DOI:
10.1039/D3DT03630H
(Paper)
Dalton Trans., 2024,
53, 3397-3406
Synthesis, characterization, and density functional theory investigation of (CH6N3)2[NpO2Cl3] and Rb[NpO2Cl2(H2O)] chain structures†
Received
30th October 2023
, Accepted 16th January 2024
First published on 17th January 2024
Abstract
The actinyl tetrachloro complex [An(V/VI)O2Cl4]2−/3− tends to form discrete molecular units in both solution and solid state materials, but related aquachloro complexes have been observed as both discrete coordination compounds and 1-D chain topologies. Subtle differences in the inner sphere coordination significantly influence the formation of structural topologies in the actinyl chloride system, but the exact reasoning for these variations has not been delineated. In the current study, we present the synthesis, structural characterization, and vibrational analysis of two 1-D neptunyl(V) chain compounds: (CH6N3)2[NpO2Cl3] (Np-Gua) and Rb[NpO2Cl2(H2O)] (Np-Rb). Bonding and non-covalent interactions (NCIs) in the systems were evaluated using periodic Density Functional Theory (DFT) to link these properties to related phases. We observed ∼6.5% and ∼3.9% weakening of Np![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in Np-Gua and Np-Rb compared to the reference Cs3[NpO2Cl4]. NCI analysis distinguished specific assembly modes, where Np-Gua was connected via hydrogen bonding (N–H⋯Cleq and N–H⋯Oyl) and Np-Rb contained both cation interactions (Rb+⋯Oyl and Rb+⋯Cleq) and hydrogen bonding (Oeq–H⋯Oyl) networks. Thermodynamically viable formation pathways for both compounds were explored using DFT methodology. The [NpO2Cl4](aq)3− and [NpO2Cl3(H2O)](aq)2− substructures were identified as precursors to Np-Gua and [NpO2Cl3(H2O)](aq)2− and [NpO2Cl2(H2O)2](aq)− were isolated as the primary building units of Np-Rb. Finally, we utilized DFT to analyze the vibrational modes for Np-Gua and Np-Rb, where we found evidence of the NpO bond weakening within the Np(V) chain structures compared to [NpO2Cl4]3−.
O bonds in Np-Gua and Np-Rb compared to the reference Cs3[NpO2Cl4]. NCI analysis distinguished specific assembly modes, where Np-Gua was connected via hydrogen bonding (N–H⋯Cleq and N–H⋯Oyl) and Np-Rb contained both cation interactions (Rb+⋯Oyl and Rb+⋯Cleq) and hydrogen bonding (Oeq–H⋯Oyl) networks. Thermodynamically viable formation pathways for both compounds were explored using DFT methodology. The [NpO2Cl4](aq)3− and [NpO2Cl3(H2O)](aq)2− substructures were identified as precursors to Np-Gua and [NpO2Cl3(H2O)](aq)2− and [NpO2Cl2(H2O)2](aq)− were isolated as the primary building units of Np-Rb. Finally, we utilized DFT to analyze the vibrational modes for Np-Gua and Np-Rb, where we found evidence of the NpO bond weakening within the Np(V) chain structures compared to [NpO2Cl4]3−.
1. Introduction
Speciation of high valent actinides in chloride rich aqueous media is complex due to the formation of mixed aquachloro compounds with the general formula of [AnO2Clx(H2O)y]−/z+.1,2 These complexes form for both pentavalent and hexavalent oxidation states of the lighter actinide elements (i.e. uranium, neptunium, and plutonium), with the nearly linear dioxo cation (An(V/VI)O2)+/2+ further coordinated around the equatorial plane with either Cl− anions or H2O molecules. In solutions with lower chloride concentrations ([Cl−] < 2 M), the [AnO2(H2O)5]+/2+ species is thought to be the most stable,3–5 but under conditions with increased [Cl−], the chloride anion can displace water molecules via ligand substitution to form aquachloro complexes with a formula of [AnO2Cl(n)(H2O)x]−n+m (x = 3 or 4, n = 1 to 4, m = 1 or 2). At higher concentrations ([Cl−] > 13 M), saturation of the halide ion is achieved within the equatorial plane to create the [AnO2Cl4]2−/3− or [AnO2Cl4(H2O)]2−/3− species.2,6–9
While the presence of aquachloro actinyl species has been systematically evaluated in solution, solid-state phases are much less well studied than the more common tetrachloro forms. Discrete U(VI), Np(VI), and Np(V) tetrachloro complexes have all been structurally characterized with a range of charge balancing cations, and they are widely used as synthons for crystal engineering or as model systems in the study of Non-Covalent Interactions (NCIs) in hybrid materials.8–16 Within the aquachloro complexes, only the [UO2Cl2(H2O)3],17 [C4H12N2]2[UO2Cl4(H2O)]Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and [NMe4][NpO2Cl(H2O)4]Cl18 coordination compounds have been synthesized and structurally characterized using X-ray diffraction techniques. These aquachloro structural building units have also been observed within extended 1-D chain topologies (e.g. UO2Cl2H2O,19 Cs[Np(V)O2Cl2(H2O)],20 K4[Np(V)O2)3Cl7(H2O)4],21 but no extended structures built from the [AnO2Cl4]2−/3− moiety have been noted in the literature. Prevalence of the isolated tetrachloro species compared to both 0-D and 1-D topologies for the aquachloro complexes suggests that subtle differences in the inner sphere coordination to the actinyl cation significantly influence the structural characteristics, but the exact reasoning for these variations has not been delineated.
8 and [NMe4][NpO2Cl(H2O)4]Cl18 coordination compounds have been synthesized and structurally characterized using X-ray diffraction techniques. These aquachloro structural building units have also been observed within extended 1-D chain topologies (e.g. UO2Cl2H2O,19 Cs[Np(V)O2Cl2(H2O)],20 K4[Np(V)O2)3Cl7(H2O)4],21 but no extended structures built from the [AnO2Cl4]2−/3− moiety have been noted in the literature. Prevalence of the isolated tetrachloro species compared to both 0-D and 1-D topologies for the aquachloro complexes suggests that subtle differences in the inner sphere coordination to the actinyl cation significantly influence the structural characteristics, but the exact reasoning for these variations has not been delineated.
In the current study, we evaluated the difference in bonding and intermolecular interactions that occur within Np(V) aquachloro species and the saturated tetrachloro complexes within solid materials. Herein, we report the synthesis, structural analysis, and Raman spectroscopy of two novel phases with Np(V) 1-D chain topologies formed by bridging chloride anions: (CH6N3)2[NpO2Cl3] (Np-Gua) and Rb[NpO2Cl2(H2O)] (Np-Rb). Periodic Density Functional Theory (DFT) was utilized to evaluate and compare the primary coordination sphere and NCIs of Np-Gua and Np-Rb to that of the isolated [NpO2Cl4]3− complex observed in Cs3[NpO2Cl4]. We then used DFT to propose a mechanism in the 1-D chain formation from individual molecular units and identify neptunyl vibrational features of these two phases.
2. Experiment
2.1 Synthesis
CAUTION
: Neptunium-237 (
237
Np) is a highly radioactive isotope that should be restricted to specialized laboratories and handled under appropriate regulatory controls. Syntheses of (CH6N3)2[NpO2Cl3] (Np-Gua) and Rb[NpO2Cl2(H2O)] (Np-Rb) were performed by mixing 20 μL of 0.27 M Np(V) in 2 M HCl with 20 μL of 0.50 M aqueous guanidinium chloride or rubidium chloride (detailed description on making the Np(V) stock solution is provided in the ESI, Section 1†). The resulting solutions were left to evaporate in a well plate, and the formation of green crystals was observed within a week. While quantitative yields were not determined because of the radioactive nature of the solid materials, qualitative yields were moderate for Np-Gua and low for Np-Rb.
2.2 Single-crystal X-ray diffraction
A small amount of the solid product was removed from the mother liquor, and a high-quality single crystal of the material was placed on a Mitigen® micromount. X-ray diffraction data were collected at 136 K using 0.5° ω scans on a Bruker® D8 Quest single X-ray diffractometer (λMoKα = 0.71073 Å) with a PHOTON detector and an Oxford® cryo-system. The Bruker software was used to integrate the data and the absorption correction was performed using the SADABS software.22,23 Structures were solved using APEX3 intrinsic phasing 1 and the refinement of the data was performed using SHELXL24 and the OLEX225 software package. Hydrogen atoms associated with guanidinium and water were modeled with AFIX 93 and AFIX 7, respectively. Selected crystallographic parameters are summarized in Table 1 and thermal ellipsoids of Np-Gua and Np-Rb are given in the ESI (Section 1, Fig. S1 and S2†).
Table 1 Selected crystallographic information of Np-Gua and Np-Rb
| Compound |
Np-Gua
|
Np-Rb
|
| Empirical formula |
NpO2Cl3C2H12N6 |
RbNpO3Cl2H2 |
| Crystal color and habit |
Green |
Green |
| Formula weight (g) |
495.53 |
443.39 |
| Crystal system |
Orthorhombic |
Monoclinic |
|
a (Å) |
8.6197(3) |
8.4988(4) |
|
b (Å) |
9.7216(4) |
11.3956(5) |
|
c (Å) |
14.5517(6) |
7.4459(6) |
|
α (°) |
90 |
90 |
|
β (°) |
90 |
114.2050(10) |
|
γ (°) |
90 |
90 |
| Unit cell volume (Å3) |
1219.39(8) |
657.73(7) |
| Temperature (K) |
136.01 |
137.76 |
| Density, ρ (g cm−3) |
2.699 |
4.478 |
| Space group |
P212121 |
C2/c |
|
Z
|
4 |
4 |
|
μ (mm−1) |
9.168 |
23.873 |
|
F(000) |
904.0 |
760.0 |
|
Θ range (°) |
5.04 to 50.68 |
6.356 to 50.698 |
| Limiting indices |
−10 ≤ h ≤ 10, −11 ≤ k ≤ 11, −17 ≤ l ≤ 17 |
−10 ≤ h ≤ 10, −13 ≤ k ≤ 13, −8 ≤ l ≤ 8 |
| Ref. collected/unique |
47334 |
12453 |
|
R
int
|
0.0315 |
0.0257 |
| Data/restraints/parameters |
2236/0/129 |
608/0/39 |
| GOF on F2 |
1.161 |
1.245 |
|
R
1 ([I > 2σ(I)]) |
0.0070 |
0.0069 |
| wR2 ([I > 2σ(I)]) |
0.0174 |
0.0157 |
|
R
1 (all data) |
0.0071 |
0.0070 |
| wR2 (all data) |
0.0174 |
0.0157 |
| Largest diff. peak/hole (e Å−3) |
0.31/−0.32 |
0.35/−0.51 |
| CCDC deposition number |
2287578
|
2287577
|
2.3 Raman spectroscopy
Solid-state Raman spectra of Np-Gua were collected using SnRI High-Resolution Sierra 2.0 Raman spectrometer outfitted with a 786 nm laser and a 2048 pixel TE-CCD. The laser intensity was set to 100 mW and spectra were obtained with a 10 s integration period. As the Np-Rb system had low yields, the Raman spectra could only be obtained using a Renishaw Raman microscope outfitted with a 786 nm laser. The laser power was set to 10 mW with a 10 s integration time and the final spectrum was obtained through 15 accumulations. In both cases, the spectral fitting was performed using the Origin Pro software.26
2.4 Computational details
All periodic DFT calculations were performed using the Vienna Ab Initio Simulation Program (VASP).27–29 The generalized gradient approximation of Perdew–Burke–Ernzerhof (GGA-PBE)30 was utilized to model exchange–correlation energy, and PAW pseudopotentials31,32 were employed for all the atoms. A planewave basis set cutoff of 550 eV and a gamma-centered Monkhorst–Pack33k-grid of at least 5 × 4 × 3 were employed. As scalar relativistic effects are included only by the employed pseudopotential, spin–orbit coupling is ignored. All structures were subjected to comprehensive geometric optimizations without symmetry constraints, and the forces and total energy converged to within 1 meV−1 and 1 × 10−8 eV, respectively. The Hubbard U correction34–36 was applied to the neptunium f-states using the approach of Dudarev et al. with a U–J value of 3.75 eV.37 In all DFT computations, the van der Waals dispersion correction methods DFT-D3 were utilized with the Becke–Johnson damping term.38,39 Bond orders (BO) and partial atomic charges were computed using the Chargemol program's Density Derived Electrostatic and Chemical 6 (DDEC6) method.40–43
The Gaussian 16 software package44 was used to perform all molecular Density Functional Theory (DFT) computations. To model exchange correlation effects, the B3LYP (Becke, 3-parameter, Lee–Yang–Parr)45,46 hybrid function was used for the calculations. Van der Waals dispersion correction method DFT-D3 was again utilized with the Becke–Johnson damping term.38,39 The polarized triple zeta (def2-TZVP)47 basis set was utilized to represent the C, N, H, and Cl atoms, while the ECP60MWB pseudopotentials and ECP60MWB-SEG valence basis set were used for Np.48,49 Scalar relativistic effects are included by employing small-core Effective Core Potentials (ECPs), while spin–orbit effects are ignored.
Solvation in water was simulated with the Integral Equation Formalism Polarizable Continuum Model (IEFPCM).50,51 All structures were optimized with no symmetry constraints to a tight convergence criterion with Root Mean Square (RMS) force criterion of 1 × 10−5 Hartrees per radians. The calculated vibrational frequencies were monitored to ensure that structures were optimized to a true minimum. Structures manifesting imaginary frequencies underwent reoptimization with a finer integration grid and stringent convergence criteria (RMS force criterion of 1 × 10−6 Hartrees per radians), ensuring their progression towards true minima without imaginary frequencies.
3. Results and discussion
3.1 Structural description and bond analysis
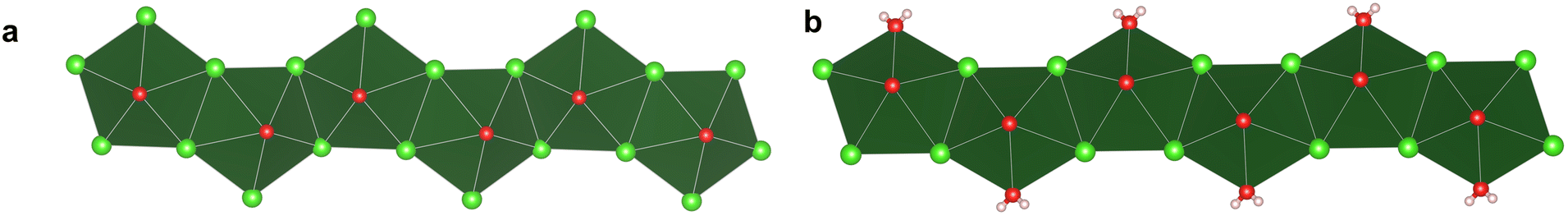
Structure determination using single crystal X-ray diffraction revealed the presence of one-dimensional chains of [NpO2(μ2-Cl)4Cl]2− in Np-Gua and [NpO2(μ2-Cl)4(H2O)]− in Np-Rb. Both Np-Gua and Np-Rb contain four crystallographically unique (Np(V)O2)+ units with average NpO bond lengths of 1.818(3) and 1.826(2) Å, respectively. The neptunyl bond in both cases was nearly linear with ONpO angles of 179.63(10) for Np-Gua and 179.10(9)° for Np-Rb. For Np-Gua, five additional Cl− anions are present in the equatorial plane at an average distance of 2.8534(7) Å to create an overall pentagonal bipyramidal coordination geometry. Np-Rb has a similar coordination geometry, but one of the Cl− anions is replaced with a ligated water molecule. Np–Cl distances for Np-Rb are similar to that of Np-Gua, but the Np–OH2 distance is shorter at 2.452(2) Å. Both chains propagate through two μ2-Cl bridges, although there are differences in the orientation of the neptunyl polyhedra along the length of the chain (Fig. 1). Within Np-Rb, the neptunyl cations along the 1-D chain remain parallel, but in the case of Np-Gua they are canted due to a change in the angle of the μ2-Cl bridge. This can also be observed based upon the ONp–NpO dihedral angles of 7.48(2)° for Np-Gua and 0.82(2)° for Np-Rb (Fig. 2).
 |
| | Fig. 1 Neptunyl chloride chains are present in (a) Np-Gua and (b) Np-Rb. The Np(V) is located at the center of the dark green polyhedra and the spheres represent Cl (green), O (red), and H (pink) atoms. (Images of chains with six neptunyl units were generated by creating 3 × 3 × 3 supercell.) | |
 |
| | Fig. 2 The bond lengths (Å) and BOs of dimeric units in (a) Np-Gua, (b) Np-Rb and (c) [NpO2Cl4]3− units in Np-Cs.53,57 The dark green, light green and red sphere represent Np, Cl, and O atoms respectively. In ball and stick representation, the dashed sticks represent extended bonding. Tables with selected crystallographic bond lengths and bond angles are provided in the ESI, Section 2.1.† | |
Within the Np(V) chloride system, there are only a handful of known 1-D chain compounds that have been structurally characterized. The 1-D chains of Cs[NpO2Cl2(H2O)] are formed by edge sharing neptunyl(V) polyhedrons that are linked via bridging Cl−.20 In contrast, K4[(NpO2)3Cl7(H2O)4] chains are formed as a consequence of bridging Cl− and neptunyl–neptunyl interactions. While the 1-D structural topology within Np-Rb is isostructural to the previously reported Cs[NpO2Cl2(H2O)] compound,20 the chain topology for the Np-Gua has not been noted in the literature for either Np or U solids. In the Np(VI) chloride system, only one 1-D chain compound, [NpO2Cl2(THF)]n, has been structurally characterized. This compound exhibits a 1-D structural topology like Np-Rb, differing primarily in the valence state of Np and the substitution of water with tetrahydrofuran.52 It is also important to note that the individual molecular components within Np-Rb ([NpO2Cl5]4−) and Np-Gua ([NpO2Cl4(H2O)]3−) have not been crystallized as isolated units.
To gain further insight into the bonding within the Np(V) chain while preserving the periodicity, we determined Bond Orders (BOs) using periodic Density Functional Theory and compared them to the related reoptimized Cs3[NpO2Cl4] phase (Np-Cs).53 Compared to Np-Gua, Np-Cs has similar experimental NpO bond lengths and has experimental Np–Cl bond lengths that are elongated by about ∼3.5%. For the Np-Rb chain, the experimental Np–Cl bond lengths on average show ∼3.0% elongation compared to (Np-Cs) and the NpO BOs of [NpO2(μ2-Cl)4Cl]2−, [NpO2(μ2-Cl)4(H2O)]− and [NpO2Cl4]3− are 1.43, 1.47 and 1.53, respectively. These BOs indicate weakening of the neptunyl bond by ∼6.5% and ∼3.9% in [NpO2(μ2-Cl)4Cl]2− and [NpO2(μ2-Cl)4(H2O)]− compared to [NpO2Cl4]3−. This observed weakening of the bond could be caused by higher steric hindrance (4 vs. 5 equatorial coordination) as well as differences in non-covalent interaction by axial oxygen in the three different crystal structures.11,14–16 The total BOs of equatorial coordination varies as follows: [NpO2Cl4]3− (BO = 1.60) > [NpO2(μ2-Cl)4(H2O)]− (BO = 1.48) ≈ [NpO2(μ2-Cl)4Cl]2− (BO = 1.46). In Np-Gua, the BO of bridging chlorides and non-bridging chlorides remain comparable, while in Np-Rb the BOs of equatorial water remain identical to the chloride anions (Fig. 2 and ESI, Section 3, Table S8†). The significantly weaker equatorial bonding compared to axial bond suggests that equatorial bonding within the chain structures remains ionic.54–56
3.2 Non-covalent interactions within the extended lattice
Our previous work with uranyl11 and neptunyl57 solid state compounds has highlighted the importance of Non-Covalent Interactions (NCIs) in driving crystallization and the overall formation energies. To analyze the NCIs of Np-Gua and Np-Rb, we adopted the methodology developed in our prior work.40–43 Total hydrogen bond energy (EtotalH) and total electrostatic interaction energy (Etotalcation) were determined with eqn (1), (2) and (3), (4), respectively, to quantify the degree of NCIs.| |  | (1) |
| |  | (2) |
| |  | (3) |
| |  | (4) |
In eqn (1) the qi+ and qi− represent the partial atomic charges of hydrogen and hydrogen bond accepter. Whereas in eqn (2), the qj+ and qj− are partial charges of the cation and anion involved in the interaction. In these equations, r represents the interaction distance, and Z represents the number of formula units in the unit cell. Additionally, we determined the sum of bond order (BO“yl”sum) of all NCIs of a single axial oxygen with eqn (5) as a measure of axial oxygen engagement in NCIs.H
| |  | (5) |
ere, the BO
“yl”i is the bond order of NCIs through the axial oxygen calculated by DDEC6 method.
11,40–43 The
Etotalcation and BO
“yl”sum values for
Np-Cs were taken from literature to compare the NCIs of NpO
2+ chains to a well-studied [NpO
2Cl
4]
2− containing system.
57 The DDEC6 technique has been applied previously to assess hydrogen bonding in natrolite and hexagonal ice crystals, yielding a BO of 0.07–0.13 for strong hydrogen bonds, thus this value will be used as a reference when assessing the strength of NCIs.
42
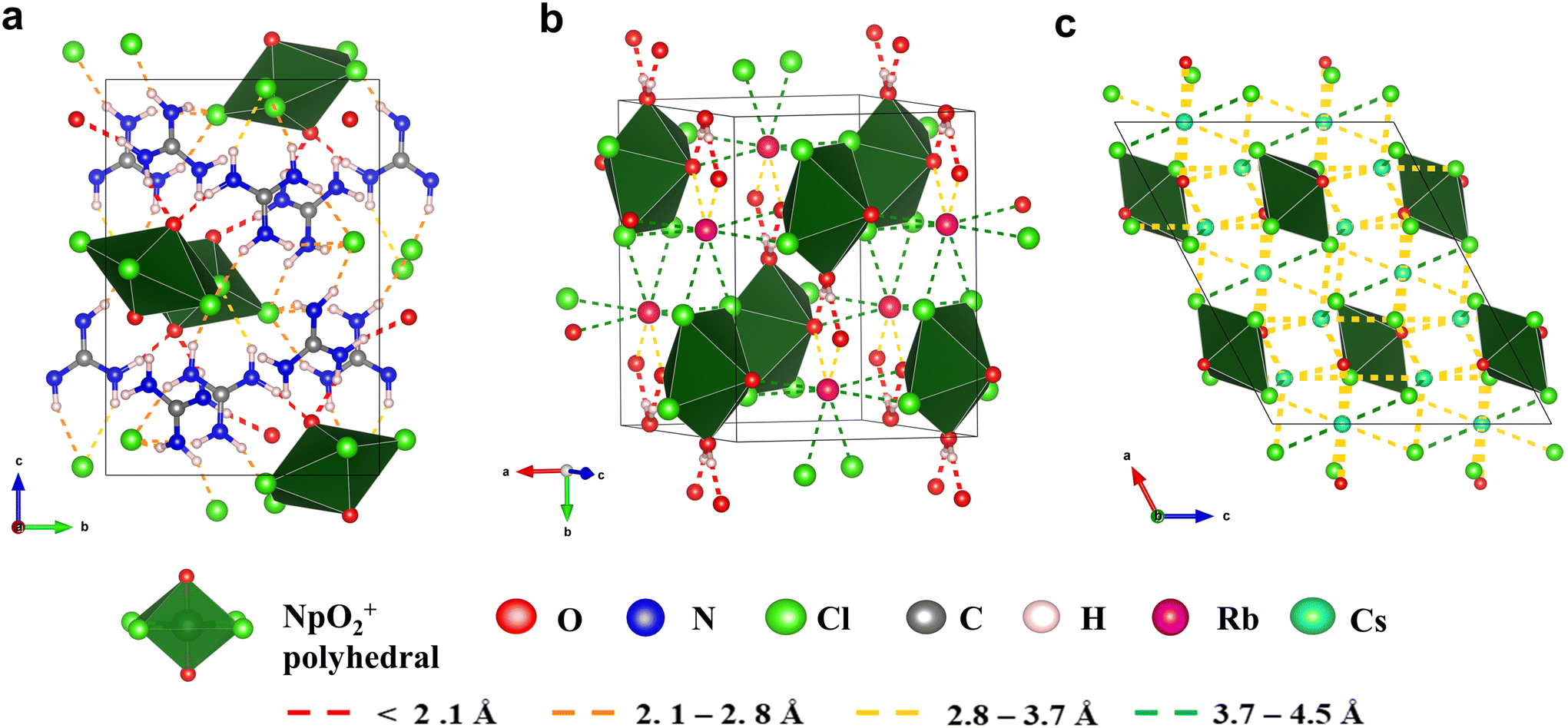
The hydrogen bonding network of Np-Gua was mapped (Fig. 3a) and shows the presence of N–H⋯Cleq and N–H⋯Oyl hydrogen bonding, with EtotalH of −2196.37 kJ mol−1. When the hydrogen bonding BOs of bridging and non-bridging Cl− are compared, non-bridging Cl− have 2.5 times the hydrogen bonding ability compared to the bridging Cl− (ESI, Section 4, Table S22†). The Np-Rb crystal structure contains both cation interactions and hydrogen bonding interactions with energies of Etotalcation = −2538.98 kJ mol−1 and EtotalH = −528.15 kJ mol−1. Rb+⋯Cleq that were ∼0.1–0.3 Å longer than Rb+⋯Oyl interactions, yet Rb+⋯Cleq has 1.14 times higher BOs compared to Rb+⋯Oyl interactions (ESI, Section 4, Table S23†). Ligated water molecules present in the Np-Rb chains form strong hydrogen bonding (BO = 0.109) with the axial oxygens on the neighboring neptunyl(V) chains. The BO“yl”sum of Np-Cs was reported as 0.374,57 which allows us to rank the BO“yl”sum of complexes as Np-Cs > Np-Rb > Np-Gua (Table 2). This trend in BO“yl”sum and interaction energies can be rationalized as being related to the overall molecular charges as it goes from −3 in the isolated Np-Cs units to −2 and −1 for Np-Gua and Np-Rb, respectively. Even though the magnitude of the negative charge for Np-Rb is smaller than that of Np-Gua, the larger number of NCIs may be a result of additional hydrogen bonding with the equatorial water of neighboring chains. Since the primary coordination sphere is different between Np-Gua, Np-Rb and Np-Cs, we could not compare the influence of NCIs on the NpO bond lengths. Our objective in analyzing the non-covalent interactions (NCIs) within Np chain structures is to emphasize the distinctions in their NCI networks when compared to an extensively studied Np-Cs system.
 |
| | Fig. 3 The NCIs networks for (a) Np-Gua, (b) Np-Rb, and (c) Np-Cs57 compounds. The color of the interaction indicates the distance. (VESTA visualization files are provided as a part of the ESI.†) A legend provided below the image defines the molecular units, atoms, and interaction distances. | |
Table 2 The number of hydrogen bonding interactions per formula unit and number of cation interactions per formula unit, EtotalH, Etotalcation and BO“yl”sum of NpO2+ compounds
| Compounds |
No. H bonding interactions |
No. cation interactions |
E
totalH (kJ mol−1) |
E
totalcation (kJ mol−1) |
BO“yl”sum |
|
Np-Gua
|
15 |
— |
−2196.37 |
— |
0.215 |
|
Np-Rb
|
4 |
8 |
−2538.98 |
−528.15 |
0.235 |
|
Np-Cs
57
|
— |
30 |
— |
−6656.86 |
0.374 |
3.3 Vibrational signatures
The linear NpO2+ moiety has D∞h symmetry that leads to three fundamental vibrational modes: symmetrical stretch (ν1: Raman active), bending mode (ν2: IR active), and asymmetrical stretch (ν3: IR active).58,59 Typically, the ν1 of [NpO2(H2O)5]+ species is centered at 765 cm−1, but the addition of ligands with significant sigma donation and pi-interactions can result in weakening of the neptunyl bond and a red-shift in the peak position.15 Interactions between the Np(V) cation and the Cl− anions bonded in the equatorial plane are typically considered ionic, leading to smaller red-shifts in the ν1 band (4–20 cm−1). The ν1 is also sensitive to NCIs, where strong interactions can weaken the NpO bond and reduce the symmetry, causing a red-shift in the peak position and the appearance of additional features.14–16,60
The experimental Raman spectrum of Np-Gua contains two peaks that are centered at 1009 cm−1 and 741 cm−1. To determine the origin of these peaks, we calculated the Raman active modes of Np-Gua using an isolated guanidinium cation and [(NpO2)2Cl8]6− dimer. The calculated guanidinium feature was observed at 1018 cm−1, while the [(NpO2)2Cl8]6− dimer had two vibrational bands at 770 cm−1 and 765 cm−1 for in-phase and out-of-phase symmetrical stretches (Fig. 4a and Table 3). Aided by this computational data, experimental features in the Np-Gua spectrum are assigned to the guanidium (1009 cm−1) and (NpO2)+ (741 cm−1) symmetric stretches. The calculation of vibrational modes of Np-Rb is done using the isolated dimer [(NpO2)2Cl6(H2O)2]4−. The Np-Rb experimental Raman spectrum has one prominent feature at 751 cm−1, and a minor feature at 716 cm−1 (Fig. 4b), while the calculated in-phase and out-of-phase ν1 vibrations are at 781 cm−1 and 776 cm−1 respectively. With the computational results, the peak at 751 cm−1 in the experimental spectrum can be assigned to (NpO2)+ symmetrical stretch, but the minor feature at 716 cm−1 did not align with any calculated features. This feature may have appeared due to distortion of the NpO2+ geometry caused by NCIs15 or due to minor impurity. According to the literature, the NaxNpIV(NpVO2)6(OH)1+xCl9(H2O)8−x (0 < x ≤ 1) phase has a feature at 717 cm−1; therefore, the impurity could be this phase or a related phase.61 Comparing the neptunyl symmetric stretch of the two compounds, we note that the ν1 in Np-Gua is 10 cm−1 redshifted compared to Np-Rb, which coincides with our prior observations regarding the trends in NpO BO for these systems.
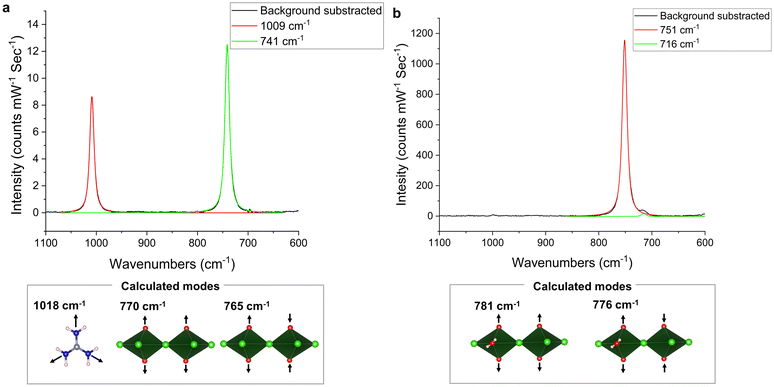 |
| | Fig. 4 The experimental Raman spectra of (a) Np-Gua and (b) Np-Rb. The spectra were fitted using a pseudo-Voigt 1 model to obtain the peak centroids. The DFT-calculated Raman active modes in the spectral window of 1100–600 cm−1 are highlighted with arrows showing motion of atoms during the vibration. | |
Table 3 The experimental and calculated neptunyl(V) vibrational features of Np-Gua and Np-Rb. The calculation of vibrational modes of Np-Gua and Np-Rb are done using isolated dimers of [(NpO2)2Cl8]6− and [(NpO2)2Cl6(H2O)2]4− respectively
| Compounds |
Experimental ν1 (cm−1) |
Calculated ν1 (cm−1) |
Calculated ν3 (cm−1) |
| In-phase |
Out-of-phase |
In-phase |
Out-of-phase |
|
Np-Gua
|
741 |
770 |
765 |
781 |
776 |
|
Np-Rb
|
751 |
798 |
796 |
812 |
810 |
3.4 Insight into chain formation
The Np(V) in aqueous media with Cl− exist as molecular units of [NpO2Cl(n)(H2O)x]−n+1 (x = 4 or 5, n = 0 to 4) where n increases as a function of the chloride concentration.2 We investigated the thermodynamic feasibility of assembling these molecular units to form dimers and trimers that can extend to generate the Np(V) chain structures found in Np-Gua and Np-Rb. We modeled [NpO2Cl(2to4)(H2O)(0to2)]−x (x = 1 to 3) complexes in aqueous media using molecular DFT. The true stationary point of these calculations was confirmed by the lack of imaginary vibrational frequencies. From these initial Np(V) aquachloro complexes, we then proposed a thermodynamically viable pathway for the chain formation to occur by assuming that these reactions occur via dissociative ligand substitution or addition, which is supported by literature precedent.6,7,62
The crystallization of Np-Gua occurs when the mother liquor is highly concentrated and has a high viscosity, where the [Cl−] > 10 M based on final concentrations. At such high chloride concentrations, NpO+ will coordinate to 3 or 4 equatorial chlorides.2 We computed the reaction enthalpy for the transformation of the [NpO2Cl(3to4)(H2O)(0to2)]2−to3− molecular species, initially forming a dimer [(NpO2)2Cl7](aq)5− in the first step. Subsequently, this dimer associates with monomeric units in the second step, resulting in the formation of [(NpO2)3Cl10](aq)7− trimers (Fig. 5). Calculations show that seven reactions are enthalpy favored to form the dimer, where the most energetically favorable is the dimerization of [NpO2Cl3(H2O)](aq)2− (Table 4, step 1, reaction (I)). None of the first step reactions were thermodynamically favorable with both monomeric species having tetrachloride coordination (ESI, Section 6, Table S28†). This may be due to this reaction leading to the cleavage of Np–Cl bonds and release of Cl− as HCl(aq). In the second step, only the reaction between [(NpO2)2Cl7](aq)5− and [NpO2Cl3(H2O)](aq)2− was energetically favored (ESI, Section 6, Table S28†), highlighting the crucial role of [NpO2Cl3(H2O)](aq)2− in Np-Gua chain formation, with potential chain propagation through its repeated occurrence.
 |
| | Fig. 5 Most energetic chain formation path for (a) Np-Gua and (b) Np-Rb. The green polyhedra represents neptunyl(V) units. NCIs energies (EtotalH and Etotalcation) are shown to highlight the driving force for crystallization in the presence of charge balancing cations. | |
Table 4 Possible formation reactions that may be involved in the formation of Np-Gua
| Step |
Possible reactions |
ΔH (kJ mol−1) |
| 1 |
(I) |
2[NpO2Cl3(H2O)](aq)2− + HCl(aq) → [(NpO2)2Cl7](aq)5− + H3O(aq)+ + H2O(aq) |
−94.00 |
| (II) |
[NpO2Cl4(H2O)](aq)3− + [NpO2Cl3(H2O)](aq)2− → [(NpO2)2Cl7](aq)5− + 2H2O(aq) |
−46.17 |
| (III) |
[NpO2Cl4](aq)3− + [NpO2Cl3(H2O)](aq)2− → [(NpO2)2Cl7](aq)5− + H2O(aq) |
−44.63 |
| (IV) |
2[NpO2Cl3(H2O)2](aq)2− + HCl(aq) → [(NpO2)2Cl7](aq)5− + H3O(aq)+ + 3H2O(aq) |
−34.50 |
| (V) |
[NpO2Cl4(H2O)](aq)3− + [NpO2Cl3(H2O)2](aq)2− → [(NpO2)2Cl7](aq)5− + 3H2O(aq) |
−16.42 |
| (VI) |
[NpO2Cl4](aq)3− + [NpO2Cl3(H2O)2](aq)2− → [(NpO2)2Cl7](aq)5− + 2H2O(aq) |
−14.88 |
| |
| 2 |
(I) |
[(NpO2)2Cl7](aq)5− + [NpO2Cl3(H2O)](aq)2− → [(NpO2)3Cl10](aq)7− + H2O(aq) |
−23.47 |
In contrast, Np-Rb crystallization occurs considerably sooner during the slow evaporation of the aqueous solution; thus, the [Cl−] is likely at the appropriate level to contain dichloride coordination. Thus, we have included [NpO2Cl(2to4)(H2O)(0to3)]−to3− in our calculations. Here, we found that the dimerization of [NpO2Cl3(H2O)](aq)2−, is the most favorable reaction of step 1 (Table 5). Additionally, the [NpO2Cl3(H2O)](aq)2− reactions with [NpO2Cl3(H2O)2](aq)2− and [NpO2Cl2(H2O)2](aq)− were also found to be thermodynamically feasible (ESI, Section 6, Table S29†). Here, the trimer formation and chain propagation can happen via subsequent addition of [NpO2Cl(2to3)(H2O)(1to2)](aq)−to2− to the end of the oligomeric unit.
Table 5 Possible formation reactions that may be involved in the formation of Np-Rb
| Step |
Possible reactions |
ΔH (kJ mol−1) |
| 1 |
(I) |
2[NpO2Cl3(H2O)](aq)2− → [(NpO2)2Cl6(H2O)2](aq)4− |
−34.27 |
| (II) |
[NpO2Cl3(H2O)](aq)2− + [NpO2Cl2(H2O)2](aq)− + HCl(aq) → [(NpO2)2Cl6(H2O)2](aq)4− + H3O(aq)+ |
−8.19 |
| (III) |
[NpO2Cl3(H2O)](aq)2− + [NpO2Cl3(H2O)2](aq)2− → [(NpO2)2Cl6(H2O)2](aq)4− + H2O(aq) |
−4.52 |
| |
| 2 |
(I) |
[(NpO2)2Cl6(H2O)2](aq)4− + [NpO2Cl3(H2O)](aq)2− + H3O(aq)+ → [(NpO2)3Cl8(H2O)3](aq)5− + H2O(aq) + HCl(aq) |
−56.66 |
| (II) |
[(NpO2)2Cl6(H2O)2](aq)4− + [NpO2Cl2(H2O)2](aq)− → [(NpO2)3Cl8(H2O)3](aq)5− + H2O(aq) |
−30.58 |
| (III) |
[(NpO2)2Cl6(H2O)2](aq)4− + [NpO2Cl3(H2O)2](aq)2− + H3O(aq)+ → [(NpO2)3Cl8(H2O)3](aq)5− + 2H2O(aq) + HCl(aq) |
−26.91 |
| (IV) |
[(NpO2)2Cl6(H2O)2](aq)4− + [NpO2Cl2(H2O)3](aq)− → [(NpO2)3Cl8(H2O)3](aq)5− + 2H2O(aq) |
−14.59 |
4. Conclusion
In this study, we synthesized two Np(V) chain structures and analyzed them using single crystal X-ray diffraction, Raman spectroscopy, and DFT calculations. Structural analysis of the solid phases revealed the presence of 1-D chain topologies built from repeating Np(V)O2+ units linked by bridging Cl− anions. The NpO bond lengths within these chains are similar to those observed for the isolated [NpO2Cl4]3− coordination complex, however BO analysis indicates that the neptunyl bond is weakened by 6.5% compared to the molecular unit. NCI analysis determined that the Np-Gua compound is primarily assembled through charge assisted hydrogen bonding, where both N–H⋯Cleq and N–H⋯Oyl interactions are present and total interaction energy was −2196.37 kJ mol−1. The total interaction energy of Np-Rb was −3067.13 kJ mol−1 and the structure contained Rb+⋯Oyl, Rb+⋯Cleq and Oeq–H⋯Oyl NCIs. Raman spectroscopy showed that there was a red-shift in the neptunyl symmetric stretch of Np-Gua compared to Np-Rb, suggesting a weakening of the NpO bond in Np-Gua. This conclusion is consistent with the trend in BO values for these structures. DFT calculations were also performed to evaluate the thermodynamic feasibility to form Np-Gua and Np-Rb from the molecular precursors, and the results suggest that propagation of the chain via dissociative ligand substitution reactions is thermodynamically favorable. Overall, this study provides a thorough investigation of the primary coordination sphere bonding, NCIs, chain formation reactions, and vibrational features for the Np(V) chloride chain systems.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
H. R., G. C. B., E. L. M., and T. Z. F. acknowledge funding support provided by the Department of Energy, Basic Energy Sciences program under DE-SC0021420. This work used the Theory and Computation facility of the Center for Functional Nanomaterials (CFN), which is a U.S. Department of Energy Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704. Computational support was provided in part by the University of Iowa.
References
- C. Nguyen Trung, G. M. Begun and D. A. Palmer, Inorg. Chem., 1992, 31, 5280–5287 CrossRef CAS.
- L. Soderholm, S. Skanthakumar and R. E. Wilson, J. Phys. Chem. A, 2011, 115, 4959–4967 CrossRef CAS PubMed.
- K. E. Knope and L. Soderholm, Chem. Rev., 2013, 113, 944–994 CrossRef CAS PubMed.
- M. Grigor'ev and N. Krot, Radiochemistry, 2010, 52, 375–381 CrossRef.
- P. Allen, J. Bucher, D. Shuh, N. Edelstein and T. Reich, Inorg. Chem., 1997, 36, 4676–4683 CrossRef CAS PubMed.
- Z. Szabó, T. Toraishi, V. Vallet and I. Grenthe, Coord. Chem. Rev., 2006, 250, 784–815 CrossRef.
- F. P. Rotzinger, J. Chem. Theory Comput., 2008, 4, 1654–1658 CrossRef CAS PubMed.
- H. Rajapaksha, S. E. Mason and T. Z. Forbes, Inorg. Chem., 2023, 62, 14318–14325 CrossRef CAS PubMed.
- R. G. Surbella, L. C. Ducati, K. L. Pellegrini, B. K. McNamara, J. Autschbach, J. M. Schwantes and C. L. Cahill, J. Am. Chem. Soc., 2017, 139, 10843–10855 CrossRef CAS PubMed.
- L. J. Augustine, H. Rajapaksha, M. M. F. Pyrch, M. Kasperski, T. Z. Forbes and S. E. Mason, Inorg. Chem., 2023, 62, 372–380 CrossRef CAS PubMed.
- H. Rajapaksha, S. E. Mason, L. J. Augustine and T. Forbes, Angew. Chem., 2023, 135, e202305073 CrossRef.
- M. B. Andrews and C. L. Cahill, Chem. Rev., 2013, 113, 1121–1136 CrossRef CAS PubMed.
- C. L. Cahill, N. P. Deifel, D. Reusser, L. Zhang and A. Navrotsky, J. Chem. Thermodyn., 2017, 114, 66–70 CrossRef CAS.
- J. L. Bjorklund, M. M. Pyrch, M. C. Basile, S. E. Mason and T. Z. Forbes, Dalton Trans., 2019, 48, 8861–8871 RSC.
- M. M. Pyrch, J. L. Bjorklund, J. M. Williams, D. L. Parr IV, S. E. Mason, J. Leddy and T. Z. Forbes, Dalton Trans., 2020, 49, 6854–6866 RSC.
- M. M. Pyrch, J. M. Williams, M. W. Kasperski, L. C. Applegate and T. Z. Forbes, Inorg. Chim. Acta, 2020, 508, 119628 CrossRef CAS.
- P. Debets, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 400–402 CrossRef CAS.
- S. L. Estes, B. Qiao and G. B. Jin, Nat. Commun., 2019, 10, 59 CrossRef CAS PubMed.
- J. C. Taylor and P. W. Wilson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 169–175 CrossRef CAS.
- S. Tomilin, Y. F. Volkov, R. Melkaya, V. Spiryakov and I. Kapshukov, Sov. Radiochem., 1987, 28, 634–639 Search PubMed.
- T. Z. Forbes and P. C. Burns, J. Solid State Chem., 2007, 180, 106–112 CrossRef CAS.
-
Bruker AXS Inc, Madison, Wisconsin, USA, 2001.
-
Bruker AXS Inc, Madison, Wisconsin, USA, 2019.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
-
Origin(Pro)(2023), OriginLab Corporation, Northampton, MA, USA, 2023.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS PubMed.
-
E. Voloshina, in Encyclopedia of Interfacial Chemistry, ed. K. Wandelt, Elsevier, Oxford, 2018, pp. 115–121. DOI:10.1016/B978-0-12-409547-2.14157-5.
- J. Voss, J. Phys. Commun., 2022, 6, 035009 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. A. Manz and N. G. Limas, RSC Adv., 2016, 6, 47771–47801 RSC.
- N. G. Limas and T. A. Manz, RSC Adv., 2016, 6, 45727–45747 RSC.
- T. A. Manz, RSC Adv., 2017, 7, 45552–45581 RSC.
- N. G. Limas and T. A. Manz, RSC Adv., 2018, 8, 2678–2707 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- X. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2004, 673, 203–209 CrossRef CAS.
- X. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS.
- R. Ramirez and D. Borgis, J. Phys. Chem. B, 2005, 109, 6754–6763 CrossRef CAS PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- S. M. Cornet, L. J. L. Häller, M. J. Sarsfield, D. Collison, M. Helliwell, I. May and N. Kaltsoyannis, Chem. Commun., 2009, 917–919, 10.1039/B818973K.
- N. W. Alcock, M. M. Roberts and D. Brown, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1805–1806 CrossRef.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- R. G. Denning, T. R. Snellgrove and D. R. Woodwark, Mol. Phys., 1976, 32, 419–442 CrossRef CAS.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- H. Rajapaksha, G. C. Benthin, D. V. Kravchuk, H. Lightfoot, S. E. Mason and T. Z. Forbes, Inorg. Chem., 2023, 62, 17265–17275 CrossRef CAS PubMed.
- G. Lu, A. J. Haes and T. Z. Forbes, Coord. Chem. Rev., 2018, 374, 314–344 CrossRef CAS PubMed.
- G. Lu, T. Z. Forbes and A. J. Haes, Anal. Chem., 2016, 88, 773–780 CrossRef CAS PubMed.
- L. J. Augustine, M. M. F. Pyrch, D. V. Kravchuk, J. M. Williams, S. E. Mason and T. Z. Forbes, Eur. J. Inorg. Chem., 2023, e202200693 CrossRef CAS.
- G. B. Jin, Inorg. Chem., 2013, 52, 12317–12319 CrossRef CAS PubMed.
- A. C. Bean, Y. Xu, J. A. Danis, T. E. Albrecht-Schmitt, B. L. Scott and W. Runde, Inorg. Chem., 2002, 41, 6775–6779 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Grant C.
Benthin
a,
Grant C.
Benthin