
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing isoprene polymerization with high activity and adjustable monomer enchainment using cyclooctyl-fused iminopyridine iron precatalysts†
Nighat
Yousuf
abc,
Yanping
Ma
 *a,
Qaiser
Mahmood
*b,
Wenjuan
Zhang
*d,
Yizhou
Wang
ac,
Hassan
Saeed
abc and
Wen-Hua
Sun
*abc
*a,
Qaiser
Mahmood
*b,
Wenjuan
Zhang
*d,
Yizhou
Wang
ac,
Hassan
Saeed
abc and
Wen-Hua
Sun
*abc
aKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: whsun@iccas.ac.cn; myanping@iccas.ac.cn
bChemistry and Chemical Engineering Guangdong Laboratory, Shantou 515031, China. E-mail: qaiser@ccelab.com.cn
cCAS Research/Education Center for Excellence in Molecular Sciences and International School, University of Chinese Academy of Sciences, Beijing 100049, China
dBeijing Key Laboratory of Clothing Materials R&D and Assessment, School of Materials Science and Engineering, Beijing Institute of Fashion Technology, Beijing 100029, China. E-mail: zhangwj@bift.edu.cn
First published on 30th November 2023
Abstract
In this study, a series of structurally rigid cyclooctyl-fused iminopyridine iron complexes, [L2FeCl][FeCl4] and [2L3Fe][Cl][3FeCl4], was synthesized via a one-pot method and investigated as precatalysts in conjunction with methylaluminoxane for isoprene (Ip) polymerization. Combined characterization through FTIR analysis, elemental analysis and single crystal XRD analysis fully verified the structure of these complexes. The most active iron complex, FeH, exhibited a trisligated nature, with its cation adopting an octahedral geometry around the metal center. In contrast, all the other iron complexes (Fe2Me, Fe2Et, Fe2iPr, Fe3Me, Fe2Et,Me) displayed bisligated configurations, with distorted trigonal bipyramidal geometry of cations. During isoprene polymerization, the extent of steric hindrance of the ligand framework exerted a significant impact on catalytic performance. The FeH precatalyst with less steric hindrance demonstrated excellent performance, producing high molecular weight polyisoprenes with conversions exceeding 99% for 4000 equiv. of monomer. Even at very low catalyst loadings, as low as 0.0025 mol% (Fe/Ip), the polymerization of isoprene could proceed smoothly with an exceptionally high activity of 4.0 × 106 gPI (molFe, h)−1. Moreover, this precatalyst exhibited good thermal stability, maintaining high activity levels (typically 105 gPI (molFe, h)−1) across a broad temperature range from −20 °C to 100 °C. Additionally, by adjusting steric substituents and the reaction temperature, the 1,4/3,4 regioselectivity could be modulated from 9/91 to 69/31 while maintaining a high stereoselectivity of cis-1,4 structures (cis/trans: >99/1).
Introduction
Metal catalytic polymerization of isoprene may afford various polyisoprene (PI) configurations, including cis-1,4, trans-1,4, 3,4 (isotactic, syndiotactic, atactic) and the less common 1,2 (isotactic, syndiotactic, atactic).1 Polyisoprenes (PI) with these distinct configurations exhibit versatile chemical, thermal and physical properties, rendering them valuable for a wide array of applications such as rubber manufacturing, shape memory technology, the pharmaceutical industry, etc.2 The catalyst structure is crucial for producing highly selective polyisoprenes. Recent advancements in the catalyst structure have even enabled the synthesis of polyisoprene structures closely resembling natural rubber.3 PI with high trans-1,4 selectivity possesses crystalline and tough characteristics comparable to balata and gutta-percha rubbers, while cis-1,4 PI, resembling Hevea brasiliensis latex, offers elasticity with lower crystallinity.4,5 Over the various polymerization methods, including radical, cationic, and anionic processes, it is possible to fine-tune the stereo- and regioselectivity of the polymer microstructure, to a certain extent, through coordination–insertion polymerization. The degree of control mainly depends on the ligand structure with fine tuning through steric and electronic factors.2,3Classical catalysts, such as TiCl4/alkylaluminum, lithium organic, and neodymium catalysts, are widely used for producing cis-1,4 PI.6 In contrast, the vanadium trichloride/triethylaluminum catalytic system is efficient for the synthesis of trans-1,4 PI.7 Over the past two decades, the development of new well-defined transition metal catalysts afforded not only highly selective PI, but also introduced new microstructures of polyisoprene featuring a diverse sequence of 1,4 and 3,4 additions.8 Among them, iron catalysts are preferred for use due to their high metal abundance, cost-effectiveness, low toxicity, ease of preparation, and stability under a wide range of polymerization conditions.9 However, there were few reports on iron precatalysts for isoprene polymerization until the 2000s.10 Iron complexes bearing aromatic N,N bidentate ligand structures in combination with alkyl aluminum cocatalysts have emerged as highly active catalysts for isoprene polymerization, with high 3,4 selectivity (A, Fig. 1).11 Thus, N,N bidentate-Fe(II) catalysts have recently gained significant attention for their remarkable activity, selectivity, living characteristics (in some cases) and the ability to polymerize various conjugated dienes.12 The discovery of α-diimine-nickel/-palladium catalysts in 1995 and bis(imino)pyridine-iron/cobalt catalysts in 1998 for ethylene polymerization marked a significant milestone in catalyst development for olefin and diene polymerization (B, Fig. 1).13 In these efforts, Ritter and co-workers reported iminopyridine iron complexes as highly active catalysts for isoprene polymerization. These complexes bearing N-octyl (C, Fig. 1) or N-aryl (D, Fig. 1) imine moieties afford trans-1,4 or cis-1,4 polyisoprene, respectively.14 Subsequently, Chen et al. demonstrated alkyl and disubstituted aryl variants of iminopyridine iron complexes for isoprene polymerization, providing high molecular weight polyisoprenes predominantly consisting of cis-1,4 units.15 More recently, Ricci and coworkers modified these catalysts with mono-substituted N-aryl units and achieved polyisoprenes with a preference for cis-1,4 and 3,4 alternating microstructures, in which cis-1,4 structures were in a sequence of 3 or 5 units.8d,16 Wang et al. examined aminopyridine iron precatalysts in which the N-aryl group was extensively modified with various alkyl and aryl substituents, aiming to fine-tune the monomer enchainment (E, Fig. 1). These iron precatalysts notably favored the addition of the 3,4-unit monomer, accounting for approximately 50% of the product, while displaying high activity levels reaching up to 1.9 × 106 gPI (molFe, h)−1.12e Visseaux and coworkers incorporated the methyl group at the imine carbon (F, Fig. 1), resulting in high polymerization activities (>2.1 × 107 gPI (molFe, h)−1), tunable regioselectivity (up to 1,4/3,4: 9/1), and stereoselectivity of polyisoprene (up to cis-1,4/trans-1,4: >99/0).17 In recent research, our group introduced a novel class of π-conjugated naphthalenyl-substituted iminopyridine iron precatalysts, which exhibited remarkable catalytic activity, reaching up to 5.5 × 105 gPI (molFe, h)−1, and high molecular weight polyisoprenes with tunable selectivity (G, Fig. 1).18 Fusion of the carbocyclic ring at C2 and C3 carbon of pyridine exhibited dramatic effects on the polymerization behaviour, significantly enhancing activities and thermal stability (H and I, Fig. 1). For instance, incorporation of cyclohexyl significantly improved the activities up to 1.0 × 108 gPI (molFe, h)−1 with high thermal stability and produced high molecular weight polyisoprenes with tunable stereochemistry (H, Fig. 1).19 These outcomes are even more pronounced when transitioning to larger carbocyclic rings, such as cycloheptyl (I, Fig. 1). In this case, the polymerization activity experiences a notable boost, reaching 1.2 × 108 gPI (molFe, h)−1 and the resulting polyisoprenes consistently maintain high molecular weights (typically 105 g mol−1) across all reaction temperatures within the range of 25 °C to 100 °C.20 These exciting results highlight the potential of iron catalysts to replace classical titanium-based and rare earth metal catalysts for isoprene polymerization. However, it is essential to simultaneously improve polymerization activities, selectivity, and polymer molecular weights and control dispersity under conditions critical for industrial applications.
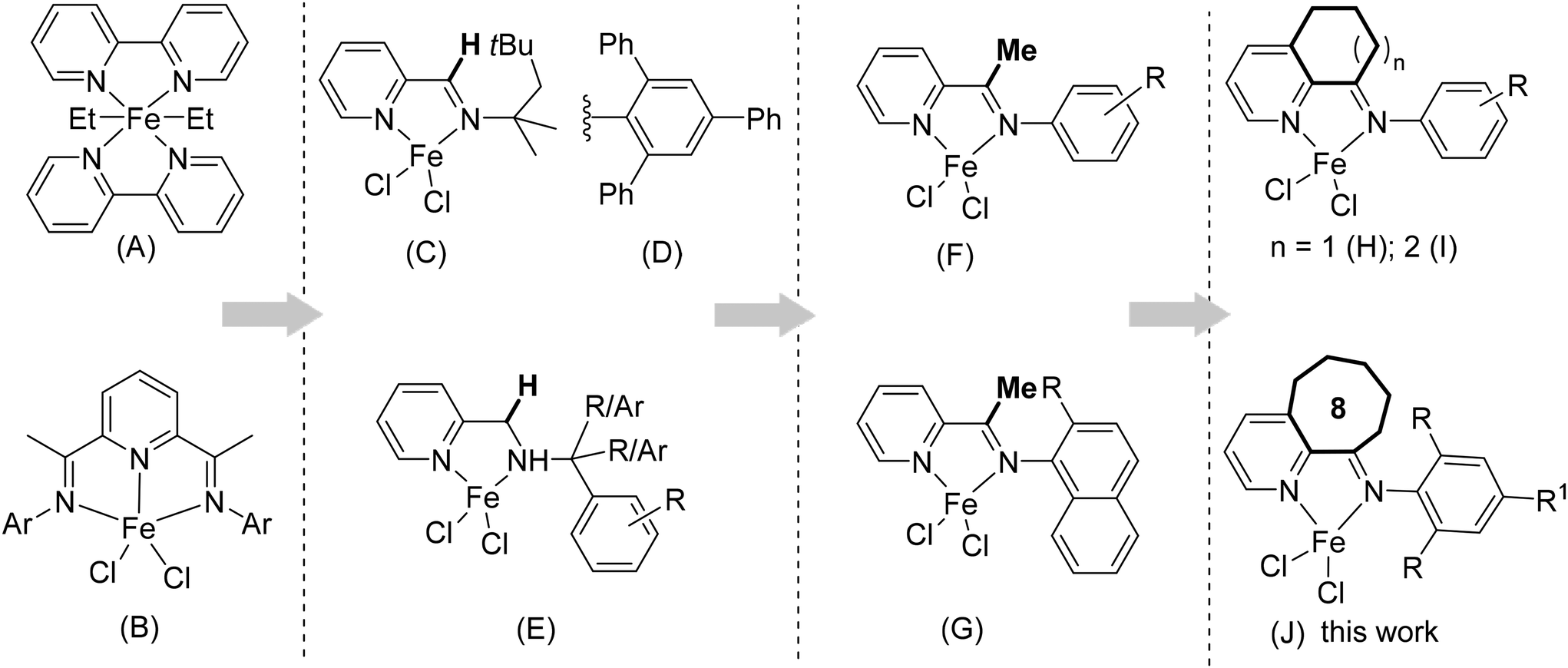 | ||
| Fig. 1 Structural variations in iminopyridine iron precatalysts for isoprene polymerization. | ||
Motivated by the results of incorporating carbocyclic rings into iminopyridine iron catalysts for ethylene polymerization, our objective was to investigate the effects of using even larger rings on the catalytic performance of iron catalysts in isoprene polymerization.21 Herein, we synthesized a series of cyclooctyl-fused iminopyridine iron precatalysts specifically designed for isoprene polymerization (J, Fig. 1) and systematically investigated the influence of various reaction conditions (cocatalyst type and amount, temperature, reaction time, catalyst and monomer quantities) and the impact of the ligand structure on the polymerization performance. The results revealed that, besides the key role of the catalyst structure in determining the polymerization activities, the monomer enchainment (predominantly cis-1,4/3,4) in the produced polyisoprene could be tuned by adjusting the steric substituents and reaction temperature. Furthermore, the high thermal stability and prolonged life of active species were clearly evident from parallel reactions conducted over a wide range of temperatures and run times. To gain a thorough understanding and characterization of the prepared complexes and the resulting polymer, a comprehensive analysis using various methods and techniques was performed.
Results and discussion
Synthesis and characterization of iron complexes
The iron complexes [L2FeCl][FeCl4], where L = 10-(alkylphenylimino)-5,6,7,8,9-pentahydrocycloocta[b]pyridine [alkyl = 2,6-dimethyl (Fe2Me), 2,6-diethyl (Fe2Et), 2,6-isopropyl (Fe2iPr), 2,4,6-trimethyl (Fe3Me), 2,6-diethyl-4-methyl (Fe2Et,Me)], and [2L3Fe][Cl][3FeCl4] where L = 10-(phenylimino)-5,6,7,8,9-pentahydrocycloocta[b] pyridine (FeH), were prepared using a straightforward one-pot template method, eliminating the need for tedious synthesis and purification of ligands, as depicted in Scheme 1.2d,18,19,22 The synthesis involved the reaction of 5,6,7,8,9-pentahydrocycloocta[b]pyridine-10-one, iron chloride, and aniline in acetic acid at boiling temperature, affording the corresponding iron complexes in high yields. Characterization of these compounds revealed an unexpected oxidation–reduction reaction, leading to ion pairs: [L2FeCl][FeCl4] and [2L3Fe][Cl][3FeCl4], where L represents the ligand structure.15,18,23 FTIR analysis, elemental analysis, and single crystal X-ray diffraction analysis verified the structures, as shown in Scheme 1. The cation of the FeH complex is trisligated, while the cations of all other complexes are bisligated. Their FTIR spectra showed the absorption of the imine functional group in the range of 1635–1621 cm−1 which are slightly lower in value than those reported for the ligands in the literature.21b Meanwhile, the results of elemental analysis data are also supportive of their structures. Similar oxidation reactions have been previously reported for iron complexes.15,18,23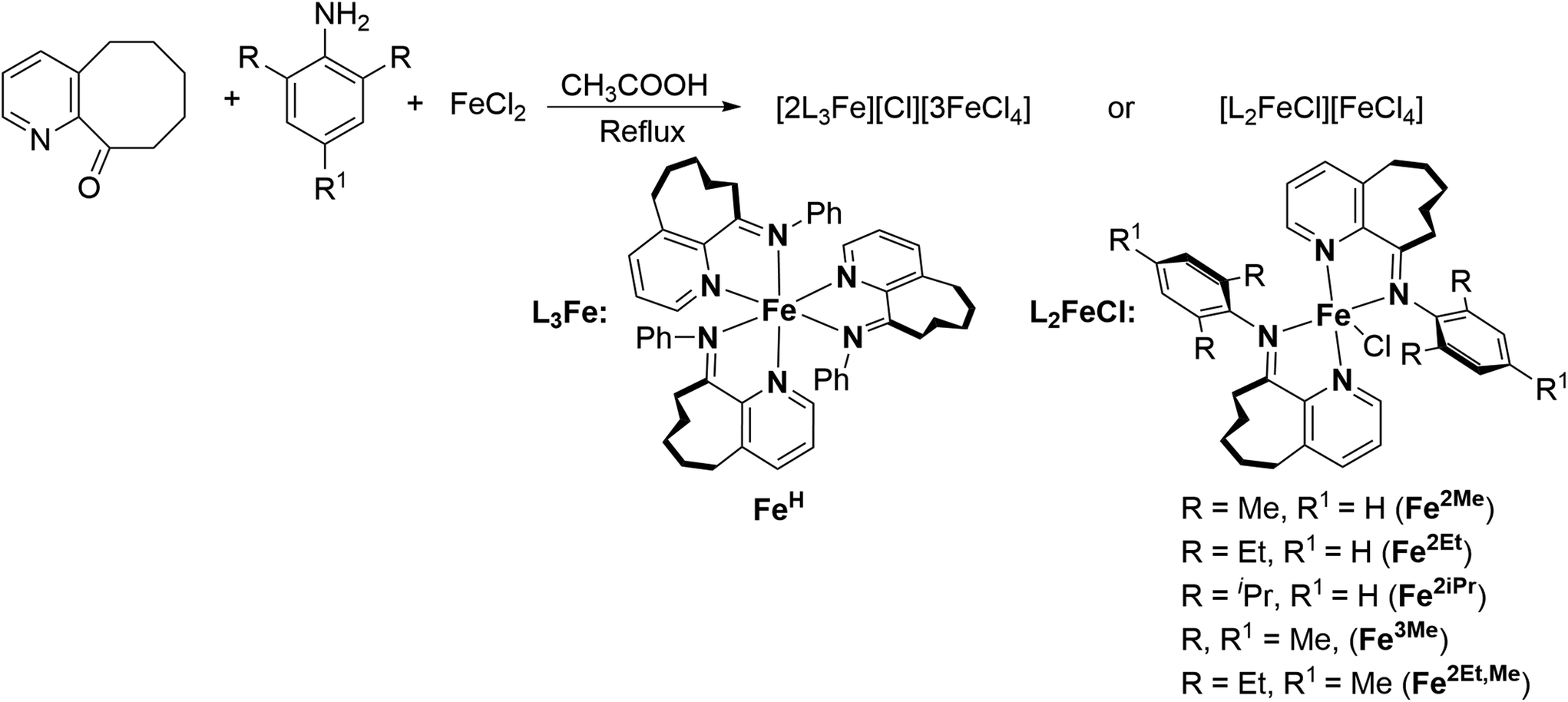 | ||
| Scheme 1 One-pot synthesis of Fe complexes using acetic acid as solvent at boiling temperature. | ||
Single crystals of FeH, Fe2Me and Fe2Et,Me, suitable for X-ray determinations, were grown through the slow diffusion of n-hexane into a solution of the corresponding complexes in dichloromethane at room temperature. The molecular structures of these complexes demonstrated an ion pair configuration, with FeH featuring a trisligated iron cation and Fe2Me and Fe2Et,Me exhibiting bisligated iron chloride cations. Perspective views of these three complexes are given in Fig. 2 and 3, respectively, along with key bond lengths and angles in the figure captions.
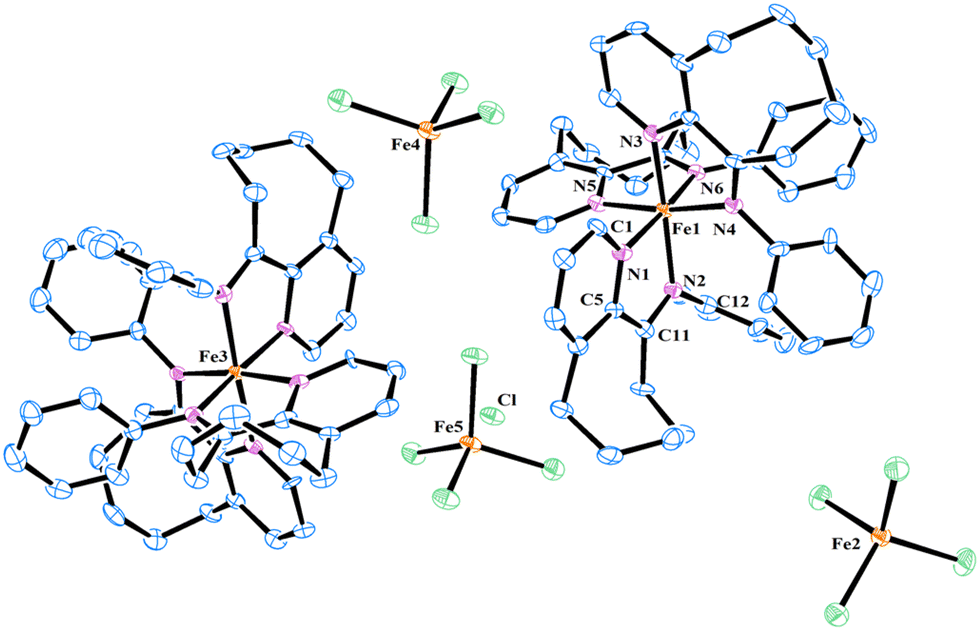 | ||
| Fig. 2 Molecular structure of FeH with thermal ellipsoids shown at a 30% probability level. All hydrogen atoms are omitted for clarity. The iron center (Fe2) exhibited disorder, which is omitted for clarity. Selected bond lengths (Å): Fe1–N1 1.969(7), Fe1–N2 1.991(6), Fe1–N3 1.969(7), Fe1–N4 1.991(6), Fe1–N5 1.969(7), Fe1–N6 1.991(6), N1–C1 1.368(10), N1–C5 1.365(11), N2–C11 1.280(11), and N2–C12 1.440(9) and bond angles (°): N1–Fe1–N2 78.9(3), N3–Fe1–N4 78.9(3), N5–Fe1–N6 78.9(3), N1–Fe1–N4 89.7(3), N2–Fe1–N5 89.7(3), N3–Fe1–N6 89.7(3), N2–Fe1–N4 98.5(3), N2–Fe1–N6, 98.5(3), N4–Fe1–N6, 98.5(3), N2–Fe1–N3, 171.7(3), N4–Fe1–N5, 171.7(3), and N1–Fe1–N6, 171.7(3). | ||
 | ||
| Fig. 3 Molecular structure of Fe2Me (a) and Fe2Et,Me (b) with thermal ellipsoids shown at a 30% probability level. All hydrogen atoms are omitted for clarity. Ethyl groups of Fe2Et,Me exhibited disorder, which are omitted for clarity. Selected bond lengths (Å) for Fe2Me: Fe1–N1 2.182(3), Fe1–N2 2.125(3), Fe1–N3 2.183(3), Fe1–N4 2.126(3), Fe1–Cl1 2.2574(10), N1–C1 1.332(4), N1–C5 1.354(4), N2–C11 1.285(4), and N2–C12 1.446(4) and bond angles (°): N2–Fe1–Cl1 119.36(8), N2–Fe1–N4 119.85(10), N4–Fe1–Cl1 120.78(8), N1–Fe1–Cl1 98.85(8), N3–Fe1–Cl1 98.13(8), N1–Fe1–N2 73.68(10), N3–Fe1–N4 73.86(10), and N1–Fe1–N3 163.01(10). Selected bond lengths (Å) for Fe2Et,Me: Fe1–N1 2.193(3), Fe1–N2 2.141(3), Fe1–N3 2.183(3), Fe1–N4 2.140(3), Fe1–Cl1 2.2536(10), N1–C1 1.335(4), N1–C5 1.352(4), N2–C11 1.287(4), and N2–C12 1.442(4) and bond angles (°): N2–Fe1–Cl1 116.05(8), N2–Fe1–N4 124.95(11), N4–Fe1–Cl1 119.00(9), N1–Fe1–Cl1 101.90(8), N3–Fe1–Cl1 103.83(8), N1–Fe1–N2 72.97(10), N3–Fe1–N4 72.83(11), and N1–Fe1–N3 154.27(11). | ||
In the molecular structure of the FeH cation, six nitrogen atoms originating from three ligands form a geometry that can be best described as an octahedron around the iron center, while the iron tetrachloride counterion adopts a typical tetrahedral geometry with characteristic bond lengths and angles. The bond distance between Npyridine and Fe is slightly shorter than the corresponding Fe–Nimine bond distance [Fe1–N1 = 1.969(7) Å vs. Fe1–N2 = 1.991(6) Å], indicating a stronger coordination of Npyridine compared to Nimine with the metal center. A similar discrepancy in the bond lengths is observed for other ligands chelated with the metal center. The bite angles N1–Fe1–N2, N3–Fe1–N4, and N5–Fe1–N6 all measure 78.9°, which are significantly smaller than other corresponding angles.23 Moreover, the N-phenyl ring in the ligand framework is oriented almost perpendicularly to the chelate ring plane.
In contrast to the FeH cation, the coordination spheres of Fe2Me and Fe2Et,Me cations adopt a geometry which can be best described as distorted trigonal bipyramidal around the iron center, characterized by bond lengths and angles typically found in bidentate iron complexes used for olefin polymerization. In the molecular structure of Fe2Me, Cl1 and nitrogen atoms from the imine functionality (N2, N4) form a basal plane around the iron center with angles approximately close to 120° [N2–Fe1–Cl1 (119.4), N4–Fe1–Cl1 (120.8) and N2–Fe1–N4 (119.8)], while the N1 and N3 atoms occupy the axial positions of this basal plane with moderate distortion from 180°. Moreover, the N-aryl rings in the ligand framework are oriented almost perpendicularly to the plane of the chelate ring. The N1–Fe1–N2 and N3–Fe1–N4 bite angles are similar [73.68° and 73.86°, respectively], but are considerably smaller than the other four angles in the chelate ring. There are modest variations in the bond lengths. The bond distance between Nimine and the iron metal is slightly smaller than that of Npyridine and the metal bond distance [Fe1–N2 = 2.125(3) Å vs. Fe1–N1 = 2.182(3) Å], indicating a stronger coordination of Nimine compared to Npyridine with the metal center. Similar structural features are noticed for Fe2Et,Me and also consistent with the previous reports.15,18,22a,23
Isoprene polymerization
| Entry | Cocat. | Con.b (%) | Act.c |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d (g mol−1) d (g mol−1) |
Đ | Microstructuree (mol%) | ||
|---|---|---|---|---|---|---|---|---|
| cis-1,4 | trans-1,4 | 1,4/3,4 | ||||||
| a General conditions: FeH amount: 5 μmol, isoprene (Ip): 20 mmol, Ip/Fe: 4000, toluene: 5 mL, cocatalyst: MAO, Al/Fe ratio: 100, reaction temperature: 25 °C, reaction time: 1 h. b Isolated yield. c 105 gPI (molFe, h)−1. d M n: 105 g mol−1; determined by GPC. e Determined by 1H and 13C NMR spectra. | ||||||||
| 1 | AlMe2Cl | Trace | — | — | — | — | — | — |
| 2 | AlEt2Cl | Trace | — | — | — | — | — | — |
| 4 | MAO | >99 | 2.74 | 3.62 | 2.23 | >51 | <1 | 52/48 |
| Entry | Al/Fe | Temp. (°C) | Time (min) | Conv.b (%) | Act.c |
M
nd (105) |
Đ | Microstructuree (mol%) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| cis-1,4 | trans-1,4 | 1,4/3,4 | ||||||||
| a General conditions: FeH amount: 5 μmol, isoprene (Ip): 20 mmol, MAO, Ip/Fe: 4000, toluene: 5 mL. b Isolated yield. c 105 gPI (molFe, h)−1. d M n: 105 g mol−1; determined by GPC. e Determined by 1H and 13C NMR spectra. | ||||||||||
| 1 | 100 | 25 | 60 | >99 | 2.74 | 3.62 | 2.23 | >51 | <1 | 52/48 |
| 2 | 40 | 25 | 60 | 95 | 2.60 | 3.88 | 2.16 | >49 | <1 | 50/50 |
| 3 | 20 | 25 | 60 | 86 | 2.36 | 3.00 | 1.97 | >48 | <1 | 49/51 |
| 4 | 100 | −20 | 60 | 69 | 1.90 | 2.01 | 2.07 | >8 | <1 | 09/91 |
| 5 | 100 | 0 | 60 | 95 | 2.60 | 2.28 | 2.29 | >14 | <1 | 15/85 |
| 6 | 100 | 40 | 60 | >99 | 2.74 | 2.71 | 2.07 | >50 | <1 | 51/49 |
| 7 | 100 | 60 | 60 | 96 | 2.64 | 2.15 | 2.60 | >51 | <1 | 52/48 |
| 8 | 100 | 80 | 60 | 82 | 2.24 | 1.97 | 2.94 | >51 | <1 | 52/48 |
| 9 | 100 | 100 | 60 | 66 | 1.80 | 0.78 | 3.67 | 50 | 4 | 54/46 |
| 10 | 100 | 25 | 45 | >99 | 3.66 | 2.92 | 2.55 | >51 | <1 | 52/48 |
| 11 | 100 | 25 | 30 | >99 | 5.48 | 2.62 | 3.19 | >50 | <1 | 51/49 |
| 12 | 100 | 25 | 15 | 87 | 9.52 | 2.46 | 2.90 | >49 | <1 | 50/50 |
| 13 | 100 | 25 | 5 | 69 | 23.8 | 2.33 | 2.22 | >49 | <1 | 50/50 |
| 14 | 100 | 25 | 2 | 48 | 39.6 | 2.19 | 2.57 | >48 | <1 | 49/51 |
| Entry | Cat. | Conv.b (%) | Act.c |
M
nd |
Đ | Microsctructurese (mol%) | ||
|---|---|---|---|---|---|---|---|---|
| cis-1,4 | trans-1,4 | 1,4/3,4 | ||||||
| a General conditions: iron precatalyst amount: 5 μmol, isoprene (Ip): 20 mmol, Ip/Fe: 4000, toluene: 5 mL, cocatalyst: MAO, Al/Fe ratio: 100, temperature: 25 °C, time: 30 min. b Isolated yield. c 105 gPI (molFe, h)−1. d M n: 105 g mol−1; determined by GPC. e Determined by 1H and 13C NMR spectra. | ||||||||
| 1 | FeH | >99 | 5.48 | 2.67 | 3.22 | >50 | <1 | 51/49 |
| 2 | Fe2Me | 72 | 3.92 | 1.39 | 3.57 | >57 | <1 | 58/42 |
| 3 | Fe2Et | 61 | 3.36 | 0.45 | 2.19 | 55 | 5 | 60/40 |
| 4 | Fe2iPr | 50 | 2.76 | 0.37 | 3.20 | 55 | 8 | 63/37 |
| 5 | Fe3Me | 70 | 3.84 | 0.88 | 2.61 | 51 | 14 | 65/35 |
| 6 | Fe2Et,Me | 58 | 3.20 | 0.43 | 4.28 | 48 | 21 | 69/31 |
Besides the cocatalyst nature, activity and molecular weights also relied on the changes in the cocatalyst amount employed. Monomer conversion and polymerization activity decreased as the Al/Fe ratio reduced from 100. Despite this decline, the activity remained notably high, even at a very low Al/Fe ratio of 20, yielding an 86% conversion with an activity of 2.36 × 105 gPI (molFe, h)−1. Remarkably, the complex FeH achieved full monomer conversion with the highest activity of 2.74 × 105 gPI (molFe, h)−1 at an Al/Fe ratio of 100 (Table 2, entry 1).12a,17,18,24 It is noteworthy that polymer molecular weights exhibited minimal variation at Al/Fe ratios of 100 or 40, suggesting negligible occurrence of chain transfer reactions to aluminium species at these cocatalyst amounts. This phenomenon is further reflected by the narrow polymer molecular weight distributions. In contrast, changing the Al/Fe ratio from 20 to 40 resulted in an increase in polymer molecular weight from 3.0 × 105 g mol−1 to 3.88 × 105 g mol−1.2d,12e Likely, chain propagation reactions were more rapid compared to chain transfer reactions, given the enhanced monomer conversion resulting from an increase in the quantity of the cocatalyst. Regarding the microstructure of the polymer, there is no significant effect of variations in the cocatalyst amount on the mode of the monomer coordination–insertion reaction as the resultant polyisoprene contained almost equal proportions of cis-1,4 and 3,4 units, in the range of 48–51% and 49–52%, respectively. Notably, similar observations have been reported previously in the literature.2d,25
As shown in Table 2, entries 1 and 4–9, the reaction temperature exerts a significant impact on monomer conversion, catalytic activity, and various polymer properties such as the molecular weight, dispersity, and microstructure. Gradually elevating the reaction temperature from −20 °C to 0 °C, 25 °C, and 40 °C resulted in a progressive improvement in both monomer conversion and activity. However, further increases in temperature resulted in a consistent decrease in yield and activity (Fig. 4a). Complete conversion with the highest activity of 2.74 × 105 gPI (molFe, h)−1 was achieved at 25 °C or 40 °C. At 80 °C and 100 °C, comparatively lower activity levels of 2.24 × 105 gPI (molFe, h)−1 and 1.8 × 105 gPI (molFe, h)−1 were obtained, indicating partial decomposition of active species. Despite the gradual decrease in activity with increasing reaction temperature, the FeH/MAO system still exhibited good activity at elevated temperatures, demonstrating high thermal stability of the active species.9 As shown in Fig. 4b, polymer molecular weights showed a gradual decrease with rising reaction temperature. The highest polymer molecular weight of 3.62 × 105 g mol−1 was observed at 25 °C, which gradually declined to 0.78 × 105 g mol−1 at 100 °C. This suggests that higher temperatures facilitate chain transfer reactions relative to chain propagation, resulting in reduced polymer molecular weights.8c,12a,25 The dispersity of polymer molecular weights became relatively broad when the temperature increased from 60 °C to 100 °C (Đ = 2.60–3.67), also indicating partial decomposition of active species at higher temperatures. Furthermore, variations in the reaction temperature significantly influenced the regioselectivity of monomer addition in the polymer chain. Lower temperatures favored 3,4 addition; for instance, polymerization at −20 °C resulted in 91% regioselectivity for 3,4 addition, decreasing to 85% at 0 °C and further to 48% at 20 °C (Table 2, entries 1, 4 and 5). However, increasing the temperature beyond this range had a negligible impact on regioselectivity. Additionally, across the temperature range (−20 to 100 °C), the stereoselectivity remained largely consistent, favoring cis-1,4 addition over trans-1,4 addition. This observation suggests that higher temperature induced rotations of the N-aryl unit could alter the steric environment around the active species, potentially modifying the coordination mode of monomer insertion from 3,4 to trans-1,4.25b,c
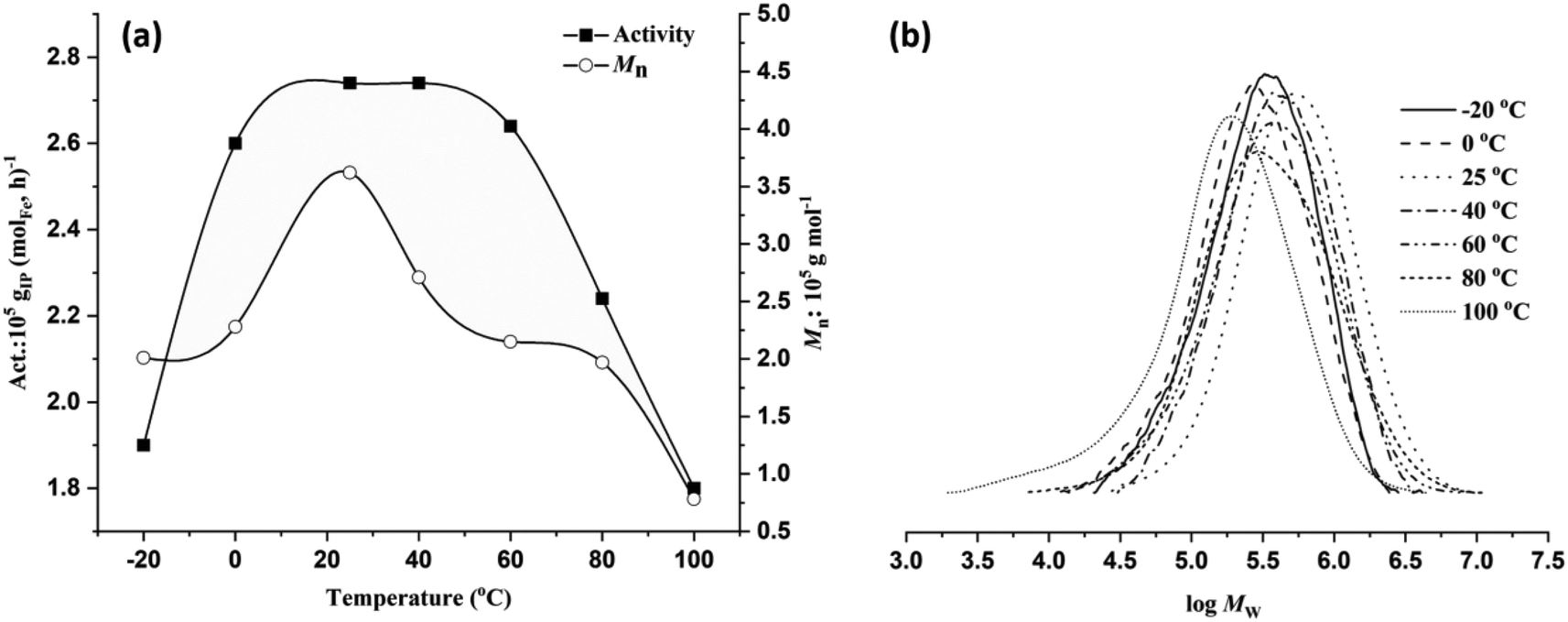 | ||
| Fig. 4 Activity and Mnversus temperature (a) and GPC curves at different reaction temperatures (b). | ||
The catalytic performance of FeH/MAO was further explored at shorter reaction run times such as 45, 30, 15, 5, or 2 minutes, while maintaining a constant temperature of 25 °C (Table 2, entries 10–14). This investigation aimed to determine the minimum time required to achieve complete monomer conversion. Apparently, complete conversion was attained within 30 min, but as the reaction time decreased beyond that point, the conversion gradually decreased (87%, 69%, and 48% for 15, 5, and 2 min, respectively), revealing that optimal run time of 30 min was necessary to achieve full conversion for an Ip/Fe ratio of 4000. The calculated polymerization activity increased as the reaction time was decreased, reaching a maximum activity of 3.96 × 106 gPI (molFe, h)−1 with a 2 min reaction time, highlighting a short incubation period for the cocatalyst to activate FeH.18,26 The number-average molecular weights of polyisoprene exhibited a gradual increase with extended reaction times but exhibited a significant deviation from linear behavior after 30 minutes. The dispersity remained narrow across different reaction times (Đ = 2.22–3.19).18,26 There was no significant change in the selectivity of monomer insertion at varying reaction times, with nearly equal proportions of 1,4 and 3,4 monomer additions (1,4/3,4 ≈ 50/50). The 1H and 13C NMR spectra of representative polyisoprene obtained over a run time of 30 min are presented in Fig. 5. Furthermore, there was a strong preference for cis-1,4 structures over trans-1,4 structures in the resulting polyisoprenes.18
 | ||
| Fig. 5 1H and 13C NMR spectra of the representative sample of polyisoprene obtained using the FeH/MAO system (Table 2, entry 11). | ||
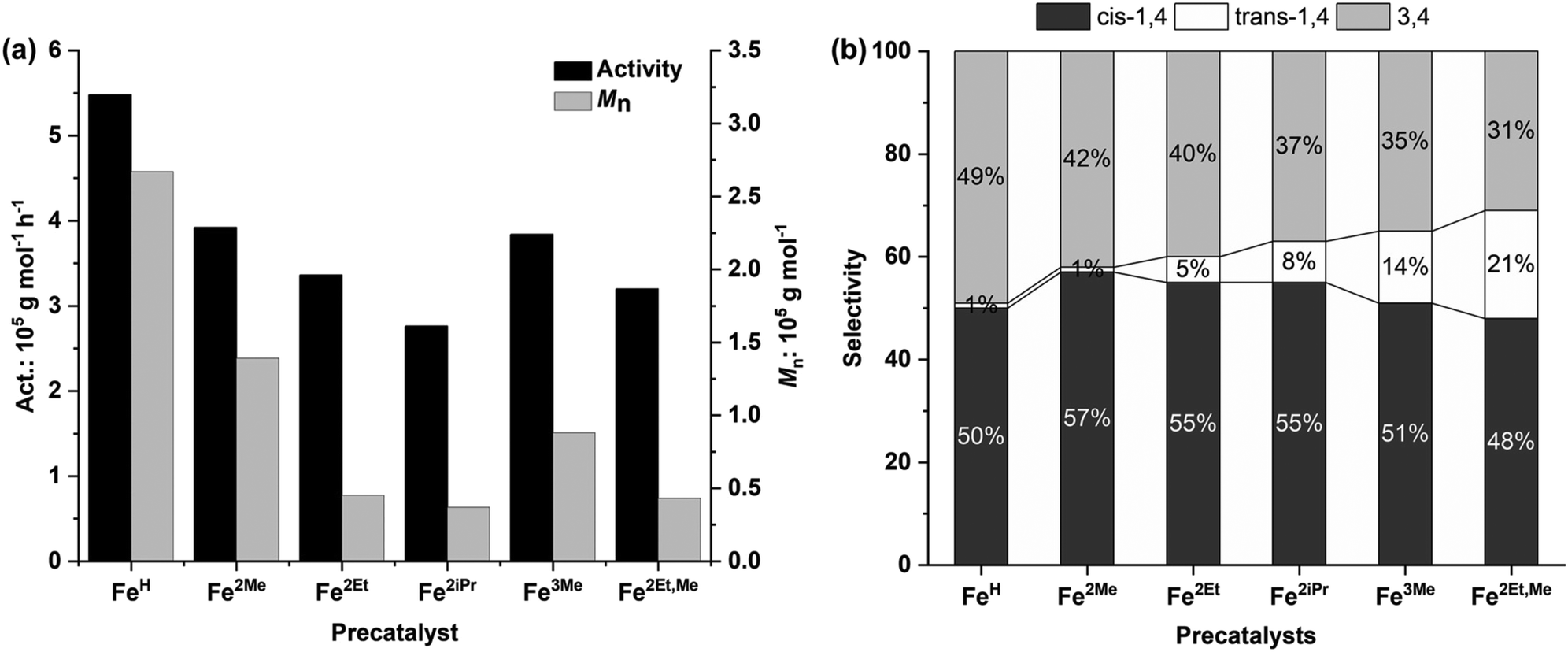 | ||
| Fig. 6 Polymerization activity and number average molecular weight (a) and selectivity (b) versus the structure of precatalysts. | ||
Additionally, the electron-donating effect of the R1 substituent at the para position of the aniline resulted in a slight decline in monomer conversion and activity. This decrease can be attributed to the electron-donating nature of R1, which decreased the Lewis acidic character of the metal and, in turn, reduced its ability to coordinate with the monomer. Consequently, Fe3Me and Fe2Et,Me displayed lower polymerization activity compared to their analogue complexes, Fe2Me and Fe2Et.2d,12a The rate of polymerization, which depends on the ligand structure, reveals that the cations of these iron complexes, after reacting with MAO, yield active species with single-site behaviour, as reflected in the narrow and unimodal molecular weight distributions of the resulting polymer. On the other hand, it is likely that the iron center of the anion, after reacting with MAO, remains inactive or intact during the initiation of polymerization.
With regard to the molecular weight of the resulting polymer, the steric hindrance induced by the R substituent of the aniline leads to a reduction in the molecular weights of the resulting polyisoprenes (Fig. 6a). FeH without steric hindrance at the ortho position of aniline produced a polyisoprene with the highest molecular weight, up to 2.67 × 105 g mol−1. Fe2Me with lower steric hindrance yielded a polyisoprene with the second highest molecular weight, while Fe2iPr, with significant steric hindrance resulted in a polyisoprene with the lowest molecular weight. These findings suggest that large steric groups hinder monomer coordination–insertion, leading to more pronounced chain transfer/termination reactions. The dispersity of polymer molecular weights ranged from unimodal to broad (Đ = 2.19–4.28), mainly depending on the steric hindrance of the R substituents of the aniline.12g,18
The influence of the precatalyst structure on polymerization selectivity was also observed. According to the results from 1H and 13C NMR spectra (Fig. 5 and 7, S14–S17†), the prepared Fe complexes exhibited a preference for 1,4 additions over 3,4 additions, in the range of 51–69%, (Fig. 6b), primarily influenced by the steric hindrance of the R substituent in the precatalyst structure (Table 3, entries 1–6). The increase in steric hindrance induced by the ortho substituents promotes 1,4 additions. Specifically, this rise in 1,4 units corresponds to an increase in the trans-1,4/cis-1,4 ratio, contrasting with the sterically less bulky FeH complex, which produces nearly no trans-1,4 units. Thus, polyisoprenes derived from Fe2Et,Me exhibited a regioselectivity of up to 69% for 1,4 addition, including 21% trans-1,4 units.12g,15,19 In summary, these catalytic systems tend to polymerize isoprene with high polymerization activity, high polymer molecular weights, moderate regioselectivity, and high stereoselectivity.
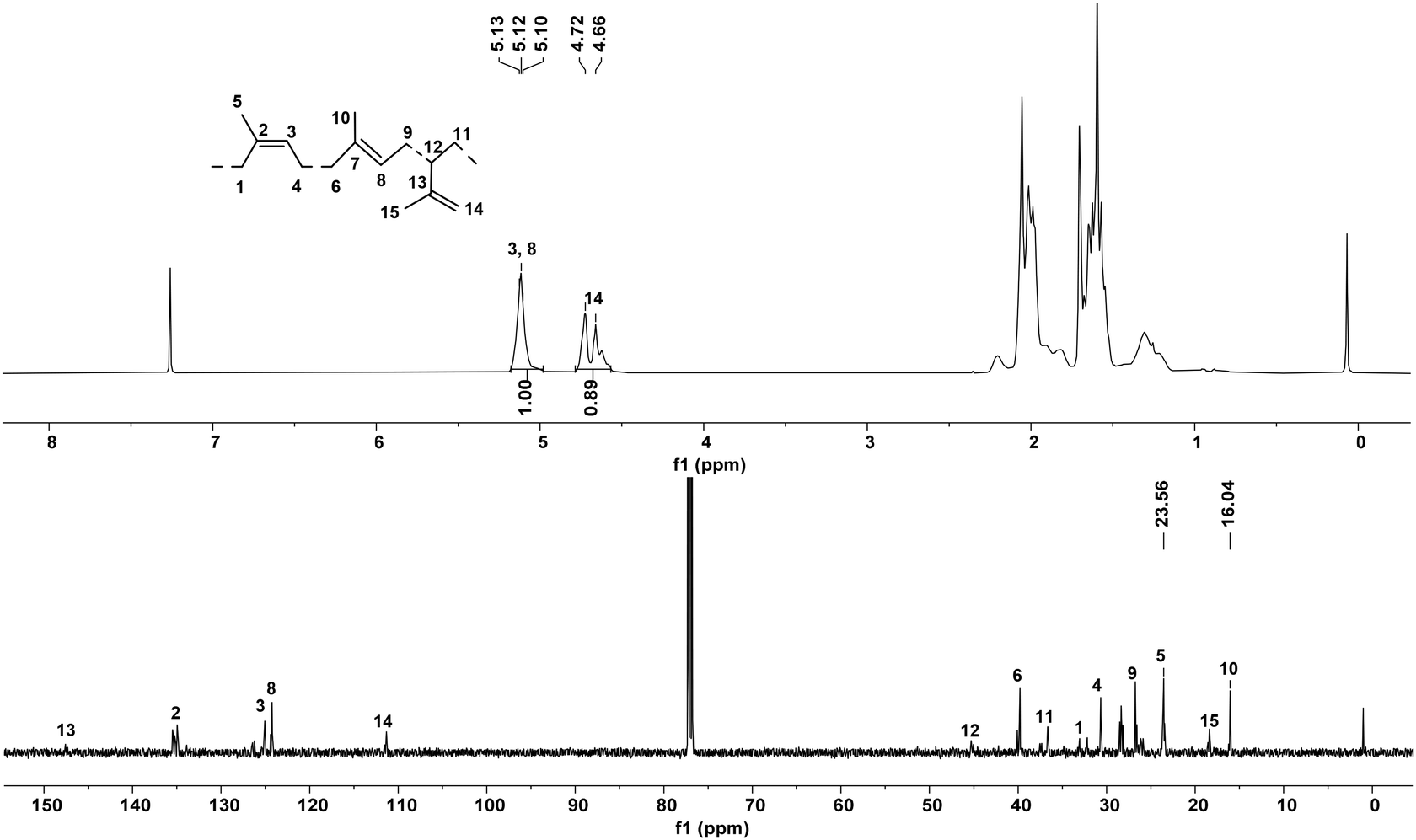 | ||
| Fig. 7 1H and 13C NMR spectra of the representative sample of polyisoprene obtained using the Fe2Et,Me/MAO system (Table 3, entry 6). | ||
Impact of reducing precatalyst loading on isoprene polymerization
FeH, at a concentration of 5 μmol, exhibited high polymerization activity, achieving complete monomer conversion within 30 min for an Ip/Fe ratio of 4000. To explore the impact of precatalyst quantity on isoprene polymerization, polymerization tests were conducted using reduced amounts of precatalyst, while maintaining a fixed quantity of MAO and isoprene (2 mL) at room temperature. As shown in Table 4, polymerization activity increased as the catalyst amount was decreased. Despite the decrease in conversions, high to excellent conversions were still achieved, along with high polymerization activities. Remarkably, even at a precatalyst loading of 0.0025 mol%, which is 20 times lower than the initially used catalyst loading (5 μmol), polymerization proceeded smoothly with a conversion of 73% and an excellent activity of 4.0 × 106 gPI (molFe, h)−1, highlighting promising efficiency of the iron catalyst for isoprene polymerization.9 The decrease in the catalyst loading demonstrated negligible impact on polymer molecular weights, with only a slight improvement observed, reaching up to 2.71 × 105 g mol−1. This is likely due to the gradual reduction in monomer conversion. On the other hand, the dispersity gradually decreased to a narrow value of 1.53 (Table 4, entry 5).12g There was no significant impact of precatalyst quantity on selectivity, with similar monomer additions (cis-1,4/3,4: 1/1) observed at different precatalyst amounts (Table 4).| Entry | Ip/Fe | Con.b (%) | Act.c |
M
nd (g mol−1) |
Đ | Microstructuree (mol%) | ||
|---|---|---|---|---|---|---|---|---|
| cis-1,4 | trans-1,4 | 1,4/3,4 | ||||||
| a General conditions: precatalyst: FeH, isoprene (Ip): 20 mmol, toluene: 5 mL, cocatalyst: MAO, Al/Fe ratio: 100, temperature: 25 °C, time: 30 min. b Isolated yield. c 105 gPI (molFe, h)−1. d M n: 105 g mol−1; determined by GPC. e Determined by 1H and 13C NMR spectra. | ||||||||
| 1 | 4000 | >99 | 5.48 | 2.62 | 3.19 | >50 | <1 | 51/49 |
| 2 | 8000 | 95 | 10.4 | 2.67 | 3.22 | >50 | <1 | 51/49 |
| 3 | 16000 |
89 | 19.5 | 2.36 | 1.95 | >49 | <1 | 50/50 |
| 4 | 20000 |
87 | 24.0 | 2.71 | 1.69 | >49 | <1 | 50/50 |
| 5 | 40000 |
73 | 40.0 | 2.63 | 1.53 | >49 | <1 | 50/50 |
Comparison with previously reported iron complexes
In the class of iminopyridine Fe and Co precatalysts investigated for isoprene polymerization, Fe precatalysts bearing N-octyl or N-aryl imine groups were particularly effective in controlling the selectivity of monomer additions. The resulting polyisoprene displayed 1,4 and 3,4 structures in the range of 2:1 to 12:1, with complete cis or trans selectivity for 1,4 structures (C and D, Fig. 1).14 The alkyl and disubstituted aryl variants of these Fe precatalysts exhibited similar catalytic performance in terms of activity (up to 8.3 × 104 gPI (molFe, h)−1 and polymer properties. The aminopyridine iron analogues (E, Fig. 1) also exhibited high activities, reaching up to 1.9 × 106 gPI (molFe, h)−1 with a significant preference for 3,4 enchainment (approximately 50%).12e The incorporation of a methyl group at the imine carbon in these Fe precatalysts had a significant positive effect on improving activity (up to 2.0 × 107 gPI (mol Fe, h)−1) along with high monomer stereoselectivity (cis-1,4/trans-1,4 = 100) (F and G, Fig. 1).8d,16,18 Cyclohexyl-fused iminopyridine Fe precatalysts further enhanced polymerization activity, reaching a peak of 1.0 × 108 gPI (molFe, h)−1, while maintaining high thermal stability and tunable 1,4-cis/trans stereoslectivity from <1/99 to 96/4 (H, Fig. 1).19 These iron catalysts with larger carbocyclic rings, such as cycloheptyl (I, Fig. 1) exhibited even higher activity, reaching 1.2 × 108 gPI (molFe, h)−1, and produced polyisoprenes with consistently high molecular weights.20 Similarly, the title Fe complexes bearing a fused cyclooctyl ring were highly active, providing activity in the level of 105 to 106 gPI (mol Fe, h)−1 with the use of very small amounts of precatalyst and cocatalyst (J, Fig. 1). The resulting polyisoprene was mainly composed of cis-1,4 structures along with a variable amount of 3,4 units in the range of 31–49%. Moreover, the confinement of the iminopyridine ligand framework with the incorporation of the cyclooctyl ring also improved thermal stability, enabling smooth polymerization at elevated temperatures (up to 100 °C). In comparison to iminopyridine cobalt precatalysts, the prepared iron complexes exhibited significantly higher polymerization activity and produced high molecular weight polyisoprenes. Overall, the prepared iron complexes proved to be highly active precatalysts for the synthesis of high molecular weight polyisoprenes.
Experimental section
Synthesis and characterization of Fe complexes
Conclusion
In summary, a novel series of iron complexes in the form of ion pairs [2L3Fe][Cl][3FeCl4] and [L2FeCl][FeCl4] was efficiently prepared using a simple, cost-effective, one-pot method and comprehensively characterized through various analytical techniques, including FTIR, elemental composition, and single crystal X-ray diffraction. Upon activation with methylaluminoxane, these iron complexes not only exhibited high activity but also generated high molecular weight polyisoprenes. Systematic variations in the steric hindrance of ortho-substituents on the ligands revealed that bulkier groups had a detrimental effect on the polymerization activity and polymer molecular weight. The precatalyst with the least steric hindrance displayed the highest activity, achieving 2.74 × 105 gPI (molFe, h)−1 and complete conversion of 4000 equiv. of monomer in just 30 min. Even at a very low precatalyst loading of 0.0025 mol%, polymerization proceeded smoothly with a remarkable activity of 4.0 × 106 gPI (molFe, h)−1 and high polymer molecular weights (Mn = 2.63 × 105 g mol−1). The impact of reaction temperature, time, and cocatalyst amount was systematically explored, revealing that FeH-MAO displayed high thermal stability and consistently yielded high polymer molecular weights over a wide temperature range (−20–100 °C). All resulting polyisoprenes exhibited mixed cis-1,4/3,4 microstructures, which were significantly influenced by the ligand structure and polymerization temperature. Polymerization at lower temperatures favored 3,4 additions (up to 91%), while more sterically hindered iron precatalysts produced polyisoprenes with cis-1,4 selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the Chemistry and Chemical Engineering Guangdong Laboratory (2111018 and 2132012), and Q. M. would like to express gratitude towards the Foreign Youth Talent Program (QN2022030008L) for their support. N. Y. would like to express gratitude towards the Chinese Government Scholarship-Belt and Road Program and the ANSO Scholarship Program for their support.References
- (a) G. Ricci, A. Sommazzi, F. Masi, M. Ricci, A. Boglia and G. Leone, Coord. Chem. Rev., 2010, 254, 661–676 CrossRef CAS; (b) G. Ricci, G. Pampaloni, A. Sommazzi and F. Masi, Macromolecules, 2021, 54, 5879–5914 CrossRef CAS.
- (a) Y. Champouret, O. H. Hashmi and M. Visseaux, Coord. Chem. Rev., 2019, 390, 127–170 CrossRef CAS; (b) H. Wang, Y. Yang, M. Nishiura, Y. I. Hong, Y. Nishiyama, Y. Higaki and Z. Hou, Angew. Chem., Int. Ed., 2022, 61, e202210023 CrossRef CAS PubMed; (c) M. Zhao, L. Wang, Q. Mahmood, C. Jing, G. Zhu, X. Zhang, X. Chen and Q. Wang, J. Polym. Sci., 2020, 58, 2708–2717 CrossRef CAS; (d) N. Yousuf, Y. Ma, Q. Mahmood, W. Zhang, M. Liu, R. Yuan and W.-H. Sun, Catalysts, 2023, 13, 1120 CrossRef CAS.
- (a) L. Zhang, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2005, 127, 14562–14563 CrossRef CAS PubMed; (b) L. Zhang, T. Suzuki, Y. Luo, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2007, 46, 1909–1913 CrossRef CAS PubMed; (c) H. Liu, J. He, Z. Liu, Z. Lin, G. Du, S. Zhang and X. Li, Macromolecules, 2013, 46(9), 3257–3265 CrossRef CAS; (d) G. Zhang, B. Deng, S. Wang, Y. Wei, S. Zhou, X. Zhu, Z. Huang and X. Mu, Dalton Trans., 2016, 45, 15445 RSC; (e) Y. Ren, J. T. Miller, S. T. Polderman, T. D. Vo, A. C. M. Wallace, J. M. O. Cue, S. T. Tran, M. C. Biewer and M. C. Stefan, RSC Adv., 2019, 9, 3345 RSC.
- S. K. De and J. R. White, Rubber Technologists Handbookm, Rapra Technology Limited, Shawbury, UK, 2001, pp. 49–51 Search PubMed.
- S. Ouardad, M.-E. Bakleh, S. V. Kostjuk, F. Ganachaud, J. E. Puskas, A. Deffieux and F. Peruch, Polym. Int., 2012, 61, 149–156 CrossRef CAS.
- (a) E. Shoenberg, H. A. Marsh, S. J. Walters and W. M. saltman, Rubber Chem. Technol., 1979, 52, 526–604 CrossRef; (b) C. W. Kamienski, Ind. Eng. Chem., 1965, 57, 38–55 CrossRef CAS; (c) L. Friebe, O. Nuyken and W. Obrecht, Adv. Polym. Sci., 2006, 204, 1–154 CrossRef CAS.
- (a) J. S. Lasky, H. K. Garner and R. H. Ewart, Ind. Eng. Chem. Prod. Res. Dev., 1962, 1, 82–85 CAS; (b) M. Zhao, Q. Mahmood, C. Jing, L. Wang, G. Zhu, X. Zhang and Q. Wang, Polymers, 2019, 11, 1122 CrossRef PubMed.
- (a) A. He, G. Wang, W. Zhao, X. Jiang, W. Yao and W.-H. Sun, Polym. Int., 2013, 62, 1758–1766 CrossRef CAS; (b) G. Wang, X. Jiang, W. Zhao, W.-H. Sun, W. Yao and A. He, J. Appl. Polym. Sci., 2013, 131, 1 Search PubMed; (c) M. N. Alnajrani and F. S. Mair, Dalton Trans., 2016, 45, 10435–10446 RSC; (d) M. Scoti, F. D. Stefano, G. Zanchin, G. Leone, C. D. Rosa and G. Ricci, Macromolecules, 2023, 56, 4629–4638 CrossRef CAS.
- L. Wang, X. Wang, H. Hou, G. Zhu, Z. Han, W. Yang, X. Chen and Q. Wang, Chem. Commun., 2020, 56, 8846–8849 RSC.
- (a) H. Noguchi and S. Kambara, J. Polym. Sci., Part B: Polym. Lett., 1964, 2, 593–596 CrossRef; (b) H. E. Swift, J. E. Bozik and C. Y. Wu, J. Catal., 1970, 17, 331–340 CrossRef CAS; (c) W. L. Hsu and A. F. Halasa, Rubber Chem. Technol., 1994, 67, 82–85 CrossRef.
- (a) C. Bazzini, A. Giarrusso and L. Porri, Macromol. Rapid Commun., 2002, 23, 922–927 CrossRef CAS; (b) C. Bazzini, A. Giarrusso, L. Porri, B. Pirozzi and R. Napolitano, Polymer, 2004, 45, 2871–2275 CrossRef CAS.
- (a) M. Zhao, L. Wang, Q. Mahmood, C. Jing, G. Zhu, X. Zhang, X. Wang and Q. Wang, Appl. Organomet. Chem., 2019, 33, e4836 CrossRef; (b) M. Zhao, X. Zhang, L. Wang, L. Wang, G. Zhu and Q. Wang, Appl. Organomet. Chem., 2022, 36, e6848 CrossRef CAS; (c) M. Zhao, Y. Ma, X. Zhang, L. Wang, G. Zhu and Q. Wang, Polymers, 2022, 14, 3612 CrossRef CAS PubMed; (d) G. Zhu, X. Zhang, M. Zhao, L. Wang, C. Jing, P. Wang, X. Wang and Q. Wang, Polymers, 2018, 10, 934 CrossRef PubMed; (e) C. Jing, L. Wang, Q. Mahmood, M. Zhao, G. Zhu, X. Zhang, X. Wang and Q. Wang, Dalton Trans., 2019, 48, 7862–7874 RSC; (f) X. Zhang, G. Zhu, Q. Mahmood, M. Zhao, L. Wang, C. Jing, X. Wang and Q. Wang, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 767–775 CrossRef CAS; (g) C. Jing, L. Wang, G. Zhu, H. Hou, L. Zhou and Q. Wang, Organometallics, 2020, 39, 4019–4026 CrossRef CAS.
- (a) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS; (b) C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664–11615 CrossRef CAS; (c) B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef CAS; (d) G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 8, 849–850 RSC; (e) G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Stromberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef CAS; (f) Y. Zhang, C. Huang, X. Wang, Q. Mahmood, X. Hao, X. Hu, C. Y. Guo, G. A. Solan and W.-H. Sun, Polym. Chem., 2017, 8, 995 RSC; (g) Y. Zeng, Q. Mahmood, X. Hao and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1910–1919 CrossRef CAS; (h) Y. Zeng, Q. Mahmood, T. Liang and W.-H. Sun, New J. Chem., 2017, 41, 3653 RSC; (i) Z. Wang, G. A. Solan, W. Zhang and W.-H. Sun, Coord. Chem. Rev., 2018, 363, 92–108 CrossRef CAS; (j) Q. Mahmood, Y. Zeng, X. Wang, Y. Sun and W.-H. Sun, Dalton Trans., 2017, 46, 6934–6947 RSC; (k) Q. Mahmood, Y. Zeng, E. Yue, G. A. Solan, T. Liang and W.-H. Sun, Polym. Chem., 2017, 8, 6416–6430 RSC; (l) Z. Wang, G. A. Solan, Q. Mahmood, Q. Liu, Y. Ma, X. Hao and W.-H. Sun, Organometallics, 2018, 37, 380 CrossRef CAS; (m) J. Guo, W. Zhang, Q. Mahmood, R. Zhang, Y. Sun and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1269 CrossRef CAS; (n) Q. Mahmood, Y. Ma, X. Hao and W.-H. Sun, Appl. Organomet. Chem., 2019, e4857 Search PubMed; (o) M. Khoshsefat, Y. Ma and W.-H. Sun, Coord. Chem. Rev., 2021, 434, 213788 CrossRef CAS; (p) Q. Mahmood, J. Guo, W. Zhang, Y. Ma, T. Liang and W.-H. Sun, Organometallics, 2018, 37, 957–970 CrossRef CAS; (q) Q. Mahmood, E. Yue, J. Guo, W. Zhang, Y. Ma, X. Hao and W.-H. Sun, Polymer, 2018, 159, 124–137 CrossRef CAS; (r) Q. Mahmood, X. Li, L. Qin, L. Wang and W.-H. Sun, Dalton Trans., 2022, 51, 14375–14407 RSC; (s) M. Qasim, M. S. Bashir, S. Iqbal and Q. Mahmood, Eur. Polym. J., 2021, 160, 110783 CrossRef CAS; (t) Z. Wang, Q. Mahmood, W. Zhang and W.-H. Sun, Adv. Org. Chem., 2023, 79, 41–86 CAS; (u) L. Wang, M. Liu, Q. Mahmood, S. Yuan, X. Li, L. Qin, S. Zou, T. Liang and W.-H. Sun, Eur. Polym. J., 2023, 194, 112112 CrossRef CAS.
- J. Raynaud, J. Y. Wu and T. Ritter, Angew. Chem., Int. Ed., 2012, 51, 11805–11808 CrossRef CAS PubMed.
- L. Guo, X. Jing, S. Xiong, W. Liu, Y. Liu, Z. Liu and C. Chen, Polymers, 2016, 8, 389 CrossRef PubMed.
- G. Ricci, G. Leone, G. Zanchin, B. Palucci, A. C. Boccia, A. Sommazzi, F. Masi, S. Zacchini, M. Guelfi and G. Pampaloni, Macromolecules, 2021, 54, 9947–9959 CrossRef CAS.
- O. H. Hashmi, Y. Champouret and M. Visseaux, Molecules, 2019, 24, 3024 CrossRef CAS PubMed.
- W. Lin, L. Zhang, H. Suo, A. Vignesh, N. Yousuf, X. Hao and W.-H. Sun, New J. Chem., 2020, 44, 8076–8084 RSC.
- G. Zhu, L. Wang, Q. Mahmood, L. Zhou and Q. Wang, Polym. Test., 2021, 102, 107317 CrossRef CAS.
- R. Yuan, G. Ren, Q. Mahmood, Y. Zeng, Y. Wang, T. Liang and W.-H. Sun, Organometallics, 2023, 42, 3307–3318 CrossRef CAS.
- (a) F. Huang, Z. Sun, S. Du, E. Yue, J. Ba, X. Hu, T. Liang, G. B. Galland and W. H. Sun, Dalton Trans., 2015, 44, 14281–14292 RSC; (b) R. Zhang, Z. Wang, Z. Flisak, X. Hao, Q. Liu and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2601–2610 CrossRef CAS; (c) J. Yu, Y. Zeng, W. Huang, X. Hao and W.-H. Sun, Dalton Trans., 2011, 40, 8436–8443 RSC; (d) Y. Zhang, C. Huang, X. Wang, Q. Mahmood, X. Hao, X. Hu, C. Y. Guo, G. A. Solan and W.-H. Sun, Polym. Chem., 2017, 8, 995–1005 RSC.
- (a) J. Li, Q. Zhang, X. Hu, Y. Ma, G. A. Solan, Y. Sun and W.-H. Sun, Appl. Organomet. Chem., 2019, 34, e5254 CrossRef; (b) L. Xua, J. Li, W. Lin, Y. Ma, X. Hua, Z. Flisak and W.-H. Sun, J. Organomet. Chem., 2021, 937, 121720 CrossRef.
- (a) M. Yankey, C. Obuah, I. A. Guzei, E. Osei-Twum, G. Hearne and J. Darkwa, Dalton Trans., 2014, 43, 13913–13923 RSC; (b) M. S. Shongwe, B. A. Al-Rashdi, H. Adams, M. J. Morris, M. Mikuriya and G. R. Hearne, Inorg. Chem., 2007, 46, 9558–9568 CrossRef CAS PubMed.
- S. Jiang, Y. Zheng, M. Liu, Z. Yu, Y. Ma, G. A. Solan, W. Zhang, T. Liang and W.-H. Sun, Organometallics, 2022, 41, 3197–3211 CrossRef CAS.
- (a) L. Fang, W. Zhao, C. Han, H. Liu, Y. Hu and X. Zhang, Eur. J. Inorg. Chem., 2019, 2019, 609–616 CrossRef CAS; (b) G. Ricci, G. Leone, A. Boglia, A. C. Boccia and L. Zetta, Macromolecules, 2009, 42, 9263–9267 CrossRef CAS; (c) B. Liu, X. Wang, Y. Pan, F. Lin, C. Wu, J. Qu, Y. Luo and D. Cui, Macromolecules, 2014, 47, 8524–8530 CrossRef CAS.
- M. N. Alnajrani and F. S. Mair, RSC Adv., 2015, 5, 46372–46385 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General consideration and materials, the procedure for isoprene polymerization, X-ray crystallographic studies, and Table S1. Crystal data and structure refinement for FeH, Fe2Me and Fe2Et,Me, and Fig. S1–S22. 1H and 13C NMR spectra for entry 3, Table 1; entries 2–10 and 12–14, Table 2; entries 2–5, Table 3; entries 2–5, Table 4. CCDC 2305303 (FeH), 2305304 (Fe2Me) and 2305305 (Fe2Et,Me). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03674j |
| This journal is © The Royal Society of Chemistry 2024 |