
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization and in vitro cytotoxicity of gallium(III)-dithiocarbamate complexes†
Nicola
Salvarese‡
*a,
Nicolò
Morellato
b,
Carolina
Gobbi
a,
Valentina
Gandin
b,
Michele
De Franco
b,
Cristina
Marzano
b,
Alessandro
Dolmella
 b and
Cristina
Bolzati‡
*a
b and
Cristina
Bolzati‡
*a
aConsiglio Nazionale delle Ricerche – Istituto di Chimica della Materia Condensata e di Tecnologie per l'Energia (CNR-ICMATE), Corso Stati Uniti 4, 35127 Padua, Italy. E-mail: nicola.salvarese@cnr.it; cristina.bolzati@cnr.it
bDipartimento di Scienze del Farmaco, Università degli Studi di Padova, Via F. Marzolo 5, 35131 Padua, Italy
First published on 31st January 2024
Abstract
A library of homoleptic mononuclear Ga(III) complexes of the general formula [Ga(DTC)3], where DTC is an alicyclic or a linear dithiocarbamate chelator, is reported. The complexes were prepared in high yields starting from Ga(NO3)3·6H2O and fully characterized by elemental analysis and IR and NMR spectroscopy. Crystals of five of these complexes were obtained. The antitumor activity of the newly synthesized compounds against a panel of human cancer cell lines was evaluated. The chemical nature of the DTC does not have a marked impact on the structural features of the final compound. X-ray crystal structure analyses revealed that all these complexes have a trigonal prismatic geometry with three identical chelating DTCs coordinating the Ga(III) ion. It is noteworthy that in complex 22, [Ga(NHEt)3] (NHEt = N-ethyldithiocarbamate), the asymmetric unit is formed by two independent and structurally different molecules. Cellular studies showed that all the synthesized Ga-DTC complexes exhibit marked cytotoxic activity, even against human colon cancer cells that are less sensitive to cisplatin. Among the tested compounds, 6 ([Ga(CEPipDTC)3], CEPipDTC = (ethoxycarbonyl)-piperidinedithiocarbamate) and 21 ([Ga(Pr-13)3], PR13 = 4 and N-(2-ethoxy-2-oxoethyl)-N-methyldithiocarbamate) are very promising derivatives, but they have no selectivity towards cancer cells. Nevertheless, the obtained data provide a foundation for developing gallium-dithiocarbamate complexes as anticancer agents.
1. Introduction
Gallium compounds – especially gallium(III) salts and coordination complexes – have been shown to possess antimicrobial,1,2 antineoplastic3,4 and antiviral5,6 properties, as well as anti-inflammatory activity.7 Moreover, the radioactive isotopes 67Ga and 68Ga are used in nuclear medicine for SPECT and PET imaging, respectively.8–12 In recent years, 68Ga-based radiopharmaceuticals have been considered increasingly attractive due to their growing clinical applications. This development has been facilitated by the advancement in the application of some corresponding 177Lu- and 90Y-tagged radiotherapeutics, as their theranostic companions in the treatment of different tumors. Combining 68Ga and 177Lu or 90Y makes diagnostic molecular imaging possible followed by personalized treatment based on the diagnostic scan.13,14With regard to gallium compounds in the chemotherapy of cancer, the first compound that proved to be active was the salt gallium(III) nitrate. It has been evaluated for the treatment of advanced bladder cancer and non-Hodgkin's lymphoma. The mechanism of action of the Ga3+ ion seems to be connected to the similarity to the Fe3+ ions in terms of valence, ionic radius, electron affinity, and ionization potentials.4 This ionic mimicry is fundamental to the biological activity of Ga(III) compounds. Early studies suggested that the antitumor activity of Ga3+ is relevant to the disruption of cellular iron homeostasis and metabolism, including cellular uptake, transport, and intracellular trafficking.15 However, gallium(III) nitrate requires long intravenous infusions and it is highly nephrotoxic (high hydration of the patient helps minimize this side effect).16 In addition, in vivo speciation of Ga3+ causes a significant reduction of the effective concentration.17,18 To overcome these issues, scientific interest in developing gallium(III) complexes as novel metal-based anticancer drugs has been increasing over the past decade. Gallium maltolate, [(tris(3-hydroxy-2-methyl-4H-pyran-4-onato)gallium(III))] (GaM),19,20 and KP46, [(tris(8-quinolonato)gallium(III))],21,22 are the two most studied complexes in this context. Both of them are sufficiently stable to be orally administered; however, the first one undergoes dissociation in vivo thus releasing Ga3+ ions,23 whereas the second one is much more stable and acts through a different mechanism, which involves the upregulation of p53 and the resulting increase of the Ca2+ and ROS intracellular levels.24 Gallium maltolate and KP46 were clinically investigated for safety, tolerance, and efficacy against various refractory malignancies (Clinical Trials.gov ID NCT00050687).21,22 Moreover, the FDA has granted an orphan drug designation to GaM for use as a potential therapeutic option in pediatric patients with glioblastoma.25
Lately, gallium(III) complexes with thiosemicarbazones,26,27 pyridine and phenolate derivatives,28,29 and phosphinoarylbisthiolato30 have been studied at the preclinical level, with interesting results toward various types of tumors. Gallium(III) complexes with planar tetradentate ligands derived from porphyrin and corrole have also been evaluated for chemo- and photodynamic therapy associated with optical imaging for a theranostic approach (see ref. 4 for an overview).
Dithiocarbamates (DTC) are organic compounds that, together with their chemical derivatives, have been proved to be useful in several fields, from the manufacturing industry to medicine.31 The medical applications of DTC derivatives encompass the treatment of cancer, bacterial infections, Alzheimer's disease, tuberculosis, glaucoma, hyperglycemia, influenza, and inflammations; some of them also show activity as sperm-immobilizing agents.32,33 Furthermore, DTCs are efficient ligands in the coordination chemistry of a wide range of main group metals and transition metals,34,35 and numerous dithiocarbamate metal complexes have shown therapeutic potential against cancer, as well as fungal, bacterial, and viral infections; in general, metal coordination tends to enhance the activity of free DTC.33,36–39 In addition, DTC complexes have been used efficiently in the radiopharmaceuticals field, for the development of homoleptic and heteroleptic technetium and rhenium complexes for SPECT imaging and radiation therapy.40–51
The DTC affinity for gallium has been well established since 1974. With gallium(III) they usually form stable mononuclear homoleptic complexes of the type [Ga(DTC)3], characterized by a GaS6 core with a trigonal prismatic or triple-helix symmetry rather than an octahedral one, due to the small bite angle of the CS2 group, though heteroleptic complexes have also been described.34,52–62 Nonetheless, the pharmaceutical properties of Ga-DTC complexes are poorly explored. Recently, the biocidal activity of some DTC complexes has been reported;63 however, to the best of our knowledge, the anticancer activity of this class of compounds has never been explored so far. The relatively low number of studies performed to date, combined with the intrinsic anticancer properties of the Ga(III) ion itself, offer an exciting possibility for research on novel gallium complexes with biologically active ligands such as DTC.
In this paper, we describe the synthesis and physicochemical characterization by elemental analysis, IR and NMR spectroscopy, and X-ray crystallography of a large library of Ga(III)-DTC complexes. A screening of the newly synthesized compounds against a panel of human cancer cell lines derived from different solid tumors was carried out with the aim of highlighting structure–activity relationships and identifying potential gallium-based anticancer candidates.
2. Experimental
2.1 Reagents
All reagents and solvents for reactions, as well as sodium N,N-dimethyldithiocarbamate (NaDMDC), ammonium N,N-diethyldithiocarbamate (NH4DEDC) and ammonium pyrrolidinedithiocarbamate (HN4PDTC) were purchased from Aldrich Chemicals (Milan, Italy) and used without further purification sodium bis(2-ethoxyethyl)dithiocarbamate (NaDBODC) was acquired from Alchemy (Bologna, Italy); sodium N-ethyldithiocarbamate (NaNHEt) was kindly provided by Dr Roberto Pasqualini of CisBio Bioassays. Sodium piperidinedithiocarbamate (NaPipDTC), sodium 4-ethylpiperazine dithiocarbamate (NaEPDTC), sodium 4-(2-methoxyphenyl)piperazine-1-dithiocarbamate (NaPIPE-1) and sodium 2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyldithiocarbamate (NaPIPE-2) were previously synthesized,64,65 as well as 4-(ethoxycarbonyl)-piperidinedithiocarbamate, 4-(ethoxycarbonyl)piperidinium salt ([CEPipH][CEPipDTC])66 and sodium 1,4-dioxa-8-azaspiro[4,5]decandithiocarbamate (NaDASD).42 4-oxopiperidine-1-dithiocarbamate (COPipDTC), N,N-(2-methoxyethyl)dithiocarbamate (DPODC), N-ethyl-N-propyldithiocarbamate (PrEt), N-ethyl-N-(2-methoxyethyl)dithiocarbamate (PoEt), N-isopropyl-N-2-methoxyethyldithiocarbamate (IsoMe) and N-(2-ethoxy-2-oxoethyl)-N-methyldithiocarbamate (Pr-13) where obtained in situ, immediately before the synthesis of the corresponding gallium(III) complexes, as detailed in the following methods (ii and iii). The other dithiocarbamates were prepared as sodium salts following the standard way of synthesis as described in the ESI.†Cisplatin (CDDP), MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide), antimycin, CCCP (carbonyl cyanide m-chlorophenyl hydrazone) and RPMI medium without phenol red were obtained from Sigma Chemical Co., St Louis, MO, USA.
2.2 Physical measurements
Carbon, hydrogen, and nitrogen analyses were performed using a Carlo Erba 1106 elemental analyzer. Infrared (IR) spectra were recorded in the range of 4000–400 cm−1 on a Perkin–Elmer 1700 FT-IR spectrometer with Spectrum v. 5.0.1 software (PerkinElmer), using KBr pellets.1H, 13C{1H} and two-dimensional NMR spectra were acquired in the indicated deuterated solvents at 298 K with a Bruker AMX 400 spectrometer with TopSpin 3.2 software. Chemical shifts are reported in ppm and referenced to the internal residual solvent signal for 1H (CD2Cl2: 5.32 ppm, CDCl3: 7.26 ppm; DMSO-d6: 2.50 ppm; D2O: 4.8 ppm) and deuterated solvent signal for 13C (CD2Cl2: 54.00 ppm; CDCl3: 77.00 ppm; DMSO-d6: 39.5 ppm); 13C NMR spectra recorded in D2O were calibrated to external tetramethylsilane. Signal assignments were confirmed by 2D experiments (1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC) where necessary. Common abbreviations for signal multiplicity were used (s = singlet, d = doublets, t = triplets, q = quartets, etc.; bs = broad singlet).
Thin-layer chromatography (TLC) analyses were performed on SiO2 F254S plates (Merck, Milan, Italy), using dichloromethane as the mobile phase.
High performance liquid chromatography (HPLC) analyses were used to evaluate the stability as the variation of absolute area of the peak of compounds 6 and 21. Complexes were analyzed on a Dionex Ultimate 3000 instrument; data were registered and elaborated by using Chromeleon 6.8 software.
2.3 Synthesis of gallium(III) complexes with dithiocarbamates, general procedures
Method i: the relevant dithiocarbamate salt (0.412 mmol, 3 eq.) and gallium nitrate hexahydrate Ga(NO3)3·6H2O (49.8 mg, 0.137 mmol, 1 eq.) were separately dissolved in water (10 mL and 2 mL, respectively). The aqueous solution of Ga(NO3)3·6H2O was added to the aqueous solution of dithiocarbamate under magnetic stirring at room temperature. The prompt formation of a white precipitate was observed. The precipitate was then collected by filtration, washed with water, ethanol, and diethyl ether, and then dried under vacuum.Method ii: NaOH (66.0 mg, 1.65 mmol, 6 eq.) was dissolved in ethanol (15 mL) adding a few mL of water to facilitate dissolution. The hydrochloride salt of the appropriate amine (1.65 mmol, 6 eq.) was dissolved in 5 mL of ethanol. The latter solution was then added to the NaOH solution under magnetic stirring. A white precipitate was observed; the mixture was filtered. The obtained solution was cooled on ice, and then chilled carbon disulfide (198.5 mL, 3.30 mmol, 12 eq.) was added dropwise under magnetic stirring. The mixture was left at room temperature under magnetic stirring. After 3 h, a solution of Ga(NO3)3·6H2O (101.8 mg, 0.280 mmol, 1 eq.) in 2 mL of ethanol was slowly added under magnetic stirring. The prompt formation of a white precipitate was observed. The precipitate was then collected by filtration, washed with water, ethanol, and diethyl ether, and then dried under vacuum.
Method iii: the chosen amine (0.822 mmol, 6 eq.) was dissolved in water (10 mL). The solution was cooled on ice, then carbon disulfide (98.6 mL, 1.64 mmol, 12 eq., dissolved in 2 mL of ethanol) was added dropwise under magnetic stirring. The mixture was left at room temperature under magnetic stirring. After 3 h, an aqueous solution of Ga(NO3)3·6H2O (49.8 mg, 0.137 mmol, 1 eq.) was slowly added under magnetic stirring. The prompt formation of a white precipitate was observed. The precipitate was then collected by filtration, washed with water, ethanol, diethyl ether, and then dried under vacuum.
2.4 Effect of pH on the synthesis of [Ga(DTC)3] complexes
The synthesis of complex 14 was selected as the pilot reaction to evaluate the effect of the pH on the reaction yield. Different buffer systems were used to stabilize the pH at the desired value in the range of 3.5–13.The general procedure is as follows.
DEDC ammonium salt (141.70 mg, 0.852 mmol, 3 eq.) was dissolved in the relevant buffer solution (5 mL). Then, Ga(NO3)3·6H2O (49.8 mg, 0.137 mmol, 1 eq.) was dissolved in water (2 mL) and added under magnetic stirring. The formation of the complex was followed by TLC analysis (SiO2, CH2Cl2, vide supra). The pure complex 14 was achieved as a white precipitate, which was collected by filtration, washed with water, and vacuum dried.
pH 3.5. The buffer solution was prepared using 1 M aqueous solution of tartaric acid; 6 M aqueous NaOH was added to adjust the pH to 3.5. No precipitate was observed. Yield: 0%.
pH 5. The buffer was prepared using 1 M aqueous solution of acetic acid; the pH was adjusted to 5 by the dropwise addition of 6 M aqueous NaOH. Pure complex (14) was obtained. Yield: 54.4%.
pH 6 (autogenous). Non-buffered solution was used. Pure complex (14) was obtained. Yield: 84.2%.
pH 6.8 The buffer solution was prepared using 1 M aqueous solution of MES (2-(n-morpholino)ethanesulfonic acid); 6 M aqueous HCl was added to adjust the pH to 6.8. The pure complex (14) was obtained. Yield: 84.6%.
pH 9. The buffer used was a 1 M aqueous solution of MES. The pure complex (14) was obtained. Yield: 39.5%.
pH 10. The buffer solution was prepared using 1 M aqueous solution of sodium monohydrogen phosphate; the pH was adjusted to 10 by the dropwise addition of 6 M aqueous NaOH. The formation of complex 14 was not detected. Yield: 0%.
pH 13. The buffer solution was obtained from a 1 M aqueous solution of sodium phosphate whose pH was adjusted to 13 by the dropwise addition of 6 M aqueous NaOH. The formation of complex 14 was not detected. Yield: 0%.
2.5 X-ray crystallography
Single crystals of complexes 1, 2, 15, 17, and 22, suitable for the X-ray investigation, were grown by slow diffusion of n-hexane into a dichloromethane solution and dried under a dinitrogen atmosphere. The selected specimens were mounted on the top of a glass capillary and fastened on the goniometer head of an Oxford Diffraction Gemini E diffractometer, equipped with a 2K × 2K EOS CCD area detector and sealed tube Enhance (Mo) and (Cu) X-ray sources, under a gentle dinitrogen stream from an Oxford Instruments CryojetXL sample chiller.Raw diffraction data were collected using the ω-scans technique at 130, 130, 143, 173, and 175 K for complexes 1, 2, 15, 17, and 22 respectively, using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å), in a 1024 × 1024 pixel mode and 2 × 2 pixel binning. Data collection, reduction and finalization were performed using the CrysAlisPro software (Agilent Technologies, Version 1.171.34.47, release 21-12-2010 CrysAlis171 .NET).
Due to laboratory environmental conditions, sample icing has become more problematic in the last three experiments despite the de-icing collection strategy; to circumvent the problem, we resolved to increase the temperature during data collection. Raw diffraction data were corrected for Lorentz and polarization effects, as well as for absorption; an empirical absorption correction was performed by means of a multi-scan approach, using the scaling algorithm SCALE3 ABSPACK, using equivalent reflections (CrysAlisPro, Agilent Technologies, Version 1.171.34.47, release 21-12-2010 CrysAlis171 .NET). Unit cell parameters were obtained by the least-squares refinement of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 340, 4496, 2839, 10470 and 10712 strongest reflections chosen throughout the data collection for 1, 2, 15, 17, and 22, respectively. The crystal and equipment stability were tested every 50 frames by monitoring two reference frames. No change in peak positions or in intensities was observed in all experiments.
340, 4496, 2839, 10470 and 10712 strongest reflections chosen throughout the data collection for 1, 2, 15, 17, and 22, respectively. The crystal and equipment stability were tested every 50 frames by monitoring two reference frames. No change in peak positions or in intensities was observed in all experiments.
The structures were solved by direct methods, using SHELXS67 and refined by standard full-matrix least squares based on Fo2 using the SHELXL-9767 program embedded within the OLEX2 program.68 Usually, non-H atoms were allowed to vibrate anisotropically in the last cycles of refinement, whereas H atoms were placed instead at calculated positions and refined as riding on the pertinent parent atom. In complex 17, two-thirds of the dithiocarbamate side chains were found to be disordered over two positions, whose calculated partial occupancies were restricted to a sum of 1.0. The involved atoms (O2, O4, C8, C9, C13, C14, C17, C18, C22, and C23) have been refined only isotropically with the position defined by a series of DFIX, DANG and SADI restraints. Introducing anisotropy and RIGU restraints when treating these atoms (not involved in the gallium coordination sphere) did not appreciably improve the final model, so we consider the refinement completed at this stage. Likewise, the final steps of refinement of compound 22 revealed a few residual electron density peaks compatible with the second arrangement of some atoms belonging to one of the dithiocarbamate ligands and of the bound gallium atoms. Again, the alternate positions of the disordered atoms (Ga1, S5, S6, C7, and N3) have been refined with calculated partial occupancies restricted to sum to unity, but in this case, we were able to introduce anisotropy, except for the alternate position of atom C7A, that could be refined only isotropically; RIGU restraints were also applied to model the involved sulfur atoms. Appropriate comments were introduced in the pertinent .cif files and other comments were also introduced (when necessary) also in the .cif file of complex 15 to address B-type and some of the C-type alerts that emerged during the checkcif procedure. Full listings of atomic coordinates, bond lengths and angles, and anisotropic thermal parameters are available in the ESI† in the form of .cif files.
2.6 Experiments with cultured human cancer cells
Gallium(III) complexes were dissolved in DMSO just before the experiment (1 mg mL−1), and a calculated amount of drug solution was added to the cell growth medium at a final solvent concentration of 0.5%, which had no detectable effects on cell viability. Cisplatin (CDDP) was dissolved in a 0.9% NaCl solution.Before the experiments, the stability of the compounds in solution was assessed over time (t = 0 min vs. 72 h) by NMR spectroscopy. Samples were prepared by dissolving 2 mg of the compound in a minimum amount of DMSO-d6, then diluted to 1 mL with deuterated physiological saline solution.
In addition, the stability of the best performing compounds, 6 and 21, was also assessed by RP-HPLC, incubating the compounds at 37 °C for 72 h, under conditions close to those of the in vitro cytotoxicity test.
Experiments were performed in triplicate.
3. Results and discussion
3.1 Synthesis
A small library of alicyclic and linear DTC ligands was evaluated and prepared adopting the procedure described in the Experimental section. For the former, progressively larger N-heterocycle rings (5, 6, 7 and 9) were considered, as well as five high-sterically hindered piperidyl dithiocarbamates and two piperazine derivatives. For the latter, the impact of the length of the backbone chain of symmetric and unsymmetric secondary amines, which also include ether and ester functions, was evaluated.A series of gallium(III) complexes with dithiocarbamate ligands, having the general formula [Ga(DTC)3] (where DTC is a generic dithiocarbamate), were synthesized in moderate to high yield (70%–90%), in water or hydroalcoholic solutions, adopting the pathways of synthesis sketched in Scheme 1.
 | ||
| Scheme 1 Synthesis of gallium(III) complexes according to methods i, ii and iii. R1, R2: see Fig. 1. i. H2O, Ga(NO3)·6H2O (1 eq.) dithiocarbamate (3 eq.), RT, few minutes. ii. (1) Ethanol/H2O, amine hydrochloride (6 eq.), NaOH (6 eq.), RT, few minutes; carbon disulfide (12 eq.), RT, 3 h; (2) Ga(NO3)3·6H2O (1 eq.). iii. (1) H2O, amine (6 eq.), carbon disulfide (12 eq.), RT, 3 h; (2) Ga(NO3)3·6H2O (1 eq.), RT, few minutes. | ||
It should be remembered that in an aqueous solution, the Ga3+ cation is subject to hydrolysis processes that lead to the formation of various hydroxide species whose nature depends on the pH of the solution. These hydrolytic equilibria govern the reactivity of aqueous Ga3+ thus affecting the reaction yield.71 Therefore, the reaction pH was considered to select the conditions to limit the formation of unreactive hydroxide species and prevent DTC degradation. Using the formation of complex 14 as an example, reactions were carried out at different pH values ranging from 3.5 to 13. From the data obtained, it is evident that the highest [Ga(DTC)3] yields are found at autogenous pH, or at pH values in the range of 6–7. Lower pH values prevent the formation of the complex. This behavior is most likely due to the intrinsic instability of the DTC ligand at such acidic pH. Even at alkaline pH values the formation of the complex is not observed, and this is probably due to the generation of the tetrahydroxylated complex ([Ga(OH)4]−), favored by the high concentration of hydroxide ions in solution.
Complexes 1–4, 6–15 and 22 and 23 were obtained by mixing the appropriate ligand with Ga(NO3)3·6H2O (stoichiometric ratio 3:1) in water at room temperature, at autogenous pH, leading to precipitation of the product as a white powder (Method i).
Purification of linear dithiocarbamates DPODC, PrEt, PoEt, and IsoMe was difficult; thus, they were isolated directly as Ga complexes. Complexes 16 and 18–20 were prepared in situ by the formation of the relevant dithiocarbamate (starting from the corresponding amine and carbon disulfide). The metal precursor Ga(NO3)3·6H2O was added to the reaction mixture to yield the formation of the corresponding complexes that precipitated as a white powder (Method iii). In this case, no strong bases, such as sodium hydroxide, were used in the initial formation of the dithiocarbamate to completely avoid the subsequent precipitation of gallium hydroxide, thus a larger amount of amine and carbon disulfide was used. Syntheses of complexes 5 and 21 were achieved through a similar procedure, but since the starting amine was provided as a hydrochloride salt, the use of a strong base was mandatory to obtain the corresponding dithiocarbamate (Method ii). Therefore, a minimum amount of sodium hydroxide was used (amine hydrochloride:sodium hydroxide molar ratio 1:1) as described in the Experimental section. Fig. 1 shows the formulae for the obtained complexes.
 | ||
| Fig. 1 Formulae of the complexes 1–23. The organization of the structures in the figure is not according to numbering, but according to structural similarity. | ||
3.2 Characterization
The obtained complexes can be classified into three groups: 1–12 are characterized by ‘alicyclic’ dithiocarbamate ligands, where dithiocarbamic nitrogen belongs to a heterocyclic structure; in contrast, in compounds 13–21, the dithiocarbamate ligands has a linear backbone; in 22 and 23 the dithiocarbamate ligands were derived from a primary amine, hence the dithiocarbamic nitrogen binds a single R group and a hydrogen atom.Most of the complexes are insoluble in water, alcohols, diethylether, and n-hexane and are soluble in chlorinated solvents; the solubility in other organic solvents can change. Complexes 1, 2, 5, 6, 9, 11–13, 17, 20, and 21 are soluble in dimethyl sulfoxide; 3, 4, 7, 10, 14–16 and 18–19 are slightly soluble in dimethylsulfoxide, whereas 8, 22 and 23 are insoluble.
All complexes were characterized by elemental analysis, IR spectroscopy (in the 4000–400 cm−1 region) and NMR spectroscopy (1H and 13C); two-dimensional homo- and heteronuclear NMR experiments (1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC) were performed to assign all proton and carbon resonances observed in one-dimensional spectra when necessary. All results were in agreement with the formulation proposed as a general formula [GaIII(DTC)3].
Mid-IR spectroscopy can be used to confirm the presence of the dithiocarbamate ligands in gallium(III) dithiocarbamate complexes. The spectra of all obtained complexes show the typical strong absorption peak due to the antisymmetric stretching of the dithiocarbamic C–N bond, which appears in the region between 1580 and 1450 cm−1, and the typical medium/strong signal due to the antisymmetric stretching of the dithiocarbamic C–S bonds, falling in the 1060–940 cm−1 region. In general, in the spectra of the complexes, the antisymmetric C–N stretching signal is observed at energies higher than those of signals observed in the free dithiocarbamate spectra, indicating that in the complex the C–N bond has a higher double bond character.72 Regarding the C–S antisymmetric stretching, the presence of a single peak or two peaks with a split lower than 20 cm−1 supports a symmetrical bidentate coordination for each ligand.72
NMR spectra of the obtained complexes show sharp peaks in a narrow window typical of diamagnetic species, in agreement with the d10 electronic configuration of gallium(III). 1H and 13C spectra are basically unexceptional, displaying the signals expected to belong to the dithiocarbamate ligands. In general, chemical shift values do not change significantly upon chelation, however, little variations are found for the resonances of protons and carbons near the NCS2 group. This is in agreement with data reported in the literature for complexes with similar ligands.66,73 Another exception is the signal of dithiocarbamic carbon NCS2 in the 13C NMR spectra of the complexes, which display a significantly lower chemical shift compared to the corresponding signal in the free ligands’ spectra. This occurrence has been connected to the higher double bond character of the C–N bond.63,74
 | ||
| Fig. 2 ORTEP drawings of complexes 1 (a), 2 (b), 15 (c), and 17 (d). Thermal ellipsoids are at the 40% probability level; hydrogen atoms are omitted for clarity. | ||
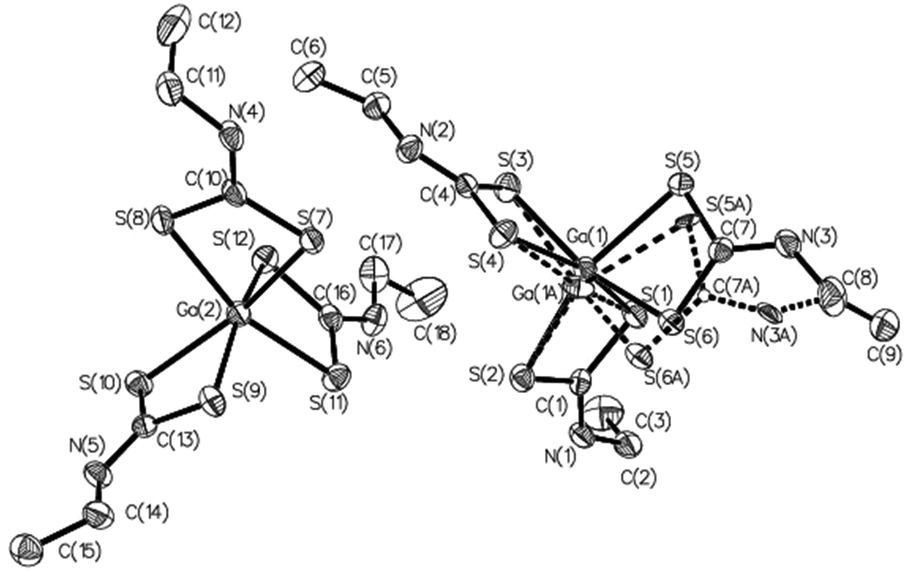 | ||
| Fig. 3 ORTEP drawing of the independent MOL 1 and MOL 2 molecules in the asymmetric unit of complex 22, also showing the alternate positions of disordered atoms in MOL 1. Thermal ellipsoids are at the 50% probability level; hydrogen atoms are omitted for clarity. | ||
| Complex | 1 | 2 | 15 | 17 | 22 |
|---|---|---|---|---|---|
| Empirical formula | C15H24N3S6Ga | C18H30N3S6Ga | C21H42N3S6Ga | C27H54N3O6S6Ga | C9H18N3S6Ga |
| Formula weight | 508.45 | 550.53 | 598.66 | 778.81 | 430.34 |
| Temperature/K | 130(9) | 129.8(3) | 142.9(2) | 172.8(5) | 175(4) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
I2/a |
| a/Å | 13.1191(3) | 13.2161(6) | 10.3567(7) | 10.03891(18) | 17.0141(4) |
| b/Å | 9.2840(2) | 11.9598(4) | 17.1715(14) | 13.4326(2) | 14.1126(3) |
| c/Å | 17.8157(5) | 15.9645(9) | 17.2231(14) | 15.1709(4) | 30.8701(8) |
| α/° | 90.00 | 90.00 | 90.00 | 102.8698(18) | 90.00 |
| β/° | 100.383(2) | 100.562(5) | 90.130(7) | 100.4319(19) | 105.711(3) |
| γ/° | 90.00 | 90.00 | 90.00 | 94.3380(14) | 90.00 |
| Volume/Å3 | 2134.37(9) | 2480.6(2) | 3063.0(4) | 1947.02(7) | 7135.4(3) |
| Crystal size/mm3 | 0.6 × 0.22 × 0.12 | 0.22 × 0.20 × 0.10 | 0.4 × 0.25 × 0.15 | 0.6 × 0.45 × 0.18 | 0.5 × 0.42 × 0.2 |
| Radiation | Mo Kα (λ = 0.7107) | Mo Kα (λ = 0.7107) | Mo Kα (λ = 0.7107) | Mo Kα (λ = 0.7107) | Mo Kα (λ = 0.7107) |
| Collected reflections | 41908 |
31922 |
19381 |
32664 |
34326 |
| Final R (R1;wR2) indexes [I ≥ 2σ(I)] | 0.0289, 0.0607 | 0.0441, 0.0946 | 0.0764, 0.1697 | 0.0385, 0.0882 | 0.0356, 0.0729 |
All complexes are of type ‘2 + 2 + 2’, each having three identical chelating dithiocarbamate ligands coordinating the Ga(III) ion, so that the coordination sphere is characterized by the S6 donor set. Notably, in complex 22 the asymmetric unit (Fig. 3) is formed by two independent and structurally different molecules, one of which is affected by disorder. The ‘inner core’ always has a trigonal prismatic geometry. The dihedral angle between the opposing triangular faces ranges from 0.1° to 8.8°, compared with 0° for a regular trigonal prism. Likewise, the twist angle between the corresponding vertices of opposite triangular faces, as measured from the dihedral angle formed by two sulfur atoms of the same dithiocarbamate ligand and by the geometric centroids of the triangular faces (e.g.: S1–CS1S3S5–CS2S4S6–S2) does not change much, oscillating between 35.8° and 41.5°. A comparison of the structures using Mercury software76 revealed that the “inner cores” of the complexes are essentially superimposable (root-mean-square value in the range of 0.07–0.22 Å, which narrows to 0.07–0.11 Å by ignoring disorder effects). This means that the variation in the molecular structure of the dithiocarbamate ligand does not affect the coordination sphere geometry appreciably (Fig. 4).
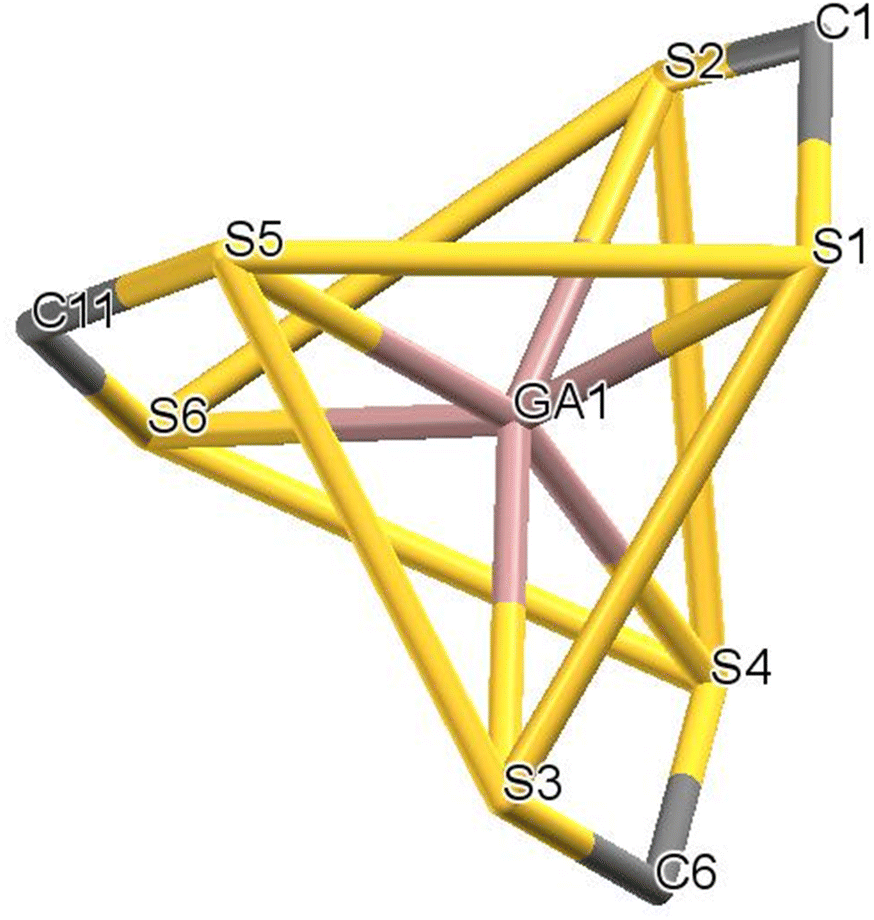 | ||
| Fig. 4 The S6 environment common to all complexes. The opposing triangular faces S1–S3–S5 and S2–S4–S6 are highlighted. | ||
With respect to the distances in the coordination sphere (Table 2), in all complexes the lengths of the six Ga–S bonds are not perfectly identical and a certain variance is allowed; there is always at least one bond that is slightly longer than the others (differences range between +0.01 and +0.09 Å, largely only due to disorder in compound 22). In contrast, the mean Ga–S bond length values for each complex are very close to each other: 2.45 or 2.44 Å for 1 and the independent molecules in the asymmetric unit of 22, and 2.43 Å for the remaining compounds. Finally, the lengths of Ga–S bonds found in this work mostly fit well within the range of distances found in the limited number of reported structures (2.256–2.599 Å, with a mean of 2.409 Å), which include four-, five and six-coordinated gallium species.53,55–57,59,77–82
| Complex | 1 | 2 | 15 | 17 | 22, Mol 1A | 22, Mol 1B | 22, Mol 2 |
|---|---|---|---|---|---|---|---|
| Cdtc = carbon atom of the dithiocarbamate moiety. MOL 1B column of complex 22 lists alternate positions for atoms with sofs = 0.069; MOL 1A column refers to atoms not affected by disorder and to atoms with sofs = 0.931. Distances involving gallium were given with four decimal digits, other ones with three decimals; those for disordered atoms with one decimal digit less. | |||||||
| Ga1–S1 | 2.4551(4) | 2.4515(7) | 2.4228(15) | 2.4633(6) | 2.4191(14) | 2.570(19) | 2.4630(6) |
| Ga1–S2 | 2.4354(4) | 2.4124(6) | 2.4336(14) | 2.4189(6) | 2.4817(11) | 2.201(14) | 2.4076(7) |
| Ga1–S3 | 2.4868(4) | 2.4134(6) | 2.4164(16) | 2.4169(6) | 2.4220(12) | 2.687(15) | 2.4299(7) |
| Ga1–S4 | 2.3973(4) | 2.4435(7) | 2.4327(16) | 2.4445(6) | 2.4340(14) | 2.317(19) | 2.4781(6) |
| Ga1–S5 | 2.4084(4) | 2.4251(6) | 2.4544(15) | 2.4264(6) | 2.4759(11) | 2.518(17) | 2.4283(7) |
| Ga1–S6 | 2.4870(4) | 2.4270(6) | 2.4131(15) | 2.4111(6) | 2.4222(12) | 2.400(18) | 2.4514(7) |
| S1–Cdtc | 1.722(2) | 1.729(2) | 1.713(5) | 1.718(2) | 1.712(2) | 1.715(3) | |
| S2–Cdtc | 1.726(2) | 1.729(2) | 1.714(6) | 1.720(2) | 1.719(2) | 1.711(2) | |
| S3–Cdtc | 1.719(2) | 1.728(2) | 1.728(6) | 1.724(2) | 1.712(2) | 1.707(2) | |
| S4–Cdtc | 1.720(2) | 1.724(2) | 1.702(5) | 1.717(2) | 1.717(2) | 1.726(3) | |
| S5–Cdtc | 1.724(2) | 1.728(2) | 1.712(5) | 1.718(2) | 1.726(3) | 1.68(3) | 1.721(2) |
| S6–Cdtc | 1.719(2) | 1.734(2) | 1.722(5) | 1.727(2) | 1.709(3) | 1.70(3) | 1.712(2) |
| N1–Cdtc | 1.313(2) | 1.319(3) | 1.315(7) | 1.332(3) | 1.319(3) | 1.322(3) | |
| N2–Cdtc | 1.318(2) | 1.323(3) | 1.328(7) | 1.328(3) | 1.316(3) | 1.320(3) | |
| N3–Cdtc | 1.321(2) | 1.324(3) | 1.338(6) | 1.321(3) | 1.320(4) | 1.39(4) | 1.315(3) |
In addition, the bite angles S–Ga–S are very similar (Table 3), both within the same complex as well as across the five complexes, with average values ranging from 73.5° (in complex 22) to 74.0° (in complex 2). These constraints imposed on the coordination sphere are certainly the origin of the common coordination geometry found in these complexes. Because of the trigonal prismatic geometry, the S–Ga–Strans angles (where S and Strans are the sulfur atoms in the reciprocal trans position) deviate significantly from the ideal value of 180° (Table 3): the pertinent mean values range from 159.7° (in complex 1) to 163.3° (in complex 2). Interestingly, the trigonal prismatic geometry is not uncommon in sulfur-rich complexes with c.n. = 6: a survey of the Cambridge database62,83 reveals more than one hundred structures with the coordination sphere having a set of angles similar to those found in this series of compounds.
| Complex | 1 | 2 | 15 | 17 | 22, Mol 1A | 22, Mol 1B | 22, Mol 2 |
|---|---|---|---|---|---|---|---|
| dtc = atom of the dithiocarbamate moiety. Angles involving the gallium atom are given with two decimal digits, other ones are reported with one decimal; and those for disordered atoms with one decimal digit less. | |||||||
| Sdtc–Ga1–Sdtc | 73.84(1) | 73.76(2) | 73.82(5) | 73.46(2) | 73.38(4) | 75.3(5) | 73.97(2) |
| 73.59(1) | 74.13(2) | 73.79(5) | 73.95(2) | 74.24(4) | 71.3(5) | 73.40(2) | |
| 73.51(1) | 74.10(2) | 73.46(5) | 74.01(2) | 73.57(4) | 73.3(6) | 73.60(2) | |
| S–Ga1–S(trans) | 158.04(2) | 164.82(2) | 161.91(6) | 164.51(2) | 164.09(5) | 159.1(7) | 159.36(2) |
| 160.42(2) | 164.41(2) | 160.65(6) | 161.90(2) | 160.63(6) | 153.5(9) | 162.90(2) | |
| 160.78(2) | 160.77(2) | 163.69(6) | 160.67(2) | 162.21(6) | 163.5(9) | 163.13(2) | |
| N1–Cdtc–Sdtc | 122.00(1) | 123.1(2) | 121.7(4) | 122.2(2) | 122.3(2) | 120.6(2) | |
| 121.1(1) | 121.8(2) | 121.6(4) | 121.6(2) | 120.6(2) | 121.8(2) | ||
| N2–Cdtc–Sdtc | 122.2(1) | 121.4(2) | 120.7(4) | 121.2(2) | 121.8(2) | 121.8(2) | |
| 121.2(1) | 122.6(2) | 123.1(4) | 122.4(2) | 120.7(2) | 120.7(2) | ||
| N3–Cdtc–Sdtc | 120.7(1) | 122.4(2) | 122.5(4) | 123.4(2) | 119.6(2) | 124(2) | 120.4(2) |
| 122.6(1) | 122.4(2) | 121.6(4) | 121.3(2) | 123.2(2) | 115(2) | 122.9(2) | |
The C–S and C–N bond lengths in the dithiocarbamate moieties show less variation, with mean values ranging between 1.71 and 1.73 Å and between 1.32 and 1.34 Å, respectively (Table 2). Bond lengths and angles within the dithiocarbamate groups strongly support the presence of extensive π electron delocalization.
With respect to intermolecular interactions, in most cases no canonical hydrogen bonds nor nonbonding interactions with contact distances shorter than 2.7 Å were found. In complex 22 a relatively large unit cell hosts 16 molecules (8 pairs). In each pair, the two molecules are held in position by a limited number of loose sulfur-based hydrogen bonds, supported by other (also loose) non-bonding contacts that help in stabilizing the molecular packing (Fig. S1†). Table S1† reports a list of the tightest nonbonding contacts.
3.3 In vitro cytotoxicity
The newly synthesized Ga-DTC complexes were tested for purity grade and stability in DMSO/deuterated physiological saline solution by NMR spectroscopy and evaluated for their cytotoxic activity against four human cancer cell lines derived from solid tumors with different sensitivity to the reference metal-based chemotherapeutic drug cisplatin: colon (HCT-15, LoVo), pancreatic (BxPC-3) and ovarian (2008) carcinoma cells. For comparison purposes, the cytotoxicity of gallium nitrate and cisplatin were assessed under the same experimental conditions.One of the prerequisites for using Ga-complexes is the stability to degradation to avoid the formation of less soluble [Ga(OH)3] species. Thus, an assessment of the stability of the newly synthesized Ga-DTC complexes in aqueous solutions is necessary to determine their biological activity. The 1H-NMR spectra of the complexes in DMSO-d6 solution and upon dilution with deuterated physiological saline solution were recorded over time (t = 0 min vs. 72 h). As an example, 1H-NMR spectra of 6 and 21 complexes are shown in Fig. S2 and S3.† The spectra did not show variation with time, confirming the stability of the complexes. The good stability of the investigated molecules was also proven by the reproducibility observed in cytotoxicity tests where measurements in the same complex solutions have been replicated over time. Moreover, the stability of the two most active complexes, 6 and 21, under conditions close to those of the in vitro cytotoxicity test has been evaluated by incubating the compounds at 37 °C in the complete RPMI cell culture medium and in phosphate buffer 0.2 M pH 7.4 and performing a suitable RP-HPLC analysis at 0, 24, 48 and 72 hours, evaluating the absolute areas of the compound peaks. No significant variations of the areas have been detected, indicating that the complexes are stable (Fig. 5 and 6).
 | ||
| Fig. 5 (A) RP-HPLC chromatogram of complex 6 (1 mg mL−1 solution in DMSO). (B) RP-HPLC chromatogram of RPMI-1640 containing 10% fetal calf serum with 10% DMSO. (C) RP-HPLC chromatogram of phosphate buffer solution, 0.2 M, pH 7.4, with 10% DMSO. (D) RP-HPLC chromatograms of complex 6 in RPMI-1640 containing 10% fetal calf serum at 37 °C after 0, 24, 48 and 72 h. (E) RP-HPLC chromatograms of complex 6 in phosphate buffer 0.2 M, pH 7.4, at 37 °C after 0, 24, 48 and 72 h. (F) Area (mAU min) of the complex 6 peaks after each incubation time in the experimental medium. | ||
 | ||
| Fig. 6 (A) RP-HPLC chromatogram of complex 21 (1 mg mL−1 solution in DMSO). (B) RP-HPLC chromatogram of RPMI-1640 containing 10% fetal calf serum with 10% DMSO. (C) RP-HPLC chromatogram of phosphate buffer solution, 0.2 M, pH 7.4, with 10% DMSO. (D) RP-HPLC chromatograms of complex 21 in RPMI-1640 containing 10% fetal calf serum at 37 °C after 0, 24, 48 and 72 h. (E) RP-HPLC chromatograms of complex 21 in phosphate buffer 0.2 M, pH 7.4, at 37 °C after 0, 24, 48 and 72 h. (F) Area (mAU min) of the complex 21 peaks after each incubation time in the experimental medium. | ||
Cytotoxicity parameters, expressed in terms of IC50 values calculated from the dose-survival curves obtained after 72 h of exposure in the MTT assay, along with the LogP values of the free DTC calculated using MOLINSPIRATION (Cheminformatics) are reported in Table 4. Compounds 8, 22 and 23 were not tested due to their insolubility in dimethyl sulfoxide.
| IC50 (μM) ± S.D. | |||||
|---|---|---|---|---|---|
| HCT-15 | BxPC-3 | LoVo | 2008 | LogP DTCa |
|
| Cells (3–8 × 103 per well) were treated for 72 h with increasing concentrations of the tested compounds. Cytotoxicity was assessed by the MTT test. The IC50 values were calculated by the four-parameter logistic model (p < 0.05).a LogP values of the ligands were calculated using MOLINSPIRATION (Cheminformatics).b LogP values of hydrolyzed species. |
|||||
| 1 | 1.2 ± 0.2 | 4.4 ± 0.8 | 0.8 ± 0.5 | 2.2 ± 0.8 | 1.47 |
| 2 | 2.5 ± 0.4 | 2.8 ± 0.6 | 1.0 ± 0.2 | 2.9 ± 1.0 | 1.98 |
| 3 | 15.6 ± 3.1 | 17.2 ± 1.5 | 9.7 ± 1.6 | 9.7 ± 3.2 | 1.34 |
| 4 | 20.3 ± 3.8 | 29.5 ± 3.8 | 14.2 ± 2.3 | 17.4 ± 2.7 | 3.17 |
| 5 | >50 | >50 | >50 | >50 | 0.64 |
| 6 | 0.8 ± 0.1 | 0.3 ± 0.1 | 1.5 ± 0.6 | 0.9 ± 0.2 | 2.08 (1.09)b |
| 7 | 5.9 ± 1.2 | 4.3 ± 0.7 | 7.2 ± 1.9 | 6.4 ± 2.3 | 1.35 |
| 8 | — | — | — | — | 1.33 |
| 9 | 3.2 ± 0.3 | 4.7 ± 1.2 | 2.5 ± 0.6 | 2.3 ± 0.4 | 2.48 |
| 10 | 2.3 ± 0.3 | 2.9 ± 0.4 | 3.8 ± 0.5 | 5.5 ± 1.1 | 2.99 |
| 11 | 27.8 ± 3.5 | 19.8 ± 3.1 | 29.3 ± 3.6 | 37.5 ± 2.9 | 3.65 |
| 12 | 2.1 ± 0.5 | 3.7 ± 1.5 | 6.1 ± 0.6 | 5.6 ± 0.9 | 2.67 |
| 13 | 3.3 ± 1.5 | 3.5 ± 0.5 | 4.1 ± 1.1 | 2.9 ± 0.4 | 1.07 |
| 14 | 2.3 ± 0.4 | 3.8 ± 1.1 | 0.7 ± 0.1 | 2.9 ± 0.9 | 1.82 |
| 15 | 7.4 ± 2.1 | 5.9 ± 1.1 | 3.2 ± 0.8 | 4.3 ± 1.0 | 2.83 |
| 16 | 2.4 ± 0.3 | 3.9 ± 1.0 | 2.4 ± 0.3 | 3.2 ± 0.7 | 1.04 |
| 17 | 5.5 ± 1.3 | 3.9 ± 0.7 | 8.4 ± 2.5 | 7.6 ± 2.2 | 1.79 |
| 18 | 3.2 ± 1.0 | 2.8 ± 0.5 | 3.1 ± 0.9 | 2.9 ± 0.3 | 2.32 |
| 19 | 4.5 ± 1.4 | 5.2 ± 1.7 | 6.1 ± 0.8 | 5.3 ± 0.6 | 1.43 |
| 20 | 11.5 ± 2.3 | 7.5 ± 1.4 | 4.9 ± 0.7 | 6.1 ± 0.9 | 1.73 |
| 21 | 1.3 ± 0.3 | 1.8 ± 0.5 | 1.2 ± 0.3 | 0.4 ± 0.2 | 1.31 (−0.60)b |
| 22 | — | — | — | — | 1.20 |
| 23 | — | — | — | — | 2.45 |
| Cisplatin | 12.8 ± 2.3 | 6.2 ± 1.1 | 9.3 ± 2.4 | 2.2 ± 1.7 | |
| Ga(NO 3 ) 3 | >100 | >100 | >100 | >100 | |
As previously observed for other classes of gallium-based compounds, bioactivity may be influenced by many factors; besides the lipophilic nature of the ligands, which may guarantee larger cancer cell accumulation through passive diffusion, other aspects should be considered, such as the presence of heteroatoms that, aside from modifying lipophilicity, may influence the charge distribution of the gallium(III) complex varying electrical binding to cell molecular targets and consequently its potential anticancer activity.84
Cytotoxicity results showed that, except compound 5, both ‘alicyclic’ and ‘linear’ DTC gallium complexes exhibited a noticeable antiproliferative activity, with IC50 values in the micromolar range against all tested cancer cell lines. Notably, most of the Ga-DTC compounds were more effective than cisplatin in inhibiting cancer cell growth, even in the case of cancer cells endowed with poor sensitivity to cisplatin, such as LoVo and HCT-15 colon cancer cells, suggesting for this class of metal complexes a mechanism of action different from that of cisplatin. Only complexes 3, 4 and 11 were less potent, showing IC50 values significantly higher than those calculated for the reference drug. A comparison with gallium nitrate was also accomplished under the same experimental conditions. Gallium nitrate was ineffective in inhibiting cancer cell growth.
However, when analyzing the results as a whole, it seems rather difficult to define straightforward structure–activity relationships for the two series of ‘alicyclic’ and ‘linear’ Ga-DTC compounds.
Among the ‘alicyclic’ Ga-DTC derivatives, the cytotoxic profiles of compounds 1, 2, 9 and 10 were on average very similar, independently of the size of the unsubstituted heterocyclic structure. Inserting an ester group at position 4 of the piperidine moiety of PipDTC, as in CEPipDTC in complex 6, increases the antiproliferative activity by 3–10 times over the parent complex 2. The insertion of a cyclic ether group at the same position (7) slightly reduces the cytotoxic effect of the complex. Except for 12, any other modification on the piperidine backbone, such as the inclusion of an additional heteroatom or a carbonyl group, as in 3 and 5, as well as the attachment of bulky and lipophilic substituents, as in 4, 8 and 11, significantly hampered the antiproliferative effect on cancer cells.
Taking into account the series of ‘linear’ Ga-DTC compounds, the cytotoxic potency is dependent on the chemical nature of the backbone chain, i.e. on the steric hindrance of the ligands. Complexes characterized by simple alkyl DTC are generally more active; the insertion of alkoxyalkyl substituents does not result in a significant increase in cytotoxic activity. The presence of an ethoxy-carbonyl moiety again leads to the most active compound (21) as observed for the alicyclic series.
From this preliminary screening, the ‘alicyclic’ Ga-DTC derivative 6, and the ‘linear’ Ga-DTC derivative 21, carrying an ethoxy-carbonyl moiety as a substituent in the N function both showed impressive activity. Compound 6 was on average approximately three times more cytotoxic than cisplatin and against HCT-15 cells was approximately 16 times more effective in inhibiting cancer cell growth.
An attempt was made to correlate cytotoxicity with the lipophilicity of the ligands (Table 4). In general, it is observed that the more active complexes are associated with less lipophilic dithiocarbamates. In this connection, 6 and 21 deviate from this behavior; however, envisioning a possible explanation for their remarkable antiproliferative activity in all investigated cell lines, we can evocate the hydrolysis of the ester group by the esterase inside the cells, and the conversion of the complexes in the most hydrophilic species. Considering that the compounds are stable in the complete medium, this event might occur inside the cells.
One of the major drawbacks of chemotherapeutics, including metal-based drugs, is the side effects originating in part from toxic effects to non-cancerous cells. We measured the cytotoxicity of the most effective derivatives against two non-cancer cell lines (CHO and HEK-293) and calculated the selectivity index (SI) defined as non-tumor/tumor cell lines. The values were compared with those obtained for cisplatin as a reference compound (Table 5). The reported data showed that 6 and 21 complexes had no selectivity towards cancer cells.
| IC50 (μM) ± S.D. | |||
|---|---|---|---|
| HEK293 | CHO | S.I. | |
| Cells (3–8 × 103 per well) were treated for 72 h with increasing concentrations of the tested compounds. The cytotoxicity was assessed by the MTT test. IC50 values were calculated using a four-parameter logistic model 4-PL (P < 0.05). *Selectivity index (SI) is defined as IC50 non-tumor/tumor cell lines. | |||
| 6 | 1.1 ± 0.1 | 2.7 ± 0.3 | 2.2 |
| 21 | 1.4 ± 0.6 | 3.0 ± 0.9 | 1.9 |
| Cisplatin | 19.1 ± 3.6 | 23.5 ± 2.3 | 2.7 |
Preliminary studies on anticancer mechanisms of the synthesized gallium compounds showed that they did not lead to increased cellular ROS production, nor mitochondrial dysfunction in treated HCT-15 cells (see the ESI, Fig. S4 and S5†). In addition, as recently disulphide isomerase (PDI) is emerging as a molecular target for some classes of gallium complexes,85 we also evaluated the ability of our complexes to act as PDI inhibitors by a biochemical colorimetric method (Proteostat kit) but no notable inhibition of PDI was observed consequent to treatment with gallium(III) compounds (see the ESI, Fig. S6†).
Further investigations, such as those that assess their ability to induce the impairment of DNA synthesis, have to be performed to gain insights into their mechanism of action.
4. Conclusion
A series of gallium(III) complexes with alicyclic and linear dithiocarbamate ligands were prepared and characterized. The obtained data support the general formulation [GaIII(DTC)3] and agree well with the existing literature. The chemical nature of the dithiocarbamate ligand does not have a marked influence over the structural features of the resulting complexes.In general, the newly synthesized Ga-DTC complexes showed a marked cytotoxic activity, even against human colon cancer cells with poor sensitivity to cisplatin. The preliminary screening allowed the identification of [Ga(CEPipDTC)3] (6) and [Ga(Pr-13)3] (21) as very promising derivatives, thus providing a foundation for developing gallium-dithiocarbamate complexes as anticancer agents. The accessibility of radioactive isotopes 67/68Ga for the preparation of the corresponding radioactive complexes may offer an important option for a precise evaluation of the pharmacokinetic properties of such compounds and for possible theranostic applications.
Author contributions
Conceptualization, design, and project administration: Cristina Bolzati and Nicola Salvarese. Product syntheses and spectroscopic characterization: Nicolò Morellato, Carolina Gobbi and Nicola Salvarese. X-ray work: Alessandro Dolmella. In vitro cytotoxicity assays: Valentina Gandin, Michele De Franco and Cristina Marzano. Analysis of results: Cristina Bolzati and Nicola Salvarese. First draft organization: Nicola Salvarese. Funding acquisition: Cristina Bolzati and Cristina Marzano.Each author contributed to drafting the paragraphs concerning the experimental parts of their competence and critically revising the manuscript drafts. All authors have given their final approval to the version of the submitted manuscript.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Dr Valentina Bacciolo for her help in the experimental work. This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC, IG 2020 ID 24528) and the University of Padova (PRID BIRD225980).References
- M. Claudel, J. V. Schwarte and K. M. Fromm, Chemistry, 2020, 2, 849–899 CrossRef CAS.
- E. Mitidieri, D. Visaggio, E. Frangipani, C. Turnaturi, D. Vanacore, R. Provenzano, G. Costabile, R. Sorrentino, F. Ungaro, P. Visca and R. d'Emmanuele di Villa Bianca, Pharmacol. Res., 2021, 170, 105698–105706 CrossRef CAS PubMed.
- F. Trudu, F. Amato, P. Vaňhara, T. Pivetta, E. M. Peña-Méndez and J. Havel, J. Appl. Biomed., 2015, 25 Search PubMed.
- X.-X. Peng, S. Gao and J.-L. Zhang, Eur. J. Inorg. Chem., 2022, 2022, e202100953–e202100953 CrossRef CAS.
- F. Kratz, B. Nuber, J. Weiß and B. K. Keppler, Synth. React. Inorg. Met.-Org. Chem., 1991, 21, 1601–1615 CrossRef CAS.
- F. Kratz, B. Nuber, J. Weiß and B. K. Keppler, Polyhedron, 1992, 11, 487–498 CrossRef CAS.
- V. de A. W. Sales, T. R. R. Timóteo, N. M. da Silva, C. G. de Melo, A. S. Ferreira, M. V. G. de Oliveira, E. de O. Silva, L. M. dos S. Mendes, L. A. Rolim and P. J. R. Neto, Curr. Med. Chem., 2021, 28, 2062–2076 CrossRef.
- M. U. Khan, S. Khan, S. El-Refaie, Z. Win, D. Rubello and A. Al-Nahhas, Eur. J. Surg. Oncol., 2009, 35, 561–567 CrossRef CAS PubMed.
- S. R. Banerjee and M. G. Pomper, Appl. Radiat. Isot., 2013, 76, 2–13 CrossRef CAS.
- P. Mikuš, M. Melník, A. Forgácsová, D. Krajčiová and E. Havránek, Main Group Met. Chem., 2014, 37, 53–65 Search PubMed.
- A. R. Jalilian, Iran. J. Nucl. Med., 2016, 24, 1–10 CAS.
- P. J. Blower, R. Cusnir, A. Darwesh, N. J. Long, M. T. Ma, B. E. Osborne, T. W. Price, J. Pellico, G. Reid, R. Southworth, G. J. Stasiuk, S. Y. A. Terry and R. T. M. de Rosales, in Advances in Inorganic Chemistry, ed. C. D. Hubbard and R. van Eldik, Academic Press, 2021, vol. 78, pp. 1–35 Search PubMed.
- M. Inubushi, H. Miura, I. Kuji, K. Ito and R. Minamimoto, Ann. Nucl. Med., 2020, 34, 879–883 CrossRef.
- K. A. Morgan and P. S. Donnelly, in Advances in Inorganic Chemistry, ed. C. D. Hubbard and R. van Eldik, Academic Press, 2021, vol. 78, pp. 37–63 Search PubMed.
- L. Todorov and I. Kostova, Molecules, 2023, 28, 1959 CrossRef CAS PubMed.
- C. R. Chitambar, Future Med. Chem., 2012, 4, 1257–1272 CrossRef CAS PubMed.
- R. A. Newman, A. R. Brody and I. H. Krakoff, Cancer, 1979, 44, 1728–1740 CrossRef CAS.
- A. Levina, D. C. Crans and P. A. Lay, Coord. Chem. Rev., 2017, 352, 473–498 CrossRef CAS.
- L. R. Bernstein, T. Tanner, C. Godfrey and B. Noll, Met.-Based Drugs, 2000, 7, 33–47 CrossRef CAS PubMed.
- C. R. Chitambar, D. P. Purpi, J. Woodliff, M. Yang and J. P. Wereley, J. Pharmacol. Exp. Ther., 2007, 322, 1228–1236 CrossRef CAS PubMed.
- A. R. Timerbaev, Metallomics, 2009, 1, 193–198 CrossRef CAS.
- R.-D. Hofheinz, C. Dittrich, M. A. Jakupec, A. Drescher, U. Jaehde, M. Gneist, N. G. V. Keyserlingk, B. K. Keppler and A. Hochhaus, Int. J. Clin. Pharmacol. Ther., 2005, 43, 590–591 CrossRef CAS PubMed.
- L. R. Bernstein, J. J. M. van der Hoeven and R. O. Boer, Adv. Anticancer Agents Med. Chem., 2011, 11, 585–590 CrossRef CAS PubMed.
- R. Gogna, E. Madan, B. Keppler and U. Pati, Br. J. Pharmacol., 2012, 166, 617–636 CrossRef CAS PubMed.
- FDA Awards Orphan Drug Designation to Gallium Maltolate for Pediatric Glioblastoma Multiforme, https://www.onclive.com/view/fda-awards-orphan-drug-designation-to-gallium-maltolate-for-pediatric-glioblastoma-multiforme, (accessed December 12, 2023).
- J. A. Lessa, M. A. Soares, R. G. dos Santos, I. C. Mendes, L. B. Salum, H. N. Daghestani, A. D. Andricopulo, B. W. Day, A. Vogt and H. Beraldo, BioMetals, 2013, 26, 151–165 CrossRef CAS PubMed.
- J. Qi, J. Deng, K. Qian, L. Tian, J. Li, K. He, X. Huang, Z. Cheng, Y. Zheng and Y. Wang, Eur. J. Med. Chem., 2017, 134, 34–42 CrossRef CAS PubMed.
- R. Shakya, F. Peng, J. Liu, M. J. Heeg and C. N. Verani, Inorg. Chem., 2006, 45, 6263–6268 CrossRef CAS PubMed.
- D. Chen, M. Frezza, R. Shakya, Q. C. Cui, V. Milacic, C. N. Verani and Q. P. Dou, Cancer Res., 2007, 67, 9258–9265 CrossRef CAS PubMed.
- E. Fischer-Fodor, A.-M. Vălean, P. Virag, P. Ilea, C. Tatomir, F. Imre-Lucaci, M. P. Schrepler, L. T. Krausz, L. B. Tudoran, C. G. Precup, I. Lupan, E. Hey-Hawkins and L. Silaghi-Dumitrescu, Metallomics, 2014, 6, 833–844 CrossRef CAS PubMed.
- T. O. Ajiboye, T. T. Ajiboye, R. Marzouki and D. C. Onwudiwe, Int. J. Mol. Sci., 2022, 23, 1317 CrossRef CAS PubMed.
- S. D. Shinde, A. P. Sakla and N. Shankaraiah, Bioorg. Chem., 2020, 105, 104346–104364 CrossRef CAS PubMed.
- L. Kaul, R. Süss, A. Zannettino and K. Richter, iScience, 2021, 24, 102092–102106 CrossRef CAS PubMed.
- P. J. Heard, in Progress in Inorganic Chemistry, John Wiley & Sons, Ltd, 2005, pp. 1–69 Search PubMed.
- G. Hogarth, in Progress in Inorganic Chemistry, John Wiley & Sons, Ltd, 2005, pp. 71–561 Search PubMed.
- G. Hogarth, Mini-Rev. Med. Chem., 2012, 12, 1202–1215 CrossRef CAS PubMed.
- C. K. Adokoh, RSC Adv., 2020, 10, 2975–2988 RSC.
- C. I. Yeo, E. R. T. Tiekink and J. Chew, Inorganics, 2021, 9, 48–73 CrossRef CAS.
- G. Hogarth and D. C. Onwudiwe, Inorganics, 2021, 9, 70–117 CrossRef CAS.
- D. J. Berry, R. T. M. de Rosales, P. Charoenphun and P. J. Blower, Mini-Rev. Med. Chem., 2012, 12, 1174–1183 CrossRef CAS PubMed.
- T. Liu, Q. Gan, J. Zhang, Z. Jin, W. Zhang and Y. Zhang, MedChemComm, 2016, 7, 1381–1386 RSC.
- N. Salvarese, D. Carta, C. Marzano, G. Gerardi, L. Melendez-Alafort and C. Bolzati, J. Med. Chem., 2018, 61, 11114–11126 CrossRef CAS PubMed.
- J. Borràs, J. Foster, R. Kashani, L. Meléndez-Alafort, J. Sosabowski, J. Suades and R. Barnadas-Rodríguez, Molecules, 2021, 26, 2373 CrossRef PubMed.
- A. Shegani, M. Ischyropoulou, I. Roupa, C. Kiritsis, K. Makrypidi, A. Papasavva, C. Raptopoulou, V. Psycharis, H. M. Hennkens, M. Pelecanou, M. S. Papadopoulos and I. Pirmettis, Bioorg. Med. Chem., 2021, 47, 116373 CrossRef CAS PubMed.
- C. Bolzati, E. Benini, M. Cavazza-Ceccato, E. Cazzola, E. Malagò, S. Agostini, F. Tisato, F. Refosco and G. Bandoli, Bioconjugate Chem., 2006, 17, 419–428 CrossRef CAS PubMed.
- C. Bolzati, M. Cavazza-Ceccato, S. Agostini, F. Refosco, Y. Yamamichi, S. Tokunaga, D. Carta, N. Salvarese, D. Bernardini and G. Bandoli, Bioconjugate Chem., 2010, 21, 928–939 CrossRef CAS PubMed.
- A. Boschi, C. Bolzati, L. Uccelli and A. Duatti, Nucl. Med. Biol., 2003, 30, 381–387 CrossRef CAS PubMed.
- S. Thieme, S. Agostini, R. Bergmann, J. Pietzsch, H.-J. Pietzsch, D. Carta, N. Salvarese, F. Refosco and C. Bolzati, Nucl. Med. Biol., 2011, 38, 399–415 CrossRef CAS PubMed.
- R. Pasqualini and A. Duatti, J. Chem. Soc., Chem. Commun., 1992, 1354–1355 RSC.
- A. Boschi, L. Uccelli, A. Duatti, P. Colamussi, C. Cittanti, A. Filice, A. H. Rose, A. A. Martindale, P. G. Claringbold, D. Kearney, R. Galeotti, H. J. Turner and M. Giganti, Nucl. Med. Commun., 2004, 25, 691 CrossRef CAS PubMed.
- M. B. Mallia, V. Chirayil and A. Dash, Appl. Radiat. Isot., 2018, 137, 147–153 CrossRef CAS PubMed.
- L. Que and L. H. Pignolet, Inorg. Chem., 1974, 13, 351–356 CrossRef CAS.
- K. Dymock, G. J. Palenik, J. Slezak, C. L. Raston and A. H. White, J. Chem. Soc., Dalton Trans., 1976, 28–32 RSC.
- S. W. Haggata, M. A. Malik, M. Motevalli, P. O'Brien and J. C. Knowles, Chem. Mater., 1995, 7, 716–724 CrossRef CAS.
- S. Bhattacharya, N. Seth, D. K. Srivastava, V. D. Gupta, H. Nöth and M. Thomann-Albach, J. Chem. Soc., Dalton Trans., 1996, 2815–2820 RSC.
- P. C. Andrews, S. M. Lawrence, C. L. Raston, B. W. Skelton, V.-A. Tolhurst and A. H. White, Inorg. Chim. Acta, 2000, 300–302, 56–64 CrossRef CAS.
- D. P. Dutta, V. K. Jain, A. Knoedler and W. Kaim, Polyhedron, 2002, 21, 239–246 CrossRef CAS.
- S.-A. Chae, O.-H. Han and W.-S. Jung, Bull. Korean Chem. Soc., 2009, 30, 2762–2764 CrossRef CAS.
- I. L. Fedushkin, A. S. Nikipelov, A. A. Skatova, O. V. Maslova, A. N. Lukoyanov, G. K. Fukin and A. V. Cherkasov, Eur. J. Inorg. Chem., 2009, 2009, 3742–3749 CrossRef.
- K. Ramalingam, G. S. Sivagurunathan and C. Rizzoli, Main Group Met. Chem., 2015, 38, 75–82 CAS.
- N. I. Gorshkov, A. Y. Murko, I. I. Gavrilova, M. A. Bezrukova, A. I. Kipper, V. D. Krasikov and E. F. Panarin, Molecules, 2020, 25, 4681–4700 CrossRef CAS PubMed.
- A. Kassymbek, J. F. Britten, D. Spasyuk, B. Gabidullin and G. I. Nikonov, Inorg. Chem., 2019, 58, 8665–8672 CrossRef CAS PubMed.
- I. P. Ferreira, G. M. de Lima, E. B. Paniago, J. A. Takahashi and C. B. Pinheiro, J. Coord. Chem., 2014, 67, 1097–1109 CrossRef CAS.
- C. Bolzati, N. Salvarese, D. Carta, F. Refosco, A. Dolmella, H. J. Pietzsch, R. Bergmann and G. Bandoli, J. Biol. Inorg. Chem., 2011, 16, 137–155 CrossRef CAS PubMed.
- C. Bolzati, M. Cavazza-Ceccato, S. Agostini, F. Refosco, Y. Yamamichi, S. Tokunaga, D. Carta, N. Salvarese, D. Bernardini and G. Bandoli, Bioconjugate Chem., 2010, 21, 928–939 CrossRef CAS PubMed.
- N. Salvarese, N. Morellato, A. Venzo, F. Refosco, A. Dolmella and C. Bolzati, Inorg. Chem., 2013, 52, 6365–6377 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. a. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, Cancer Res., 1988, 48, 589–601 CAS.
- M. Pellei, C. Santini, L. Bagnarelli, C. Battocchio, G. Iucci, I. Venditti, C. Meneghini, S. Amatori, P. Sgarbossa, C. Marzano, M. De Franco and V. Gandin, Int. J. Mol. Sci., 2022, 23, 9397 CrossRef CAS PubMed.
- S. A. Wood and I. M. Samson, Ore Geol. Rev., 2006, 28, 57–102 CrossRef.
- D. A. Brown, W. K. Glass and M. A. Burke, Spectrochim. Acta, Part A, 1976, 32, 137–143 CrossRef.
- A. Z. Halimehjani, K. Marjani, A. Ashouri and V. Amani, Inorg. Chim. Acta, 2011, 373, 282–285 CrossRef CAS.
- H. L. M. Van Gaal, J. W. Diesveld, F. W. Pijpers and J. G. M. Van der Linden, Inorg. Chem., 1979, 18, 3251–3260 CrossRef CAS.
- C. K. Jhonson, ORTEP Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, TN, 1976 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- E. M. Gordon, A. F. Hepp, S. A. Duraj, T. S. Habash, P. E. Fanwick, J. D. Schupp, W. E. Eckles and S. Long, Inorg. Chim. Acta, 1997, 257, 247–251 CrossRef CAS.
- X. Zhou, M. L. Breen, Stan A. Duraj and A. F. Hepp, Main Group Met. Chem., 1999, 22, 35–40 CAS.
- A. Keys, S. G. Bott and A. R. Barron, J. Chem. Crystallogr., 1998, 28, 629–634 CrossRef CAS.
- W. Uhl, M. Willeke, A. Hepp, D. Pleschka and M. Layh, Z. Anorg. Allg. Chem., 2017, 643, 387–397 CrossRef CAS.
- A. Keys, S. G. Bott and A. R. Barron, Chem. Mater., 1999, 11, 3578–3587 CrossRef CAS.
- I. L. Fedushkin, O. V. Kazarina, A. N. Lukoyanov, A. A. Skatova, N. L. Bazyakina, A. V. Cherkasov and E. Palamidis, Organometallics, 2015, 34, 1498–1506 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- J. Qi, T. Liu, W. Zhao, X. Zheng and Y. Wang, RSC Adv., 2020, 10, 18553–18559 RSC.
- H.-Y. Yin, J.-J. Gao, X. Chen, B. Ma, Z.-S. Yang, J. Tang, B.-W. Wang, T. Chen, C. Wang, S. Gao and J.-L. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20147–20153 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full listings of atomic coordinates, bond lengths and angles and anisotropic thermal parameters for the crystallographic structures are available in the ESI in the form of .cif files. Procedures for the synthesis of dithiocarbamates sodium 4-(4-clorophenyl)-4-hydroxypiperidine dithiocarbamate (NaCPHPDTC), sodium 4-morpholinepiperidine dithiocarbamate (NaMPipDTC), sodium hexamethyleneimine dithiocarbamate (NaAzepamDTC), sodium heptamethyleneimine dithiocarbamate (NaAzocanDTC), sodium 4-benzylpiperidine dithiocarbamate (NaBzPipDTC) and sodium dipropyldithiocarbamate (NaDPDC); 1H NMR spectra of gallium(III) complexes, crystal data of complex 22 (Fig. S1 and Table S1), over time stability of complexes 6 and 21 in DMSO/saline solution (Fig. S2–S3), and figures related to anticancer mechanism tests (Fig. S4–S6). CCDC 2303003–2303007. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03552b |
| ‡ Present address: Dipartimento di Scienze del Farmaco, Università degli Studi di Padova, Via Marzolo 5, 35131 Padua, Italy. |
| This journal is © The Royal Society of Chemistry 2024 |