
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterobimetallic 3d–4f complexes supported by a Schiff-base tripodal ligand†
Till
Neumann‡
 a,
Benedict C.
Thompson‡
a,
Denny
Hebron
a,
Daniel M.
Graycon
a,
Alberto
Collauto
b,
Maxie M.
Roessler
b,
Daniel W. N.
Wilson
a and
Rebecca A.
Musgrave
*a
a,
Benedict C.
Thompson‡
a,
Denny
Hebron
a,
Daniel M.
Graycon
a,
Alberto
Collauto
b,
Maxie M.
Roessler
b,
Daniel W. N.
Wilson
a and
Rebecca A.
Musgrave
*a
aDepartment of Chemistry, King's College London, Britannia House, 7 Trinity Street, London, SE1 1DB, UK. E-mail: rebecca.musgrave@kcl.ac.uk
bMolecular Sciences Research Hub, Department of Chemistry, Imperial College London, 82 Wood Lane, London, W12 0BZ, UK
First published on 30th April 2024
Abstract
Complexes featuring multiple metal centres are of growing interest regarding metal–metal cooperation and its tuneability. Here the synthesis and characterisation of heterobimetallic complexes of a 3d metal (4: Mn, 5: Co) and lanthanum supported by a (1,1,1-tris[(3-methoxysalicylideneamino)methyl]ethane) ligand is reported, as well as discussion of their electronic structure via electron paramagnetic resonance (EPR) spectroscopy, electrochemical experiments and computational studies. Competitive binding experiments of the ligand and various metal salts unequivocally demonstrate that in these heterobimetallic complexes the 3d metal (Mn, Co) selectively occupies the κ6-N3O3 binding site of the ligand, whilst La occupies the κ6-O6 metal binding site in line with their relative oxophilicities. EPR spectroscopy supported by density functional theory analysis indicates that the 3d metal is high spin in both cases (S = 5/2 (Mn), 3/2 (Co)). Cyclic voltammetry studies on the Mn/La and Co/La bimetallic complexes revealed a quasi-reversible Mn2+/3+ redox process and poorly-defined irreversible oxidation events respectively.
Introduction
Cooperation between two or more metal atoms to achieve properties that are “greater than the sum of their parts” is of growing interest to the inorganic chemistry community.1 Featuring two metal centres in close proximity to one another introduces a wide range of tuneable parameters including bond length, bond order, charge distribution, and electronic coupling (Fig. 1a).2–4 Judicious selection of the metal centres and the ligand framework can influence the overall properties of the complex, for example by tuning redox potentials, or allowing cooperative reactivity between the metals to achieve otherwise inaccessible chemical transformations.4 Additionally, the tuneability of each of the parameters outlined in Fig. 1a (ligand sterics and electronics, bond polarity/order/length, and magnetic coupling) results in a wide area of chemical space for inorganic chemists to explore.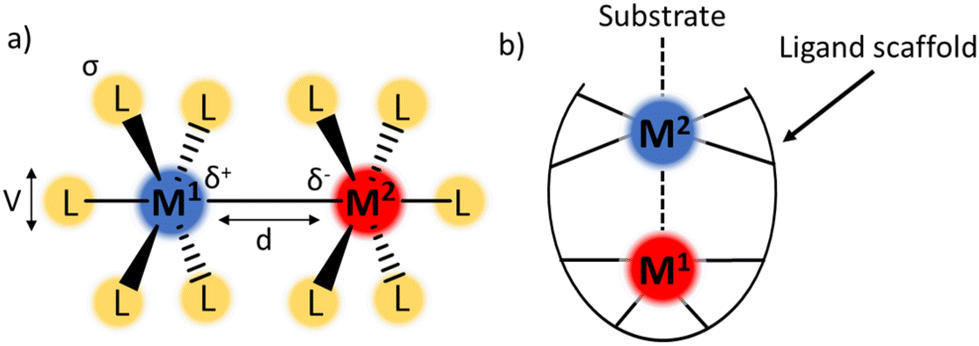 | ||
| Fig. 1 (a) Tuneable parameters in bimetallic complexes including ligand sterics (V), bond polarity (δ), M–M bond order, M–M bond length (d), magnetic coupling, ligand electronics (σ). (b) Example structure of a bimetallic complex with a single-metal active site. | ||
The complexes discussed in this report belong to a class of bimetallic complexes where one of the metal centres is buried within a multidentate ligand framework and coordinatively saturated (M1, Fig. 1b). The second metal (M2) is coordinatively unsaturated and available for potential substrate binding. In many cases, the redox profile of the bimetallic combination is richer than for either metal in isolation.5 Complexes fitting this archetype have been realised with a wide range of metal–metal combinations including main group elements,6,7 rare-earth metals,8–10 early–late transition metal (TM) pairings,11–14 and TMs with alkali metals.15,16
A common structural motif in heterobimetallic complexes is the tripodal ligand structure, which provides the requisite steric and coordinative saturation around M1, whilst leaving M2 coordinatively unsaturated. Many examples of heterobimetallic complexes supported by tripodal ligands feature a buried Lewis acidic metal centre to inductively modulate the properties of a catalytically active transition metal. The tuning of properties of a TM centre with either an electrophilic main group element or a lanthanide (4f) element has enabled N2 activation (Fig. 2a and c),6,17–21 and oxidative cleavage of H2 (Fig. 2b).5,22,23 The bimetallic complex NiLa[iPr2PCH2NPh]3 (Fig. 2d) was also recently shown to catalyse alkyne semihydrogenation.10
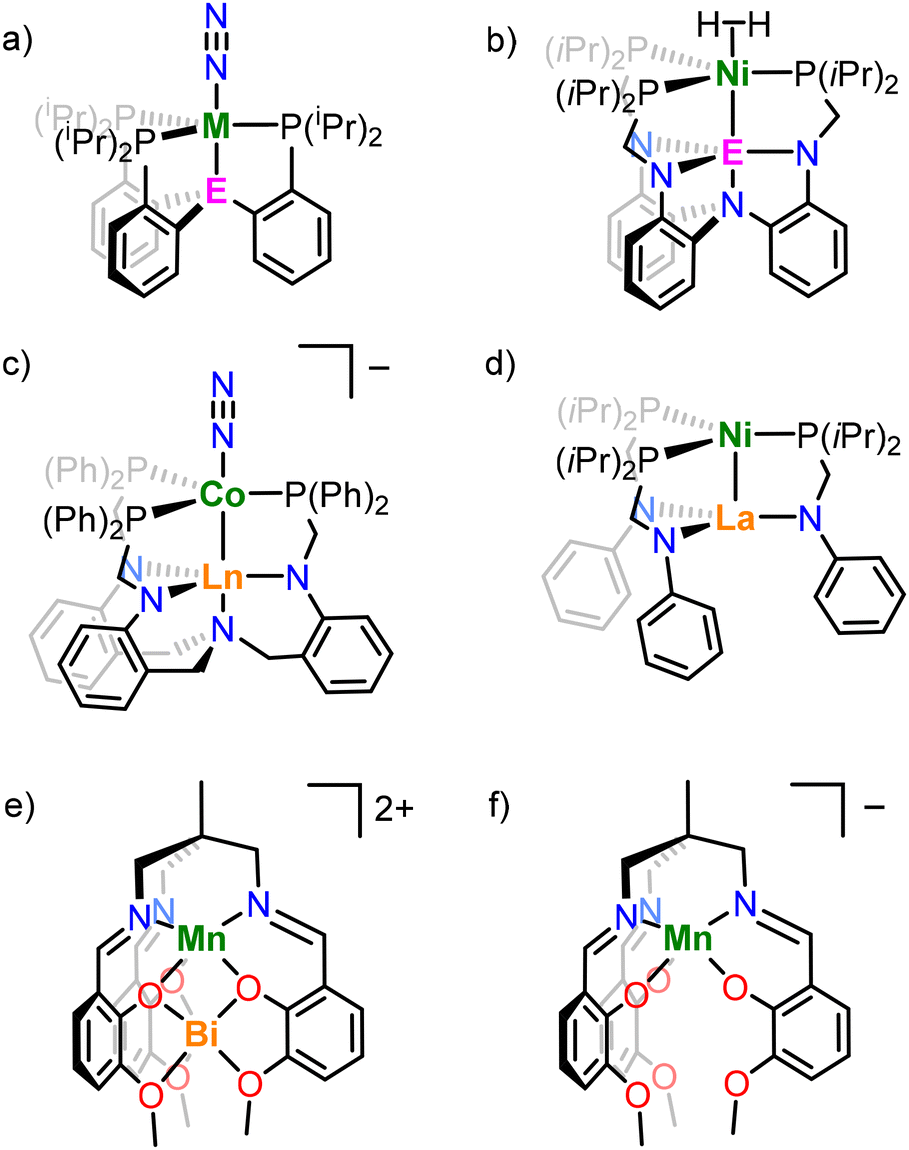 | ||
| Fig. 2 Example metal complexes, where (a) M = Fe, Co; E = B, Al, Ga, (b) E = Al, Ga, In, (c) Ln = Sc, Lu, Yb, Y, Gd, and those based on the (1,1,1-tris[(3-methoxysalicylideneamino)methyl]ethane ligand with (e) Mn/Bi (triflate counterions excluded), and (f) Mn (ammonium counterion excluded). | ||
Pairings of TM and lanthanide metals in heterometallic complexes are of interest for their magnetic properties,24–31 cooperative and frustrated Lewis pair-like reactivity,12,32 and polymerisation and transesterification catalysis.33,34 Although examples of bimetallic complexes with a buried electrophilic metal centre are numerous, the reverse case, in which the transition metal is buried in the ligand scaffold (i.e. at the M1 site) is less well-explored. Multidentate Schiff base ligands35 are well-known to support combinations of TM and lanthanide ions, with common motifs including bipodal ligand systems,36–40 clusters of higher nuclearity: trinuclear41–45 and hexanuclear46 systems, as well as coordination polymers.47
The tripodal Schiff base ligand 1,1,1-tris[(3-methoxysalicylideneamino)methyl]ethane (LH3) has been previously shown to support both manganese and bismuth ions in the κ6-N3O3 and κ6-O6 binding sites respectively (Fig. 2e) in order to study the effect of diamagnetic Bi3+ on the magnetic properties of the Mn2+ centre.48,49 Other work has demonstrated the ability of this ligand to stabilise multiple oxidation states of manganese (e.g. in [NH4][MnL], Fig. 2f),50 support heterotrimetallic clusters,51 and act as an ionophore to selectively extract alkali metals from solution.52 In this work, we employ the same ligand to support heterobimetallic complexes of a divalent 3d metal (Co, Mn) and trivalent lanthanum, and explore their structure, bonding, and spectroscopic and electrochemical properties.
Results and discussion
The synthesis of the heterobimetallic 3d–4f complexes 4 and 5 described herein was adapted from Pearson et al.48 (Scheme 1). Additionally, analogues in which only Mn, Co or La ions are present (accompanied by an Na+ ion in some cases) were prepared to demonstrate the site-selectivity of the metalation reactions (complexes 1–3). The synthesis of complexes 1–5 is outlined in Scheme 1.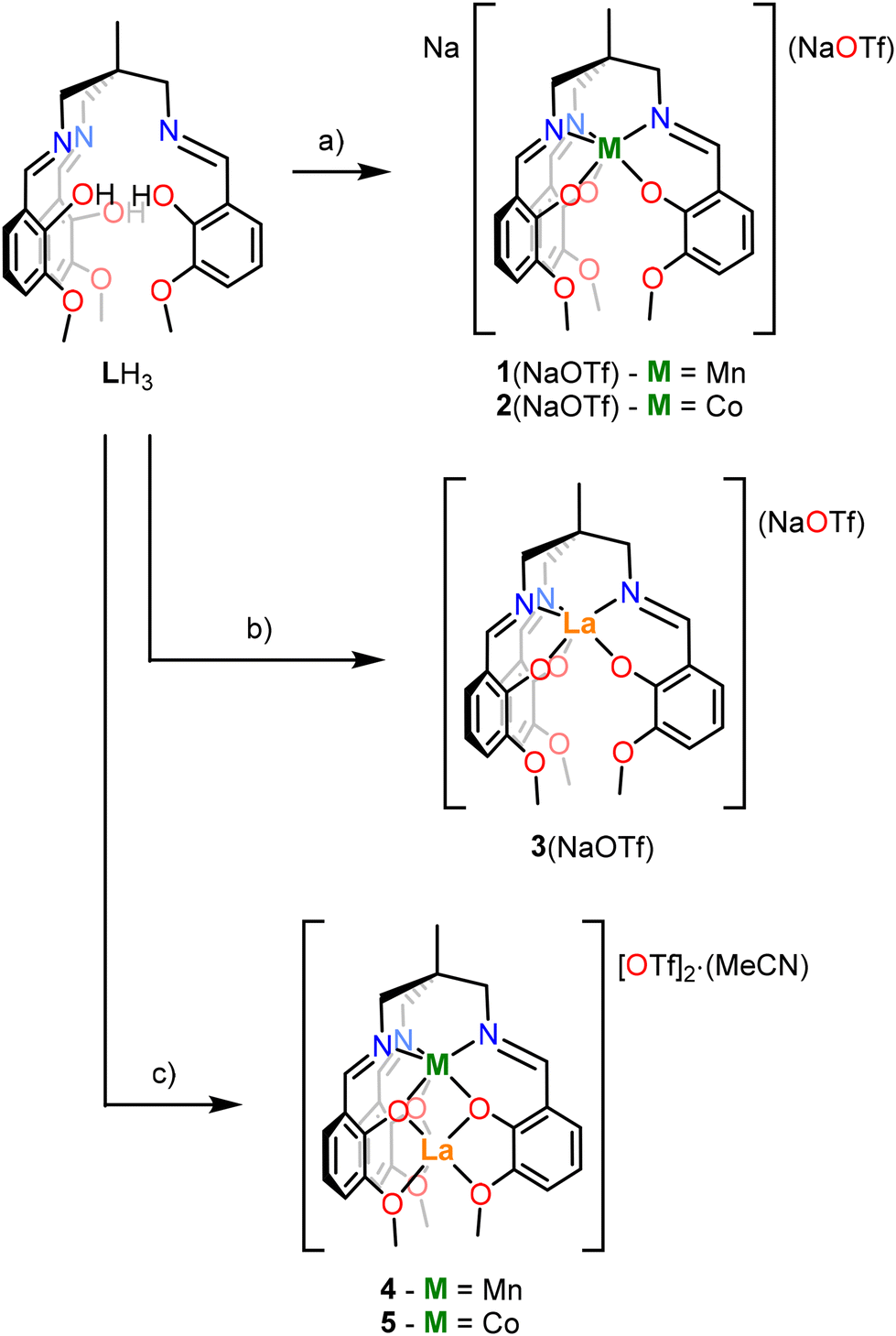 | ||
| Scheme 1 General synthetic scheme for complexes 1–5. LH3 = 1,1,1-tris[(3-methoxysalicylideneamino)methyl]ethane. Conditions: (a) (i) 3.2 NaOMe, THF, 0.5 h, RT, −3 MeOH; (ii) M[OTf]n, THF, 16 h, RT; (b) (i) 3.2 NaOMe, THF, 0.5 h, RT, −3 MeOH; (ii) La[OTf]3, THF, 16 h, RT; (c) (i) 3.2 NaOMe, THF, 0.5 h, RT, −3 MeOH; (ii) M[OTf]n, THF, 4 h, RT; (iii) La[OTf]3, THF, 16 h, RT, –Na[OTf]. | ||
Complexes 1(NaOTf) and 2(NaOTf) were synthesised by deprotonation of LH3 with NaOMe in tetrahydrofuran (THF) at room temperature (RT), addition of the resulting solution to a THF slurry of Mn[OTf]2 or Co[OTf]2 respectively, and stirring at room temperature for 16 hours (Scheme 1a). The resulting products were extracted into CH2Cl2, filtered, and single crystals suitable for X-ray diffraction (XRD) were grown via the slow diffusion of hexanes into a CH2Cl2 solution of each product. The resulting isomorphous structures, [1(NaOTf)]2 and [2(NaOTf)]2, which are dimeric ‘ate’ complexes of 1 and 2 respectively, are depicted in Fig. 3. In each structure, a central eight-membered ring is apparent, involving two sodium and two bridging triflate ions, with a centre of inversion lying at the centre. The metal ion (Mn, Co) occupies the κ6-N3O3 binding pocket of the ligand, coordinated by three imine nitrogen atoms (N1, N2, N3) and three phenolate oxygen atoms (O1, O3, O5) (Fig. 3). The Mn–Navg and Co–Navg bond lengths (2.253(3) and 2.135(2) Å respectively) are slightly longer than the Mn–Oavg and Co–Oavg bond lengths (2.123(2) and 2.080(2) Å respectively) (Table 1). The two sodium cations are structurally inequivalent, with Na1 coordinated by two phenolate oxygen atoms (O3 and O5), two methoxy oxygen atoms (O4 and O6), and two triflate oxygen atoms (O8 and O9) in κ2 fashion. Na2 is coordinated by two phenolate oxygen atoms (O1 and O5), one methoxy oxygen atom (O2), and oxygen atoms (O7 and O9) of two different triflate ions in κ1 fashion.
 | ||
| Fig. 3 Molecular structures of (a) [1(NaOTf)]2 and (b) [2(NaOTf)]2. Thermal ellipsoids are displayed at the 50% probability level, and hydrogen atoms have been omitted for clarity. Grey = carbon, blue = nitrogen, red = oxygen, yellow = sulphur, green = fluorine. Throughout, solid bonds denote interatomic distances <3 Å and dashed bonds denote distances in the range 3–4 Å. | ||
| Complex | Distances (computational)a (Å) | Distances (crystallographic)b (Å) | Angles (crystallographic)c (°) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| κ6-N3O3 binding site | κ6-O6 binding site | ||||||||
| La–Oavg | θ | θ | θ | ||||||
| M⋯La | M⋯La | M–Navg | M–Oavg | phenolate | methoxy | C2–N1–M–O1 | C2–N2–M–O3 | C2–N3–M–O5 | |
| a B3LYP-GD3/(Mn/Co/La: def2TZVPP; N/O/F/S: def2TZVP; C/H: def2SVPP basis sets). b Where ESD values are quoted for average distances, the highest value is reported. c Dihedral angle, θ (C2–Nx–M–Oy) at the κ6-N3O3 binding site for complexes 1(NaOTf), 2(NaOTf), 4, & 5 describes the coordination environment at the metal centre (defined in Fig. 6). | |||||||||
| 1(NaOTf) | — | — | 2.253(3) | 2.123(2) | — | — | 15.1 | 8.1 | 31.3 |
| 2(NaOTf) | — | — | 2.135(2) | 2.080(2) | — | — | 37.7 | 16.6 | 20.6 |
| 3(NaOTf) | — | — | 2.721(2) | 2.479(2) | — | — | — | — | — |
| 4 | 3.41 | 3.398(1) | 2.192(3) | 2.157(2) | 2.446(2) | 2.725(3) | 10.9 | 10.4 | 6.4 |
| 5 | 3.38 | 3.363(1) | 2.103(2) | 2.097(2) | 2.432(2) | 2.715(2) | 22.0 | 22.5 | 19.9 |
To investigate the ability of LH3 to bind larger metal ions, the synthetic procedure was repeated with a La3+ salt (Scheme 1b). Following deprotonation of the ligand LH3, the resulting solution was added to 1 equivalent of La[OTf]3 to affect the metalation. The product was extracted into CH2Cl2 and filtered, and single crystals suitable for XRD were grown from MeCN/Et2O.
The resulting solid state structure (Fig. 4) reveals the metalated ligand, [LaL] (3), and Na[OTf] have formed an ‘ate’ complex [3(NaOTf)]2. Both halves of the dimer are structurally equivalent, with La1 occupying the κ6-N3O3 binding site of one L3− ligand, as well as being coordinated by the phenolate (O3′, O5′) and methoxy (O4′, O6′) oxygens of the second L3− ligand, with an overall coordination number of 10. The three (3-methoxysalicylideneamino)methyl ‘arms’ of the ligand L3− are not equivalent: in two cases the phenolate oxygens (O3, O5) bridge the two La atoms in μ2 fashion, but in one case (O1) the phenolate oxygen is coordinated to only one La atom. The La⋯La distance in [3(NaOTf)]2 is 3.6656(2) Å.
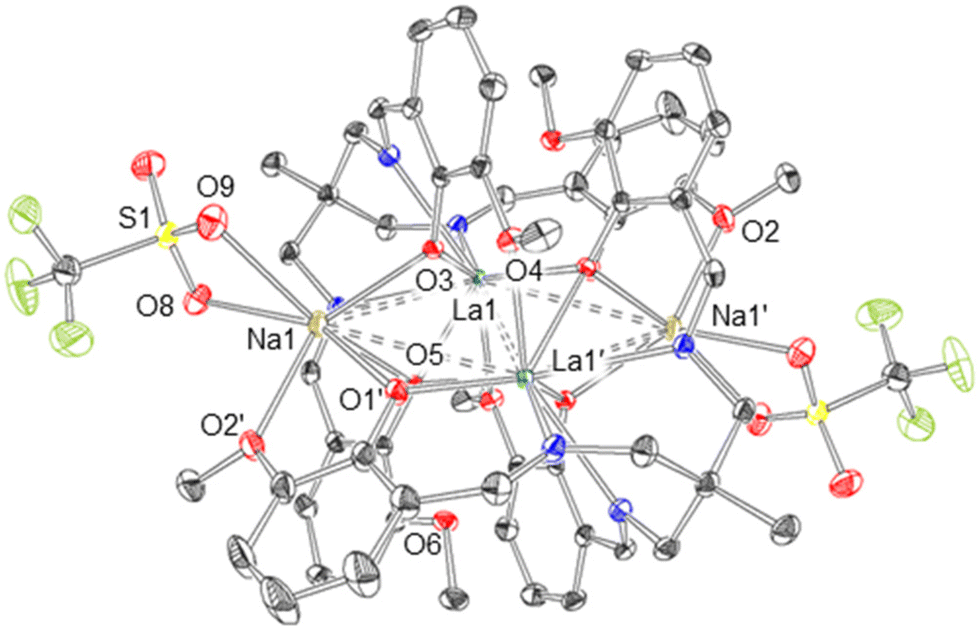 | ||
| Fig. 4 Molecular structure of [3(NaOTf)]2. Thermal ellipsoids are displayed at the 50% probability level, and hydrogen atoms have been omitted for clarity. Grey = carbon, blue = nitrogen, red = oxygen, yellow = sulphur, green = fluorine. Solid bonds denote interatomic distances <3 Å and dashed bonds denote distances in the range 3–4 Å. | ||
Although compound 1(NaOTf) is 1H NMR-silent (Fig. S1†), 1H NMR spectroscopic data for compound 2(NaOTf) (Fig. S2†) reveals six paramagnetically broadened and shifted resonances, implying effective C3-symmetry. This suggests that upon dissolution, the ‘ate’ complex observed in the solid state is not preserved in solution, and [CoNaL] is likely the observable species.
Following this, the ability of this ligand to support both 3d and 4f metals was investigated, where an initial metalation reaction was carried out at the κ6-N3O3 binding site with a 3d metal, followed by addition of La[OTf]3 to form a mixed 3d-La bimetallic complex. Specifically, it was of interest to determine whether this second metalation reaction with La3+ occurs at the κ6-O6 binding site or whether the metal atoms (a) are able to ‘swap’ coordination sites according to thermodynamic preferences, or (b) form a statistical mixture of metal ions at each binding site. The synthetic procedure outlined in Scheme 1c was used to prepare the heterobimetallic complex [MnLaL][OTf]2·MeCN (4) via deprotonation of LH3 with NaOMe in THF and addition of Mn[OTf]2, followed by addition of La[OTf]3. After extraction of the product into CH2Cl2, single crystals suitable for XRD were grown via slow diffusion of Et2O into an MeCN solution.56 The resulting crystal structure of 4 is depicted in Fig. 5a. At the κ6-N3O3 binding site, manganese is coordinated by the three Schiff base nitrogen atoms (N1, N2, N3) with an average bond length of 2.192(3) Å, and the three phenolate oxygens (O1, O3, O5) with an average bond length of 2.157(2) Å (Table 1). This Mn–Navg value for 4 is found to be a little longer than that reported by Pearson et al. for the Mn2+–Bi3+ complex (2.157(5) Å), although the Mn–Navg bond lengths were found to be statistically identical.
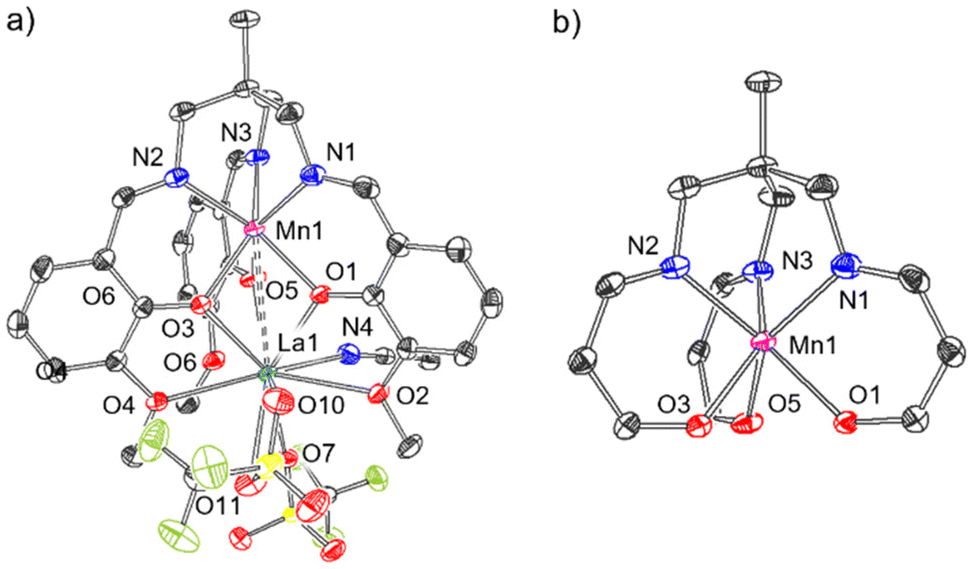 | ||
| Fig. 5 Molecular structure of (a) [MnLaL][OTf]2·MeCN (4). (b) Selected fragment of the molecular structure of 4 showing the coordination environment about Mn1. Thermal ellipsoids are displayed at the 50% probability level, and hydrogen atoms have been omitted for clarity. Grey = carbon, blue = nitrogen, red = oxygen, yellow = sulphur, green = fluorine. Solid bonds denote interatomic distances <3 Å and dashed bonds denote distances in the range 3–4 Å. | ||
The dihedral angle, θ (Nx–C2–Mn–Oy, x = 1, 2, 3, y = 1, 3, 5), between each N-donor and its corresponding O-donor of the same (3-methoxysalicylideneamino)methyl arm is a useful metric to describe this environment, where θ = 60° corresponds to an octahedron and θ = 0° corresponds to a trigonal prism (Fig. 6). The dihedral θ angles C2–N1–Mn1–O1, C2–N2–Mn1–O3, C2–N3–Mn1–O5 in 4 are 10.9, 10.4 and 6.4° respectively (Table 1), demonstrating the imine donors (N1, N2, N3) and the phenolate donors (O1, O3, O5) are close to eclipsed, and that the coordination environment at Mn (shown in more detail in Fig. 5b) is closer to trigonal prismatic than octahedral.
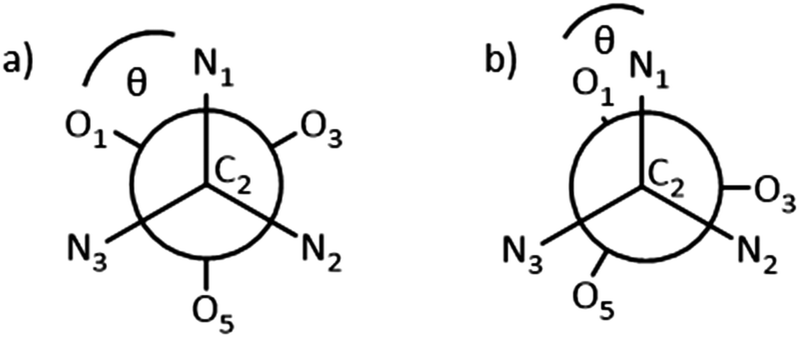 | ||
| Fig. 6 Newman projection viewed along the C2⋯M axis (M = Mn, Co). θ defined as dihedral angle Nx–C2–M–Oy (in case shown x = 1). (a) Staggered arrangement, θ = 60°, corresponding to an idealised octahedral M environment. (b) Eclipsed arrangement, θ = 0°, corresponding to a trigonal prismatic M environment. | ||
Lanthanum occupies the κ6-O6 site in 4 and is coordinated by the three phenolate oxygen atoms (O1, O3, O5), the three methoxy oxygen atoms (O2, O4, O6) and two charge-balancing triflate ions. These two anions are structurally inequivalent: one binds to La1 through O7 in a κ1 coordination mode whilst the other binds to La1 through both O10 and O11 in κ2 fashion, bringing the total coordination number at La to 10. This is typical of La3+ complexes, which commonly exhibit coordination numbers at La of 9 or 10.29,33,34,57,58 The Mn⋯La distance is 3.398(1) Å (Table 1), which is significantly longer than the Mn⋯Bi distance (3.2163(5) Å) reported by Pearson et al. in their analogous Mn2+/Bi3+ complex (Fig. 2e).48 The formal shortness ratio (FSR), defined as the ratio of the interatomic distance (d) to the sum of the atomic radii as defined by Pauling (R1,A, R1,B)53 (eqn (1)), is a useful metric to describe metal–metal bonding between A and B,54 and in general for metal–metal bonds, interactions with an FSR value above 1.2 are not considered covalent in nature.55
The FSR in 4 was calculated as 1.19, which suggests this interaction is at the limit of what can be considered a covalent interaction.55
Eqn (1): Formal shortness ratio (FSR).
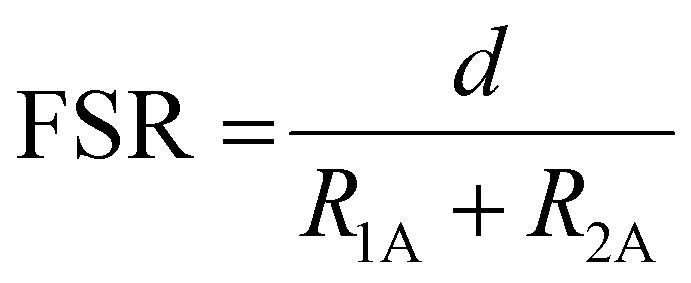 | (1) |
An identical procedure to that of complex 4 (Scheme 1c) was applied with Co[OTf]2 to produce the heterobimetallic complex [CoLaL][OTf]2·MeCN (5). Single crystals were grown from MeCN/Et2O,56 and the resulting crystal structure is shown in Fig. 7a, and the structure and bonding in complex 5 is very similar to 4. Co1 is coordinated by N1, N2, N3 and O1, O3, O5, with dihedral angles, (θ as defined in Fig. 6) of 22.5, 22.0, and 19.9° (Table 1), suggesting that, like 4, the coordination environment about Co1 lies somewhere between octahedral and trigonal prismatic, but is rather closer to octahedral than 4. Two charge-balancing triflate ions are coordinated to La, but unlike in complex 4, both of the triflate ions are coordinated via one oxygen (O7 and O10 respectively) in κ1 fashion, resulting in a coordination number of 9 at La1 (cf. 10 for 4).
 | ||
| Fig. 7 Molecular structure of (a) [CoLa(L)][OTf]2·MeCN (5). (b) Selected fragment of the molecular structure of 5 showing the coordination environment about Co1. Thermal ellipsoids are displayed at the 50% probability level, and hydrogen atoms have been omitted for clarity. Grey = carbon, blue = nitrogen, red = oxygen, yellow = sulphur, green = fluorine. Solid bonds denote interatomic distances <3 Å and dashed bonds denote distances in the range 3–4 Å. | ||
1H NMR spectroscopic data (Fig. S5†) for complex 5 reveals six paramagnetically broadened and shifted resonances (similar in nature to complex 2(NaOTf), Fig. S2†), suggesting that in the solution phase at room temperature, complex 5 is effectively C3-symmetric. In a similar fashion to 4, The La ion is coordinated by the phenolate oxygens O1, O3, O5 and the methoxy oxygens O2, O4, O6. The Co⋯La distance is 3.363(1) Å, which corresponds to a FSR of 1.19, almost identical to complex 4, and again at the limit of what can be considered a covalent interaction.
Comparing complexes 4 and 5, the mean Mn–N bond length (2.192(3) Å) is slightly longer than the mean Co–N bond length (2.103(2) Å), which is expected due to the difference in atomic radii of Mn and Co.59 The same trend is observed for the Mn–O/Co–O bond lengths. For both 4 and 5 the X-type phenolate O donors have shorter La–O bond lengths (around 2.4 Å) than the L-type methoxy O donors (around 2.7 Å). However, both the mean phenolate and methoxy La–O bond lengths are slightly longer for 4 (2.446(2) and 2.725(3) Å) than 5 (2.432(2) and 2.715(2) Å). This is likely due to the shorter M–N bond lengths in 5 (M = Co) vs.4 (M = Mn), which effectively bring the three relatively rigid (3-methoxysalicylideneamino)methyl ‘arms’ of the L3− ligand closer together around the La centre.
Comparing between the cobalt-containing complexes 2(NaOTf) and 5, the Co–Navg bond lengths are found to be slightly shorter in complex 5 (2.103(2) Å) than in complex 2(NaOTf) (2.135(2) Å). Conversely, the Co–Oavg bond lengths are slightly longer in 5 (2.097(2) Å) relative to those in 2(NaOTf) (2.080(2) Å). This can be rationalized by considering that in the case of 5 the phenolate O donor atoms (O1, O3, O5) are bridging Co2+ and trivalent La3+, rather than Co2+ and Na+ in 2(NaOTf).
Given that the synthesis of the bimetallic complexes 4 and 5 proceeds with apparent site selectivity, it was of interest to determine whether this is due to (a) the non-labile nature of the chelating ligand, or (b) thermodynamic preferences derived from the relative oxophilicity of the La3+ ion.60 To investigate this, synthesis of an “inverse” bimetallic complex, [LaCoL][OTf]2, was attempted (Scheme 2), where complex 3(NaOTf) was treated with Co[OTf]2 in THF at two different temperatures, followed by purification and recrystallisation. The resulting products were characterized by 1H NMR spectroscopy and single crystal XRD. When this reaction is conducted at room temperature, single crystals of the product grown from MeCN/Et2O were shown to have identical unit cell parameters to the crystalline sample of [3(NaOTf)]2, demonstrating that the Co metalation does not proceed, and the starting material (3(NaOTf)) is retained. The presence of 3(NaOTf) was further corroborated by 1H NMR spectroscopic analysis (Fig. S6†). However, when the same reaction is conducted at 70 °C, single crystal XRD analysis of crystals grown from MeCN/Et2O was instead consistent with the formation of [CoLaL][OTf]2·MeCN (5) (by lattice parameter comparison). In this case, it is evident that cobalt and lanthanum have exchanged binding sites so that Co occupies the κ6-N3O3 site and La the κ6-O6 site. This observation was corroborated by 1H NMR spectroscopy, for which the spectrum of the product was identical to that of complex 5 (Fig. S7†). The intended product of this reaction [LaCo(L)][OTf]2 was not isolated, with the formation of either 3(NaOTf) at room temperature, or 5 when sufficient heat is applied to overcome the kinetic barrier to metal exchange.
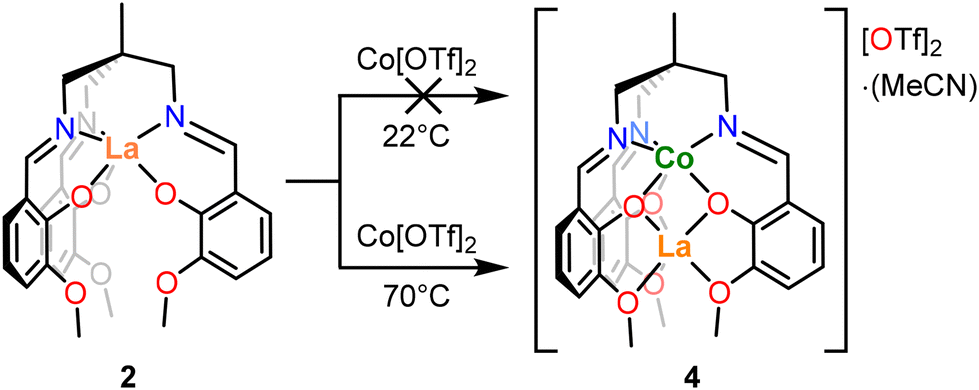 | ||
| Scheme 2 Reaction scheme for reversal of metalations. Conditions: THF, 16 h. | ||
Electronic structure
Continuous-wave electron paramagnetic resonance (CW-EPR) spectra at two microwave frequencies enabled determination of the spin state of 4 and its key magnetic parameters. Both the Q-band and X-band spectra (Fig. 8a) could be simulated as an S = 5/2 spin system with a zero field splitting (ZFS) parameter |D| ≈ νmw (|D| = 7.2 GHz) and a very low rhombicity of the ZFS interaction (E/D = 0.07). Remarkably, no hyperfine coupling to the 55Mn nucleus (I = 5/2, 100% natural abundance) could be resolved in the experimental spectra, which may be attributed to the presence of intermolecular couplings.61 Mn2+ complexes typically exhibit values for |D| in the range of 0–1.3 cm−1 (where magnetic anisotropy increases as |D| increases).62,63 The obtained D value for 4 (|D| = 0.24 cm−1) lies between those reported for the analogous S = 5/2 Mn/Bi complex [MnBiL][OTf]2 (Fig. 2e, D = 0.74 cm−1)48 and the S = 5/2 monometallated complex [NH4][MnL] (Fig. 2f, D = 0.17 cm−1).50 Upon raising the temperature between 10 K and 100 K the signal intensity of the X-band CW-EPR spectrum of 4 becomes weaker, but no additional broadening is observed; moreover, the overall shape of the spectrum remains unchanged (see Fig. S8†).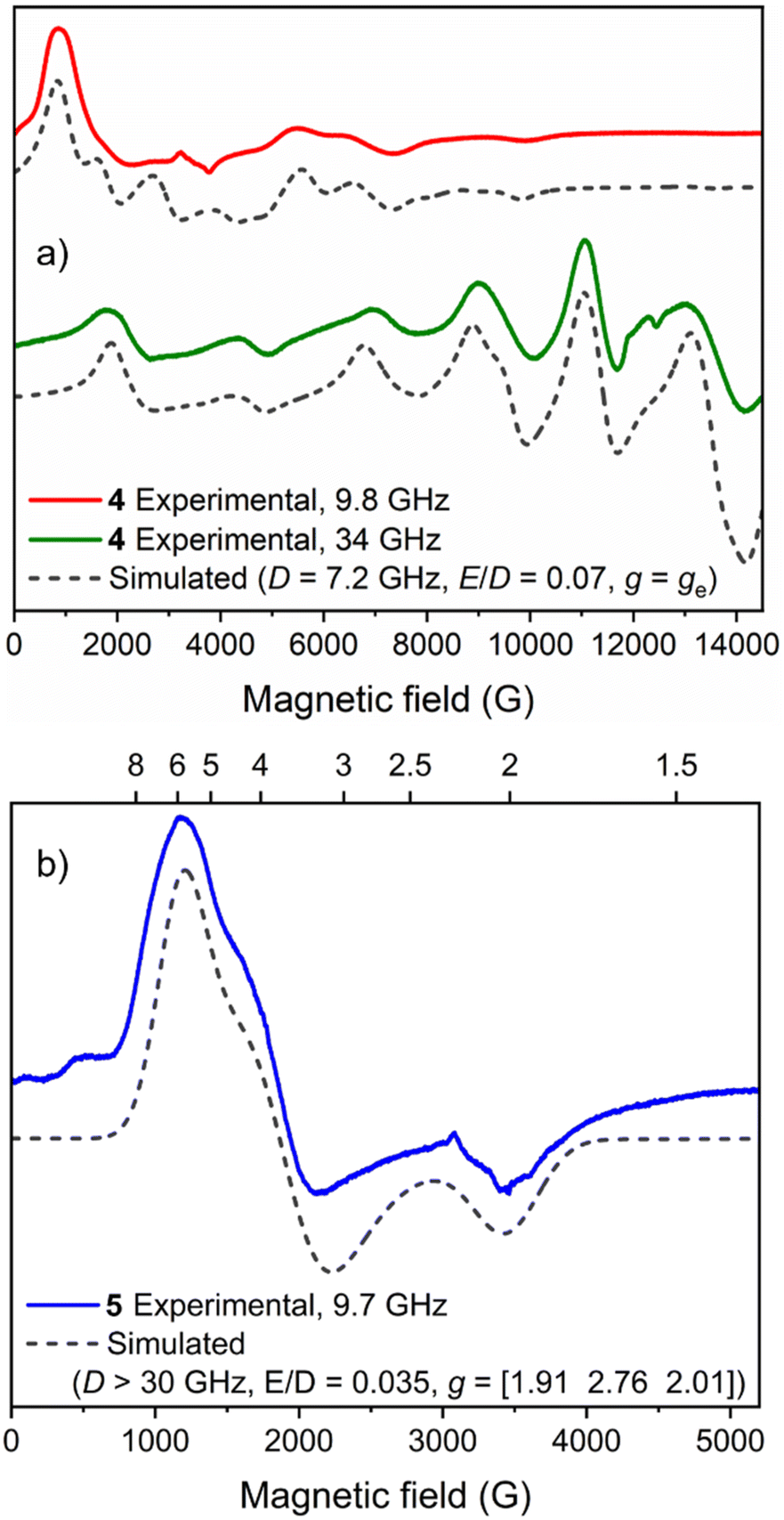 | ||
| Fig. 8 EPR spectra of 5 mM MeCN solutions of (a) 4 and (b) 5 at 10 K. | ||
The major features of the X-band EPR spectrum of 5 at 10 K could be assigned to transitions within the MS = ±1/2 Kramers’ doublet of a S = 3/2 high-spin Co2+ centre with D > 0 and can be accounted for by assuming a sizeable ZFS interaction (|D| > νmw) with a very low rhombicity (E/D = 0.035). Based on the absence of transitions between Kramers’ doublets in the field range up to 5200 G (see Fig. 8b), a lower boundary D > 1 cm−1 (30 GHz) can be set. This leads to ‘true’ g values of 1.91, 2.76 and 2.01 (see Materials and methods), in line with other high-spin Co2+ centres.64 We note that within the formalism of a fictious spin-1/2 Hamiltonian (i.e. a ‘pseudo’ S = ½ system),65 the effective g values (as observed in Fig. 8b) are 5.8, 3.6 and 2.0. Similar to that observed for 4, no hyperfine coupling to the 59Co nucleus (I = 7/2, 100% natural abundance) could be resolved. The CW-EPR spectrum of 5 broadens upon raising the temperature between 10 K and 50 K (Fig. S9†), with no detectable signal at 100 K, ruling out the presence of low-spin Co2+ species and providing further evidence for a high-spin configuration (S = 3/2) because this is typically faster-relaxing than low-spin (S = 1/2) species.64 The EPR spectrum of 2(NaOTf) at 10 K (Fig. S10†) is characterised by a very broad, shapeless feature at 6000 G, indicating significant change in the electronic structure of Co2+, and additional measurements recorded at higher temperatures (≤80 K) showed prominent line broadening consistent with the expected behaviour of a high-spin metal centre with large ZFS interaction parameters.
The Evans method was also employed to determine the magnetic moments of the paramagnetic compounds 4 and 5, and the results (μeff = 6.09 and 4.31μB respectively) are consistent with the S = 5/2 and S = 3/2 spin state assignments by EPR and computational analysis (see ESI Section 6 for details†).
Computational studies
Complementary DFT studies were conducted to analyse the bonding in complexes 4 and 5, with a particular interest in analysing interaction between the heterometals. Geometry optimizations were performed from the atomic coordinates obtained via single crystal XRD analysis and found to be in reasonable agreement with experimental data (Table S5†). Possible spin states were optimized with unrestricted single-point density functional theory (DFT) calculations with various multiplicities (see ESI Section 8 for details†). Of these, only the high spin configurations (4: S = 5/2 and 5: S = 3/2) converged. Multiple basis sets and functionals were screened, with the best reproduction of bonding parameters given by the hybrid functional B3LYP in combination with the basis sets def2SVPP(C, H)/def2TZVP(N, O, F, S)/def2TZVPP(La, Mn/Co) and Grimme's empirical dispersion correction (see ESI Section 8 for further details†). Natural bond orbital analysis was performed to interrogate the nature of the bonding interactions. In both complexes there were no discernible bonding interactions between the La and transition metal atoms, and the Wiberg bond orders (WBO) of 0.15 (4) and 0.26 (5) are effectively negligible (Fig. S24 and S25†). Overall, the analysis is in agreement with the formal shortness ratios of 1.19 obtained from the solid-state structures in both cases (i.e., negligible metal–metal interaction). The La-ligand interactions are highly ionic in both cases (WBOLa–O = 0.17–0.31), with no covalent interactions between La and the surrounding light atoms. Similarly, the ligand interactions with the 3d metal give comparable WBO values (WBOMn–N/O = 0.20–0.27 (4) WBOCo–N/O = 0.21–0.31 (5)). Thus, the best description of both 4 and 5 involves charge-separated L3− with internally coordinated M2+ (M = Mn, Co) and La3+ cations, charge-balanced by triflate anions.Electrochemistry
The electrochemistry of LH3, 2(NaOTf), 4 and 5 were investigated in CH2Cl2 solution under nitrogen atmosphere to preclude oxidation of Mn and Co by atmospheric oxygen.Scanning oxidatively, the cyclic voltammogram for 4 shows a quasi-reversible oxidation centred at 0.33 V vs. Fc+/Fc (Fc = [Fe(η5C5H5)2]; Fig. 9), assigned to the one-electron oxidation of Mn2+ to Mn3+, with near-equivalent ia (peak anodic current) and ic (peak cathodic current) values (Fig. S22 and S23†). However, when the maximum voltage applied was increased to 1.29 V (vs. Fc+/Fc), irreversible oxidation was observed and the oxidation peak at 0.33 V decayed upon repeated cycles (Fig. S20†). This is thought to be due to sample decomposition, and the deposition of a solid on the surface of the electrode was observed during this process. Furthermore, thorough polishing of the working electrode led to recovery of the initial voltammogram. Comparison of the measured Mn2+/3+ redox couple in compound 4 to similar Mn compounds of the same ligand (Table 2) indicates that the presence of the La3+ ion in 4 makes oxidation of the Mn2+ ion much more difficult, as the overall charge state of the complex increases (0 in [MnL] vs. +2 in compound 4). Kojima et al. also noted that the presence of a K+ cation showed a pronounced effect on the redox potentials of trivalent manganese in [MnL] and [MnKL][PF6].50 This effect of the La3+ ion may also explain why the Mn3+/4+ couple was not observed for compound 4, as it would involve the further removal of electrons from an already tricationic complex.
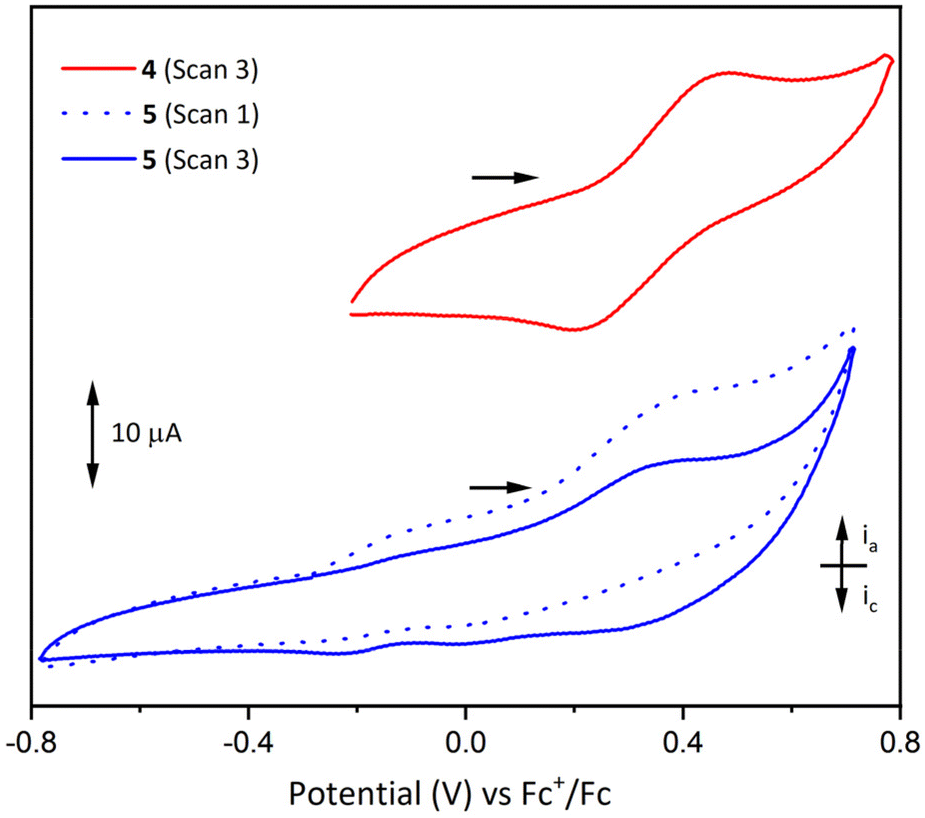 | ||
| Fig. 9 Cyclic voltammograms for 4 (7.1 mM in CH2Cl2, with 0.20 M [(nBu)4N][PF6] supporting electrolyte, 0.2 V s−1) and 5 (1 mM in CH2Cl2 with 0.25 M [(nBu)4N][PF6] supporting electrolyte, 0.1 V s−1). Glassy carbon working and counter electrodes were used, and the cyclic voltammograms were referenced to the Fc+/Fc redox couple, scanning oxidatively in both cases (ia = peak anodic current, ic = peak cathodic current). A Savitzky–Golay smooth was applied to even out periodic noise in the trace. Scan numbers denote the chronological order in which full cycles were conducted. Scan 1 for compound 4 (not shown) exhibits identical behaviour to scan 3. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 66
66
Analogous cyclic voltammetry experiments on complex 5 showed poorly-resolved redox processes in the range −0.4 to 0.6 V and evidence of sample decomposition and deposition on the surface of the electrode when the applied potential was increased above 0.75 V (vs. Fc+/Fc). An example voltammogram is shown in Fig. 9, exhibiting irreversible oxidation around 0.35 V and concordant loss of the reductive signal upon repeated cycles (the first and third cycles are depicted). It is proposed that this decomposition process is similar to that seen in 4 for potentials above 0.75 V, and this oxidative event may be assigned to a one-electron oxidation of Co2+, or oxidation of the ligand. The cyclic voltammograms of LH3 and 2(NaOTf) showed only indistinct and irreversible redox processes that gradually disappeared upon repeated cycles (scanning oxidatively) (Fig. S18 and S19†). As in the above cases, it is thought that the samples undergo decomposition upon application of a positive potential leading to deposition on the surface of the electrode.
UV/Visible spectroscopy
The UV/Vis spectra of complexes 1–5 and LH3 are presented in Fig. 10 and S11–S16,† and their distinct absorption bands are presented in Table 3. For LH3 the two higher energy bands at 223 and 265 nm respectively were assigned as ligand π–π* transitions, and the broader, lower energy transition at 332 nm as a π–π transition. The UV/Vis spectra for bimetallic complexes 4 and 5 exhibit similar general features to LH3; an absorption at around 230 nm, and a shoulder at lower energy (258 and 271 nm respectively). The lower energy band is centred around 340 nm in both 4 and 5, redshift relative to ligand LH3. Time-dependent DFT (TD-DFT) calculations performed on LH3, 4 and 5 produce calculated UV/Vis spectra in excellent agreement with the experimental data (Fig. S26–28†), and excited states contributing to this low energy transition (Peak 3, Table 3) were investigated. For LH3, excited state 3 (Fig. S26†) comprises a π–π electron transfer into the higher energy, zwitterionic resonance structure of the arylimine units (Fig. S29†). For each bimetallic complex, two excited states were found to contribute to the observed absorption bands: excited states 19 and 21 (complex 4, Fig. S27†) and excited states 21 and 24 (complex 5, Fig. S28†). For complex 4, these excited states comprise both metal-to-ligand and ligand-to-metal charge transfers between the S = 5/2 Mn centre and the arylimine units (Fig. S30 and S31†). In complex 5 however, only metal-to-ligand charge transfer is evident for the S = 3/2 Co centre (Fig. S32 and S33†). As expected, the La centre in both 4 and 5 does not contribute to any of these transitions, and the redshift of this band relative to LH3 is due contributions of the 3d metal orbitals to electron transfer events.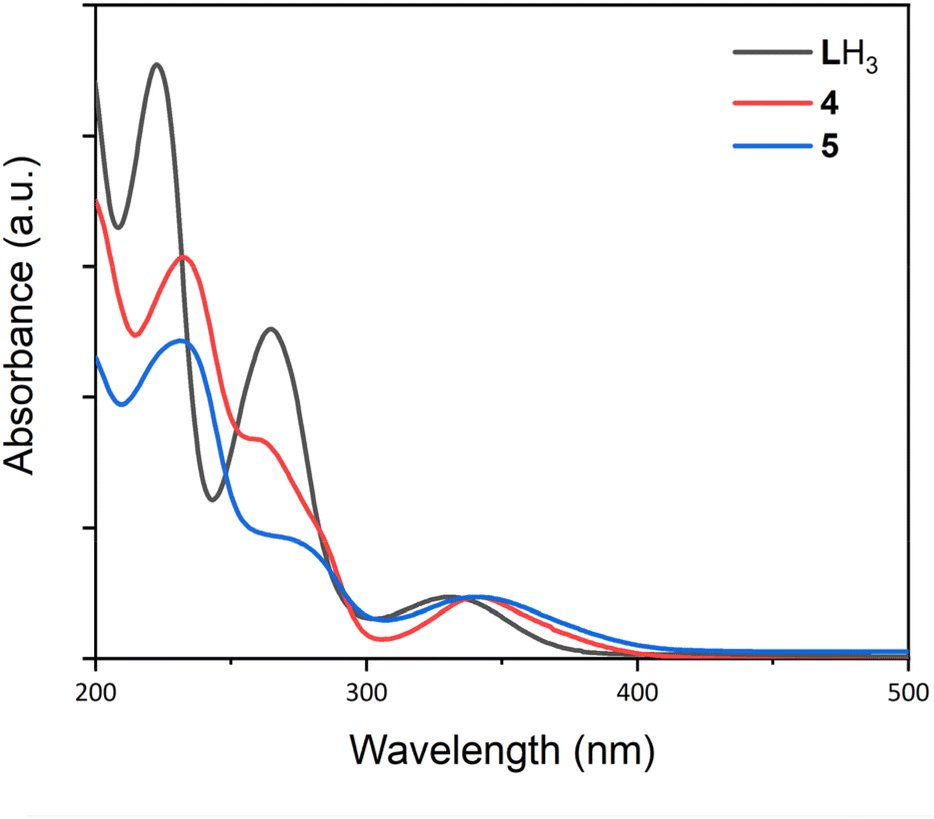 | ||
| Fig. 10 UV/Vis spectra for LH3, and complexes 4 and 5. Spectra were recorded in MeCN using a sealed quartz cuvette. LH3: c = 1.6 × 10−5 M, 4: c = 6.4 × 10−5 M, 5: c = 1.0 × 10−5 M. | ||
| Compound | Peak 1 (nm) | Peak 2 (nm) | Peak 3 (nm) |
|---|---|---|---|
| LH3 | 223 | 265 | 332 |
| 1(NaOTf) | 227 | 270 | 356 |
| 2(NaOTf) | 239 | — | 359 (sh) |
| 3(NaOTf) | 233 | 273 | 364 |
| 4 | 232 | 258 (sh) | 341 |
| 5 | 231 | 271 (sh) | 340 |
Comparison of the UV/Vis spectra of complexes 5 and 2(NaOTf) indicates that the proximal La3+ installed at the κ6-O6 site in 5 has a minor tuning effect on the energy of this transition, shifting it to a slightly longer wavelength from 340 to 359 nm. This effect is similarly observed in the comparison of 4 and 1(NaOTf) (341 and 356 nm respectively).
Conclusions
Two heterobimetallic complexes of 3d metals and lanthanum were prepared and their structural and electronic properties investigated. Upon reaction of the tripodal ligand framework LH3 with metal salts, the first metalation is directed towards the κ6-N3O3 binding site for the metals Mn,50 Co, and La, and upon a second metalation reaction, lanthanum is installed selectively at the κ6-O6 binding site to yield complexes 4 and 5. This synthetic procedure provides a route towards site-selective 3d-La heterobimetallic complexes with flexibility in the choice of transition metal used. There is an apparent thermodynamic preference for the 3d transition metal to occupy the κ6-N3O3 site and La the outer κ6-O6 site as demonstrated by the reaction of complex 3(NaOTf) with Co[OTf]2, in which the cobalt centre displaces the lanthanum centre from the κ6-N3O3 site upon heating. EPR spectroscopy and the Evans method indicated that the 3d metal in complexes 4 and 5 is high spin in both cases (S = 5/2 (Mn), 3/2 (Co)), and this was supported by density functional theory analysis. Complex 4 exhibits quasi-reversible redox activity through oxidation of Mn2+ to Mn3+, although the redox capability of these complexes is limited by apparent oxidative decomposition. Future work will be directed at expanding the redox chemistry of these systems by incorporating redox-active f-block metals at the κ6-O6 coordination site in the place of the closed-shell La3+ ion.Materials and methods
All reactions and product manipulations of molecular species were carried out under an inert atmosphere of dinitrogen or argon using standard Schlenk line or glovebox techniques (MBraun glovebox MB150G-B maintained at <0.1 ppm H2O and <0.1 ppm O2), unless otherwise stated. Glassware for metalation reactions was oven dried at 200 °C overnight prior to use. Dry tetrahydrofuran (THF), Et2O, CH2Cl2 and MeCN were obtained from a Grubbs-type solvent system employing alumina columns, stored over 4 Å molecular sieves (3 Å for MeCN), and degassed with N2 prior to use. The deuterated solvents d3-MeCN and d6-DMSO, and CDCl3 was purchased from Sigma-Aldrich, distilled from calcium hydride, and stored over 4 Å molecular sieves (3 Å for d3-MeCN). Silica gel (for chromatography) was purchased from VWR and used as received. Celite 521 was obtained from Fluorochem and heated to 200 °C for 16 h prior to use. NaOMe and La[OTf]3 were purchased from Alfa Aesar and used without further purification. Mn[OTf]2 and Co[OTf]2 were purchased from Apollo Scientific and Strem respectively, and used without further purification.1H NMR spectra were recorded at ambient temperature on a Bruker NMR 400 MHz spectrometer. All spectra are referenced to the most downfield residual solvent resonance. In all 1H NMR spectra of paramagnetic species, backward linear prediction from 0 to 15 data points was employed to remove baseline distortion, phase correction was addressed manually, and an ablative baseline correction applied. 1H NMR spectra were collected between +250 and −250 ppm to observe signals at both low and high field (number of scans 2048).
UV/Vis spectra were recorded on an Agilent Technologies Cary Series UV-Vis spectrophotometer. Samples were dissolved in MeCN and placed in a sealed quartz cuvette suitable for air- and moisture-sensitive compounds. Attenuated total reflectance infrared (ATR-IR) spectra were recorded on a Shimadzu IRAffinity-1S Fourier Transform Infrared Spectrophotometer.
Cyclic voltammetry (CV) studies were carried out using a Metrohm Autolab PGSTAT204 potentiostat and Metrohm’s Nova 2.1 software, with glassy carbon working and counter electrodes, and a platinum wire reference electrode. Measurements were conducted in CH2Cl2, with 0.20−0.25 M [(nBu)4N][PF6] as the supporting electrolyte. Sample concentrations were typically in the range 1−10 mM. All measurements were referenced against the Fc+/Fc redox couple as an internal standard.
X-ray crystallography
Suitable single crystals of 1–5 were coated with Fomblin® Y oil, and mounted onto a Hampton Research nylon loop (20 μm thickness, 0.3 mm diameter). All experiments were performed at 150 K using Cu Kα radiation (λ = 1.54184 Å). Measurements for complexes 3(NaOTf) and 5 were made using a twin-source SuperNova diffractometer with a micro-focus Cu Kα X-ray beam (50 kV, 0.8 mA), an Atlas (135 mm CCD) detector. Measurements for complexes 2(NaOTf) and 4 were made using a twin-source Oxford Xcalibur Gemini diffractometer with a Sapphire 3 CCD plate. Cell refinement, data collection and data reduction for all experiments were performed using Rigaku CrysAlisPro 1.171.40.57a. Crystal structures were solved and refined by least-squares within the Olex2 program suite67 using the ShelXT structure-solution program68 and the ShelXL 2014 refinement program.68 Positions of hydrogen atoms were identified as the strongest peaks in Fourier difference maps and were either refined when appropriate or constrained with AFIX commands within SHELX. Crystallographic details are provided in Tables S2 and S3.† For detailed computational methods and calculated molecular coordinates see Section 7 in the ESI.†EPR spectroscopy
The samples were prepared under anaerobic conditions by dissolving 4 and 5 in MeCN to a concentration of 5 mM. The solutions were loaded in quartz tubes (4: 2.0 mm ID, 2.8 mm OD, Wilmad LabGlass; 5: 3.0 mm ID, 4.0 mm OD, Wilmad LabGlass); the resulting samples were closed with an NMR tip-off manifold (Wilmad LabGlass), removed from the glovebox, degassed by means of repeated freeze–pump–thaw cycles and sealed under vacuum. When not in use, the EPR samples were kept frozen under liquid nitrogen.CW-EPR measurements were performed on a Bruker Elexsys E580 spectrometer equipped with an ER 4118X-MD5 dielectric-ring resonator (Bruker) for X-band measurements (νMW ≈ 9.5 GHz) or an ER 5106QT-2W cylindrical resonator for Q-band measurements (νMW ≈ 34 GHz). The sample temperature was set by using a closed-circuit Helium cryostat (Cryogenic Ltd) equipped with a Lakeshore 350 temperature controller (Lake Shore Cryotronics, Inc.).
X-band CW-EPR spectra of 4 were recorded at 10 K, 20 K, 50 K and 100 K using a microwave power of 15 μW, a field modulation amplitude of 10 G at 100 kHz, a conversion time of 40.96 ms and a field sweep rate of 43.2 G s−1; 10 scans per spectrum were accumulated and averaged. The Q-band CW-EPR spectrum of 4 was recorded at 10 K using a microwave power of 100 μW, a field modulation amplitude of 5 G at 100 kHz, a conversion time of 81.92 ms and a field sweep rate of 21.6 G s−1; the spectrum was recorded as a single scan. X-band CW-EPR spectra of 5 were recorded at 10 K, 20 K and 50 K using a microwave power of 150 μW, a field modulation amplitude of 10 G at 100 kHz, a conversion time of 40.96 ms and a field sweep rate of 43.2 G s−1; 10 scans per spectrum were accumulated and averaged. An additional measurement performed at a temperature of 100 K did not give any observable signal. For both samples, tests were performed at 10 K to check for the absence of microwave power saturation effects. Simulations of the CW-EPR spectra were performed using EasySpin 5.2.3569 running on MATLAB 2021a (The MathWorks, Inc.). Due to the lack of resolved features arising from the hyperfine coupling with the 55Mn (100% natural abundance; I = 5/2) and 59Co nuclei (100% natural abundance; I = 7/2), the corresponding interaction was not included in the spin Hamiltonian. Furthermore, single D and E/D values were considered (D and E strains were not introduced in the model).61 For the simulation of the CW-EPR spectrum of 4, g = ge was assumed based on the nature of the electronic ground state (6S: S = 5/2, L = 0). The simulation of the CW-EPR spectrum of 5 was performed by imposing a constraint on the ‘true’ g values, namely (g1 + g2 + g3)/3 was forced to be equal to geff obtained from the Evans method (geff = 2.226).64
Synthesis
(1,1,1-tris[(3-Methoxysalicylideneamino)methyl]ethane) (LH3) was synthesised according to literature procedure, by first preparing 1,1,1-tris(aminomethyl)ethane trihydrochloride,70 and subsequently reacting onwards with o-vanillin.48,50 Yield: 98%, 1H NMR (400 MHz, d6-DMSO) δ (ppm): 13.70 (s, 3 H, OH), 8.56 (s, 3 H, CHN), 7.02 (m, 6 H, ArH), 6.75 (m, 3 H, ArH), 3.77 (s, 9 H, OCH3), 3.59 (s, 6 H, CH2), 1.03 (s, 3 H, CH3). 13C NMR (101 MHz, d6-DMSO) δ (ppm): 166.92 (ArCHN), 154.78 (Ar), 151.89 (Ar), 148.33 (Ar), 123.00 (Ar), 118.64 (Ar), 114.54 (Ar), 63.94 (CH2N), 55.67 (OCH3), 39.62 (C(CH3)(CH2)3), 20.14 (CH3). UV/Vis: (MeCN, 1.6 × 10−5 mol dm−3) 223, 265, 332 nm (Fig. S11†).[MnNaL](NaOTf) (1(NaOTf))
LH3 (519 mg, 0.999 mmol), and NaOMe (173 mg, 3.202 mmol) were dissolved in THF (10 cm3) and stirred at RT for 30 min. This solution was transferred by cannula to a suspension of Mn(OTf)2 (353 mg, 1.000 mmol) in THF (10 cm3). The reaction mixture was stirred for 16 h at RT. The solvent was removed in vacuo, and the resultant solid was dissolved in CH2Cl2 (20 cm3), filtered through Celite, and the filtrate collected. The volume of solvent was reduced to 5 cm3in vacuo and the solution was filtered and layered with hexanes (10 cm3). 1(NaOTf) was isolated as orange crystals (275 mg, 0.359 mmol) in 36% yield. Compound 1(NaOTf) is 1H NMR inactive (Fig. S1†). UV/Vis: (MeCN, c = 1.4 × 10−5 M), 227, 270 (sh), 356 nm (Fig. S12†). Elemental analyses were consistently (and reproducibly) found to be low in nitrogen. Anal. calcd for C30H30F3MnN3Na2O9S: C, 47.00 H, 3.94; N, 5.48. Found: C, 47.05; H, 4.48; N, 4.14.[CoNaL](NaOTf) (2(NaOTf))
LH3 (205 mg, 0.395 mmol), and NaOMe (68 mg, 1.259 mmol) were dissolved in THF (10 cm3) and stirred at RT for 30 min. This solution was transferred by cannula to a suspension of Co(OTf)2 (141 mg, 0.395 mmol) in THF (5 cm3). The reaction mixture was stirred for 16 h at RT. The solvent was removed in vacuo, and the resultant solid was dissolved in CH2Cl2 (20 cm3) and filtered through Celite, and the filtrate collected. The volume of solvent was reduced to 5 cm3in vacuo and the solution was filtered and layered with hexanes (10 cm3). 2(NaOTf) was isolated as dark orange crystals (86 mg, 0.112 mmol) in 28% yield. 1H NMR (400 MHz, d6-DMSO) δ (ppm): 183.02 (s, 6 H), 75.05 (s, 3 H), 30.61 (s, 3 H), 25.28 (s, 9 H), −15.98 (s, 3 H), −21.96 (s, 3 H) (Fig. S2†). UV/Vis: (MeCN, c = 4.2 × 10−5 M) 239, 359 (shoulder) nm (Fig. S13†). Anal. calcd for C30H30CoF3N3Na2O9S: C, 46.76 H, 3.92; N, 5.45. Found: C, 46.38; H, 3.75; N, 5.04.[LnL](NaOTf) (3(NaOTf))
LH3 (100 mg, 0.192 mmol), and NaOMe (34 mg, 0.629 mmol) were dissolved in THF (10 cm3) and stirred at RT for 30 min. This solution was transferred by cannula to a suspension of La[OTf]3 (113 mg, 0.193 mmol) in THF (5 cm3). The reaction mixture was stirred for 16 h at RT. The solvent was removed in vacuo, and the resultant solid was dissolved in CH2Cl2 (10 cm3) and filtered through Celite, and the filtrate collected. The solvent was removed in vacuo and the residue was dissolved in MeCN (5 cm3). The solution was filtered and layered with Et2O (10 cm3). 3(NaOTf) was isolated as a dark yellow/orange crystalline powder (125 mg, 0.151 mmol) in 79% yield. 1H NMR (400 MHz, d6-DMSO) δ (ppm): 8.16 (s, 3 H), 6.80 (ddd, J = 7.9, 6.4, 1.7 Hz, 6 H), 6.34 (t, J = 7.7 Hz, 3 H), 3.72 (s, 9 H), 3.55 (s, 6 H), 1.10 (s, 3 H) (Fig. S3†). UV/Vis: (MeCN, c = 3.0 × 10−5 M) 233, 273, 364 nm (Fig. S14†). Anal. calcd for C32H33F3LaN4NaO9S: C, 44.25; H, 3.83; N, 6.45.* Found: C, 44.05; H, 3.80; N, 6.22. *Includes one molecule of non-coordinating MeCN per asymmetric unit in the crystal structure.[MnLaL][OTf]2·MeCN (4)
LH3 (200 mg, 0.385 mmol), and NaOMe (67 mg, 1.240 mmol) were dissolved in THF (10 cm3) and stirred at RT for 30 min. This solution was transferred by cannula to a suspension of Mn[OTf]2 (136 mg, 0.385 mmol) in THF (5 cm3). The reaction mixture was stirred for 4 h at RT and then added to a suspension of La[OTf]3 (226 mg, 0.386 mmol), and stirred for 16 h at RT. The solvent was removed in vacuo, and the resultant solid was dissolved in CH2Cl2 (20 cm3) and filtered through Celite, and the filtrate collected. The solvent was removed in vacuo and the residue was dissolved in MeCN (5 cm3). This solution was filtered and subsequently layered with Et2O (10 cm3). Complex 4 was isolated as yellow needle-like crystals (147 mg, 0.140 mmol) in 36% yield. Compound 4 is 1H NMR inactive (Fig. S4†). UV/Vis: (MeCN, c = 6.4 × 10−5 M) 232, 258 (shoulder), 341 nm (Fig. S15†). Anal. calcd for C33H33F6LaMnN4O12S2: C, 37.76; H, 3.17; N, 5.34. Found: C, 37.90; H, 3.16; N, 5.13.[CoLaL][OTf]2·MeCN (5)
LH3 (304 mg, 0.585 mmol), and NaOMe (101 mg, 1.870 mmol) were dissolved in THF (10 cm3) and stirred at RT for 30 min. This solution was transferred by cannula to a suspension of Co[OTf]2 (209 mg, 0.585 mmol) in THF (5 cm3). The reaction mixture was stirred for 4 h at RT and then added to a suspension of La[OTf]3 (343 mg, 0.585 mmol) in THF (5 cm3), and stirred for 16 h at RT. The solvent was removed in vacuo, and the resultant solid was dissolved in CH2Cl2 (10 to 20 cm3) and filtered through Celite, and the filtrate collected. The solvent was removed in vacuo and the residue was dissolved in MeCN (5 cm3). This solution was filtered and subsequently layered with Et2O (10 cm3). Complex 5 was isolated as dark orange block-like crystals (199 mg, 0.189 mmol) in 32% yield. 1H NMR (400 MHz, d6-DMSO) δ (ppm): 198.04 (s, 6 H), 81.60 (s, 3 H), 26.05 (s, 3 H), 25.80 (s, 9 H), −15.38 (s, 3 H), −25.55 (s, 3 H) (Fig. S5†). UV/Vis: (MeCN, c = 1 × 10−5 M) 231, 271 (shoulder), 340 nm (Fig. S16†). Anal. calcd for C33H33CoF6LaN4O12S2: C, 37.62; H, 3.16; N, 5.32. Found: C, 38.24; H, 3.17; N, 5.32.Reactions of [3(NaOTf)]2 with Co[OTf]2
[3(NaOTf)]2 (44 mg, 0.0532 mmol), and Co[OTf]2 (19 mg, 0.0532 mol) were dissolved in THF (10 cm3) and stirred at RT for 16 h. The solvent was removed in vacuo and the residue redissolved in CH2Cl2 (10 cm3) and filtered. The solvent was removed in vacuo and the residue was dissolved in d6-DMSO for 1H NMR spectroscopic analysis, which showed retention of 3(NaOTf) (Fig. S6†).This procedure was repeated but stirred at 70 °C for 16 h, and the residue was dissolved in CH2Cl2 (10 cm3) and filtered through Celite, and the filtrate collected. The solvent was removed in vacuo and the residue was dissolved in MeCN (5 cm3), filtered, and layered with Et2O (10 cm3). This yielded dark orange crystals. A unit cell check was performed by XRD on a single crystal sample and the remaining product was dissolved in d6-DMSO for 1H NMR spectroscopic analysis, both of which showed formation of 5, [CoLaL][OTf]2·MeCN.
Author contributions
TN and BCT contributed equally. TN, BCT, DH and DMG performed the synthesis and characterisation. BCT drafted the manuscript with assistance from DWNW and RAM. RAM designed the research with contributions from DWNW. AC and MMR performed EPR experiments and data analysis. TN, DWNW and RAM carried out the computational chemistry. RAM performed crystallographic experiments. All authors provided input and agreed on the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The EPR measurements were performed at the Centre for Pulse EPR at Imperial College London (PEPR), supported by the EPSRC grant EP/T031425/1. RAM gratefully acknowledges the Royal Society (RGS\R2\212323) for funding. TN thanks the École normale supérieure (ENS-PSL) and the Studienstiftung des deutschen Volkes for generous support. We also thank Sara Belazregue, Dr Richard Collins and Dr F Mark Chadwick for assistance in EPR sample preparation. Crystallographic data for complexes 1–5 can be accessed via the CCDC 2336119 and 2307060–2307063.†References
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- J. Campos, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS PubMed.
- M. Navarro, J. J. Moreno, M. Pérez-Jiménez and J. Campos, Chem. Commun., 2022, 58, 11220–11235 RSC.
- Q. R. Wang, S. H. Brooks, T. C. Liu and N. C. Tomson, Chem. Commun., 2021, 57, 2839–2853 RSC.
- R. C. Cammarota, L. J. Clouston and C. C. Lu, Coord. Chem. Rev., 2017, 334, 100–111 CrossRef CAS.
- J. Fajardo and J. C. Peters, Inorg. Chem., 2021, 60, 1221–1228 Search PubMed.
- P. A. Rudd, S. S. Liu, L. Gagliardi, V. G. Young and C. C. Lu, J. Am. Chem. Soc., 2011, 133, 20724–20727 CrossRef CAS PubMed.
- P. Cui, C. J. Wu, J. Du, G. Luo, Z. M. Huang and S. L. Zhou, Inorg. Chem., 2021, 60, 9688–9699 CrossRef CAS PubMed.
- K. Y. Shi, I. Douair, G. F. Feng, P. L. Wang, Y. Zhao, L. Maron and C. Q. Zhu, J. Am. Chem. Soc., 2021, 143, 5998–6005 CrossRef CAS PubMed.
- B. L. Ramirez and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 5396–5407 CrossRef CAS PubMed.
- H. Braunschweig, K. Radacki and K. Schwab, Chem. Commun., 2010, 46, 913–915 RSC.
- J. Du, Y. N. Zhang, Z. M. Huang, S. L. Zhou, H. Y. Fang and P. Cui, Dalton Trans., 2020, 49, 12311–12318 RSC.
- B. P. Greenwood, S. I. Forman, G. T. Rowe, C. H. Chen, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2009, 48, 6251–6260 CrossRef CAS PubMed.
- L. H. Gade, H. Memmler, U. Kauper, A. Schneider, S. Fabre, I. Bezougli, M. Lutz, C. Galka, I. J. Scowen and M. McPartlin, Chem. – Eur. J., 2000, 6, 692–708 CrossRef CAS PubMed.
- A. J. Gaston, Z. Greindl, C. A. Morrison and J. A. Garden, Inorg. Chem., 2021, 60, 2294–2303 CrossRef CAS PubMed.
- A. H. Reath, J. W. Ziller, C. Tsay, A. J. Ryan and J. Y. Yang, Inorg. Chem., 2017, 56, 3713–3718 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- T. J. Del Castillo, N. B. Thompson, D. L. M. Suess, G. Ung and J. C. Peters, Inorg. Chem., 2015, 54, 9256–9262 CrossRef CAS PubMed.
- M. E. Moret and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118–18121 CrossRef CAS PubMed.
- Y. Zhang, X. W. Pan, M. Xu, C. Y. Xiong, D. J. Hong, H. Y. Fang and P. Cui, Inorg. Chem., 2023, 62, 3836–3846 CrossRef CAS PubMed.
- P. Cui, C. Y. Xiong, J. Du, Z. M. Huang, S. J. Xie, H. Wang, S. L. Zhou, H. Y. Fang and S. W. Wang, Dalton Trans., 2020, 49, 124–130 RSC.
- J. T. Moore and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 11641–11646 CrossRef CAS PubMed.
- R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12486–12489 CrossRef CAS PubMed.
- Z. Y. Li, Y. Dai, H. Zhang, J. Zhu, J. J. Zhang, S. Q. Liu and C. Y. Duan, Eur. J. Inorg. Chem., 2014, 2014, 384–391 CrossRef CAS.
- W. K. Dong, J. C. Ma, Y. J. Dong, L. C. Zhu and Y. Zhang, Polyhedron, 2016, 115, 228–235 CrossRef CAS.
- J. J. Jesudas, C. T. Pham, A. Hagenbach, U. Abram and H. H. Nguyen, Inorg. Chem., 2020, 59, 386–395 CrossRef CAS PubMed.
- T. Shiga, H. Ōkawa, S. Kitagawa and M. Ohba, J. Am. Chem. Soc., 2006, 128, 16426–16427 CrossRef CAS PubMed.
- S. M. T. Abtab, M. C. Majee, M. Maity, J. Titiš, R. Boca and M. Chaudhury, Inorg. Chem., 2014, 53, 1295–1306 CrossRef CAS PubMed.
- D. Basak, J. van Leusen, T. Gupta, P. Kögerler, V. Bertolasi and D. Ray, Inorg. Chem., 2020, 59, 2387–2405 CrossRef CAS PubMed.
- N. Ahmed and K. U. Ansari, Dalton Trans., 2022, 51, 4122–4134 RSC.
- J. P. Costes, T. Yamaguchi, M. Kojima and L. Vendier, Inorg. Chem., 2009, 48, 5555–5561 CrossRef CAS PubMed.
- J. Du, Z. M. Huang, Y. N. Zhang, S. W. Wang, S. L. Zhou, H. Y. Fang and P. Cui, Chem. – Eur. J., 2019, 25, 10149–10155 CrossRef CAS PubMed.
- H. Asaba, T. Iwasaki, M. Hatazawa, J. Y. Deng, H. Nagae, K. Mashima and K. Nozaki, Inorg. Chem., 2020, 59, 7928–7933 CrossRef CAS PubMed.
- O. V. Amirkhanov, O. V. Moroz, K. O. Znovjyak, T. Y. Sliva, L. V. Penkova, T. Yushchenko, L. Szyrwiel, I. S. Konovalova, V. V. Dyakonenko, O. V. Shishkin and V. M. Amirkhanov, Eur. J. Inorg. Chem., 2014, 3720–3730 CrossRef CAS.
- X. Liu and J. R. Hamon, Coord. Chem. Rev., 2019, 389, 94–118 CrossRef CAS.
- S. S. Zhao, X. R. Liu, W. X. Feng, X. Q. Lü, W. Y. Wong and W. K. Wong, Inorg. Chem. Commun., 2012, 20, 41–45 CrossRef CAS.
- V. Béreau, H. Bolvin, C. Duhayon and J. P. Sutter, Eur. J. Inorg. Chem., 2016, 4988–4995 CrossRef.
- T. D. Pasatoiu, J. P. Sutter, A. M. Madalan, F. Z. C. Fellah, C. Duhayon and M. Andruh, Inorg. Chem., 2011, 50, 5890–5898 CrossRef CAS PubMed.
- W. K. Lo, W. K. Wong, J. P. Guo, W. Y. Wong, K. F. Li and K. W. Cheah, Inorg. Chim. Acta, 2004, 357, 4510–4521 CrossRef CAS.
- W. Y. Bi, X. Q. Lü, W. L. Chai, J. R. Song, W. Y. Wong, W. K. Wong and R. A. Jones, J. Mol. Struct., 2008, 891, 450–455 CrossRef CAS.
- T. Fatima, Imtiaz-ud-Din, M. K. Rauf, M. S. Anwar, A. Raheel, M. N. Tahir and M. Ashfaq, Polyhedron, 2020, 186, 114605 CrossRef CAS.
- M. Karmakar, S. Roy and S. Chattopadhyay, Polyhedron, 2022, 215, 115652 CrossRef CAS.
- H. L. Wang, D. P. Zhang, Z. H. Ni, X. Y. Li, L. J. Tian and J. Z. Jiang, Inorg. Chem., 2009, 48, 5946–5956 CrossRef CAS PubMed.
- H. B. Zhou, C. Chen, Y. S. Liu and X. P. Shen, Inorg. Chim. Acta, 2015, 437, 188–194 CrossRef CAS.
- J. P. Costes, J. Garcia-Tojal, J. P. Tuchagues and L. Vendier, Eur. J. Inorg. Chem., 2009, 3801–3806 CrossRef CAS.
- T. Shiga, N. Hoshino, M. Nakano, H. Nojiri and H. Oshio, Inorg. Chim. Acta, 2008, 361, 4113–4117 CrossRef CAS.
- M. Yadav, A. Bhunia, S. K. Jana and P. W. Roesky, Inorg. Chem., 2016, 55, 2701–2708 CrossRef CAS PubMed.
- T. J. Pearson, M. S. Fataftah and D. E. Freedman, Chem. Commun., 2016, 52, 11394–11397 RSC.
- S. C. Coste, T. J. Pearson and D. E. Freedman, Inorg. Chem., 2019, 58, 11893–11902 CrossRef CAS PubMed.
- Y. Sunatsuki, Y. Kishima, T. Kobayashi, T. Yamaguchi, T. Suzuki, M. Kojima, J. Krzystek and M. R. Sundberg, Chem. Commun., 2011, 47, 9149–9151 RSC.
- T. Yamaguchi, J. P. Costes, Y. Kishima, M. Kojima, Y. Sunatsuki, N. Bréfuel, J. P. Tuchagues, L. Vendier and W. Wernsdorfer, Inorg. Chem., 2010, 49, 9125–9135 CrossRef CAS PubMed.
- S. Katsuta, Y. Shimizu, R. Takahashi, N. Kanaya, T. Imoto and Y. Takeda, New J. Chem., 2012, 36, 1445–1448 RSC.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- P. Angardis, Chromium Compounds, in Multiple Bonds Between Metal Atoms, ed. F. A. Cotton, C. A. Murillo and R. A. Walton, Springer Science and Business Media, Inc., New York, 3rd edn, 2005, pp. 35–68 Search PubMed.
- J. A. Chipman and J. F. Berry, Chem. Rev., 2020, 120, 2409–2447 CrossRef CAS PubMed.
- The low yields of compounds 4 and 5 (36% and 32% respectively) are a result of mass loss during crystallisation from MeCN/Et2O, which was necessary for satisfactory elemental analysis results.
- T. D. Pasatoiu, A. M. Madalan, M. U. Kumke, C. Tiseanu and M. Andruh, Inorg. Chem., 2010, 49, 2310–2315 CrossRef CAS PubMed.
- S. A. Cotton and P. R. Raithby, Coord. Chem. Rev., 2017, 340, 220–231 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- D. Berger, C. Matei, F. Papa, D. Macovei, V. Fruth and J. P. Deloume, J. Eur. Ceram. Soc., 2007, 27, 4395–4398 CrossRef CAS.
- M. Azarkh, L. V. Penkova, S. V. Kats, O. A. Varzatskii, Y. Z. Voloshin and E. J. J. Groenen, J. Phys. Chem. Lett., 2014, 5, 886–889 CrossRef CAS PubMed.
- C. Duboc, T. Phoeung, S. Zein, J. Pecaut, M. N. Collomb and F. Neese, Inorg. Chem., 2007, 46, 4905–4916 CrossRef CAS PubMed.
- G. A. Craig and M. Murrie, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC.
- M. Cirulli, E. Salvadori, Z. H. Zhang, M. Dommett, F. Tuna, H. Bamberger, J. E. M. Lewis, A. Kaur, G. J. Tizzard, J. van Slageren, R. Crespo-Otero, S. M. Goldup and M. M. Roessler, Angew. Chem., Int. Ed., 2021, 60, 16051–16058 CrossRef CAS PubMed.
- T. Yamane, K. Sugisaki, T. Nakagawa, H. Matsuoka, T. Nishio, S. Kinjyo, N. Mori, S. Yokoyama, C. Kawashima, N. Yokokura, K. Sato, Y. Kanzaki, D. Shiomi, K. Toyota, D. H. Dolphin, W.-C. Lin, C. A. McDowell, M. Tadokoro and T. Takui, Phys. Chem. Chem. Phys., 2017, 19, 24769–24791 RSC.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr. C, 2015, 71, 3–8 CrossRef PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- C. J. Qin, L. James, J. D. Chartres, L. J. Alcock, K. J. Davis, A. C. Willis, A. M. Sargeson, P. V. Bernhardt and S. F. Ralph, Inorg. Chem., 2011, 50, 9131–9140 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2336119 and 2307060–2307063. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03760f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |