
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkaline earth metals: homometallic bonding
Josef T.
Boronski

Chemistry Research Laboratory, Department of Chemistry, Oxford, OX1 3TA, UK. josef.boronski@sjc.ox.ac.uk
First published on 30th November 2023
Abstract
The study of alkaline earth metal complexes is undergoing a renaissance. Stable molecular species featuring Mg–Mg bonds were reported in 2007 and their reactivity has since been intensively investigated. Motivated by this work, efforts have also been devoted to the synthesis of complexes featuring Be–Be and Ca–Ca bonds. These collective endeavours have revealed that the chemistry of the group 2 metals is richer and more complex than had previously been appreciated. Here, a discussion of the nature of homometallic alkaline earth bonding is presented, recent synthetic advances are described, and future directions are considered.
 Josef T. Boronski | Josef completed his undergraduate degree at the University of York in 2017 and subsequently undertook a PhD with Prof. S. T. Liddle at the University of Manchester. In 2021, Josef began a Junior Research Fellowship at St John's College, University of Oxford, hosted in the laboratories of Prof. S. Aldridge. Josef's research interests include the synthesis and reactivity of main group and actinide complexes, which feature low-valent element centres or unusual bonding linkages. In 2023, he received the Dalton Emerging Researcher Award by the Royal Society of Chemistry. |
Introduction
Group 2 of the periodic table – the alkaline earth (Ae) elements – comprises a unique cast of characters. The lightest group member, beryllium, is one of the rarest elements in the universe as it is consumed by fusion reactions in stellar interiors.1 Given their remarkably low density and high stiffness, beryllium alloys (and the metal itself) find applications in aerospace engineering.2 Radium, the heaviest (reported) Ae element, is radioactive and has no stable isotopes.3 As a result, 223Ra (t1/2 = 11.43 days), which is a potent α-particle emitter, has found use in radiopharmaceuticals.4 In contrast to these two exotic metals, the other alkaline earth elements – magnesium, calcium, strontium, and barium – are all rather abundant and cheap.5 Hence, from the perspective of sustainability, the use of Ae elements to facilitate industrially relevant chemical transformations is attractive, and has received considerable attention in recent years.5,6Decamethyldizincocene – the first stable complex of zinc(I) with a Zn–Zn bond – was reported in 2004.7 Due to the diagonal relationship between zinc and magnesium, this discovery inspired both synthetic and theoretical chemists to investigate whether the isolation of complexes with homometallic alkaline earth metal–metal (Ae–Ae) bonds might be possible.8 Consequently, in 2007, the frontiers of molecular Ae chemistry were redefined by the report of [(DippNacnac)Mg]2 (1; DippNacnac = {[(Dipp)NC(Me)]2CH}−, Dipp = 2,6-diisopropylphenyl) and [(DippPriso)Mg]2 (2; DippPriso = {[(Dipp)N]2C[NiPr2]}−) – stable complexes of magnesium(I) which feature Mg–Mg bonds (Fig. 1).9 In subsequent years, numerous complexes with Mg–Mg bonds have been prepared, their reactivity has been intensively investigated, and the nature of the Mg–Mg linkage has been probed by a range of computational and analytical methods.10–12 Indeed, magnesium(I) dimers have been described as “quasi-universal” reductants for chemical synthesis.13 However, despite the increasing ubiquity of complexes with Mg–Mg bonds, efforts to prepare homometallic bonds involving other Ae elements have been, until very recently, unsuccessful.14–16
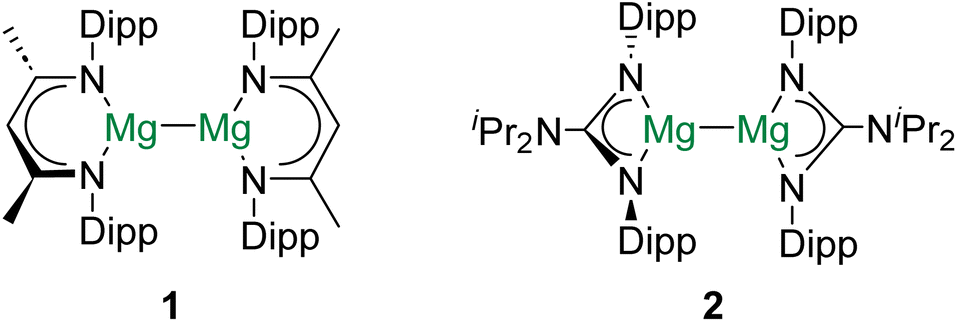 | ||
| Fig. 1 Magnesium(I) dimers 1 and 2. The first isolated complexes that feature Ae–Ae bonds. | ||
This article will review recent advances in the field of alkaline earth homometallic bonding. It also seeks to highlight lessons from theoretical investigations into Ae–Ae bonding, as well as those from other blocks of the Periodic Table. Thus, it is hoped it will inspire new approaches to the synthesis of complexes with Ae–Ae bonds.
Bonding considerations
Numerous theoretical investigations into the stability of complexes of the type XAeAeX, with two alkaline earth metals in the +1-oxidation state and an Ae–Ae bond, have been published.8,17–21 Many highlight the need for bulky X-groups, which kinetically stabilize the Ae–Ae bond and inhibit disproportionation to AeX2 and Ae. Data for the dimetallocenes, CpAeAeCp (3Ae; Ae = Be, Mg, Ca, Sr, Ba), are presented in Table 1.19 In all cases, in the gas phase, the disproportionation of 3Ae into divalent MCp2 and atomic M (ΔEDp) is endothermic. However, ΔEDp for 3Mg (58.2 kJ mol−1) is around 22% of that for 3Be (259 kJ mol−1). The value of ΔEDp for all other 3Ae complexes is between 9–15% of that of 3Be. Although the endothermic ΔEDp value for all 3Ae species might suggest that these complexes would be stable in the condensed phase, this assumption does not account for the vaporization energies of the metals (ΔEV) or 3Ae/AeCp2. Indeed, when ΔEV is considered, only 3Be is predicted to be amenable to isolation.18 Considering limited kinetic stabilization afforded to the Ae–Ae bond by the Cp ligands, it seems probable that the other 3Ae complexes would spontaneously disproportionate in the solid state. The homolytic Ae–Ae bond dissociation energies (BDEs) for the 3Ae complexes decrease down the Ae group and are by far the highest for 3Be (300 kJ mol−1). The BDE for 3Mg (202 kJ mol−1) is only around 2/3 that of 3Be.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19,22
19,22
| M | d(Ae–Ae)/Å | ΔEDp/kJ mol−1 | ΔEV/kJ mol−1 | BDE/kJ mol−1 |
|---|---|---|---|---|
| a ΔEDp is for the gas phase reaction, CpAeAeCp → AeCp2 + Ae. b BDE is calculated via the gas phase reaction, CpAeAeCp → 2 AeCp, where AeCp is a neutral radical. | ||||
| Be | 2.06 | 259 | 309 | 300 |
| Mg | 2.81 | 58.2 | 132 | 202 |
| Ca | 3.75 | 38.2 | 153 | 123 |
| Sr | 4.11 | 22.3 | 141 | 106 |
| Ba | 4.52 | 33.6 | 149 | 90.9 |
Initial theoretical studies of Ae–Ae bonds assumed that these interactions would resemble conventional (weak) covalent, homopolar bonds.11 Quantum theory of atoms-in-molecules (QTAIM) would describe such a conventional bond as possessing a (3, −1) bond critical point (BCP; a maximum of electron density in two dimensions and a minimum in the third dimension) between the two Ae centres.12,23 However, more recent computational studies indicate that this is not the case. Instead, Ae–Ae bonds are calculated to feature a (3, −3) critical point, or non-nuclear attractor (NNA), which corresponds to a large region of negative Laplacian.19,21 In other words, a NNA is a local maximum in electron density that is not associated with the position of a specific nucleus. Hence, rather than being directly bonded to one another, each electropositive Ae atom is “bonded” to the area of electron density associated with the NNA (Fig. 2). Indeed, QTAIM finds (3, −1) BCPs between each Ae atom and the NNA.19,21 Experimental evidence for this phenomenon comes from high-resolution X-ray diffraction experiments performed on magnesium(I) dimers.12 The implications of this bonding motif are wide-ranging. For example, the presence of the NNA is used to explain the highly reducing nature of complexes with Ae–Ae bonds.13 Additionally, Mg–Mg bonds have been shown to be deformable, with a shallow bond potential energy surface, and have been crystallographically characterized over a wide range of distances (2.8 to >3.2 Å).24–27
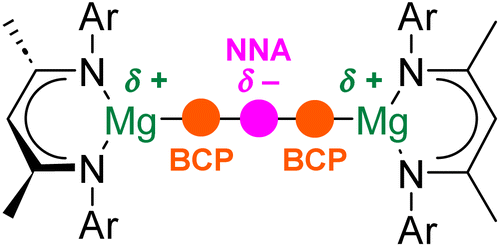 | ||
| Fig. 2 An illustration of the role of the non-nuclear attractor (NNA) in Ae–Ae bonding. | ||
From a molecular orbital standpoint, Be–Be and Mg–Mg bonds are calculated to be composed principally of contributions from the valence (n)s-orbitals, with a small degree of valence (n)p-orbital character, which varies depending on the supporting ligand set.13 This is due to the relatively large energetic gap between the (n)s- and (n)p-orbitals. By contrast, a degree of d-orbital participation might be anticipated for the Ae–Ae bonds of heavier members of the group.28–30 This is due to the fact that the (n − 1)d-orbitals are lower in energy than the (n)p-orbitals and the energy required to promote an electron from the (n)s-orbital to the (n − 1)d-orbitals is relatively small.
Recent Advances
Beryllium
Beryllium is the periodic table's second lightest metal, yet its chemistry is very poorly developed due to its extreme toxicity.31–33 The Be2+ ion is highly polarizing due to its enormous charge density (charge/radius ratio for Be2+: 6.45 Å−1; cf., Mg2+: 3.08 Å−1). Additionally, the element possesses extremely spatially concentrated frontier orbitals, which generally overlap effectively with the orbitals of other atoms in bonding interactions. As a result of these two factors, beryllium forms bonds with a significant degree of covalent character, which contrasts with the other Ae elements.20 Consequently, of all the Ae elements, the potential for homometallic bonding would seem the greatest for beryllium. This is, indeed, consistent with the data presented in Table 1 and discussed above.19 As a result, numerous synthetic and computational investigations into Be–Be bonding have been conducted over the past century.Around 1930, Herzberg made the first attempts to prepare diberyllium (Be2) in the gas phase, which were unsuccessful.34 The first spectroscopic identification of Be2 was carried out as late as 2009.35,36 The synthesis of complex 3Be, diberyllocene, was first examined in the 1970s; a benzene solution of CpBeH was heated in the presence of a platinum sheet in the hope of generating H2 and 3Be.37 However, no reaction was observed. Notably, several subsequent theoretical studies suggested that it might be possible to isolate 3Be in the condensed phases.8,18 More recently, inspired by the report of the first magnesium(I) dimers, the reduction of a range of beryllium(II) complexes furnished with β-diketiminate or diamide ligands was attempted.24,38,39 However, only products resulting from the activation of the ligand or the solvent were isolated.
In 2023 the first complex with a Be–Be bond was reported.40 Reduction of beryllocene (BeCp2) with [(MesNacnac)Mg]2 (DippNacnac = {[(Mes)NC(Me)]2CH}−, Mes = 2,4,6-trimethylphenyl) produced (MesNacnac)MgCp and 3Be in quantitative yield (Scheme 1). It is likely that (MesNacnac)Mg–BeCp, with a Mg–Be bond, is an intermediate in the formation of 3Be.23 Similarly to decamethyldizincocene, 3Be exhibits D5h symmetry.7 The crystallographically determined Be–Be bond distance in 3Be (2.0545(18) Å) is within the range predicted by previous theoretical studies (2.041 to 2.077 Å).8,18,19,21 Quantum chemical calculations indicate that the highest occupied molecular orbital (HOMO) of 3Be corresponds to the Be–Be σ-bonding orbital.40 The lowest unoccupied molecular orbital (LUMO) is principally ligand based, but the LUMO + 1 represents the Be–Be σ*-antibonding combination. QTAIM calculations indicate that 3Be also features a NNA in the Be–Be internuclear region.19 Evidence for the expected reducing character of 3Be was obtained through its reactions with aluminium- and zinc-iodides, which yield complexes with Be–Al and Be–Zn bonds, respectively, and CpBeI.40
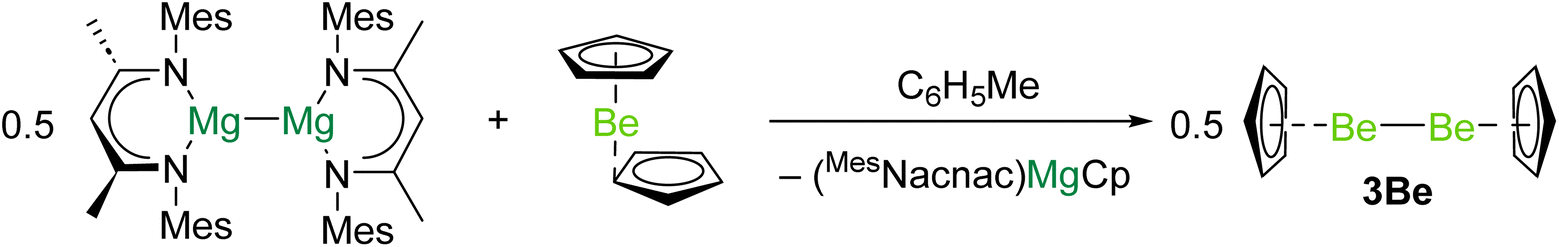 | ||
| Scheme 1 Synthesis of diberyllocene (3Be), through reduction of beryllocene with a magnesium(I) dimer. | ||
After the H–H σ-bond of H2, the Be–Be bond is the simplest reported homoelemental bond with which synthetic chemistry could be performed. Comparisons with beryllium's neighbour in the Periodic Table, boron (and B–B bonds), can also be drawn. In this context, experimental studies of the Be–Be bond are of fundamental importance. For example, the oxidative addition chemistries of H–H and B–B bonds have been extremely intensively studied due to their relevance to catalytic hydrogenation and borylation, respectively.41 Is the oxidative addition of the Be–Be bond possible? Can σ-complexes of the Be–Be bond be prepared? Moreover, considering the relationship between beryllium and boron, is it possible to isolate complexes featuring Be–Be multiple bonds?42 The one- and two-electron reduction of diborane(4) compounds, which are isoelectronic to beryllium(I) dimers, has been reported to yield species with B–B bond orders >1. Clearly, this is not likely to be a successful strategy in the case of 3Be, as the lowest energy unoccupied orbital associated with the Be–Be bond is antibonding in character.40 However, beryllium(I) dimers with alternative supporting ligands may possess Be–Be π-symmetry bonding orbitals that are of appropriate energy for population. Alternatively, theoretical studies have proposed that carbene-stabilized beryllium(0) dimers may be stable and would feature a Be![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Be double bond with σ- and π-components.20,42
Be double bond with σ- and π-components.20,42
Magnesium
Magnesium is vital to life on earth. It is the seventh most abundant element in the Earth's crust and is present in all cells of all organisms.43 The magnesium(0) dimer, Mg2, was first spectroscopically observed in 1970, sparking countless investigations into the properties of Ae diatomics.44 Significantly, species with Mg–Mg bonds are implicated as intermediates in the formation of Grignard reagents, the mechanism for which remains uncertain.45,46The study of complexes with Mg–Mg bonds has advanced rapidly over the past 15 years. Recent studies have focused on the design of complexes furnished with very bulky ligands to access new reactivity modes and structural motifs. For example, in 2021 it was reported that reduction of (DipepNacnac*)MgI (DipepNacnac* = {[(Dipep)NC(tBu)]2CH}−, Dipep = 2,6-diisopentylphenyl) with Na/NaCl gave the tetrametallic di-magnesium(0) complex {[(DipepNacnac*)Mg]Na}2 (4).47 When 4 is dissolved in benzene and heated, sodium is extruded to yield {[(DipepNacnac*)Mg]2Mg} (5), which features a linear tri-magnesium MgI–Mg0–MgI core, formally consisting of a central magnesium(0) atom bonded to two magnesium(I) centres (Scheme 2). QTAIM calculations performed on 5 indicate that both Mg–Mg interactions feature a NNA, and yield charges of +1.00 for the central magnesium(0) atom and +1.22 for the flanking magnesium(I) centres. Additionally, quantum chemical calculations indicate that the extreme steric bulk of the [DipepNacnac*]− ligand is key to the stability of 5; the extrusion of magnesium(0) from this complex to yield [(DipepNacnac*)Mg]2 is calculated to be rather endothermic (141 kJ mol−1). With the smaller [DippNacnac]− ligand the energetic barrier to this process is much lower (10.5 kJ mol−1). It is postulated by the authors that complex 5 may resemble an intermediate in Grignard reagent formation.
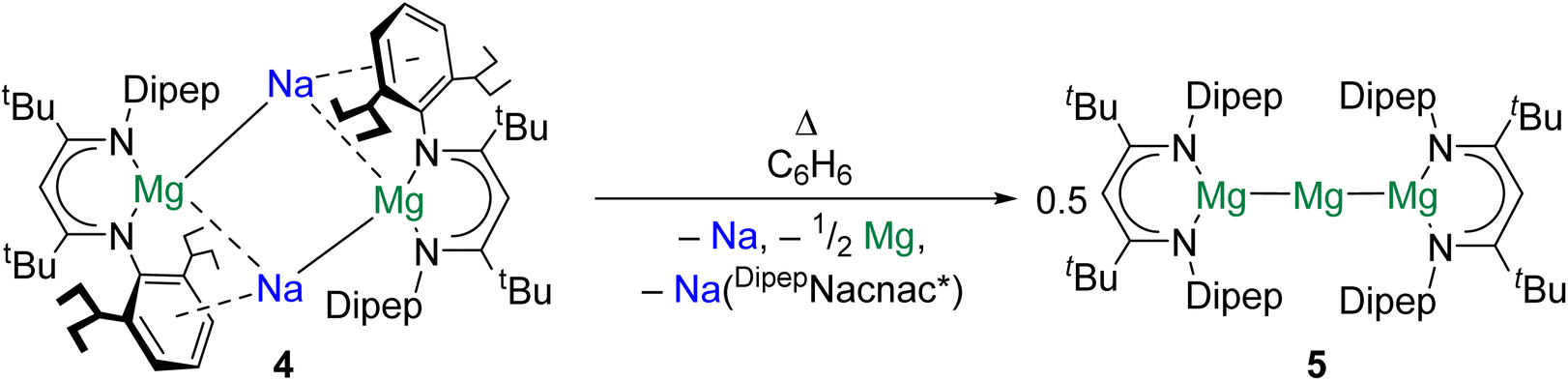 | ||
| Scheme 2 Formation of tri-magnesium complex 5, which features a MgI–Mg0–MgI core. | ||
The extrusion of Na0 in the formation of 5 is testament to the reducing power of molecular magnesium(0) species.47 Remarkably, similar behaviour has recently been described for magnesium(I) dimers. It was reported that addition of redox inert Lewis bases (e.g., THF) to magnesium(I) complex [{SiNDipp}MgNa]2 (6; [SiNDipp] = {[CH2SiMe2N(Dipp)]2}2−) induces the reduction of both Na+ ions to Na0, with the concomitant oxidation of both MgI centres (Scheme 3).48 It is proposed that the [NaI2MgI2]2+ tetrametallic core of 6 is perturbed by coordination of THF to one of the Na+ centres in this complex. This precipitates the subsequent reduction of the unsolvated Na+ cation to Na0 by the electron density in the Mg–Mg bond. The energetic barrier to THF coordination at Na+ (21.3 kJ mol−1) is far lower than that associated with coordination of the donor solvent to Mg+ (108 kJ mol−1). Notably, the Mg–Mg bond in complex 6 is very long (>3.2 Å).26 Perhaps as a consequence, QTAIM calculations performed on 6 do not locate a NNA for the Mg–Mg interaction, but find instead a single BCP between the magnesium centres and BCPs between this point and the Na+ cations. Remarkably, although the electron density at the (Mg–Mg)–Na BCP is very low (ρ = 0.003), these interactions are calculated to play an important stabilizing role in 6 (105 kJ mol−1 each). Notably, the largely electrostatic nature of the (Mg–Mg)–Na interaction in 6 differs from the more covalent Mg–Na bonding in 4 (ρ = 0.010–0.015).47 QTAIM calculations return very low charges for both magnesium centres in 6 (+0.97), which are comparable to that of the formal magnesium(0) centre in 5 (+1.00).47 Complex 6 is reported to activate H2 and reduce CO to ethynediolate.49
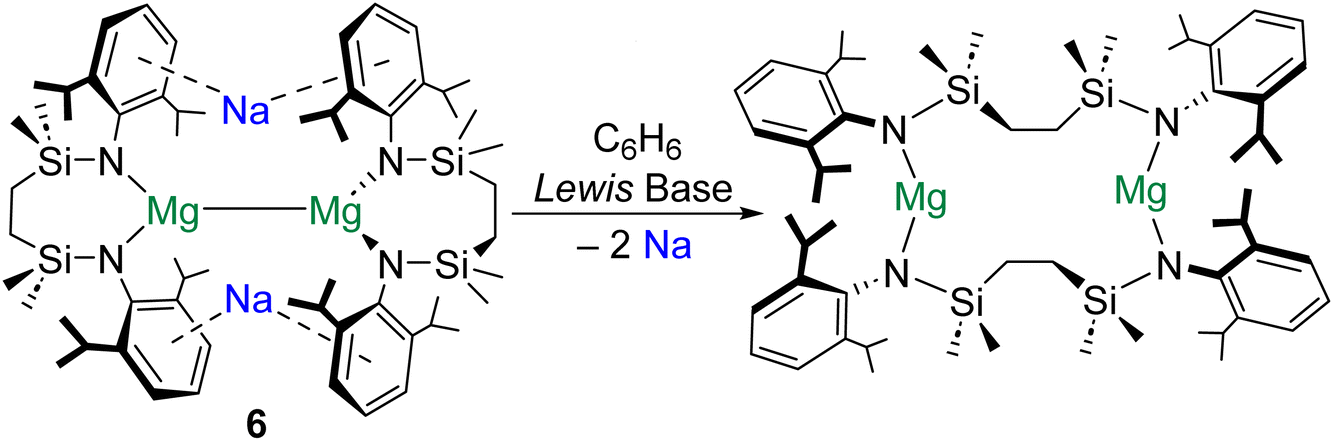 | ||
| Scheme 3 Extrusion of metallic sodium though addition of Lewis bases to magnesium(I) dimer complex 6. | ||
The preparation of 4 (and its reactivity to form 5), as well as the reactivity of 6, highlight the importance of counterion and reductant.50 In the case of 4 and 6, Na+ cations play key structural roles (through Na–aryl interactions), which contribute to the kinetic stabilization of the reactive sites in these complexes. Additionally, Mg–Na interactions lessen the accumulation of negative charge at the Mg centres in 4 and the Mg–Mg bond of 6, also stabilizing both complexes. It seems likely that future progress in this area will rely upon the synergy of low-valent Ae centres/Ae–Ae bonds and alkali metal counterions to engender novel reactivity.
Ongoing investigations into β-diketiminate-ligated magnesium(I) dimers continue to yield remarkable results. For example, coordination of a Lewis base to one of the magnesium(I) centres within 1 lengthens the Mg–Mg linkage and increases its reactivity. These singly “donor-activated” complexes possess a three-coordinate magnesium(I) centre that can coordinate a substrate. Resultantly, they are more reactive than symmetrically doubly donor-coordinated complexes in which both magnesium(I) centres are coordinatively saturated. In evidence of this, [(DippNacnac)(D)Mg–Mg(DippNacnac)] (D = 4-dimethylaminopyridine or 1,3,4,5-tetramethyl-imidazol-2-ylidene) is capable of facilitating the reductive cyclotrimerization of CO, forming the deltate dianion, [C3O3]2−.51 In other work, it has also been demonstrated that photolysis of 1 leads to Mg–Mg bond cleavage and generation of highly reactive magnesium(I) radicals, which have evaded isolation.52 However, these radicals are capable of the “Birch-like” two-electron reduction and C–H bond activation of benzene, the regio- and chemo-selective C–H bond activation of toluene and xylene, and the defluorination of fluorobenzene. Although several other fleeting, highly reactive magnesium(I) radical complexes have been reported, none are believed to possess Mg–Mg bonds, or be generated from species with Mg–Mg bonds.27,53–55
Future work will likely continue to leverage the deformability of the Mg–Mg interaction in order to engender new reactivity. For example, there is scope for the deployment of bridging ligands that enforce a specific Mg–Mg separation in order to modulate reactivity of the linkage.
Calcium and the heavier alkaline earth elements (Sr–Ra)
Calcium is the fifth most abundant element in the Earth's crust and the most abundant metal in the human body, with a range of crucial biochemical roles. Strontium and barium are also both abundant elements – the 15th and 14th most abundant in the Earth's crust, respectively. Due to its similarities with Ca, Sr is rather biologically benign.43 However, it is believed Ba2+ blocks K+ channels in the body, meaning that water-soluble Ba compounds are toxic.43 Due to their large ionic radii and polarized bonding, many coordination complexes of heavier Ae elements are labile and prone to Schlenk-type equilibria.Several attempts to prepare stable complexes featuring homometallic bonds of the heavier Ae elements have been described, although none have been successful.24 Perhaps most notably, it was recently reported that reduction of [(DipepNacnac)CaI]2, in methylcyclohexane and under an atmosphere of dinitrogen, yields the complex {[(DipepNacnac)Ca]2(μ-N2)} (7), which could be crystallographically characterized as its bis(tetrahydrofuran) and bis(tetrahydropyran) adducts (Scheme 4).56 Although a complex with a Ca–Ca bond was the target of these investigations, there is no evidence to suggest such a species is an intermediate in the formation of 7. Indeed, given the low BDE of a hypothetical Ca–Ca bond (e.g., 123 kJ mol−1 for 3Ca), other possibilities should be considered.19 For example, the similarities between rare-earth elements and heavier Ae elements are manifold – the elements are powerful reductants and their bonding interactions are highly polarized. Indeed, divalent rare-earth complexes that do not feature M–M bonds are known to activate N2.57 Furthermore, computational evidence suggests that partially occupied calcium-based d-orbitals may be responsible for the binding of N2, as is believed to be key for rare-earth complexes.56,57 Thus, it is possible that the reduction of N2 is carried out by a mononuclear calcium(I) radical, or a molecular calcium(0) species analogous to complex 4.
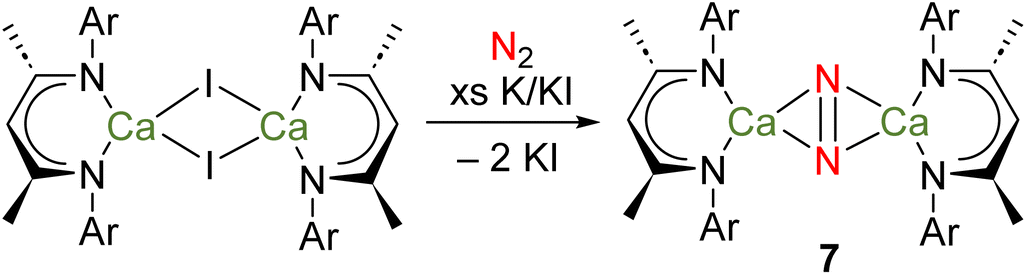 | ||
| Scheme 4 Attempted preparation of a complex with a Ca–Ca bond, instead leading to formation of di-calcium dinitrogen complex 7. | ||
Actinide-actinide and lanthanide-lanthanide bonds are predicted to be very weak, similarly to homometallic bonds between heavy alkaline earth elements (Table 1). However, the first complexes with actinide-actinide and lanthanide-lanthanide bonds, which are supported by bridging ligands, have been reported recently.58,59 Therefore, analogous strategies for the synthesis of heavier Ae–Ae bonds might be attempted. It has been demonstrated that a set of six bridging halide ligands – a pair between each pair of metal centres – will hold three thorium ions within a proximity which enables three-centre two-electron tri-thorium bonding (8, Fig. 3).58 This approach was subsequently applied for the preparation of a range of complexes with halide-supported lanthanide-lanthanide bonds (9, Fig. 3).59 Significantly, (supported) two-centre one-electron (10, Fig. 3) and three-centre one-electron homometallic bonding of the rare-earth element yttrium, which is adjacent to strontium in the Periodic Table, has been described.60 The homometallic rare-earth element bonds are all calculated to principally comprise contributions from metal d-orbitals, as might be predicted for Ca, Sr, and Ba.28 In the case of Ra, however, relativistic effects destabilize the 6d-orbital manifold.61 As a result, Ra–Ra bonding might be expected to principally comprise contributions from the 7s- and 7p-orbitals.4 This contrasts with the Th–Th bonding of 8, which is calculated to be primarily of Th 6d-orbital character (80%).58 Considering the proximity of Ra (element 88) and Th (element 90) within the Periodic Table, this dissimilarity is striking.
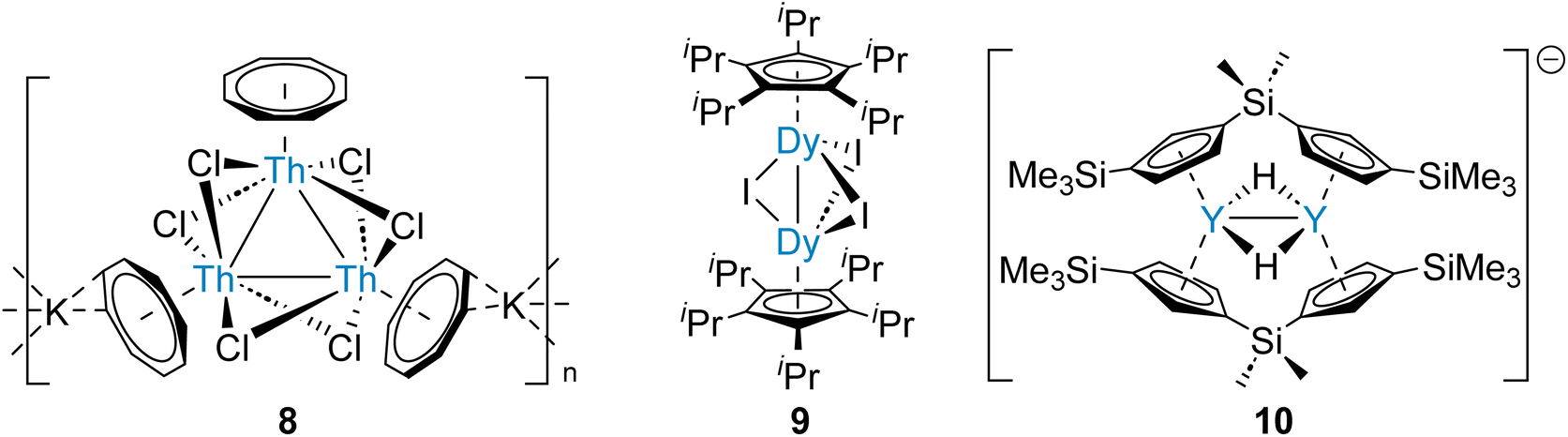 | ||
| Fig. 3 Rare-earth and actinide complexes that feature supported M–M bonds – a strategy which may be employed for the preparation of stable complexes that feature heavier Ae–Ae bonds. | ||
Successful preparation of heavier Ae–Ae bonds may also depend on judicious selection of starting materials and synthetic methodology. For example, reduction of higher-valent elements with alkali metals can lead to the formation of ill-defined product mixtures. Thus, the +1-oxidation state of the heavier Ae elements might be accessed more straightforwardly by the one-electron oxidation of the metal (in the presence of a suitable supporting ligand), rather than reduction of a divalent Ae precursor.62,63
Future directions
The synthesis of 5 demonstrates that clusters comprising several Ae–Ae bonds might be isolable.64 In this context, electrospray mass spectrometry has previously been used to detected an ion with a likely composition of [Mg16Cp*8Br4K]− from the reaction of metastable “MgBr” with KCp*.65 Although no crystallographic data for this species could be obtained, a possible structure was derived from quantum chemical calculations, featuring a Mg14 core, consisting of 27 Mg–Mg bonds, resembling hexagonal close-packed Mg metal. The structural characterization of these clusters would serve to further elucidate the possible pathways for Grignard reagent formation.66 In this context, with the report of 3Be and the relatively high strength of Be–Be bonds, it may be possible to prepare catenated chains of beryllium atoms. This has previously been suggested to be feasible by theoretical investigations of multi-beryllocenes (i.e., CpBenCp; n = 3,4,5).18 Alternatively, rings of Be atoms connected by Be–Be bonds may be synthesised. This also presents the possibility of all-Ae metal aromaticity.As previously mentioned, species that feature Ae–Ae multiple bonding interactions are feasible synthetic targets, particularly in the case of Be and Mg.20 Indeed, the LUMO and LUMO + 1 of many magnesium(I) dimers correspond to π-symmetry Mg–Mg bonding orbitals.9 Thus, shrewd ligand design may allow for the one- or two-electron population of these orbitals.
Conclusions and outlook
Considering the apparent modularity and high reactivity of complexes that feature Mg–Mg bonds, it seems likely that research into these species will continue to flourish. Indeed, such complexes have already been shown to be capable of reactivity that is currently unknown for transition metals, such as the cyclotrimerization of CO.51 Although the synthetic study of Be–Be bonding is in its infancy, this will likely yield many striking discoveries.40 Certainly, a comprehensive understanding of beryllium's chemistry is necessary in order to test models of bonding, which are primarily based on the properties of the simplest elements.The isolation of complexes with homometallic actinide and rare-earth bonding suggests new approaches for the preparation of Ae–Ae bonds for the heavier elements of the group.58–60 It seems possible that, to access many of these highly reactive species, ligands that impart sufficient solubility to facilitate their preparation in inert alkane solvents, or the use of mechanochemical synthetic methods, will be necessary.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. T. B. thanks St John's College, Oxford, for a Junior Research Fellowship, as well as the John Fell Fund (0011792) and the Royal Society of Chemistry (Research Fund R23-3176939355) for financial support. Mr L. P. Griffin, Dr L. R. Thomas-Hargreaves, and Prof. S. Aldridge are thanked for constructive and insightful discussions.References
- E. Vangioni-flam and M. Cassé, Astrophys. Space Sci., 1999, 11, 77–86 CrossRef.
- S. Freeman, Encyclopedia of Inorganic and Bioinorganic Chemistry, Wiley, 2015, pp. 1–11 Search PubMed.
- P. Curie, M. Curie and G. Bémont, C. R. Acad. Sci., 1898, 127, 1215–1217 CAS.
- F. D. White, N. A. Thiele, M. E. Simms and S. K. Cary, Nat. Chem., 2023 DOI:10.1038/s41557-023-01366-z.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- I. Resa, E. Carmona, E. Gutierrez-Puebla and A. Monge, Science, 2004, 305, 1136–1138 CrossRef CAS PubMed.
- Y. Xie, H. F. Schaefer and E. D. Jemmis, Chem. Phys. Lett., 2005, 402, 414–421 CrossRef CAS.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- A. Stasch and C. Jones, Dalton Trans., 2011, 40, 5659 RSC.
- J. Overgaard, C. Jones, A. Stasch and B. B. Iversen, J. Am. Chem. Soc., 2009, 131, 4208–4209 CrossRef CAS.
- J. A. Platts, J. Overgaard, C. Jones, B. B. Iversen and A. Stasch, J. Phys. Chem. A, 2011, 115, 194–200 CrossRef CAS.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- B. Rösch and S. Harder, Chem. Commun., 2021, 57, 9354–9365 RSC.
- C. Jones, Commun. Chem., 2020, 3, 8–11 CrossRef PubMed.
- L. A. Freeman, J. E. Walley and R. J. Gilliard, Nat. Synth., 2022, 1, 439–448 CrossRef.
- S. Li, X.-J. Yang, Y. Liu, Y. Zhao, Q.-S. Li, Y. Xie, H. F. Schaefer and B. Wu, Organometallics, 2011, 30, 3113–3118 CrossRef CAS.
- A. Velazquez, I. Fernández, G. Frenking and G. Merino, Organometallics, 2007, 26, 4731–4736 CrossRef CAS.
- M. A. Gosch and D. J. D. Wilson, Organometallics, 2023, 42, 2185–2196 CrossRef CAS.
- S. A. Couchman, N. Holzmann, G. Frenking, D. J. D. Wilson and J. L. Dutton, Dalton Trans., 2013, 42, 11375–11384 RSC.
- X. Li, S. Huo, Y. Zeng, Z. Sun, S. Zheng and L. Meng, Organometallics, 2013, 32, 1060–1066 CrossRef CAS.
- Y. Zhang, J. R. G. Evans and S. Yang, J. Chem. Eng. Data, 2011, 56, 328–337 CrossRef CAS.
- J. T. Boronski, L. R. Thomas-Hargreaves, M. A. Ellwanger, A. E. Crumpton, J. Hicks, D. F. Bekiş, S. Aldridge and M. R. Buchner, J. Am. Chem. Soc., 2023, 145, 4408–4413 CrossRef CAS PubMed.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938–955 CrossRef CAS.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079–9083 CrossRef CAS.
- H.-Y. Liu, R. J. Schwamm, S. E. Neale, M. S. Hill, C. L. McMullin and M. F. Mahon, J. Am. Chem. Soc., 2021, 143, 17851–17856 CrossRef CAS PubMed.
- T. X. Gentner, B. Rösch, G. Ballmann, J. Langer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 607–611 CrossRef CAS PubMed.
- M. Zhou and G. Frenking, Acc. Chem. Res., 2021, 54, 3071–3082 CrossRef CAS PubMed.
- X. Wu, L. Zhao, D. Jiang, I. Fernández, R. Berger, M. Zhou and G. Frenking, Angew. Chem., Int. Ed., 2018, 57, 3974–3980 CrossRef CAS.
- I. Fernández, N. Holzmann and G. Frenking, Chem. – Eur. J., 2020, 26, 14194–14210 CrossRef PubMed.
- M. R. Buchner, Chem. Commun., 2020, 56, 8895–8907 RSC.
- M. R. Buchner, Chem. – Eur. J., 2019, 25, 12018–12036 CrossRef CAS.
- D. Naglav, M. R. Buchner, G. Bendt, F. Kraus and S. Schulz, Angew. Chem., Int. Ed., 2016, 55, 10562–10576 CrossRef CAS.
- G. Herzberg, Z. Med. Phys., 1929, 57, 601–630 CAS.
- J. M. Merritt, V. E. Bondybey and M. C. Heaven, Science, 2009, 324, 1548–1551 CrossRef CAS PubMed.
- K. Patkowski, V. Špirko and K. Szalewicz, Science, 2009, 326, 1382–1384 CrossRef CAS PubMed.
- H. Schmidbaur, Be Organoberyllium Compounds, Springer Berlin Heidelberg, Berlin, Heidelberg, 1987, vol. 21 Search PubMed.
- M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall, M. F. Mahon and I. Mallov, Inorg. Chem., 2012, 51, 13408–13418 CrossRef CAS PubMed.
- K. G. Pearce, M. S. Hill and M. F. Mahon, Chem. Commun., 2023, 59, 1453–1456 RSC.
- J. T. Boronski, A. E. Crumpton, L. L. Wales and S. Aldridge, Science, 2023, 380, 1147–1149 CrossRef CAS PubMed.
- J. A. Labinger, Organometallics, 2015, 34, 4784–4795 CrossRef CAS.
- H. Braunschweig and R. D. Dewhurst, Organometallics, 2014, 33, 6271–6277 CrossRef CAS.
- R. Crichton, Biological Inorganic Chemistry, Elsevier, 2019 Search PubMed.
- W. J. Balfour and A. E. Douglas, Can. J. Phys., 1970, 48, 901–914 CrossRef CAS.
- Y. Imizu and K. J. Klabunde, Inorg. Chem., 1984, 23, 3602–3605 CrossRef CAS.
- L. A. Tjurina, V. V. Smirnov, D. A. Potapov, S. A. Nikolaev, S. E. Esipov and I. P. Beletskaya, Organometallics, 2004, 23, 1349–1351 CrossRef CAS.
- B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–721 CrossRef.
- H. Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and E. Richards, Angew. Chem., Int. Ed., 2022, e202213670, DOI:10.1002/anie.202213670.
- H. Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and B. L. Morrison, Chem. Commun., 2023, 59, 3846–3849 RSC.
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810–4813 CrossRef CAS.
- K. Yuvaraj, I. Douair, A. Paparo, L. Maron and C. Jones, J. Am. Chem. Soc., 2019, 141, 8764–8768 CrossRef CAS.
- D. D. L. Jones, I. Douair, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2021, 60, 7087–7092 CrossRef CAS.
- D. Jędrzkiewicz, J. Mai, J. Langer, Z. Mathe, N. Patel, S. DeBeer and S. Harder, Angew. Chem., Int. Ed., 2022, e202200511, DOI:10.1002/anie.202200511.
- R. Mondal, M. J. Evans, T. Rajeshkumar, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2023, e202308347, DOI:10.1002/anie.202308347.
- D. Jędrzkiewicz, J. Langer and S. Harder, Z. Anorg. Allg. Chem., 2022, 648, 16–19 CrossRef.
- B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef.
- A. J. Ryan, S. Ganesh Balasubramani, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2020, 142, 9302–9313 CrossRef CAS.
- J. T. Boronski, J. A. Seed, D. Hunger, A. W. Woodward, J. van Slageren, A. J. Wooles, L. S. Natrajan, N. Kaltsoyannis and S. T. Liddle, Nature, 2021, 598, 72–75 CrossRef CAS.
- Z. Zhu and J. Tang, Chem. Soc. Rev., 2022, 51, 9469–9481 RSC.
- J. C. Wedal, L. M. Anderson-Sanchez, M. T. Dumas, C. A. Gould, M. J. Beltrán-Leiva, C. Celis-Barros, D. Páez-Hernández, J. W. Ziller, J. R. Long and W. J. Evans, J. Am. Chem. Soc., 2023, 145, 10730–10742 CrossRef CAS PubMed.
- U. Dammalapati, K. Jungmann and L. Willmann, J. Phys. Chem. Ref. Data, 2016, 45, 013101 CrossRef.
- M. Schorpp and I. Krossing, Chem. Sci., 2020, 11, 2068–2076 RSC.
- G. Wang, J. E. Walley, D. A. Dickie, S. Pan, G. Frenking and R. J. Gilliard, J. Am. Chem. Soc., 2020, 142, 4560–4564 CrossRef CAS.
- R. Köppe, P. Henke and H. Schnöckel, Angew. Chem., Int. Ed., 2008, 47, 8740–8744 CrossRef PubMed.
- T. Kruczyński, F. Henke, M. Neumaier, K. H. Bowen and H. Schnöckel, Chem. Sci., 2016, 7, 1543–1547 RSC.
- T. Kruczyński, N. Pushkarevsky, P. Henke, R. Köppe, E. Baum, S. Konchenko, J. Pikies and H. Schnöckel, Angew. Chem., Int. Ed., 2012, 51, 9025–9029 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |