
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A copper-containing analog of the biomineral whitlockite: dissolution–precipitation synthesis, structural and biological properties†
Diana
Griesiute
a,
Agne
Kizalaite
a,
Arita
Dubnika
 bc,
Vytautas
Klimavicius
d,
Vidmantas
Kalendra
e,
Vaclav
Tyrpekl
f,
Sung Hun
Cho
g,
Tomoyo
Goto
gh,
Tohru
Sekino
g and
Aleksej
Zarkov
*a
bc,
Vytautas
Klimavicius
d,
Vidmantas
Kalendra
e,
Vaclav
Tyrpekl
f,
Sung Hun
Cho
g,
Tomoyo
Goto
gh,
Tohru
Sekino
g and
Aleksej
Zarkov
*a
aInstitute of Chemistry, Vilnius University, Naugarduko 24, LT-03225 Vilnius, Lithuania. E-mail: aleksej.zarkov@chf.vu.lt
bRudolfs Cimdins Riga Biomaterials Innovations and Development Centre of RTU, Faculty of Materials Science and Applied Chemistry, Institute of General Chemical Engineering, Riga Technical University, LV-1007 Riga, Latvia
cBaltic Biomaterials Centre of Excellence, Headquarters at Riga Technical University, LV-1658 Riga, Latvia
dInstitute of Chemical Physics, Vilnius University, Sauletekio 3, LT-10257 Vilnius, Lithuania
eInstitute of Applied Electrodynamics and Telecommunications, Vilnius University, Sauletekio 3, LT-10257 Vilnius, Lithuania
fDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43 Prague, Czech Republic
gSANKEN (The Institute of Scientific and Industrial Research), Osaka University, 8–1 Mihogaoka, Ibaraki, Osaka 567–0047, Japan
hInstitute for Advanced Co-Creation Studies, Osaka University, 1-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 13th December 2023
Abstract
In the present work, copper whitlockite (Cu-WH, Ca18Cu2(HPO4)2(PO4)12) was successfully synthesized and comprehensively characterized, founding the base knowledge for its future studies in medicine, particularly for bone regeneration. This material is a copper-containing analog of the well-known biomineral magnesium whitlockite (Mg-WH, Ca18Mg2(HPO4)2(PO4)12). The synthesis of powders was performed by a dissolution–precipitation method in an aqueous medium under hydrothermal conditions. Phase conversion from brushite (CaHPO4·2H2O) to Cu-WH took place in an acidic medium in the presence of Cu2+ ions. Optimization of the synthesis conditions in terms of medium pH, temperature, time, Ca/Cu molar ratio and concentration of starting materials was performed. The crystal structure of the synthesized products was confirmed by XRD, FTIR and Raman spectroscopy, 1H and 31P solid-state NMR, and EPR. Morphological features and elemental distribution of the synthesized powders were studied by means of SEM/EDX analysis. The ion release in SBF solution was estimated using ICP-OES. Cytotoxicity experiments were performed with MC3T3-E1 cells. The study on thermal stability revealed that the synthesized material is thermally unstable and gradually decomposes upon annealing to Cu-substituted β-Ca3(PO4)2 and Ca2P2O7.
1. Introduction
Synthetic calcium phosphates (CPs) are widely used in medicine for bone regeneration purposes due to their structural and chemical similarities to natural bone.1 Although CPs are highly biocompatible and have already demonstrated high efficiency, one of the directions of research aimed at achieving the superior performance of CP-based materials considers the partial replacement of Ca by other ions.2 The ion substitution strategy allows combining the characteristics of CPs with newly obtained substitution-induced properties: antibacterial, optical, magnetic, etc.3 This approach leads to the development of biocompatible multifunctional materials with a wider application range.4One of the most promising substituting ions is Cu2+. Being an essential element in the human body, Cu is involved in numerous physiological functions; however, its relatively high local concentration is toxic.5 This reason limited the development and application of Cu-containing biomaterials for a long time. Nevertheless, in recent years the number of publications on Cu-modified biomaterials has grown constantly.6 The reason for this increased interest is the nature and specific biological properties of Cu2+ ions. It was confirmed that Cu is exceptionally useful in the development of materials for orthopedic applications. Cu-doped CPs demonstrate antibacterial properties, provide angiogenic ability and favor osteogenesis; these properties represent the key points for ideal biomaterial integration and the healing process.6
The majority of the works investigating Cu-substituted CPs are focused on the most popular synthetic phases including calcium hydroxyapatite (HA, Ca10(PO4)6(OH)2),7 beta-tricalcium phosphate (β-TCP, Ca3(PO4)2),8–10 brushite (DCPD, CaHPO4·2H2O),11,12 and biphasic CPs (HA + β-TCP).13 For instance, Cu-doped HA demonstrated an antimicrobial effect against E. coli and C. albicans.7 Cu-containing TCP was determined to be biocompatible, revealing no cytotoxic effect regardless of the studied Cu substitution level; moreover, it showed antibacterial properties against E. coli, E. faecalis, S. aureus and P. aeruginosa.10 Cu-loaded DCPD cement was recognized as a suitable material for bone tissue defect-related repair in orthopedic surgeries demonstrating good antibacterial properties and cell affinity.11
Magnesium whitlockite (Mg-WH, Ca18Mg2(HPO4)2(PO4)12) is an ion-substituted member of the CP family naturally occurring in the human body. Due to the similarity of the crystal structures of WH and β-TCP, these two names are often used interchangeably and synonymously; however, the structural and compositional differences are well discussed in the literature.14,15 The data on the presence of Mg-WH in the human body are rather contradictory. While some authors claim that depending on the age Mg-WH can constitute a quite significant part of human hard tissues – up to 25–35%,16 some research studies show that Mg-WH is found exceptionally in pathological tissues.17 Nevertheless, synthetic Mg-WH demonstrated superior biological properties and suitability for bone regeneration applications.18,19 Considering an ion-substitution strategy and the peculiarities of the WH structure, it looks reasonable to investigate synthetic materials with the WH structure, where Mg ions are substituted by other biologically active small cations. In this light, the first row transition metal ions appear to be suitable candidates to form WH structures since their ionic radii are close enough to that of Mg2+ ions.20 To the best of our knowledge, currently there are only a few published reports regarding the synthesis and characterization of transition metal WH, including some on Zn-WH,21–23 Mn-WH,24,25 and Co-WH26 and a recently published report on the feasibility of the formation of Cu-WH.27
The main aim of this work was to comprehensively investigate the peculiarities of the formation of Cu-WH and to study its structural, morphological and biological properties in detail. For this purpose, nanodimensional Cu-WH powders were synthesized via a dissolution–precipitation process under hydrothermal conditions. The crystal structure and phase purity were confirmed by powder X-ray diffraction (XRD), vibrational and 1H and 31P nuclear magnetic resonance (NMR) spectroscopy. Electron paramagnetic resonance (EPR) was employed to investigate in depth the structural characteristics. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to study the morphology of the synthesized materials. The ion release in SBF solution was estimated using inductively coupled plasma optical emission spectrometry (ICP-OES). Finally, the cytotoxicity of the developed Cu-WH samples was evaluated using the MC3T3-E1 preosteoblastic cell line.
2. Experimental
2.1. Synthesis
Cu-WH powders were synthesized in hydrothermal autoclave reactors equipped with PTFE liners. For the synthesis, calcium hydrogen phosphate dihydrate (DCPD, CaHPO4·2H2O, 99.1%, Eurochemicals) and copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, ≥98%, Roth) were used as starting materials. In a typical procedure, appropriate amounts of metal salts were transferred into 90 mL PTFE liners and dissolved at room temperature in 50 mL of deionized water by addition of 10 mL of 1 M phosphoric acid (H3PO4, 75%, Roth). After the complete dissolution of precursors under constant mixing on a magnetic stirrer, a clear blue colored solution was obtained. In a standard synthesis, the total concentration of metal ions in the reaction mixture was 0.08 M. Next, concentrated ammonia solution was added to the reaction mixture in order to adjust the pH to a certain value. After the addition of ammonia solution, the instantaneous formation of precipitates was observed. The PTFE liners were transferred into stainless steel jackets, screwed tightly, placed into a preheated oven and heated at different temperatures (110–230 °C) for different periods of time (1–24 h). In this work, the synthesis time was calculated from the moment the hydrothermal reactors were placed in the oven to the moment they were removed to room temperature. After the synthesis, the reactors were cooled down and the resulting blue powders were separated from the solution by vacuum filtering. Afterwards, the powders were washed with deionized water and dried in the oven at 70 °C.2.2. Characterization
Powder X-ray diffraction (XRD) data of the synthesized specimens were obtained using a Rigaku MiniFlex II diffractometer (Cu-Kα, λ = 1.5419 Å) working in Bragg–Brentano (θ/2θ) geometry. The data were collected within the 10–100 2θ angle range at a speed of 1° min−1. The FullProf Suite was used for structural refinement (FullProf Suite software version September-2020).Fourier transform infrared (FTIR) spectra were obtained in the range of 4000–400 cm−1 with a Bruker ALPHA-FTIR spectrometer.
Raman spectra were recorded using a combined Raman and scanning near field optical microscope WiTec Alpha 300 R equipped with a 532 nm excitation laser source.
Thermal decomposition of the synthesized powders was analyzed by thermogravimetric analysis (TGA) using a PerkinElmer STA 6000 Simultaneous Thermal Analyzer. A dried sample of about 20 mg was heated from 25 to 900 °C at a heating rate of 10 °C min−1 under dry flowing air (20 mL min−1).
Solid-state nuclear magnetic resonance (NMR) experiments were carried out at 14.1 T on a Bruker Avance Neo 600 NMR spectrometer operating at 600.3 and 243.0 MHz for 1H and 31P, respectively, using a 2.5 mm Trigamma MAS probe and 2.5 mm zirconia rotors. The temperature was stabilized at 298 K and the MAS rate was set to 20 kHz. For 1H MAS experiments, a pulse sequence containing two π refocusing pulses with a delay of 2 μs was employed to eliminate the signal from the probe background. The 1H 90° excitation pulse was 2.1 μs and 16 scans were accumulated using a repetition delay of 10 s. For 31P MAS measurements, a saturation recovery pulse sequence was used. The saturation pulse train consisted of 10 π/2 pulses followed by 100 s delay. The π/2 excitation pulse was equal to 3.6 μs, and 1024 scans were accumulated. 1H → 31P CP MAS measurements were performed with 2 ms CP contact time employing a 100–50% ramp on the 1H channel, and 4096 scans were accumulated using a repetition delay of 10 s. 1H and 31P spectra were referenced respectively to TMS using adamantane (δ (1H) = 1.85 ppm) and 85% H3PO4 using ADP (ammonium dihydrogen phosphate, NH4H2PO4, δ (31P) = 0.8 ppm).
The electron paramagnetic resonance (EPR) spectra were recorded on a Bruker ELEXSYS E580 EPR spectrometer equipped with a Bruker ER4118X-MD5 (pulsed X-band) microwave resonator. High-power pulses were obtained using a 1 kW TWT (X-band) microwave amplifier. A helium flow cryostat was used to stabilize the temperature. The operating microwave frequency was set to 9.7 GHz to best match the broad band of the strongly over-coupled pulsed resonator. All the pulsed EPR spectra were acquired at 4.5 K. The echo-detected field sweep (EDFS) spectra were recorded using a Hahn echo pulse sequence (π/2–τ–π–τ–echo) with an interpulse delay τ of 140 ns. The echo was integrated over a 60 ns time window that was centered at the maximum of the echo signal. The lengths of the π/2- and π-pulses were 8 ns and 16 ns, respectively. For the two-dimensional (2D) Hyperfine Sublevel Correlation (HYSCORE)28 spectra, the echo amplitude was measured using the pulse sequence (π/2–τ–π/2–t1–π–t2–π/2–echo) with 8 ns and 16 ns lengths for the π/2- and π-pulses, respectively, and a 12 ns detector gate (that is centered at the maximum of the echo signal). The delays in the pulse sequence are defined as the differences in the starting points of the pulses. The echo intensity was measured as a function of t1 and t2, where t1 and t2 were increased in steps of 16 ns from an initial value of 40 ns and 32 ns, respectively. 256 steps were used for each dimension. The 8 ns time difference between the initial values of t1 and t2 was set to account for the difference in the lengths between the π/2- and π-pulses. This provided symmetric spectra in both dimensions. The unwanted echoes were eliminated by applying a 16-step phase cycling procedure. The HYSCORE spectra of the Cu-WH powder sample were recorded at magnetic field of 334.5 mT with an inter-pulse delay, τ, of 174 ns.
The time domain 2D HYSCORE data were processed using MATLAB R2023a. A third-order polynomial baseline was subtracted from the resulting time-domain spectra. The corrected spectra were zero-filled to obtain a [2048 × 2048] matrix and Fourier transformed using a Fast Fourier Transformation (FFT) algorithm. The frequency domain spectra were plotted as the amplitude (absolute value) of the 2D frequency components.
The morphology of the synthesized powders and elemental distribution were analyzed by scanning electron microscopy (SEM) using a Hitachi SU-9000 microscope equipped with an energy dispersive X-ray spectrometer (EDX).
The elemental composition of the synthesized compounds was determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using a PerkinElmer Optima 7000 DV spectrometer.
For the ion release experiment, 0.6 g of Cu-WH powders was soaked in 30 mL of simulated body fluid (SBF), and the samples were investigated in triplicate. An SBF solution was prepared according to the procedure reported by Kokubo et al.29 The samples were kept in closed containers in a shaker incubator (BioSan ES-20/80) at 37 °C for 14 days under constant mixing at 250 rpm. Every day, 1 mL of the solution was taken from the sample for elemental analysis and replaced with the same amount of fresh SBF solution to maintain a constant volume. Quantitative determination of the released Cu2+ ions was performed by ICP-OES.
2.3. In vitro cytotoxicity studies
The cytotoxicity of Cu-WH powders was assessed using the MC3T3-E1 cell line (preosteoblasts). In the extract test, 4500 cells per well were seeded in a 96-well plate with 100 μL of the cell medium, composed of 89% Alpha Modified Eagle's Medium (α-MEM), 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). The plates with the cells were incubated overnight at 37 °C under 5% CO2 (New Brunswick™ S41i CO2 Incubator Shaker, Eppendorf, Hamburg, Germany).The Cu-WH powder sample was suspended in 5 ml of fresh cell medium, resulting in a concentration of 10 mg mL−1. After 24 and 48 hours, all the solution was collected from the samples and replaced with an additional 5 ml of fresh cell medium. The collected solution was then filtered through a 0.2 μm syringe filter and subsequently used for dilution with fresh medium – at extract to cell medium ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 and 1:100. The tested concentrations were 0.1 mg mL−1 and 0.01 mg mL−1. Extract dilutions were immediately applied onto the preincubated cells (100 μL for each well). Untreated cells were used as a positive control and 5% dimethylsulfoxide (DMSO) solution in the medium was applied to cells as a negative control. There were six replicates for each sample and controls.
:10 and 1:100. The tested concentrations were 0.1 mg mL−1 and 0.01 mg mL−1. Extract dilutions were immediately applied onto the preincubated cells (100 μL for each well). Untreated cells were used as a positive control and 5% dimethylsulfoxide (DMSO) solution in the medium was applied to cells as a negative control. There were six replicates for each sample and controls.
Cell Counting Kit-8 (CCK-8) assay was used to assess the cytotoxicity of Cu-WH extracts. For both time points, samples were incubated for 24 h and then 10 μL of CCK-8 solution was added to the cultivation media in each well and incubated for 1 h at 37 °C under 5% CO2. Absorption at 450 nm was measured using an Infinite M Nano microplate reader (Tecan, Männedorf, Switzerland).
3. Results and discussion
The crystal structure of WH is closely related to that of β-TCP,14,30 which crystallizes in a rhombohedral structure with the space group R3c (#161).30 The crystal lattice of β-TCP contains 5 inequivalent Ca sites and 3 inequivalent P sites. These crystallographic positions are distributed between two types of columns arranged along the c axis: A and B. The A column has a form of P(1)O4–Ca(4)–Ca(5), whereas in the B column calcium atoms and phosphate tetrahedra are arranged in the following sequence: P(3)O4–Ca(1)–Ca(3)–Ca(2)–P(2)O4. The Ca(1), Ca(2), Ca(3) and Ca(5) sites are fully occupied by calcium, while the Ca(4) site shows an occupancy factor of 0.5. Each A column is surrounded by six B columns, and each B column is surrounded by two A columns on the opposite sides and four B columns. The Ca2+ ions in β-TCP can be substituted by a variety of other cations. Foreign ions depending on their ionic radii and substitution levels tend to occupy one or several distinct Ca sites.31 The crystal structures of natural and synthetic Mg-WH (Fig. 1) are comprehensively investigated by means of XRD analysis and neutron diffraction and the difference between WH and β-TCP structures is explained.14,15,32 | ||
| Fig. 1 Unit cell of Mg-WH (a); arrangement of columns in the unit cell (b); arrangement of atoms in structural columns (c). | ||
To some extent, WH can be considered as an ion-substituted β-TCP; however, other structural and compositional differences include metal ion to phosphorus ratio, the presence of a protonated phosphate group (HPO42−) and cationic vacancies. The B columns in both WH and non-substituted β-TCP are identical; the difference occurs in the A column. The small cation (e.g. Mg2+, Zn2+, and Mn2+) in the WH structure occupies the most energetically favorable six-fold coordinated Ca(5) site, while the Ca(4) site remains vacant. The protons are bonded to the P(1)O4 tetrahedra, being close to the Ca(4) site.33
It is known that in an aqueous medium, CPs can transform from one phase into another phase via a dissolution–precipitation process.22,34,35 In this process, the pH value, temperature and presence of foreign ions are among the most influencing factors;36 therefore, the optimization of the synthesis procedure was performed in a few steps. The XRD patterns of the synthesized products obtained at different pH values are given in Fig. 2.
 | ||
| Fig. 2 XRD patterns of the synthesis products obtained at different pH values (T = 200 °C, t = 3 h, Ca/Cu = 9). | ||
It was found that Cu-WH with no visible secondary phases can be obtained in a very narrow pH range from 6.4 to 6.5. At higher pH values, a secondary HA phase was detected; however, it is worth noting that due to the low crystallinity its detection was challenging. The obtained HA does not give any sharp peak and its presence can be observed by the increase of the background at around 32°. The formation of the additional HA at higher pH values was expected, since WH, compared to HA, is a more stable CP phase exactly at relatively low pH.37 At lower pH values, a copper-rich Ca3Cu3(PO4)4 phase was observed along with Cu-WH. The percentage of this impurity gradually increased with decreasing pH of the synthesis medium. It is surprising that the optimal pH range was so narrow, when compared to the similar synthesis of Zn-WH, where the optimal pH was determined to be in the range from 5.6 to 6.2.22
The influence of the reaction time on the formation of Cu-WH was further investigated; the XRD patterns of the products obtained by performing the synthesis for different periods of time are shown in Fig. 3.
 | ||
| Fig. 3 XRD patterns of Cu-WH powders synthesized for different periods of time (pH = 6.4, T = 200 °C, Ca/Cu = 9). | ||
The as-precipitated powder (without hydrothermal treatment) was identified as DCPD. The gradual phase transformation into Cu-WH was observed by increasing the reaction time. Although after 1 h of the reaction, Cu-WH was identified as the major crystalline phase and there were not even traces of DCPD, the co-existing HA phase was detected instead. The percentage of HA in the synthesis product decreased with increasing reaction time; after 3 h of the reaction, there were no visible signals corresponding to HA. Longer reaction times did not result in any detectable changes. Thus, it can be concluded that the synthetic procedure requires at least 3 h. This value is significantly lower compared to those reported for the solvothermal synthesis of Mn-WH24 or Zn-WH23 and comparable to the data reported for hydrothermally synthesized Mg-WH.38,39 On the other hand, previously it was shown that in an aqueous medium, Zn-WH can be formed just in 1 h.22 The observed time-dependent phase evolution from DCPD to Cu-WH suggests that the transformation via a dissolution–precipitation reaction could occur not directly, but in two steps with an intermediate HA phase. Previously, it was shown by Jang et al.37 that under certain conditions HA can be converted into Mg-WH via a multistep process; however, it is difficult to compare the phase transformations in certain systems due to plenty of influencing factors such as pH, temperature, pressure, chemical composition of the reaction mixture, etc. Future investigation on the observed phase transformation process employing in situ X-ray analysis could show the exact reaction pathway.
Next, the effect of the synthesis temperature on the phase purity of Cu-WH was studied. The XRD patterns of the products obtained in the temperature range from 110 to 200 °C are depicted in Fig. S1.† It is evident that Cu-WH was obtained as the major crystalline phase at all temperatures; however, a closer look reveals the presence of a small amount of HA as the secondary phase at lower temperatures. The amount of HA decreased with an increase of the temperature, indicating that the synthesis should be performed at a relatively higher temperature, namely 200 °C.
To further investigate the peculiarities of the formation of Cu-WH, the influence of the concentration of starting materials was studied. In this experiment, the total metal ions’ concentration in the reaction vial was varied in the range of 0.02–0.24 M, while in a typical synthetic procedure it was fixed at 0.08 M. The results of XRD analysis (Fig. S2†) revealed that the concentration had only a limited effect on the phase purity of the obtained products. Decreasing the concentration to 0.04 M led to the formation of secondary phases (both HA and Ca3Cu3(PO4)4). There was no significant variation in the width of diffraction peaks, suggesting that the crystallite size of the obtained Cu-WH powders was comparable in all cases.
We were also interested if the variable ratio of metal cations in the initial reaction mixture could lead to the formation of Cu-WH with a variable metal content. Fig. 4 shows the XRD patterns of the products obtained using different initial Ca/Cu ratios.
 | ||
| Fig. 4 XRD patterns of Cu-WH powders synthesized with different Ca/Cu ratios (pH = 6.4, T = 200 °C, t = 3 h). | ||
The results showed 2 different trends. The Cu-rich reaction mixture (Ca/Cu < 9) resulted in the formation of the secondary Ca3Cu3(PO4)4 phase; its percentage in the obtained product increased with increasing initial Cu concentration. In the case of Cu-lean reactions (Ca/Cu > 9), the formation of HA along with Cu-WH was observed. It was observed that the amount of HA increased gradually with the decrease of the Cu concentration. Based on the results of these experiments, it can be concluded that the synthesis of Cu-WH with a variable metal content is impossible under selected conditions. Similar limitations were previously observed for Zn-WH synthesized under hydrothermal conditions.22
Rietveld refinement was used to calculate the cell parameters of the synthesized Cu-WH (Fig. 5). The refinement was carried out using a rhombohedral structure (space group R3c) adopted from Mg-WH.
 | ||
| Fig. 5 Rietveld refinement of the XRD data obtained for Cu-WH (T = 200 °C; t = 3 h; pH = 6.4, Ca/Cu = 9) refined in the space group R3c. The red circle symbols and the black solid line represent the experimental and calculated intensities, respectively, and the blue line below is the difference between them. The green tick marks indicate the positions of the Bragg peaks. | ||
The results revealed a good agreement between the calculated and experimental data. The parameter a was calculated as 10.330 Å, while the parameter c was 37.077 Å. These values are slightly lower compared to those previously reported for Mg-WH,38,39 Zn-WH22 and Cu-WH.27 The Rietveld analysis did not indicate the presence of secondary crystalline phases. Analogically, as in Mg-WH, small Cu2+ ions in the WH structure fully occupy the Ca(5) crystallographic site, while the Ca(4) position remains vacant. Although the occupation of the Ca(4) site by small cations is not typical of the WH structure,32 Konishi and Watanabe reported that Cu2+ ions in Cu-WH occupy both Ca(5) and Ca(4) sites.27 On the other hand, the authors also observed a significant contamination of their product with Mg2+ ions. Recent investigations on a closely related Cu-substituted β-TCP structure did not indicate the presence of Cu2+ ions at the Ca(4) site. According to Spaeth et al.,8 at the maximal substitution level, Cu2+ occupies Ca(5), Ca(2) and Ca(3) sites. These conclusions were confirmed by a recent work by Deyneko et al.10 By contrast, Mg2+ ions were shown to occupy both Ca(5) and Ca(4) sites in a heavily doped β-TCP.40
As was mentioned above, the crystal structures of WH and β-TCP (or substituted TCP) are in a close relationship, which results in the nearly identical XRD patterns of these two materials; therefore, the reliable identification of WH is impossible without employing vibrational spectroscopy. In this context, FTIR and Raman spectroscopy are superior methods for the characterization of compounds with the WH structure, since both these methods enable the observation of the signals from the HPO42− group. Only the crystal structure of WH (not β-TCP) contains this structural unit, which can be used as a reliable spectral marker.39 The FTIR spectrum of the synthesized Cu-WH is given in Fig. 6a; the spectral region from 1500 to 400 cm−1 is shown as the most representative. The shape of the spectrum is similar to those of natural and synthetic WH.22,32 The spectrum exhibits several absorption bands: the bands located in the range from 1170 to 930 cm−1 are ascribed to ν3 and ν1 stretching modes of PO4, while the bands ranging from 650 to 410 cm−1 correspond to ν2 and ν4 bending modes. The band centered at ca. 920 cm−1 indicates the presence of the HPO42− group, confirming that the synthesized material is Cu-WH and not Cu-substituted β-TCP.
 | ||
| Fig. 6 FTIR spectrum (a) and Raman spectrum (b) of Cu-WH powders (pH = 6.4, T = 200 °C, t = 3 h, Ca/Cu = 9). | ||
The Raman spectrum of the same sample is provided in Fig. 6b. In this case, a few bands can also be seen and identified. The bands located in the ranges of 370–495, 520–640 and 990–1170 cm−1 correspond to the ν2, ν4 and ν3 vibrational modes of the phosphate group, respectively. The most intense signal peaked at 964 cm−1 is ascribed to the ν1 vibrational mode. Finally, the signal located at 920 cm−1 is attributed to HPO42−, which agrees well with the results of the FTIR analysis. Overall, both spectroscopic techniques agree very well with the results reported in the literature in terms of the spectral shape and band wavenumbers.22,32,39,41
The 1H MAS spectra obtained for the Cu-WH sample before and after annealing at 500 °C are shown in Fig. 7. The spectrum of the as-prepared Cu-WH powder looks similar to those previously reported for Zn- and Mg-WH.22,39 The 1H spectrum clearly contains a major spectral line at 9.9 ppm, which is attributed to the HPO42− moiety in the Cu-WH crystal structure (Fig. 1). Additional lines attributed to surface HxPO4 and distorted HPO4 moieties are seen at 12.3 ppm and 6.2 ppm, respectively.42 Water in the form of bulk and surface species is detected at 4.5–1.5 ppm. As can be seen from Fig. 7, annealing of the sample significantly reduces the amount of the side phosphate species and water. The linewidth of a line attributed to Cu-WH decreases two-fold after annealing, which indicates the formation of the Cu-WH phase with higher crystallinity. The presence of a relatively higher amount of water could be explained by the trapping of water molecules during the dissolution–precipitation process.
 | ||
| Fig. 7 1H MAS spectra of Cu-WH before and after annealing at 500 °C. Spectral components attributed to Cu-WH are shown in orange, water in blue, and additional phosphate species in green. | ||
Fig. 8 depicts the 31P MAS and 1H–31P CP MAS spectra of Cu-WH before and after annealing. It is seen that annealing significantly reduces the amount of the amorphous/distorted phase in the sample: before annealing, the signal from the amorphous component highly overlaps with the P(3) signal, which prevents the accurate spectral assignment. Similarly to 1H data, the lines in 31P data after annealing show narrower linewidths, indicating the formation of the Cu-WH phase of higher crystallinity. The 31P MAS spectrum after annealing consists of lines at 2.2, 1.3, and −0.5 attributed to P(2), HP(1)O42− and P(3) moieties, which are typical in the WH structure,22,39 and a line at 0.4 ppm, which is an impure phosphate phase. One more remark has to be made that in comparison with Zn- and Mg-WH, Cu-WH possesses significantly broader lines which is due to the paramagnetic nature of the Cu2+ environment. In the 1H–31P CP MAS spectra, the line assigned to HPO42− is significantly more enhanced, which corroborates our assignment that the proton is in close vicinity. The line at −2.8 ppm is assigned to the side phase of hydrated PO4, which vanishes after annealing.
 | ||
| Fig. 8 31P MAS (upper) and 1H–31P CP MAS (lower) spectra of Cu-WH before and after annealing at 500 °C. Spectral components attributed to Cu-WH are shown in orange and additional phosphate phases in green. | ||
Taking into account the results of X-ray analysis and Rietveld refinement (Fig. 5), it can be concluded that the impurities detected by NMR possess an amorphous nature, since no secondary crystalline phases were detected by XRD. This observation also indicates that ss-NMR is a highly useful method for the comprehensive structural analysis of low-crystalline CPs synthesized at low temperatures. Unfortunately, this technique is rarely employed for the characterization of materials with the WH structure;22,38,39 this makes the comparison of the experimental results almost impossible. Konishi and Watanabe38 also observed some additional signals in the 1H NMR spectra of Mg-WH, while XRD did not indicate the presence of secondary phases. The signals in the 31P NMR spectra were better resolved in their study; however, this might be caused by different particle sizes and degrees of crystallinity, and the absence of paramagnetic Cu2+ ions. In comparison, in a work by the same group focusing on Cu-WH,27 the NMR linewidths were comparable to the ones obtained in the present study.
Since Cu-WH possesses copper ions (Cu2+) in the crystal structure, the powders were also characterized by EPR spectroscopy. We recorded the spectrum of Cu-WH powders by using pulsed EPR at low temperature. Fig. 9a shows the field swept spectrum of Cu2+.
 | ||
| Fig. 9 Electron spin-echo magnetic-field-sweep EPR spectrum of Cu-WH powder. The arrow indicates the magnetic field position of 334.5 mT that was used for the acquisition of the 2D HYSCORE spectrum (a). HYSCORE spectrum of the (+, +) quadrant of the Cu-WH powders. Black dot lines on the top of 31P peaks are obtained from simulation. The spectra were acquired at a temperature of 4.5 K (b). | ||
Line broadening appears due to hyperfine interaction with nearby copper nuclei. To get more information about the surroundings of the Cu2+ paramagnetic center, we used a more advanced pulsed EPR technique – Hyperfine Sublevel Correlation (HYSCORE). The basic advantage of the HYSCORE technique is the formation of the 2D spectra of off-diagonal cross-peaks, whose coordinates are nuclear frequencies from opposite electron spin manifolds. The cross-peaks simplify significantly the analysis of congested spectra by correlating and spreading out the nuclear frequencies. In addition, the HYSCORE experiment separates overlapped peaks along a second dimension and enhances the signal-to-noise ratio through the application of a second Fourier transform. Hyperfine interactions are comprised of the isotropic Fermi contact interaction, Aiso, and anisotropic through-space dipolar interaction, T; where Aiso provides a measure of the electron spin density at the nucleus, while T reflects the dipole–dipole interaction between the electron and nuclear spins and depends on the distance and orientation of the nucleus relative to the electron spin.43
In HYSCORE spectrum (Fig. 9b), we observed a pair of off-diagonal cross-peaks, 31P, at (2.6, 9) and (9, 2.6) MHz in the (+, +) quadrant of the HYSCORE spectrum. The cross-peaks are centered at a 31P Zeeman frequency of 5.77 MHz (at a magnetic field of 334.5 mT) and arise from the hyperfine interactions of the 31P atom (nuclear spin, I = 1/2) with the Cu2+ paramagnetic center. The separation between the pair of cross-peaks 31P is ∼6.4 MHz, which is proportional to the isotropic hyperfine coupling, Aiso, of the 31P atom. Simulation of peak positions and shape allowed us to obtain an anisotropic through-space dipolar interaction, T, value of ∼1.3 MHz. As dipolar interaction T depends on the distance r between the two centers, we also calculated the r value, which is equal to ∼0.3 nm. This value is close to that between the Cu and P(2) atoms in the WH crystal lattice obtained from the XRD data. Moreover, Cu2+ ions were observed in only one crystallographic site, which agrees well with the results of Rietveld refinement. Additionally, the experimental spectrum in Fig. 9b also reveals a cross-peak, 1H, on the diagonal (14.3 MHz) which arises from very weak hyperfine couplings (Aiso < 0.1 MHz) of the effective electron spin (S = 1/2) with surrounding 1H nuclei.
The challenging control of the stoichiometry is a well-known disadvantage of the wet precipitation method, when employed for the synthesis of multicomponent materials. In order to check the chemical composition of the synthesized Cu-WH, the elemental analysis was carried out by means of ICP-OES. The results are summarized in Table 1. It is evident that the experimentally obtained values agree well with the theoretical ones. The Ca/Cu and (Ca + Cu)/P molar ratios were determined to be very close to the theoretical values, being only slightly lower.
| Sample | Determined Ca/Cu molar ratio | Determined (Ca + Cu)/P molar ratio | Theoretical Ca/Cu molar ratio | Theoretical (Ca + Cu)/P molar ratio |
|---|---|---|---|---|
| Cu-WH | 8.79 | 1.39 | 9.00 | 1.43 |
The morphological features of Cu-WH were investigated by SEM and STEM. It is known from previous works that the WH structure tends to form particles of a distinctive rhombohedral, hexagonal or octahedral shape.21,22,37,44 Varying the synthesis conditions in terms of the concentration of starting materials allows one to control the size and shape of WH particles.22 The SEM images of Cu-WH powders synthesized using different concentrations of starting materials are shown in Fig. 10a and b.
 | ||
| Fig. 10 SEM images of the Cu-WH powders synthesized when the initial concentration of metal ions was 0.08 M (a) and 0.24 M (b); STEM image of Cu-WH synthesized at concentration of 0.08 M (c); SEM image (d) and EDS mapping (e–h) of Cu-WH (pH = 6.4, T = 200 °C, t = 3 h, Ca/Cu = 9). | ||
It is seen that the variation of the concentration had only a limited effect on the morphology of the material. The sample synthesized using a 3-fold higher concentration of the starting material consisted of slightly smaller particles (Fig. 10b), compared to those obtained using the standard synthetic conditions (Fig. 10a). Both samples were composed of agglomerated particles of nanoscale dimensions. While most of the particles showed an irregular shape, a closer look allows us to see some particles of well-defined rhombohedral and hexagonal plate-like shapes. The EDS mapping (Fig. 10e–h) indicated a homogeneous distribution of the constituting elements. There were no visible agglomerations of some elements in the absence of others.
It is known that the WH structure is thermally unstable and decomposes upon annealing,21,39 which is a limiting factor for the fabrication of WH ceramics by conventional routes considering the annealing at elevated temperatures. Thermally induced degradation occurs by the transformation of WH into the mixture of β-TCP and Ca2P2O7 (CPP), accompanied by the release of water:
| Ca18M2(HPO4)2(PO4)12 → 6Ca2.67M0.33(PO4)2 + Ca2P2O7 + H2O | (1) |
Although the thermally induced decomposition of Mg-WH was previously investigated in detail,39 the stability of the synthesized Cu-WH was studied here in order to determine if the origin of small cations in the WH structure could influence the degradation temperature. The results of thermogravimetric analysis in the range from room temperature to 900 °C are depicted in Fig. 11.
 | ||
| Fig. 11 TG/DSC curves of the Cu-WH powder. | ||
It can be seen that the weight loss occurs gradually up to ca. 700 °C without any drastic drops. Above 700 °C, the residual mass was nearly constant. The total mass loss was calculated as 4 wt%, while the theoretical weight loss according to eqn (1) was only 0.8 wt%. This mismatch indicates that the as-prepared material contains some adsorbed water and/or other relatively volatile species, which could be trapped during the dissolution–precipitation process. In general, the behavior of the TG curve and the total weight loss are comparable to those observed for Mg-WH39 and Zn-WH.22
In order to investigate the structural changes and the phase evolution upon annealing, the as-prepared Cu-WH powders were annealed at different temperatures in the 500–1000 °C range with steps of 100 °C. The XRD patterns of the obtained samples are presented in Fig. 12a.
 | ||
| Fig. 12 XRD patterns (a) and FTIR spectra (b) of Cu-WH powders annealed at different temperatures. | ||
It is worth noting that the detection of the transformation of WH into β-TCP is impossible by means of XRD due to their nearly identical patterns;39 however, XRD is a suitable method for the monitoring of the formation of secondary phases, which can serve as an indicator of the thermal decomposition. The appearance of the secondary crystalline phase starts after annealing at 600 °C. The newly formed phase was identified as α-CPP. The formation of this CPP polymorph was rather unexpected, since α-CPP is assumed to be a high-temperature polymorph;45 nevertheless, its formation could be explained by the Ostwald step rule.46 It was demonstrated previously that the amorphous CP (Ca/P ratio 1:1) crystallizes as the α-CPP polymorph first; the crystallization temperature was determined as 600 °C.47 The start of the thermal decomposition of Cu-WH was found to be around 100 °C lower compared to that of Mg-WH,39 but in good agreement with that of Zn-WH.21 The thermal treatment at 800 °C resulted in the formation of both α- and β-CPP, while only β-CPP secondary phase was detected in the samples annealed at 900 °C and 1000 °C.
FTIR spectroscopy was employed in parallel with the XRD analysis to monitor the structural changes, since this technique allows for the detection the HPO42− group, which serves as a spectral marker for the identification of WH. The presence or absence of this structural unit cannot be detected by XRD. The FTIR spectra are shown in Fig. 12b. After annealing at 500 °C, there were no obvious spectral changes in terms of the appearance or disappearance of absorption bands; however, sharpening of the bands was observed, indicating the increase of the crystallinity of the material. The formation of α-CPP was confirmed after the thermal treatment at 600 °C and 700 °C. The presence and identification of CPP polymorphs can be easily detected by observing the absorption bands at 726 and 754 cm−1,48 since the FTIR spectra of WH and β-TCP do not have any bands in this region. The co-existence of two CPP polymorphs was evident after annealing at 800 °C, while only β-CPP was observed after the thermal treatment at higher temperatures. Overall, the results of FTIR spectroscopy agree very well with the results of XRD analysis; both techniques confirm that Cu-WH is thermally unstable at temperatures above 600 °C.
The ion release experiment was performed in order to estimate the release of Cu2+ ions into the SBF solution during the soaking of the as-prepared Cu-WH powder (Fig. 13). The concentration of Cu2+ ions monotonously increased during the soaking. It should be highlighted that the drastic release of Cu2+ was observed just after 1 day. Despite the fact that before the experiment the powder was washed with deionized water, the possible reason for such a behavior is the release of soluble trapped Cu-containing species. The maximal concentration of Cu2+ ions was determined after 14 days as 25.8 mg L−1. Although the ion release from Cu-containing biomaterials was investigated in various works,10,49,50 it is difficult to compare the released amounts of Cu because of the highly varying experimental conditions in terms of Cu content in materials, the sample form, the ratio of sample mass to solution volume, the soaking medium, etc.
 | ||
| Fig. 13 Cu2+ ion release into the SBF solution. | ||
The comparison of our results with other studies on Cu-WH is currently limited to one report;27 however, in that study the experiment was performed under significantly different conditions in acetate buffer (pH = 5.5) with powder mass to solution volume ratio being 400 times lower and duration of monitoring of only 180 min. Recalculating the data indicates a noticeably higher release reported in ref. 27. In another work, Deyneko et al.10 investigated the Cu2+ release from Cu-doped β-TCP in Tris-HCl buffer solution. The concentration of Cu2+ released from the samples with a similar Cu content (9.5 mol%) revealed the same trend upon increasing in time; however, after 30 days of soaking, the concentration was significantly lower compared to our study. It is worth noting that the authors tested the pellets instead of powders, the ion-release medium of different composition and the mass-to-volume ratio (0.4 g in 50 mL) was 2.5 times lower compared to ours. Moreover, the particle size of the initial powders in the mentioned paper was significantly larger as a result of high-temperature synthesis. All these factors could influence the difference in the obtained data. Gomes et al.13 studied the Cu2+ release from pelletized biphasic CPs consisting of HA and β-TCP. Depending on the phase composition of the tested samples, the authors determined a comparable or even higher amount of released Cu2+ ions, although the doping level of starting materials was lower compared to that in our work. Moreover, the ion release was observed only in the SBF solution, but not in deionized water. Compared to Zn-WH prepared and treated under similar conditions, the amount of Cu2+ ions observed in this work was around 25 times higher than that of Zn2+.22
The cell viability of Cu-WH samples was assessed using an indirect method by collecting extract solutions. Following the ISO 10993-5:2009 guidelines, the study revealed that none of the tested Cu-WH extract dilutions demonstrated cytotoxic effects after 48 h sample incubation, as these experiments showed cell viability more than 70% (Fig. 14).
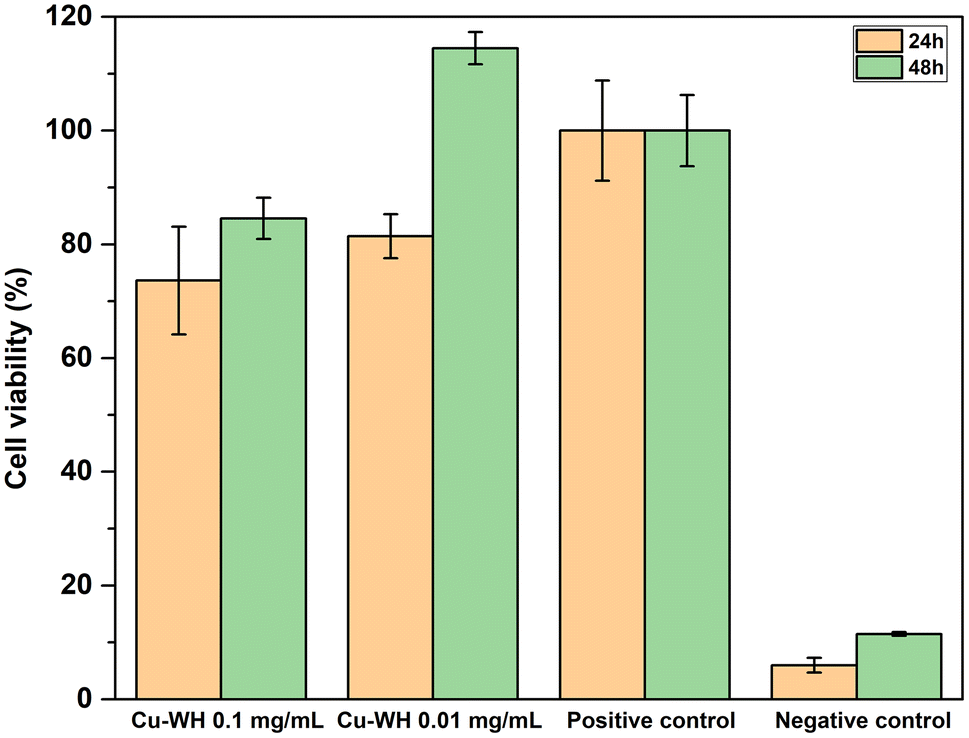 | ||
| Fig. 14 Cu-WH extract's effect on MC3T3-E1 cell proliferation. | ||
Lower cell viability (73.64 ± 9.48%) was noted for Cu-WH extracts at a concentration of 0.1 mg mL−1 after 24 h incubation period with a slight increase at 48-hour incubation timepoint. A similar trend of viability increase after 48 h was observed for the extract dilution at a lower concentration of 0.01 mg mL−1. Decreased cell viability in the first 24 hours could be explained by the rapid Cu2+ ion release in the medium. Due to its transition between different oxidation states, Cu2+ plays a role in numerous physiological processes. Duan et al.51 investigated the effect of Cu-containing alloy on osteoblast proliferation and differentiation, indicating that Cu2+ induced the proliferation of osteoblasts at 24 to 72 h timepoints, explaining the increase of cell viability after 48 h. Furthermore, Zhang et al.52 observed that the Cu ion concentration is one of the key factors for switching the biological effects of Cu2+ and Cu+ from toxicity to activity.
4. Conclusions
In this work, nanodimensional copper whitlockite (Cu-WH) powders were successfully synthesized by a dissolution–precipitation method under hydrothermal conditions. The conversion from brushite to Cu-WH occurred in a slightly acidic medium in the presence of Cu2+ ions and required at least 3 h at 200 °C. The optimal pH range was found to be very narrow being from 6.4 to 6.5. The variation of the chemical composition in terms of the Ca/Cu ratio was impossible under the selected synthesis conditions; the initial Ca/Cu ratio of 9, corresponding to the theoretical metal ratio in the WH structure, was determined as optimal. While XRD and vibrational spectroscopy did not indicate the presence of any impurity phase in the as-prepared Cu-WH powders, the NMR studies revealed the co-existence of amorphous/distorted phosphate species. The amount of distorted phosphates was reduced significantly by annealing the powder at a moderate 500 °C temperature. The Cu2+ release from the Cu-WH powder monitored for 14 days showed a gradually increasing trend with a drastic release during the first day of soaking. The study on thermal stability revealed that Cu-WH is thermally unstable and gradually decomposes to Cu-substituted β-Ca3(PO4)2 and Ca2P2O7 upon annealing above 600 °C. The cell viability study revealed that none of the tested Cu-WH extract dilutions demonstrated cytotoxic effects, as all experiments demonstrated cell viability exceeding 70%.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by a grant WHITCERAM (No. S-LJB-22-1) from the Research Council of Lithuania and by the Joint Research Projects under the Bilateral Program (JSPS), RCL JSPS, No. 120224202. The authors acknowledge the access to the infrastructure and expertise of the BBCE – Baltic Biomaterials Centre of Excellence (European Union's Horizon 2020 research and innovation programme under the grant agreement no. 857287). The authors acknowledge the Center of Spectroscopic Characterization of Materials and Electronic/Molecular Processes (‘Spectroversum’, https://spectroversum.ff.vu.lt) for the use of NMR and EPR equipment.References
- W. Habraken, P. Habibovic, M. Epple and M. Bohner, Mater. Today, 2016, 19, 69–87 CrossRef CAS.
- N. V. Bulina, O. B. Vinokurova, I. Y. Prosanov, A. M. Vorobyev, K. B. Gerasimov, I. A. Borodulina, A. Pryadko, V. V. Botvin, M. A. Surmeneva and R. A. Surmenev, Ceram. Int., 2022, 48, 35217–35226 CrossRef CAS.
- M. Gu, W. Li, L. Jiang and X. Li, Acta Biomater., 2022, 148, 22–43 CrossRef CAS PubMed.
- V. H. M. Doan, D. D. Vu, S. Mondal, T. M. T. Vo, C. D. Ly, V. T. Nguyen, S. Park, J. Choi, T. P. Nguyen, B. Lee and J. Oh, ACS Biomater. Sci. Eng., 2023, 9, 4607–4618 CrossRef CAS PubMed.
- S. Jin, L. Ren and K. Yang, J. Mater. Sci. Technol., 2016, 32, 835–839 CrossRef CAS.
- A. Jacobs, G. Renaudin, C. Forestier, J.-M. Nedelec and S. Descamps, Acta Biomater., 2020, 117, 21–39 CrossRef CAS.
- V. Stanić, S. Dimitrijević, J. Antić-Stanković, M. Mitrić, B. Jokić, I. B. Plećaš and S. Raičević, Appl. Surf. Sci., 2010, 256, 6083–6089 CrossRef.
- K. Spaeth, F. Goetz-Neunhoeffer and K. Hurle, J. Solid State Chem., 2020, 285, 121225 CrossRef CAS.
- D. V. Deyneko, I. V. Fadeeva, E. Y. Borovikova, P. B. Dzhevakov, P. V. Slukin, Y. Zheng, D. Xia, B. I. Lazoryak and J. V. Rau, Ceram. Int., 2022, 48, 29770–29781 CrossRef CAS.
- D. V. Deyneko, Y. F. Zheng, K. Barbaro, V. N. Lebedev, S. M. Aksenov, E. Y. Borovikova, M. R. Gafurov, I. V. Fadeeva, B. I. Lazoryak, G. Di Giacomo, C. Cicione, V. Tilotta, F. Russo, G. Vadala and J. V. Rau, Ceram. Int., 2023, 49, 21308–21323 CrossRef CAS.
- X. Li, G. Li, K. Zhang, Z. Pei, S. Zhao and J. Li, J. Biomed. Mater. Res., Part B, 2021, 109, 877–889 CrossRef CAS PubMed.
- M. Schamel, A. Bernhardt, M. Quade, C. Würkner, U. Gbureck, C. Moseke, M. Gelinsky and A. Lode, Mater. Sci. Eng., C, 2017, 73, 99–110 CrossRef CAS PubMed.
- S. Gomes, C. Vichery, S. Descamps, H. Martinez, A. Kaur, A. Jacobs, J.-M. Nedelec and G. Renaudin, Acta Biomater., 2018, 65, 462–474 CrossRef CAS.
- R. Gopal and C. Calvo, Nat. Phys. Sci., 1972, 237, 30–32 CrossRef CAS.
- R. Gopal, C. Calvo, J. Ito and W. K. Sabine, Can. J. Chem., 1974, 52, 1155–1164 CrossRef CAS.
- H. Cheng, R. Chabok, X. Guan, A. Chawla, Y. Li, A. Khademhosseini and H. L. Jang, Acta Biomater., 2018, 69, 342–351 CrossRef CAS PubMed.
- F. A. Shah, Acta Biomater., 2021, 125, 72–82 CrossRef CAS.
- H. D. Kim, H. L. Jang, H.-Y. Ahn, H. K. Lee, J. Park, E.-S. Lee, E. A. Lee, Y.-H. Jeong, D.-G. Kim, K. T. Nam and N. S. Hwang, Biomaterials, 2017, 112, 31–43 CrossRef CAS PubMed.
- H. L. Jang, G. B. Zheng, J. Park, H. D. Kim, H.-R. Baek, H. K. Lee, K. Lee, H. N. Han, C.-K. Lee, N. S. Hwang, J. H. Lee and K. T. Nam, Adv. Healthcare Mater., 2016, 5, 128–136 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- A. Kizalaite, I. Grigoraviciute-Puroniene, D. R. C. Asuigui, S. L. Stoll, S. H. Cho, T. Sekino, A. Kareiva and A. Zarkov, ACS Biomater. Sci. Eng., 2021, 7, 3586–3593 CrossRef CAS.
- A. Kizalaite, V. Klimavicius, J. Versockiene, E. Lastauskiene, T. Murauskas, R. Skaudzius, T. Yokoi, M. Kawashita, T. Goto, T. Sekino and A. Zarkov, CrystEngComm, 2022, 24, 5068–5079 RSC.
- M. Wang, J. Yao, S. Shen, C. Heng, Y. Zhang, T. Yang and X. Zheng, Nano Res., 2023, 16, 757–770 CrossRef CAS.
- C. Heng, W. Liu, X. Zheng, X. Ma, J. Hui and D. Fan, Colloids Surf., B, 2023, 222, 113120 CrossRef CAS PubMed.
- E. Kostiner and J. R. Rea, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 250–253 CrossRef.
- S. Y. Stefanovich, B. I. Lazoryak, A. M. Antipin, A. S. Volkov, A. I. Evdokimov, O. A. Gurbanova, O. V. Dimitrova and D. V. Deyneko, Z. Kristallogr. - Cryst. Mater., 2023, 238, 301–309 CrossRef CAS.
- T. Konishi and S. Watanabe, Phosphorus Res. Bull., 2022, 38, 18–24 CrossRef CAS.
- P. Höfer, A. Grupp, H. Nebenführ and M. Mehring, Chem. Phys. Lett., 1986, 132, 279–282 CrossRef.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- M. Yashima, A. Sakai, T. Kamiyama and A. Hoshikawa, J. Solid State Chem., 2003, 175, 272–277 CrossRef CAS.
- K. Matsunaga, T. Kubota, K. Toyoura and A. Nakamura, Acta Biomater., 2015, 23, 329–337 CrossRef CAS PubMed.
- F. Capitelli, F. Bosi, S. C. Capelli, F. Radica and G. Della Ventura, Crystals, 2021, 11, 225 CrossRef CAS.
- T. Debroise, E. Colombo, G. Belletti, J. Vekeman, Y. Su, R. Papoular, N. S. Hwang, D. Bazin, M. Daudon, P. Quaino and F. Tielens, Cryst. Growth Des., 2020, 20, 2553–2561 CrossRef CAS.
- L. Sinusaite, A. Popov, E. Raudonyte-Svirbutaviciene, J.-C. Yang, A. Kareiva and A. Zarkov, Ceram. Int., 2021, 47, 12078–12083 CrossRef CAS.
- G. Montes-Hernandez and F. Renard, J. Phys. Chem. C, 2020, 124, 15302–15311 CrossRef CAS.
- E. Raudonyte-Svirbutaviciene, L. Lukaviciute, Z. Moravec, J. Pinkas, T. Goto, T. Sekino, A. Zarkov and A. Kareiva, Ceram. Int., 2023, 49, 32816–32825 CrossRef CAS.
- H. L. Jang, H. K. Lee, K. Jin, H.-Y. Ahn, H.-E. Lee and K. T. Nam, J. Mater. Chem. B, 2015, 3, 1342–1349 RSC.
- T. Konishi and S. Watanabe, Phosphorus Res. Bull., 2021, 37, 21–25 CrossRef CAS.
- A. Kizalaite, V. Klimavicius, V. Balevicius, G. Niaura, A. N. Salak, J. C. Yang, S. H. Cho, T. Goto, T. Sekino and A. Zarkov, CrystEngComm, 2023, 25, 4370–4379 RSC.
- R. Enderle, F. Götz-Neunhoeffer, M. Göbbels, F. A. Müller and P. Greil, Biomaterials, 2005, 26, 3379–3384 CrossRef CAS.
- H. Zhou, L. Yang, U. Gbureck, S. B. Bhaduri and P. Sikder, Acta Biomater., 2021, 127, 41–55 CrossRef CAS PubMed.
- M. Ben Osman, S. Diallo-Garcia, V. Herledan, D. Brouri, T. Yoshioka, J. Kubo, Y. Millot and G. Costentin, J. Phys. Chem. C, 2015, 119, 23008–23020 CrossRef CAS.
- A. Schweiger and G. Jeschke, Principles of pulse electron paramagnetic resonance, Oxford university press, 2001 Search PubMed.
- X. Guo, X. Liu, H. Gao, X. Shi, N. Zhao and Y. Wang, J. Mater. Sci. Technol., 2018, 34, 1054–1059 CrossRef CAS.
- D. Griesiute, E. Garskaite, A. Antuzevics, V. Klimavicius, V. Balevicius, A. Zarkov, A. Katelnikovas, D. Sandberg and A. Kareiva, Sci. Rep., 2022, 12, 7116 CrossRef CAS PubMed.
- R. A. Van Santen, J. Phys. Chem., 1984, 88, 5768–5769 CrossRef CAS.
- Z. Zyman, A. Goncharenko and D. Rokhmistrov, J. Cryst. Growth, 2017, 478, 117–122 CrossRef CAS.
- D. Griesiute, E. Raudonyte-Svirbutaviciene, A. Kareiva and A. Zarkov, CrystEngComm, 2022, 24, 1166–1170 RSC.
- M. M. Erol, V. Mouriňo, P. Newby, X. Chatzistavrou, J. A. Roether, L. Hupa and A. R. Boccaccini, Acta Biomater., 2012, 8, 792–801 CrossRef CAS PubMed.
- K. M. Weiss, S. K. Kucko, S. Mokhtari, T. J. Keenan and A. W. Wren, J. Biomed. Mater. Res., Part B, 2023, 111, 295–313 CrossRef CAS.
- J.-Z. Duan, Y. Yang and H. Wang, Orthop. Surg., 2022, 14, 758–768 CrossRef PubMed.
- J. C. Zhang, Y. P. Li, K. N. Yang and X. H. Hao, Chin. J. Inorg. Chem., 2010, 26, 2251–2258 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt03756h |
| This journal is © The Royal Society of Chemistry 2024 |