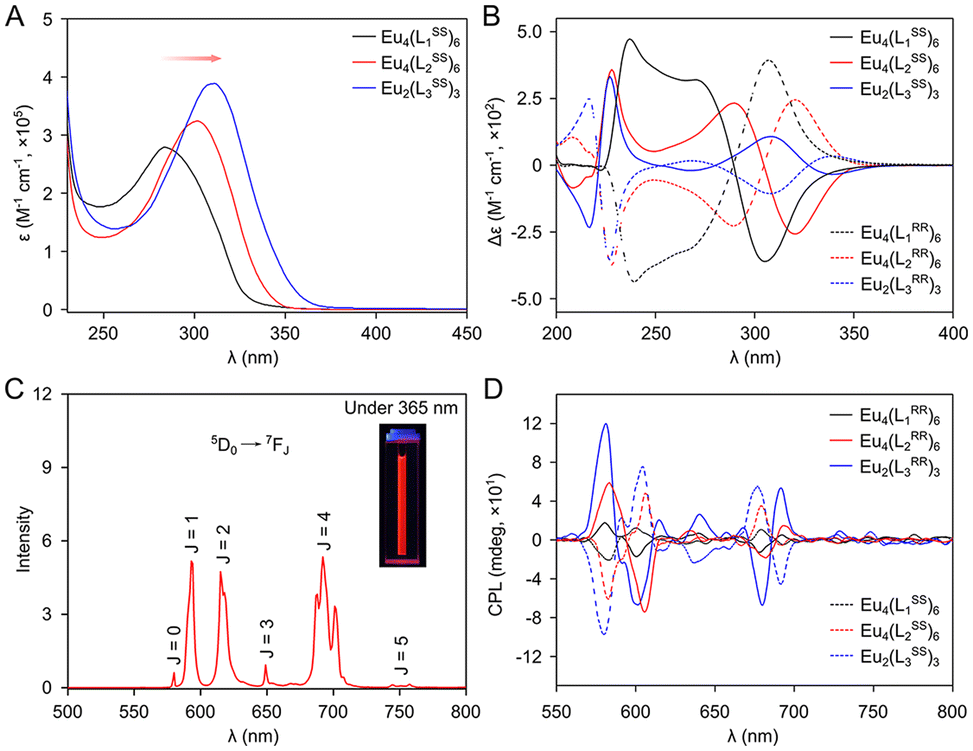 | ||
| Fig. 4 (A) UV-Vis adsorption and (B) CD spectra of the assemblies in MeCN (1 × 10−5 M). (C) Emission spectrum of Eu4(L2SS)6 excited at 331 nm with a photograph inset taken under 365 nm lamp. (D) CPL spectra of the assemblies in MeCN (1.0 × 10−5 M). | ||
To analyze the origin of different quantum yields, the phosphorescence spectra of Gd4(L1SS)6, Gd4(L2SS)6, and Gd2(L3SS)3 were tested at 77 K under an N2 atmosphere using a gating technique, from which the ligand triplet state energy levels E0–0(3T) of LnSS (n = 1–3) were estimated to be 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 920, 20790 and 19342 cm−1, respectively (Fig. S74†). The corresponding energy gaps of E0–0(3T)-E(5D0) were determined to be 3720, 3520, and 2142 cm−1, respectively. Generally, a larger energy gap implies a less favorable energy transfer from the 3T to 5D0 energy level. Consequently, the sensitization capability of ligands follows the sequence L3SS > L2SS > L1SS. To further quantitatively compare the sensitization capabilities of ligands, the sensitization efficiency of ligands (ηsens) and the intrinsic quantum yield of europium (QLnLn) are calculated (see the ESI† for details). The europium complexes demonstrate comparable intrinsic quantum yields: Eu4(L1SS)6, 42.4%; Eu4(L2SS)6, 43.3%; and Eu2(L3SS)3, 42.2%. Nonetheless, the ηsens of ligand L1SS (4%) is significantly lower than those of L2SS (85.9%) and L3SS (∼100%). Therefore, the difference in the overall quantum yields (QLLn) is primarily ascribed to the disparate sensitization efficiencies exhibited by the ligands.
920, 20790 and 19342 cm−1, respectively (Fig. S74†). The corresponding energy gaps of E0–0(3T)-E(5D0) were determined to be 3720, 3520, and 2142 cm−1, respectively. Generally, a larger energy gap implies a less favorable energy transfer from the 3T to 5D0 energy level. Consequently, the sensitization capability of ligands follows the sequence L3SS > L2SS > L1SS. To further quantitatively compare the sensitization capabilities of ligands, the sensitization efficiency of ligands (ηsens) and the intrinsic quantum yield of europium (QLnLn) are calculated (see the ESI† for details). The europium complexes demonstrate comparable intrinsic quantum yields: Eu4(L1SS)6, 42.4%; Eu4(L2SS)6, 43.3%; and Eu2(L3SS)3, 42.2%. Nonetheless, the ηsens of ligand L1SS (4%) is significantly lower than those of L2SS (85.9%) and L3SS (∼100%). Therefore, the difference in the overall quantum yields (QLLn) is primarily ascribed to the disparate sensitization efficiencies exhibited by the ligands.
The enantiomeric nature of the assemblies was verified through the analysis of their circularly polarized luminescence (CPL) spectra (Fig. 4D), suggesting the presence of a chiral environment surrounding the EuIII center. Among these complexes, the largest luminescence dissymmetry factor (glum) for Eu4(L2SS)6 was calculated to be +4.5 × 10−2 (Fig. S78†), corresponding to the 5D0 → 7F1 transition. This value is comparable to those
of previous mononuclear lanthanide complexes.59,60 It can be inferred that the synergistic effect of ligands within the multinuclear rare earth cage contributes to an increase in the rigidity of the coordination environment of lanthanide ions, effectively increasing the magnitude of glum.61
Conclusions
In summary, subtle adjustments in the chelating arm not only enable the controllable synthesis of high-nuclear LOP edifices constructed by linear ligands but also lead to the efficient sensitization of lanthanide ion luminescence. The strategy of regulating the chelating angle and introducing auxiliary groups can effectively overcome the shortcoming of variable coordination orientations of lanthanide ions, providing a feasible pathway to construct multi-nuclear LOPs.Experimental section
General
Unless otherwise stated, all chemicals and solvents were purchased from commercial companies and used as received. Deuterated solvents were obtained from Admas, J&K scientific, and Sigma-Aldrich. Triethylamine (TEA) was dried using CaH2. The 1D and 2D-NMR spectra were measured on a Bruker Biospin Avance III (400 MHz) and JEOL ECZ600S (600 MHz) spectrometers. 1H NMR chemical shifts were determined with respect to residual signals of the deuterated solvents used. ESI-TOF-MS spectra were recorded on an Impact II UHR-TOF mass spectrometer from Bruker, with tuning mix as the internal standard. Data analysis was conducted using the Bruker Data Analysis software (Version 4.3), and simulations were performed with the Bruker Isotope Pattern software. UV-Vis spectra were recorded on a UV-2700 UV-Visible spectrophotometer from SHIMADZU Corporation. Excitation and emission spectra were recorded on the FS5 spectrofluorometer from Edinburg Photonics. The microsecond emission lifetimes of solution samples were measured on an Edinburgh Instrument FLS980 spectrometer. Spectra were corrected for the experimental functions. Circular dichroism (CD) spectra were recorded on a MOS-450 circular dichroism spectrometer. Circularly polarized luminescence (CPL) measurements were performed with a JASCO CPL-200 spectrometer.Synthesis of ligands LnAC (n = 1–3)
1,4-Diazidobenzene (108 mg, 1.0 mmol, 1.0 eq.), 6-ethynyl-N-isopropylpicolinamide (395 mg, 2.1 mmol, 2.1 eq.), sodium ascorbate (411 mg, 2.1 mmol, 2.1 eq.), and CuSO4·5H2O (225 mg, 0.9 mmol, 0.9 eq.) were added to a solution of DMF (60 mL). The reaction mixture was stirred at 60 °C for 48 h. After that, the reaction solution was cooled to room temperature, and the solvent was removed under reduced pressure. Then, 50 mL of EDTA-saturated aqueous solution was added and stirred for 1 h. The solution was extracted with an organic solvent mixture (50 mL × 3, DCM/MeOH, v/v 10/1), and the organic phase was washed with distilled water (30 mL × 2) and dried with anhydrous Na2SO4. The solvents were removed in vacuo to afford a crude product, which was further purified chromatographically (SiO2, DCM/MeOH, v/v 100/1). The white powder L1AC was obtained after drying in vacuum (236 mg, 43.9%). 1H NMR (400 MHz, CDCl3/CD3OD v/v 5/1, 298 K) δ = 8.79 (s, 1H), 8.22 (d, J = 8.3 Hz, 1H), 8.19 (d, J = 8.8 Hz, 1H), 8.10 (d, J = 6.9 Hz, 1H), 8.07 (s, 2H), 7.94 (t, J = 7.8 Hz, 1H), 4.25 (m, J = 13.2, 6.6 Hz, 1H), 1.28 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3/CD3OD v/v 5/1, 298 K) δ = 163.60, 150.07, 148.40, 147.96, 138.68, 136.94, 123.09, 121.95, 120.53, 41.74, 22.54. ESI-TOF-MS for C28H28N10O2 [M + Na]+: calcd, m/z = 559.2290; found, 559.2289.L2AC was synthesized using a similar procedure, starting from 4,4′-diazidobiphenyl (80 mg, 0.34 mmol, 1.0 eq.). The white powder L2AC was obtained with a yield of 61.0% (127 mg). 1H NMR (400 MHz, CDCl3/CD3OD v/v 5/1, 298 K) δ = 8.68 (s, 2H), 8.27–8.14 (m, 4H), 8.11 (d, J = 7.5 Hz, 2H), 8.00–7.89 (m, 6H), 7.80 (d, J = 8.6 Hz, 4H), 4.25 (m, J = 13.2, 6.6 Hz, 2H), 1.28 (d, J = 6.6 Hz, 12H). 13C NMR (101 MHz, CDCl3/CD3OD v/v 5/1, 298 K) δ = 163.50, 149.97, 148.05, 140.42, 138.55, 136.40, 128.48, 123.00, 121.83, 121.16, 120.37, 41.55, 22.46. ESI-TOF-MS for C34H32N10O2 [M + Na]+: calcd, m/z = 635.2605; found, 635.2602.
L3AC was synthesized with using a similar procedure, starting from 4,4′-diazidoterphenyl (235 mg, 1.25 mmol, 2.5 eq.). The white powder L3AC was obtained with a yield of 45.3% (156 mg). 1H NMR (600 MHz, CDCl3/CD3OD v/v 5/1, 298 K) δ = 8.67 (s, 1H), 8.16 (d, J = 7.8 Hz, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.93 (t, J = 6.5 Hz, 1H), 7.90 (d, J = 8.5 Hz, 2H), 7.80 (d, J = 10.9 Hz, 2H), 7.72 (s, 2H), 4.36–4.11 (m, 6.6 Hz, 1H), 1.27 (d, J = 6.6 Hz, 6H). 13C NMR (151 MHz, CDCl3/CD3OD v/v 5/1, 298 K) δ = 163.64, 149.97, 148.15, 147.97, 141.44, 139.19, 138.61, 136.04, 128.39, 127.71, 123.04, 121.82, 121.12, 120.55, 41.62, 22.44. ESI-TOF-MS for C40H36N10O2 [M + Na]+: calcd, m/z = 711.2951; found, 711.2968.
Synthesis of ligands LnSS/RR (n = 1–3)
1,4-Diazidobenzene (160 mg, 1.0 mmol, 1.0 eq.) and 4S (901 mg, 3 mmol, 3.0 eq.) were added to a mixture of SA/sodium ascorbate (411 mg, 2.1 mmol, 2.1 equiv.) and CuSO4·5H2O (225 mg, 0.9 mmol, 0.9 eq.), and the mixture was stirred at 60 °C for 24 h. After filtration, the solvent was removed under vacuum distillation, and the crude product was purified chromatographically (SiO2, DCM/MeOH 100/1) to afford L1SS as a pale yellow solid (456 mg, 59.9%). 1H NMR (400 MHz, CDCl3/CD3OD v/v 5/1, 298 K) δ = 8.59 (d, J = 8.5 Hz, 2H), 8.46 (s, 2H), 8.21 (d, J = 7.6 Hz, 4H), 8.11 (d, J = 8.4 Hz, 2H), 7.95 (t, J = 7.8 Hz, 2H), 7.80 (d, J = 7.9 Hz, 2H), 7.77 (s, 4H), 7.74 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 7.1 Hz, 2H), 7.50–7.43 (m, 2H), 7.43–7.35 (m, 4H), 6.05 (p, J = 6.9 Hz, 2H), 1.77 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3/CD3OD v/v 5/1, 298 K) δ = 163.24, 149.82, 148.14, 148.08, 138.53, 138.37, 136.58, 133.88, 131.15, 128.78, 128.33, 126.49, 125.82, 125.13, 123.38, 123.12, 122.84, 122.16, 121.54, 120.25, 45.21, 20.93; ESI-TOF-MS for C57H51N15O3 [M + Na]+: calcd, m/z = 783.2915; found, 783.2910. L1RR was synthesized in the same procedure as above, starting from 4R.L2SS was synthesized using a similar procedure, starting from 4,4′-diazidobiphenyl 4,4′-diazido-1,1′-biphenyl (236 mg, 1.0 mmol, 1.0 eq.). Pale yellow L2SS was obtained with a yield of 75.1% (628 mg). 1H NMR (400 MHz, CDCl3, 298 K) δ = 8.41 (s, 2H), 8.37 (dd, J = 7.8, 0.8 Hz, 2H), 8.32 (d, J = 8.6 Hz, 2H), 8.27 (dd, J = 7.8, 0.8 Hz, 2H), 8.20 (d, J = 8.4 Hz, 2H), 8.00 (t, J = 7.8 Hz, 2H), 7.87 (d, J = 7.8 Hz, 2H), 7.82 (dd, J = 8.2, 6.3 Hz, 6H), 7.72 (d, J = 8.6 Hz, 4H), 7.64 (d, J = 7.1 Hz, 2H), 7.53 (t, J = 7.0 Hz, 2H), 7.47 (dd, J = 14.7, 7.2 Hz, 4H), 6.17–6.09 (m, 2H), 1.84 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3, 298 K) δ = 163.04, 149.76, 148.57, 148.11, 140.14, 138.44, 138.36, 136.40, 134.00, 131.27, 128.84, 128.45, 128.30, 126.60, 125.90, 125.20, 123.56, 123.18, 122.91, 122.04, 121.00, 119.99, 45.23, 21.11; ESI-TOF-MS for C57H51N15O3 [M + Na]+: calcd, m/z = 859.3228; found, 859.3213. L2RR was synthesized in the same procedure as above, starting from 4R.
L3SS was synthesized using a similar procedure, starting from 4,4′′-diazido-1,1′:4′,1′′-terphenyl (312 mg, 1.0 mmol, 1.0 eq.). Pale yellow L3SS was obtained with a yield of 55.0% (502 mg). 1H NMR (400 MHz, CDCl3, 298 K) δ = 8.40 (d, J = 8.6 Hz, 2H), 8.38 (s, 2H), 8.33 (d, J = 7.8 Hz, 2H), 8.24 (t, J = 8.1 Hz, 2H), 8.18 (d, J = 8.4 Hz, 2H), 7.95 (t, J = 7.8 Hz, 2H), 7.84 (d, J = 7.7 Hz, 2H), 7.78 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.5 Hz, 4H), 7.70–7.59 (m, 10H), 7.50 (dd, J = 11.2, 4.1 Hz, 2H), 7.44 (dd, J = 16.3, 8.2 Hz, 4H), 6.16–6.06 (m, 2H), 1.83 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3, 298 K) δ = 163.11, 149.74, 148.61, 148.00, 140.89, 138.98, 138.41, 136.00, 133.97, 131.26, 128.83, 128.40, 128.09, 127.54, 126.57, 125.88, 125.19, 123.53, 123.12, 122.93, 121.98, 120.85, 120.05, 45.20, 29.72, 21.11. ESI-TOF-MS for C57H51N15O3 [M + Na]+: calcd, m/z = 935.3541; found, 935.3531. L3RR was synthesized in the same procedure as above, starting from 4R.
Synthesis of self-assembled complex of L1AC
To a white suspension of L1AC (2.0 mg, 3.7 μmol, 1.0 eq.) in a solution of CD3CN/CD3OD (v/v 4/1, 500 μL), Eu(OTf)3 (1.5 mg, 2.5 μmol, 0.67 eq.) was added and then stirred at 50 °C for 1 h. The light-yellow suspension gradually turned into a homogeneous yellow solution. This solution was characterized without further treatment. 1H NMR (400 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 9.02 (s, 2H), 8.97 (s, 4H), 6.21 (t, J = 7.9 Hz, 2H), 5.97 (s, 2H), 5.28 (d, J = 7.5 Hz, 2H), 5.05 (d, J = 7.7 Hz, 2H), 3.97 (s, 1H), 3.06 (d, J = 4.6 Hz, 6H), 0.90 (d, J = 4.6 Hz, 6H). 13C NMR (101 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 173.83, 152.37, 141.05, 136.20, 131.08, 122.91, 119.81, 95.58, 86.25, 44.84, 23.95, 22.19. ESI-TOF-MS for Eu4(L1AC)6(OTf)12: the following picked signals are those at the highest intensities. m/z calcd for [Eu4(L1AC)6(OTf)4]8+ 552.8660, found 552.8663; calcd for [Eu4(L1AC)6(OTf)5]7+ 653.1258, found 653.1260; calcd for [Eu4(L1AC)6(OTf)6]6+ 787.8055, found 787.8057; calcd for [Eu4(L1AC)6(OTf)7]5+ 974.1573, found 974.1572; calcd for [Eu4(L1AC)6(OTf)8]4+ 1254.6845, found 1254.6846; [Eu4(L1AC)6(OTf)9]3+ 1722.8971, found 1722.8948.Synthesis of self-assembled complex of L2AC
To a white suspension of L2AC (2.0 mg, 3.3 μmol, 1.0 eq.) in a solution of CD3CN/CD3OD (v/v 4/1, 500 μL), Eu(OTf)3 (1.3 mg, 2.2 μmol, 0.67 eq.) was added and then stirred at 50 °C for 1 h. The light-yellow suspension gradually turned into a homogeneous yellow solution. This solution was characterized without further treatment. 1H NMR (400 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 8.78 (s, 2H), 7.58 (d, J = 7.6 Hz, 4H), 7.53 (d, J = 7.6 Hz, 4H), 7.27 (t, J = 8.0 Hz, 2H), 6.32 (d, J = 7.2 Hz, 2H), 5.88 (d, J = 8.0 Hz, 2H), 4.23 (s, 2H), 1.55 (d, J = 4.4 Hz, 6H), 0.98 (d, J = 4.4 Hz, 6H). The 13C NMR signals were too weak to be measured due to inhibition of structural transformation via low concentration of ligand. ESI-TOF-MS for Eu2(L2AC)3(OTf)6: the following picked signals are those at the highest intensities. m/z calcd for [Eu2(L2AC)3(OTf)2]4+ 610.1398, found 610.1405; calcd for [Eu2(L2AC)3(OTf)3]3+ 863.1706, found 863.1716; calcd for [Eu2(L2AC)3(OTf)4]2+ 1369.2320, found 1369.2327. ESI-TOF-MS for Eu4(L2AC)6(OTf)12: m/z calcd for [Eu4(L2AC)6(OTf)6]6+ 863.0038, found 863.0052; calcd for [Eu4(L2AC)6(OTf)7]5+ 1065.3951, found 1065.3927; calcd for [Eu4(L2AC)6(OTf)8]4+ 1368.9819, found 1368.9825.Synthesis of self-assembled complex of L3AC
To a white suspension of L3AC (4.0 mg, 5.8 μmol, 1.0 eq.) in a solution of CD3CN/CD3OD (v/v 4/1, 500 μL), Eu(OTf)3 (2.3 mg, 3.9 μmol, 0.67 eq.) was added and then stirred at 50 °C for five minutes. The white suspension gradually turned into a homogeneous yellow solution. This solution was characterized without further treatment. 1H NMR spectra showed the quantitative formation of Eu2(L3AC)3(OTf)6. 1H NMR (600 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 8.76 (s, 1H), 7.58 (d, J = 10.8 Hz, 2H), 7.53 (d, J = 10.2 Hz, 2H), 7.33 (s, 3H), 6.38 (d, J = 6.0 Hz 1H), 5.96 (d, J = 9.6 Hz, 1H), 4.16 (s, 1H), 1.51 (s, 3H), 1.00 (s, 3H). 13C NMR (151 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 174.51, 160.79, 153.88, 145.89, 142.57, 138.64, 134.03, 132.66, 128.10, 128.02, 120.85, 104.86, 96.88, 88.10, 44.12, 22.08, 21.90. ESI-TOF-MS for Eu2(L3AC)3(OTf)6: the following picked signals are those at the highest intensities. m/z calcd for [Eu2(L3AC)3(OTf)1]5+ 503.7401, found 503.7407; calcd for [Eu2(L3AC)3(OTf)2]4+ 666.9133, found 666.9136; calcd for [Eu2(L3AC)3(OTf)3]3+ 939.2020, found 939.2018; calcd for [Eu2(L3AC)3(OTf)4]2+ 1483.2793, found 1483.2794.Synthesis of self-assembled complex of L1SS
To a white suspension of L1SS (2.0 mg, 2.6 μmol, 1.0 eq.) in a solution of CD3CN/CD3OD (v/v 4/1, 500 μL), Eu(OTf)3 (1.0 mg, 1.7 μmol, 0.67 eq.) was added and then stirred at 50 °C for 1 h. The white suspension turned into a homogeneous yellow solution. This solution was characterized without further treatment. 1H NMR (400 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 9.57 (d, J = 8.1 Hz, 12H), 9.37 (s, 12H), 8.85 (s, 24H), 8.65 (s, 12H), 8.12 (t, J = 7.3 Hz, 12H), 7.57–7.41 (m, 24H), 6.94 (d, J = 8.3 Hz, 12H), 6.14 (t, J = 7.7 Hz, 12H), 5.53 (t, J = 7.9 Hz, 12H), 5.40 (d, J = 6.7 Hz, 12H), 4.34–4.16 (m, 24H), 2.36 (d, J = 5.5 Hz, 36H); 13C NMR (101 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 171.02, 159.78, 150.93, 149.81, 140.40, 140.22, 135.96, 135.67, 133.57, 130.33, 129.98, 129.70, 127.68, 127.65, 126.78, 124.86, 123.36, 122.67, 122.54, 119.87, 119.36, 113.20, 103.93, 94.60, 91.46, 85.37, 82.09, 22.79. ESI-TOF-MS for Eu4(L1SS)6(OTf)12: for [Eu4(L1SS)6(OTf)4]8+ calcd, m/z = 721.1636, found, 721.1652; for [Eu4(L1SS)6(OTf)5]7+ calcd, m/z = 845.4659, found, 845.4669; for [Eu4(L1SS)6(OTf)6]6+ calcd, m/z = 1011.2022, found, 1011.2031; for [Eu4(L1SS)6(OTf)7]5+ calcd, m/z = 1243.2332, found, 1243.2347; for [Eu4(L1SS)6(OTf)8]4+ calcd, m/z = 1591.2795, found, 1591.2808.Complex of L1RR was prepared using the same procedure. Both 1H NMR and ESI-TOF-MS demonstrated the formation of a pure-phase Eu4(L1RR)6 tetrahedron.
Synthesis of self-assembled complex of L2SS
To a white suspension of L2SS (2.0 mg, 2.4 μmol, 1.0 eq.) in a solution of CD3CN/CD3OD (v/v 4/1, 500 μL), Eu(OTf)3 (1.0 mg, 1.6 μmol, 0.67 equiv.) was added and then stirred at 50 °C for 1 h. The white suspension turned into a homogeneous yellow solution. This solution was characterized without further treatment. 1H NMR (400 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 9.57 (d, J = 8.1 Hz, 12H), 9.37 (s, 12H), 8.85 (s, 24H), 8.65 (s, 12H), 8.12 (t, J = 7.3 Hz, 12H), 7.57–7.41 (m, 24H), 6.94 (d, J = 8.3 Hz, 12H), 6.14 (t, J = 7.7 Hz, 12H), 5.53 (t, J = 7.9 Hz, 12H), 5.40 (d, J = 6.7 Hz, 12H), 4.34–4.16 (m, 24H), 2.36 (d, J = 5.5 Hz, 36H); 13C NMR (101 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 170.71, 150.74, 142.02, 140.41, 135.85, 134.18, 133.65, 130.01, 129.75, 129.11, 127.75, 127.60, 126.75, 124.94, 123.35, 122.47, 122.16, 119.90, 114.28, 103.42, 94.54, 85.13, 22.79. ESI-TOF-MS for Eu4(L2SS)6(OTf)12 and Eu2(L2SS)3(OTf)12: for [Eu4(L2SS)6(OTf)2]10+ calcd, m/z = 592.7593, found, 592.7594; for [Eu4(L2SS)6(OTf)3]9+ calcd, m/z = 675.1717, found, 675.1723; for [Eu4(L2SS)6(OTf)4]8+ calcd, m/z = 778.1872, found, 778.1869; for [Eu4(L2SS)6(OTf)5]7+ calcd, m/z = 910.6357, found, 910.6357; for [Eu4(L2SS)6(OTf)6]6+ calcd, m/z = 1087.2337, found, 1087.2337; for [Eu4(L2SS)6(OTf)7]5+ calcd, m/z = 1334.4709, found, 1334.4706; for [Eu2(L2SS)3(OTf)0]6+ calcd, m/z = 469.1406, found, 469.1415; for [Eu2(L2SS)3(OTf)1]5+ calcd, m/z = 592.7590, found, 592.7594; for [Eu2(L2SS)3(OTf)2]4+ calcd, m/z = 778.1871, found, 778.1869; for [Eu2(L2SS)3(OTf)3]3+ calcd, m/z = 1087.2336, found, 1087.2337; for [Eu2(L2SS)3(OTf)4]2+ calcd, m/z = 1705.3267, found, 1705.3273.Complex of L2RR was prepared using the same procedure. Both 1H NMR and ESI-TOF-MS demonstrated the formation of a mixture of Eu2(L2RR)3 and Eu4(L2RR)6, which coexisted in the solution. Increasing the concentration of L2RR led to a clearly identifiable pure-phase Eu4(L2RR)6 being observed in the 1H NMR spectrum.
Synthesis of self-assembled complex of L3SS
To a yellow suspension of L3SS (3.0 mg, 3.3 μmol, 1.0 eq.) in a solution of CD3CN/CD3OD (v/v 4/1, 500 μL), Eu(OTf)3 (1.3 mg, 2.2 μmol, 0.67 eq.) was added and then stirred at 50 °C for 1 h. The yellow suspension turned into a homogeneous yellow solution. This solution was characterized without further treatment. 1H NMR (400 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 8.48 (s, 6H), 8.09 (d, J = 8.2 Hz, 6H), 7.97 (d, J = 8.1 Hz, 6H), 7.75 (t, J = 7.3 Hz, 6H), 7.72–7.66 (m, 6H), 7.63 (d, J = 8.3 Hz, 6H), 7.42 (d, J = 8.2 Hz, 12H), 7.30 (d, J = 8.1 Hz, 12H), 7.20 (s, 12H), 7.05 (t, J = 7.7 Hz, 6H), 6.89 (d, J = 7.0 Hz, 6H), 6.63 (t, J = 8.0 Hz, 6H), 6.36 (s, 6H), 5.40 (d, J = 7.8 Hz, 6H), 5.26 (d, J = 7.8 Hz, 6H), 1.86 (d, J = 5.6 Hz, 18H); 13C NMR (101 MHz, CD3CN/CD3OD v/v 4/1, 298 K) δ = 167.53, 160.20, 153.35, 142.78, 142.55, 140.18, 138.83, 134.67, 132.72, 130.58, 130.21, 128.58, 128.25, 128.13, 127.70, 127.10, 126.06, 123.01, 120.93, 32.50, 30.19, 22.26, 14.17. ESI-TOF-MS for Eu2(L3SS)3(OTf)6: for [Eu2(L3SS)3(OTf)0]6+ calcd, m/z = 507.1563, found, 507.1578; for [Eu2(L3SS)3(OTf)1]5+ calcd m/z = 638.3781, found, 638.3789; for [Eu2(L3SS)3(OTf)2]4+ calcd m/z = 835.2107, found, 835.2121; for [Eu2(L3SS)3(OTf)3]3+ calcd m/z = 1163.2651, found, 1163.2667. Complex L3RR was prepared using the same procedure. Both 1H NMR and ESI-TOF-MS demonstrated the formation of a pure-phase Eu2(L3RR)3 helicate.Author contributions
Methodology: Xiao-Qing Guo and Qing-Fu Sun; investigation: Xiao-Qing Guo and Qing-Fu Sun; visualization: Xiao-Qing Guo; funding acquisition: Qing-Fu Sun; project administration: Qing-Fu Sun; supervision: Shao-Jun Hu and Li-Peng Zhou; writing – original draft: Xiao-Qing Guo and Qing-Fu Sun; writing – review and editing: Li-Peng Zhou, Shao-Jun Hu and Qing-Fu Sun.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the staff of the BL17B1 beamline at the National Centre for Protein Sciences Shanghai and Shanghai Synchrotron Radiation Facility (SSRF), Shanghai, People's Republic of China, for assistance during data collection. This work was supported by the National Key Research and Development Program of China (Grants 2021YFA1500400 and 2022YFA1503300), the National Natural Science Foundation of China (Grants 21825107, 22171264, and 22201285), the Science Foundation of Fujian Province (Grant 202lJ02016), and the Open Fund of Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications (2020B121201005).References
- J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565 CrossRef CAS PubMed.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed.
- P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2010, 254, 1760 CrossRef CAS.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed.
- D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626 RSC.
- F.-F. Zhu, L.-J. Chen, S. Chen, G.-Y. Wu, W.-L. Jiang, J.-C. Shen, Y. Qin, L. Xu and H.-B. Yang, Chem, 2020, 6, 2395 CAS.
- S.-J. Bao, Z.-M. Xu, Y. Ju, Y.-L. Song, H. Wang, Z. Niu, X. Li, P. Braunstein and J.-P. Lang, J. Am. Chem. Soc., 2020, 142, 13356 CrossRef CAS PubMed.
- Y. Li, J. Dong, W. Gong, X. Tang, Y. Liu, Y. Cui and Y. Liu, J. Am. Chem. Soc., 2021, 143, 20939 CrossRef CAS PubMed.
- Y. Katagiri, Y. Tsuchida, Y. Matsuo and M. Yoshizawa, J. Am. Chem. Soc., 2021, 143, 21492 CrossRef CAS PubMed.
- K. Omoto, S. Tashiro and M. Shionoya, J. Am. Chem. Soc., 2021, 143, 5406 CrossRef CAS PubMed.
- L. J. Wang, S. Bai and Y.-F. Han, J. Am. Chem. Soc., 2022, 144, 16191 CrossRef CAS PubMed.
- S. M. Bierschenk, J. Y. Pan, N. S. Settineri, U. Warzok, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2022, 144, 11425 CrossRef CAS PubMed.
- R. Banerjee, D. Chakraborty and P. S. Mukherjee, J. Am. Chem. Soc., 2023, 145, 7692 CrossRef CAS PubMed.
- A. C. Pearcy, L. S. Lisboa, D. Preston, N. B. Page, T. Lawrence, L. J. Wright, C. G. Hartinger and J. D. Crowley, Chem. Sci., 2023, 14, 8615 RSC.
- H.-N. Zhang and G.-X. Jin, Angew. Chem., Int. Ed., 2023, e202313605 CAS.
- I. Regeni, R. Chowdhury, K. Terlinden, S. Horiuchi, J. J. J. Holstein, S. Feldmann and G. H. H. Clever, Angew. Chem., Int. Ed., 2023, 62, e202308288 CrossRef CAS PubMed.
- X. Z. Li, C. B. Tian and Q. F. Sun, Chem. Rev., 2022, 122, 6374 CrossRef CAS PubMed.
- Y. Zhou, H. Li, T. Zhu, T. Gao and P. Yan, J. Am. Chem. Soc., 2019, 141, 19634 CrossRef CAS PubMed.
- Y. B. Tan, Y. Okayasu, S. Katao, Y. Nishikawa, F. Asanoma, M. Yamada, J. Yuasa and T. Kawai, J. Am. Chem. Soc., 2020, 142, 17653 CrossRef CAS PubMed.
- H. L. Zhang, Y. Q. Zhai, H. Nojiri, C. Schroeder, H. K. Hsu, Y. T. Chan, Z. D. Fu and Y. Z. Zheng, J. Am. Chem. Soc., 2022, 144, 15193 CrossRef CAS PubMed.
- X.-F. Duan, L.-P. Zhou, H.-R. Li, S.-J. Hu, W. Zheng, X. Xu, R. Zhang, X. Chen, X.-Q. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2023, 145, 23121 CrossRef CAS PubMed.
- B. Bocquet, G. Bernardinelli, N. Ouali, S. Floquet, F. Renaud, G. Hopfgartner and C. Piguet, Chem. Commun., 2002, 930 RSC.
- C. Piguet, Chem. Commun., 2010, 46, 6209 RSC.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729 CrossRef PubMed.
- D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244 RSC.
- S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652 RSC.
- M. Bottrill, L. Kwok and N. J. Long, Chem. Soc. Rev., 2006, 35, 557 RSC.
- T. K. Ronson, H. Adams, L. P. Harding, S. J. A. Pope, D. Sykes, S. Faulkner and M. D. Ward, Dalton Trans., 2007, 1006 RSC.
- B. E. Aroussi, S. Zebret, C. Besnard, P. Perrottet and J. Hamacek, J. Am. Chem. Soc., 2011, 133, 10764 CrossRef PubMed.
- J. Wang, C. He, P. Wu, J. Wang and C. Duan, J. Am. Chem. Soc., 2011, 133, 12402 CrossRef CAS PubMed.
- G. Bozoklu, C. Gateau, D. Imbert, J. Pécaut, K. Robeyns, Y. Filinchuk, F. Memon, G. Muller and M. Mazzanti, J. Am. Chem. Soc., 2012, 134, 8372 CrossRef CAS PubMed.
- J. Hamacek, D. Poggiali, S. Zebret, B. El Aroussi, M. W. Schneider and M. Mastalerz, Chem. Commun., 2012, 48, 1281 RSC.
- S. Han, H. Zhang, Y. J. Xie, L. L. Liu, C. F. Shan, X. K. Li, W. S. Liu and Y. Tang, Appl. Surf. Sci., 2015, 328, 368 CrossRef CAS.
- A. M. Johnson, C. A. Wiley, M. C. Young, X. Zhang, Y. Lyon, R. R. Julian and R. J. Hooley, Angew. Chem., Int. Ed., 2015, 54, 5641 CrossRef CAS PubMed.
- J. A. Kitchen, Coord. Chem. Rev., 2017, 340, 232 CrossRef CAS.
- T. Y. Bing, T. Kawai and J. Yuasa, J. Am. Chem. Soc., 2018, 140, 3683 CrossRef CAS PubMed.
- J. J. Lu, V. Montigaud, O. Cador, J. F. Wu, L. Zhao, X. L. Li, M. Guo, B. Le Guennic and J. K. Tang, Inorg. Chem., 2019, 58, 11903 CrossRef CAS PubMed.
- J. K. Zhong, L. Zhang, D. P. August, G. F. S. Whitehead and D. A. Leigh, J. Am. Chem. Soc., 2019, 141, 14249 CrossRef CAS PubMed.
- Z. W. Yao, Y. Y. Zhou, T. Gao, P. F. Yan and H. F. Li, RSC Adv., 2021, 11, 10524 RSC.
- P. R. Su, T. Wang, P. P. Zhou, X. X. Yang, X. X. Feng, M. N. Zhang, L. J. Liang, Y. Tang and C. H. Yan, Natl. Sci. Rev., 2022, 9, nwab016 CrossRef CAS PubMed.
- X.-Q. Guo, L.-P. Zhou, S.-J. Hu, L.-X. Cai, P.-M. Cheng and Q.-F. Sun, J. Am. Chem. Soc., 2021, 143, 6202 CrossRef CAS PubMed.
- X. Yang, Z. Li, S. Wang, S. Huang, D. Schipper and R. A. Jones, Chem. Commun., 2014, 50, 15569 RSC.
- S. Zebret, C. Besnard, G. Bernardinelli and J. Hamacek, Eur. J. Inorg. Chem., 2012, 2012, 2409 CrossRef CAS.
- Y. Y. Zhou, H. F. Li, T. Y. Zhu, T. Gao and P. F. Yan, J. Am. Chem. Soc., 2019, 141, 19634 CrossRef CAS PubMed.
- L.-L. Yan, C.-H. Tan, G.-L. Zhang, L.-P. Zhou, J.-C. Bunzli and Q.-F. Sun, J. Am. Chem. Soc., 2015, 137, 8550 CrossRef CAS PubMed.
- S.-J. Hu, X.-Q. Guo, L.-P. Zhou, L.-X. Cai and Q.-F. Sun, Chin. J. Chem., 2019, 37, 657 CrossRef CAS.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975 CrossRef CAS.
- X.-Z. Li, L.-P. Zhou, L.-L. Yan, D.-Q. Yuan, C.-S. Lin and Q.-F. Sun, J. Am. Chem. Soc., 2017, 139, 8237 CrossRef CAS PubMed.
- T. K. Ronson, H. Adams, L. P. Harding, S. J. A. Pope, D. Sykes, S. Faulkner and M. D. Ward, Dalton Trans., 2007, 1006 RSC.
- C.-T. Yeung, W. T. K. Chan, S.-C. Yan, K.-L. Yu, K.-H. Yim, W.-T. Wong and G.-L. Law, Chem. Commun., 2015, 51, 592 RSC.
- C.-L. Liu, L.-P. Zhou, D. Tripathy and Q.-F. Sun, Chem. Commun., 2017, 53, 2459 RSC.
- K.-H. Yim, C.-T. Yeung, M. R. Probert, W. T. K. Chan, L. E. Mackenzie, R. Pal, W.-T. Wong and G.-L. Law, Commun. Chem., 2021, 4, 116 CrossRef CAS PubMed.
- A. M. Castilla, M. A. Miller, J. R. Nitschke and M. M. J. Smulders, Angew. Chem., Int. Ed., 2016, 55, 10616 CrossRef CAS PubMed.
- J. Hamacek, S. Blanc, M. Elhabiri, E. Leize, A. Van Dorsselaer, C. Piguet and A.-M. Albrecht-Gary, J. Am. Chem. Soc., 2003, 125, 1541 CrossRef CAS PubMed.
- M. Elhabiri, J. Hamacek, J. Bünzli, G. Claude and A. M. Albrecht-Gary, Eur. J. Inorg. Chem., 2004, 2004, 51 CrossRef.
- C. O. Kappe and E. Van der Eycken, Chem. Soc. Rev., 2010, 39, 1280 RSC.
- J. A. Peters, J. Huskens and D. J. Raber, Prog. Nucl. Magn. Reson. Spectrosc., 1996, 28, 283 CrossRef CAS.
- P. W. Atkins and A. J. MacDermott, J. Chem. Educ., 1982, 59, 359 CrossRef CAS.
- J. P. Leonard, P. Jensen, T. McCabe, J. E. O'Brien, R. D. Peacock, P. E. Kruger and T. Gunnlaugsson, J. Am. Chem. Soc., 2007, 129, 10986 CrossRef CAS PubMed.
- K. T. Hua, J. Xu, E. E. Quiroz, S. Lopez, A. J. Ingram, V. A. Johnson, A. R. Tisch, A. de Bettencourt-Dias, D. A. Straus and G. Muller, Inorg. Chem., 2012, 51, 647 CrossRef CAS PubMed.
- J. I. Bruce, D. Parker, S. Lopinski and R. D. Peacock, Chirality, 2002, 14, 562 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2304348–2304351. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03791f |
| This journal is © The Royal Society of Chemistry 2024 |