
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceATRP catalysts of tetradentate guanidine ligands – do guanidine donors induce a faster atom transfer?†‡
Konstantin W.
Kröckert
 ,
Felix
Garg
,
Joshua
Heck
,
Michel V.
Heinz
,
Justin
Lange
,
Regina
Schmidt
,
Alexander
Hoffmann
and
Sonja
Herres-Pawlis
*
,
Felix
Garg
,
Joshua
Heck
,
Michel V.
Heinz
,
Justin
Lange
,
Regina
Schmidt
,
Alexander
Hoffmann
and
Sonja
Herres-Pawlis
*
Institute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1a, 52074 Aachen, Germany. E-mail: sonja.herres-pawlis@ac.rwth-aachen.de
First published on 8th January 2024
Abstract
Tripodal tetradentate N donor ligands stabilise the most active ATRP catalyst systems. Here, we set out to synthesise the new guanidine ligand TMG-4NMe2uns-penp, inspired by p-substituted tris(2-pyridylmethyl)amine (TPMA) ligands. The impact of changing pyridine against guanidine donors was examined through solid state and solution experiments and density functional theory (DFT) calculations. In the solid state, the molecular structures of copper complexes based on the ligands TMG-4NMe2uns-penp, TMG-uns-penp and TMG3tren were discussed concerning the influence of a NMe2 substituent at the pyridines and the guanidine donors. In solution, the TMG-4NMe2uns-penp system was investigated by several methods, including UV/Vis, EPR and NMR spectroscopy indicating similar properties to that of the highly active TPMANMe2 system. The redox potentials were determined and related to the catalytic activity. Besides the expected trends between these and the ligand structures, there is evidence that guanidine donors in tripodal ligand systems lead to a better deactivation and possibly a faster exchange within the ATRP equilibrium than TPMA systems. Supported by DFT calculations, it derives from an easier cleavable Cu–Br bond of the copper(II) deactivator species. The high activity was stated by a controlled initiator for continuous activator regeneration (ICAR) ATRP of styrene.
Introduction
Since 1995,1–4 atom transfer radical polymerisation (ATRP) has become a widespread method for controlled polymerisation owing to good tolerance to air using advanced ATRP techniques and the ability to synthesise polymers with a large variety of architectures.5–7 In the ATRP mechanism exists a reversible equilibrium between the dormant and active radical species, which is usually mediated by a transition metal complex with a metal centre that is stable in at least two adjacent oxidation states. Many systems have been used, but copper complexes with multidentate N-donor ligands are the most successful and widely used ATRP catalysts.8–12 The copper(I) activator complex (CuILn) reacts with the dormant radical chain or initiator (P–X) to yield the copper(II) deactivator complex (X–CuIILn) and the active radical (P˙) by homolytic cleavage of the C–X bond.13 The radical can polymerise until the deactivator complex shifts the equilibrium back to the dormant side.14 To suppress side reactions and maintain chain-end functionality, low radical concentrations and a reversible exchange of the equilibrium are necessary.15,16 For standard ATRP, this can be achieved by the position of the equilibrium (KATRP) on the dormant side.1,2 Contemporary ATRP variations such as mechanoATRP,17,18 eATRP,19,20 supplemental activators and reducing agents (SARA) ATRP,21 photoATRP,22–24 activators regenerated by electron transfer (ARGET) ATRP25–27 and initiators for continuous activator regeneration (ICAR) ATRP27–30 (Scheme 1) allow good polymerisation control combined with a low catalyst loading. An increased ratio of the deactivator to activator complex is necessary to achieve this.31,32 The reason for this is that a higher concentration of the deactivator complex improves the deactivation reaction, which has an effect on the dispersity.31,32 Therefore, in contrast to standard ATRP, high KATRP values are essential for these methods. Because of this interrelation, KATRP is an expression for the catalyst activity.33,34 Furthermore, the activator complex concentrations during the polymerisation should be low. This suppresses the formation of organometallic intermediates and the resulting radical termination (RT) reactions such as reductive radical terminations (RRT) and catalysed radical terminations (CRT), which increase the dispersity.35,36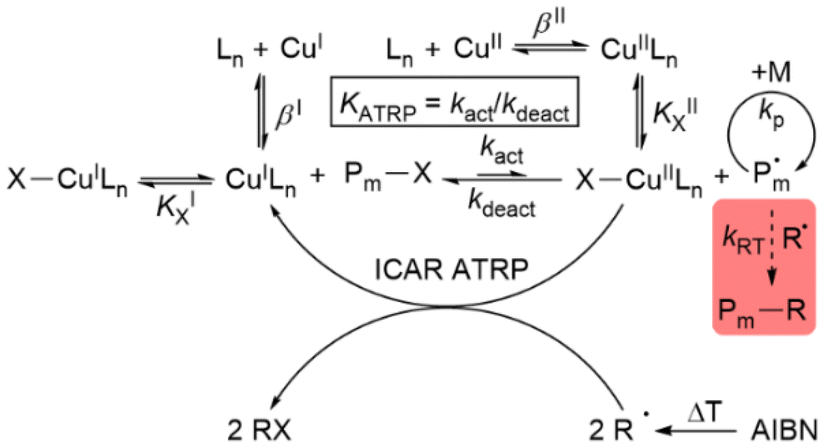 | ||
| Scheme 1 Mechanism for ICAR ATRP with halidophilicity constants (KX), stability constants (β) and unwanted radical side reactions (shaded red).27 | ||
Combining these properties with the listed activator regeneration techniques allows low amounts of catalysts down to parts per million levels and good control.27,30 This simplifies purification steps and lowers costs. KATRP, the key value for catalyst activity, is correlated with the redox potential (E1/2) for copper complexes with N donor ligands.37,38 Low redox potentials lead to high KATRP values meaning a better stabilisation of the copper(II) complex. The activity can further be correlated with the stability constants β of the copper complexes. Here, it was found that KATRP values scale with the ratio of βII/βI. Large values for both constants are necessary, but βII must be larger than βI to induce a thermodynamic driving force for radical formation.37,39,40 Additionally, the halidophilicity constant KIIX is preferred to be large for efficient deactivation, and KIX should be low since CuILn is required for activation.41–44 Several studies highlighted trends of how the coordination sphere around the copper influences the catalyst activity.45 The topology of the ligand, bridging units, denticity, the nature of the N donors, and here especially a high donor strength lead to an improvement.38,46,47 These efforts have resulted in the most active ATRP systems to date, which are based on tetradentate NR2 substituted tris(2-pyridylmethyl)amine (TPMA) ligands (an overview of the used abbreviations and the corresponding ligand structures can be found in ESI Fig. S18†).27,30
Guanidines exhibit excellent N donor properties and versatility.48 Complexes with such ligands are not just suitable for ATRP49–53 but also for ring-opening polymerisation,53–55 oxygen activation,56–58 model complexes for the entatic state of electron transfer proteins59 and photochemistry.60 Recently, we reported the most active ATRP catalyst based on a bidentate ligand, consisting of a NMe2 substituted pyridine and a tetramethylguanidine (TMG) moiety (TMGm4NMe2py).61 It was found that structural features also caused by the guanidine function are the reason for the unexpected low redox potential and, therefore, high catalytic activity.
This study presents a novel guanidine ligand inspired by the most active tetradentate systems, but one pyridine donor is exchanged by one guanidine donor. Besides the aim to synthesise a highly active catalyst, we set out to investigate the influence of guanidine donors in tetradentate, tripodal ligands on the ATRP equilibrium.
Results and discussion
Ligand synthesis
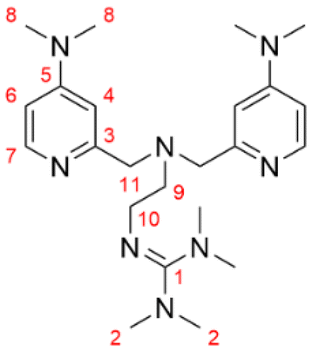
Formal exchange of a pyridine unit of TMPANMe2 with the guanidine unit leads to the novel hybrid guanidine ligand TMG-4NMe2uns-penp (L1). The synthesis is accomplished in three steps (Scheme 2). The literature-known compound 2-(1,3-dioxoisoindolin-2-yl)ethan-1-aminium 2,2,2-trifluoroacetate (1) was synthesised according to partly modified literature procedures from Franchini et al.,62 Borovik et al.,63 and Botta et al.64 (Scheme S1†). By a reductive amination with the aldehyde (2) inspired by Britovsek et al.,65 the new compound 2-(2-(bis((4-chloropyridin-2-yl)methyl)amino)ethyl)-isoindoline-1,3-dione (3) was received. In the next step, dimethylamine was inserted by nucleophilic aromatic substitution, and the phthalimide moiety was removed under basic conditions. The synthesis of amine 4 was inspired by a reaction of Matyjaszewski et al.27 Finally, the guanidine ligand L1 was obtained by a reaction of the amine precursor 4 with the Vilsmeier salt N,N,N′,N′-tetramethylchloroformamidinium chloride (TMG-VS).66,67 The ligands TMG-uns-penp (L2, uns ≙ unsymmetric, penp ≙ pyridinyl-decorated ethylenediamine, uns-penp was originally reported by Mandel et al.68) and TMG3tren (L3) were additionally selected for parts of this study to investigate the influence of electron density donating substituents and the number of guanidine donors on the catalyst structures and their redox potentials. These ligands were resynthesised according to the literature.69,70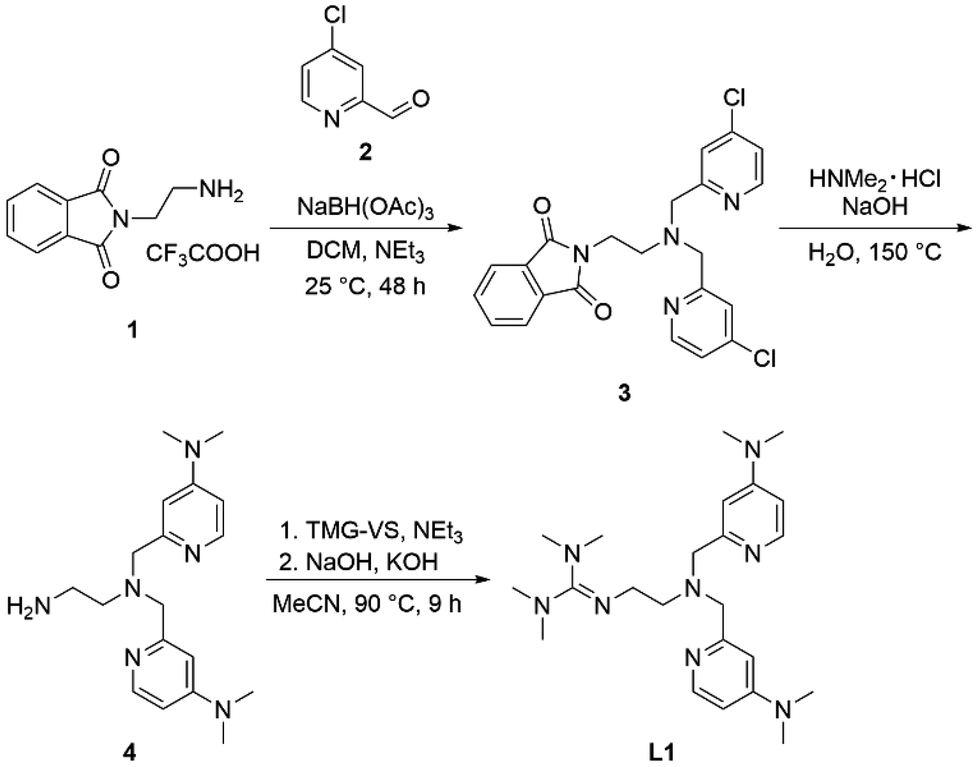 | ||
| Scheme 2 Synthetic pathway for the ligand TMG-4NMe2uns-penp (L1). | ||
Molecular structure in the solid state
The copper complexes [Cu(TMG-4NMe2uns-penp)Br]Br (C1), [Cu(TMG-4NMe2uns-penp)Cl]Cl (C2), [Cu(TMG-uns-penp)Br]Br·MeCN (C3), [Cu(TMG3tren)Br]Br (C4) and [Cu(TMG3tren)]Br·toluene (C5) were synthesised (Scheme 3) and their molecular structure in the solid state were examined via single crystal X-ray diffraction analysis. The molecular structures of the cation units in the solid state are presented in Fig. 1 and selected bond lengths, angles, and geometrical factors are denoted in Table 1. Additional data can be found in the ESI (Fig. S1, S2 and Tables S1–S3†). The Cu(II) ions in the complex cations in C1–C4 are fivefold coordinated by four N donor atoms and one halide atom, whereas the Cu(I) ion in the complex cation in C5 is fourfold coordinated without a coordinating halide atom. The τ5 value can be used to quantify the degree of distortion for fivefold coordinated systems and indicate ideal trigonal–bipyramidal coordination geometry for a value of 1 and an ideal square-pyramidal coordination geometry for a value of 0.71 In the complex cations in C1–C3, the copper atoms are distorted square-pyramidal coordinated (τ5 = 0.36–0.40) and the copper atom in the complex cation in C4 is trigonal–bipyramidal coordinated (τ5 = 1.01). The geometry of the fourfold coordinated copper atom can be described with the τ4 value and the tetrahedral characteristic parameter (THCDA).72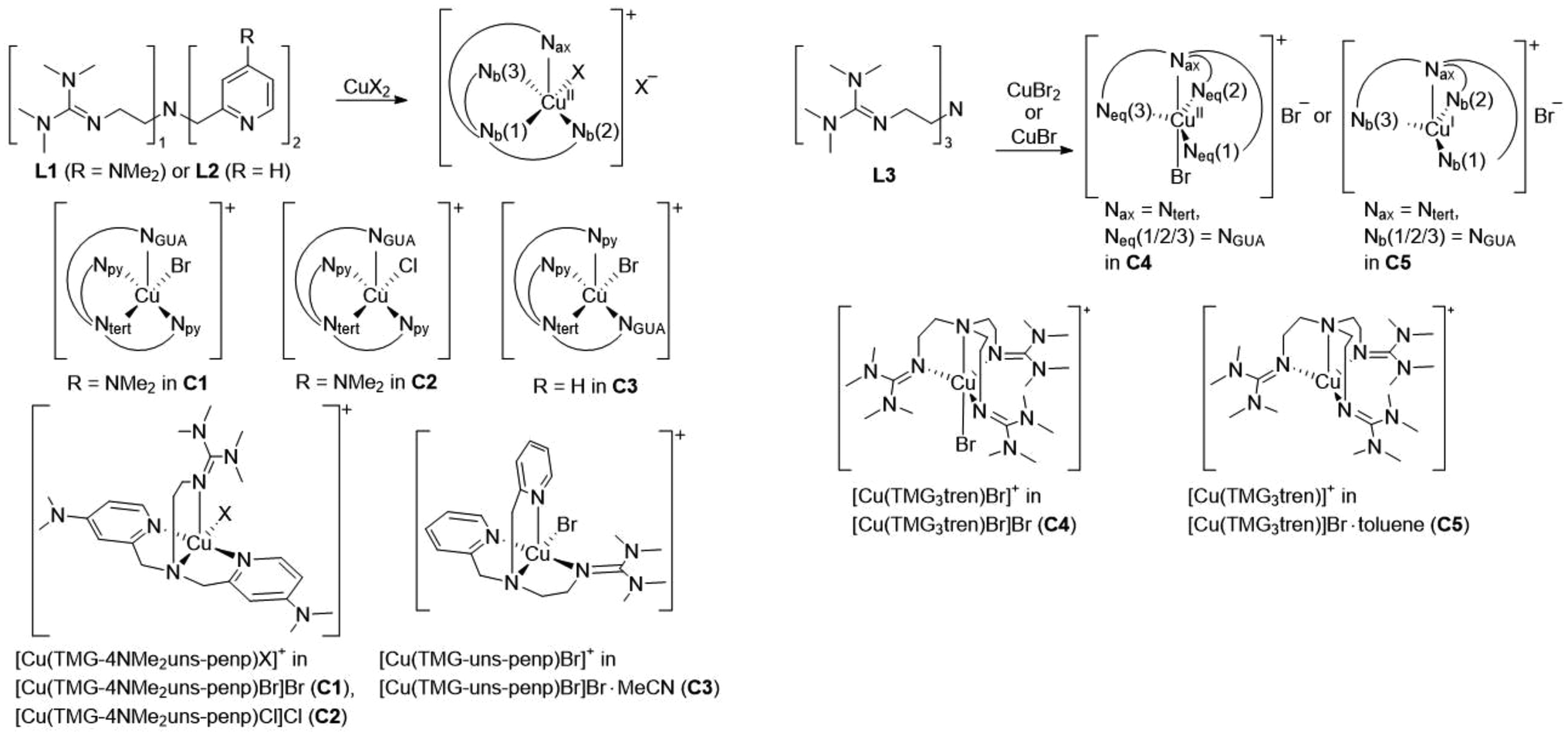 | ||
| Scheme 3 Synthesis and schematic view of the molecular structure in the solid state of various copper complexes starting from the corresponding ligands L1, L2 and L3. To clearly specify the N donors in axial, basal or equatorial position, the N donors were defined as follows: pyridine donor = Npy, TMG donor = NGUA, tertiary N donor = Ntert. | ||
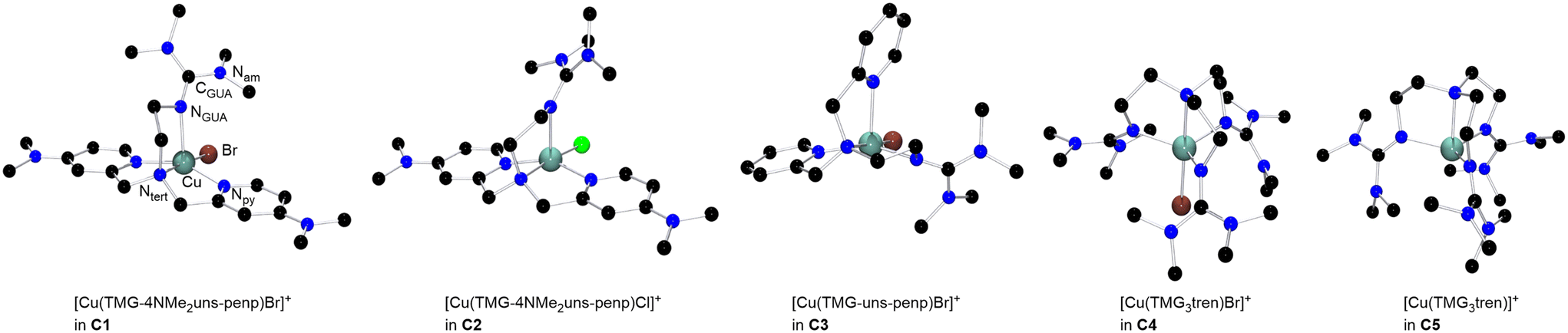 | ||
| Fig. 1 Molecular structure of the cationic complex units in crystals of [Cu(TMG-4NMe2uns-penp)Br]Br (C1), [Cu(TMG-4NMe2uns-penp)Cl]Cl (C2), [Cu(TMG-uns-penp)Br]Br·MeCN (C3), [Cu(TMG3tren)Br]Br (C4) and [Cu(TMG3tren)]Br·toluene (C5) in the solid state. Key atoms are exemplarily marked in one complex and H atoms as well as non coordinating anions or solvent molecules are omitted for clarity. | ||
| Complex | C1 | C2 | C3 |
C4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
C5 |
|---|---|---|---|---|---|
a Two crystallographic independent molecules are present in the asymmetric unit. Values for both are denoted in the ESI (Table S3†).
b
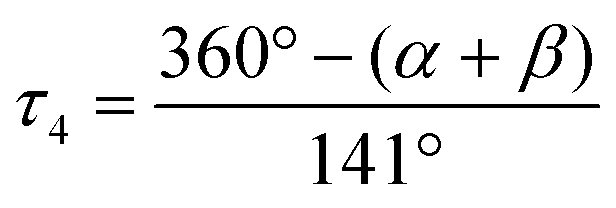 . Ideal square-planar complexes generate a τ4 value of 0 whereas ideal tetrahedral complexes generate a τ4 value of 1.72
c . Ideal square-planar complexes generate a τ4 value of 0 whereas ideal tetrahedral complexes generate a τ4 value of 1.72
c
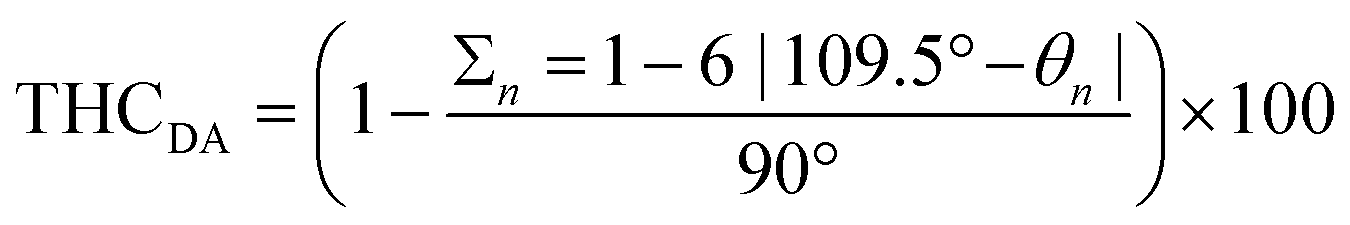 . The THCDA value reaches 100 for ideal tetrahedral complexes and 0 for ideal trigonal–pyramidal complexes.72
d . The THCDA value reaches 100 for ideal tetrahedral complexes and 0 for ideal trigonal–pyramidal complexes.72
d
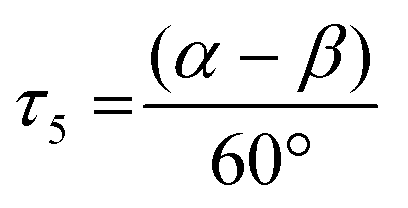 . Ideal square-pyramidal complexes generate a τ5 value of 0, whereas ideal trigonal–bipyramidal complexes generate a τ5 value of 1.71
e . Ideal square-pyramidal complexes generate a τ5 value of 0, whereas ideal trigonal–bipyramidal complexes generate a τ5 value of 1.71
e
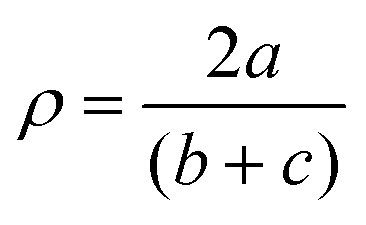 with a = d(CGUA with a = d(CGUA![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NGUA) and b and c = d(CGUA–Nam).76 NGUA) and b and c = d(CGUA–Nam).76
|
|||||
| Bond lengths [Å] | |||||
| Cu–Nax | 2.188(2); Nax = NGUA | 2.201(2); Nax = NGUA | 2.231(4); Nax = Npy | 2.112(3); Nax = Ntert | 2.200(2); Nax = Ntert |
| Cu–Nb/eq (1) | 2.088(2); Nb = Ntert | 2.089(2); Nb = Ntert | 2.082(4); Nb = Ntert | 2.061(3); Neq = NGUA | 2.053(2); Nb = NGUA |
| Cu–Nb/eq (2) | 2.010(2); Nb = Npy | 2.009(2); Nb = Npy | 1.970(4); Nb = NGUA | 2.065(3); Neq = NGUA | 2.053(2); Nb = NGUA |
| Cu–Nb/eq (3) | 2.020(2); Nb = Npy | 1.999(2); Nb = Npy | 2.031(4); Nb = Npy | 2.138(3); Neq = NGUA | 2.053(2); Nb = NGUA |
| Cu–X | 2.438(1); X = Br | 2.282(1); X = Cl | 2.394(1); X = Br | 2.447(1); X = Br | — |
| CGUANGUA |
1.308(4) | 1.307(2) | 1.328(5) | 1.311 (av.) | 1.305(2) |
| CGUA–Nam1 | 1.367(4) | 1.361(2) | 1.339(6) | 1.361 (av.) | 1.380(2) |
| CGUA–Nam2 | 1.366(4) | 1.373(2) | 1.360(5) | 1.360 (av.) | 1.374(2) |
| Bond angles [°] | |||||
| Nax–Cu–X | 101.9(1) | 102.7(1) | 99.4(1) | 179.4(1) | — |
| Nb/eq (1)–Cu–X | 174.7(1) | 174.5(1) | 177.2(1) | 98.8(1) | — |
| Nax–Cu–Nb/eq (1) | 83.1(1) | 82.7(1) | 80.1(2) | 81.8(2) | 84.1(1) |
| Nax–Cu–Nb/eq (2) | 107.7(1) | 91.7(1) | 95.6(2) | 82.3(2) | 84.1(1) |
| Nax–Cu–Nb/eq (3) | 92.5(1) | 108.0(1) | 102.8(2) | 81.8(2) | 84.1(1) |
| Nb/eq (1)–Cu–Nb/eq (2) | 82.2(1) | 82.5(1) | 84.0(2) | 122.3(2) | 119.0(1) |
| Nb/eq (1)–Cu–Nb/eq (3) | 82.2(1) | 82.0(1) | 80.3(2) | 113.1(2) | 119.0(1) |
| Nb/eq (2)–Cu–Nb/eq (3) | 152.6(1) | 153.0(1) | 153.3(2) | 118.8(2) | 119.0(1) |
| Geometrical factors | |||||
| τ 4 /THCDAc | — | — | — | — | 0.86/−16.2 |
| τ 5 | 0.37 | 0.36 | 0.40 | 1.01 | — |
| ρ | 0.96 | 0.96 | 0.98 | 0.96 | 0.95 |
A τ4 value of 1 indicates square-planar and a value of 0 indicates a tetrahedral coordination geometry. In contrast, the THCDA value reaches 100 for tetrahedral structures and a value of 0 for trigonal-pyramidal structures. Therefore, the copper atom in the cationic complex unit in C5 has a trigonal-pyramidal environment (τ4 = 0.86, THCDA = −16.2). The negative value can be explained with a smaller angle than 90° between the axial and basal donors.
In the complex cations in C1 and C2, the NMe2-substituted pyridine moieties, the tertiary amines, and the halides coordinate in the basal positions, and the guanidine is axially located. The Cu–Nb bond lengths to the same kind of donor in both complex cations in C1 (Nb = Npy with 2.010(2) Å and 2.020(2) Å and Nb = Ntert with 2.088(2) Å) and in C2 (Nb = Npy with 2.009(2) Å and 1.999(2) Å and Nb = Ntert with 2.089(2) Å) are equal in the 3 σ confidence interval. The bond lengths to basal positions are shorter than the Cu–Nax (Nax = NGUA) bond lengths equal in both complexes (in C1: 2.188(3) Å and in C2: 2.201(2) Å). The Cu–X bond length is larger for the bromide complex than for the chloride complex due to the larger ion radius. In the copper(II) bromide complex C3 with the unsubstituted pyridine moieties, one of the pyridine donors is located in the axial position in contrast to C1 where both pyridine donors coordinated in the basal position and the guanidine donor is located in the axial position. The other basal positions in C3 are located with the other pyridine moiety, guanidine, bromide and the tertiary amine donor. All Cu–N bond lengths to basal located donors differ significantly due to the different donor characteristics but the Cu–Ntert bond lengths and the Cu–Npy,b bond lengths are equal in C1–C3. The Cu–Br bond length in C3 is shorter (Cu–Br = 2.394(1) Å) relative to the analogous bond lengths in C1 (Cu–Br = 2.439(1) Å). The reason is the stronger electron density donating NMe2 substituted pyridine unit that leads to a higher electron density at the copper centre (see DFT section).73–75 Furthermore, the Cu–Nax bond lengths for both substituted complexes are elongated compared to Cu–Nb bond lengths of the unsubstituted complex C3, indicating better coordination of the basal N donors. This is in accordance with a larger ρ value in C3 (ρ = 0.98) than in C1 (ρ = 0.96) due to the stronger coordination of the guanidine moiety. The ρ value reveals the delocalisation of the electrons within the guanidine function and is 1 for ideal delocalised systems.76
Compared to the complex cations in C1–C3, in the copper(II) bromide complex cation in C4 the copper atom is trigonal–bipyramidal coordinated, despite the comparable ligand design. Two crystallographic independent molecules are present in the asymmetric unit (see Table S3†). Here the guanidine N donors coordinate in the equatorial positions (molecule 1: Cu–Neq (1) = 2.061(3), Cu–Neq (2) = 2.065(3), Cu–Neq (3) = 2.138(3)); Molecule 2: Cu–Neq (1) = 2.110(3), Cu–Neq (2) = 2.100(3), Cu–Neq (3) = 2.097(3) and the tertiary amine (molecule 1: Cu–Nax = 2.112(3); molecule 2: Cu–Nax = 2.113(3)) and the bromide (molecule 1: Cu–Br = 2.447(1); molecule 2: Cu–Br = 2.440(1)) are axially located. Three equal donors may be the reason for the change in the coordination geometry in the tripodal ligand structure, which was also observed in other copper complexes with three equal N donors.77,78 The trigonal–bipyramidal structure with the tertiary amine and the bromide at the axial positions enables comparison to the structures of the copper(II) bromide complexes based on various TPMA ligands.27,78,79 The average bond length to the equatorially located atoms is slightly increased for C4 but more significant is the elongated bond length to the axially located atoms (Cu–Br = 2.447(1) Å, Cu–Ntert = 2.112(3) in C4vs. Cu–Br = 2.390(1) Å, Cu–Ntert = 2.047(3) in TPMANMe2 and Cu–Br = 2.384(6) Å, Cu–Ntert = 2.040(3) in TPMA).27,78,79 On the one hand, the longer Cu–Br bond length could indicate a lower halidophilicity of C4 compared to the copper(II) TPMA systems, although it is required to form the deactivator complex in ATRP. On the other hand, a weaker and longer Cu–Br bond length could lead to an enhanced deactivation reaction. Furthermore, the only stable copper(I) bromide complex (C5) was received based on the ligand L3. The other systems tend to disproportionate and have a high sensitivity to air, similar to the highly active TPMANMe2-system.27 The guanidine N donors in the Cu(I) complex C5 coordinate in the basal positions (Cu–Nb (1) = 2.053(2), Cu–Nb (2) = 2.053(2), Cu–Nb (3) = 2.053(2)) and the bond lengths are shorter than of the axially located tertiary amine (Cu–Nax = 2.200(2)). Analogous to previous studies for copper(I) complexes, the guanidine functions here are less delocalised (ρ = 0.95) compared to that in the copper(II) complexes.49,80 The significant structural difference of the complex C5 without a coordinating halide is notable to previously investigated ATRP systems based on the ligand TPMA where a halide is coordinating.78 This can be interpreted as a better stabilisation of the required CuILn activator species induced by the guanidine moieties.42
Structural studies in solution
In order to clarify the structure of the copper(I) and copper(II) complexes in solution, a titration experiment and UV/Vis, EPR, and variable temperature NMR spectroscopic experiments were exemplarily conducted based on ligand L1. Since the copper(I) bromide complex tends to disproportionate to elemental copper and the copper(II) complex, the titration experiment was only conducted for the copper(II) system. The intention is to show that in solution as well as in the solid-state the formation of a monochelate species is preferred. Copper(II) bromide was dissolved in MeCN, and aliquots of L1 were added. Afterwards, the UV/Vis spectrum was obtained (Fig. S3†). The species formation was followed at 750 nm since the complex C1 exhibits one maximum at this wavelength (Fig. 2). It increases linearly up to adding a half equivalent of L1, then decreases until one equivalent is reached. The course indicates that species distributions are present using insufficient amounts of ligand (less than 1 eq.). Presumably, there is the formation of a binuclear species with either two N donors of a ligand and two bromide ligands coordinating the copper or a species with two adjacent halogen-bridged copper centres. Nevertheless, a slight excess of ligand (more than 1 eq.) may ensure the presence of the monochelate complex. Thus, an equimolar amount or a slight excess of ligand is recommended for experiments with in situ formed complexes (Table 2).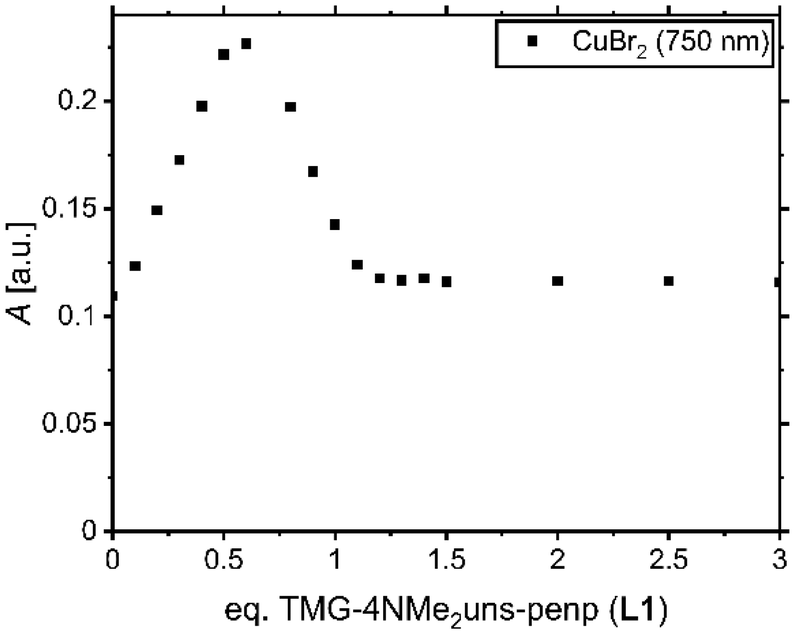 | ||
| Fig. 2 UV/Vis spectroscopic titration of 5 mM CuBr2 in MeCN with L1. | ||
| Complex | λ max [nm] (εmax [L(mol cm)−1]) | |
|---|---|---|
| 500–800 nm | 800–1100 nm | |
| [Cu(TMG-4NMe2uns-penp)Br]Br (C1) | 750 (112) | 942 (118) |
| [Cu(TMG-4NMe2uns-penp)Cl]Cl (C2) | 757 (104) | 945 (127) |
| [Cu(TMG-4NMe2uns-penp)](OTf)2 | 632 (106) | 911 (110) |
| [Cu(TPMANMe2)Br]Br27 |
776 (n. d.) | 1038 (n. d.) |
| [Cu(TPMANMe2)](OTf)227 |
725 (n. d.) | 980 (n. d.) |
UV/Vis spectra of the copper(II) bromide, chloride, and triflate complexes based on L1 were recorded, and for all three complexes, two d–d transitions are obtained (Fig. 3). This is typical for copper(II) complexes coordinated by tetradentate ligand systems and has especially been observed for trigonal–bipyramidal copper(II) complexes. For these complexes, the transitions were related to the transitions dx2−y2 ≈ dxy → dz2 and dxz ≈ dyz → dz2.81,82 The transitions for C1 are centred at 750 nm and 924 nm, for C2 at 769 nm and 938 nm, and for [Cu(TMG-4NMe2uns-penp)](OTf)2 at 632 nm and 902 nm. There is a blue-shift depending on the anion for both transitions in the order OTf < Br < Cl. This is in accordance with the transitions of the published copper(II) bromide (776 nm and 1138 nm) and triflate (725 nm and 980 nm) complexes based on the ligand TPMANMe2.27 The transitions presented here are blue-shifted; in particular, the first transition of the triflate complex is very different. This indicates a significant influence of the guanidine moiety on the optical properties of these complexes and a larger gap in the orbital energy levels.
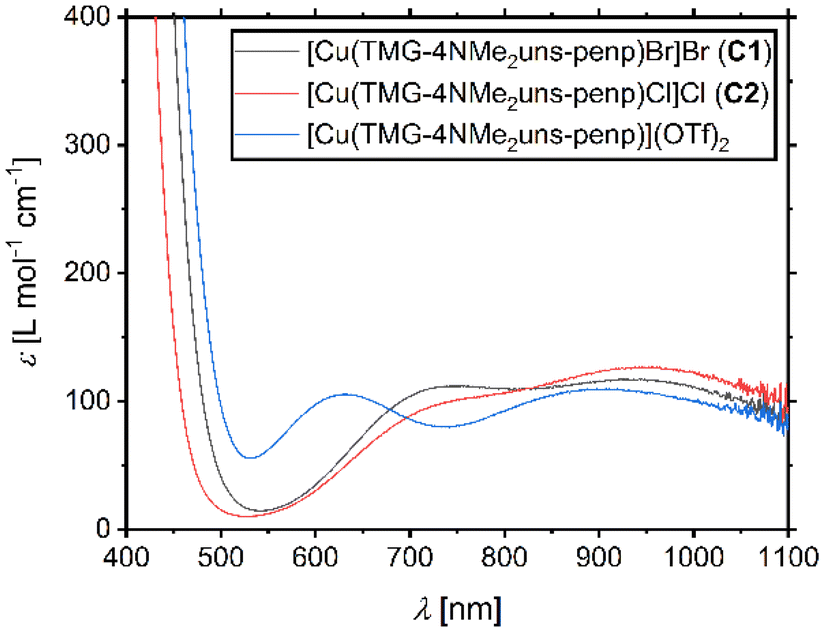 | ||
| Fig. 3 UV/Vis spectra for 2.5 mM complex solutions in MeCN of [Cu(TMG-4NMe2uns-penp)Br]Br (C1), [Cu(TMG-4NMe2uns-penp)Cl]Cl (C2), and [Cu(TMG-4NMe2uns-penp)](OTf)2. | ||
The complex C1 was further exemplarily investigated by EPR spectroscopic measurements. In a DCM/toluene = 1/1 mixture, an axial spectrum was obtained (Fig. S5†). The EPR parameters could be simulated with the MATLAB tool EasySpin:83g1 = 2.088, A1 = 139 MHz, g2 = 2.237, A2 = 426 MHz, g3 = 1.999 and A3 = 193 MHz. The g and A parameters are similar to the analogue TPMANMe2 complex in a DCM/toluene = 1/1 mixture: g1 = 2.173, A1 = 255.3 MHz, g2 = 2.198, A2 = 336.5 MHz, g3 = 1.95 and A3 = 260.1 MHz.27 The slight differences could be explained by different structures analogous to the structures in the solid state. In MeCN it exhibits an isotropic spectrum (Fig. S4†) with a calculated cubic tensor of giso = 2.242. This is in an expected range compared to the tables of Peisach and Blumberg.84 It is also similar to the measurement of the TPMANMe2 based copper(II) bromide complex in MeCN with giso = 2.123.27
Furthermore, the properties of the catalyst system based on L1 were characterised by variable temperature NMR spectroscopy. The intention is to show that this system undergoes fast ligand exchange in the presence of ligand excess relative to copper like other multidentate pyridine-based systems (Fig. 4 and Fig. S13†).85 Analogous to the TPMANMe2-system, by using equimolar amounts of the ligand, the solution turned to a dark suspension due to a disproportionation reaction from [Cu(TMG-4NMe2uns-penp)Br] to elemental copper, free ligand, and [Cu(TMG-4NMe2uns-penp)Br]Br in acetone-d6.27 Acetone is a polar solvent that can interfere with the formation of the copper(II) deactivator complex and, thus, with the disproportionating properties of the copper(I) complex.37 The reasons for this are its polarity and its weak coordination ability with copper(I). However, the main reason is the good stabilisation of the copper(II) centre by L1. Under ligand excess (L1/CuBr = 3/1), the disproportionation equilibrium is shifted to the copper(I) side. The previously paramagnetic dark suspension changed to a yellow diamagnetic solution, suitable for NMR spectroscopic investigations after a few minutes. At a temperature of 20 °C, the resonances are sharp and can be assigned in the aromatic region analogously to the resonances in the free ligand. The shifts vary slightly compared to the free ligand, but the coupling pattern of the protons is very similar. This indicates a fast exchange between the coordinated and the free ligand. By a decrease of temperature to −40 °C, the first broadening in the aromatic region was observed. This is reasoned by a slower rate of ligand exchange that continues to slow down at −80 °C. Here, the resonance of the proton c splits into separated resonance of free and coordinated ligand in an intensity ratio of 2/1. This behaviour is similar to that in previous investigated TPMA systems.27,85
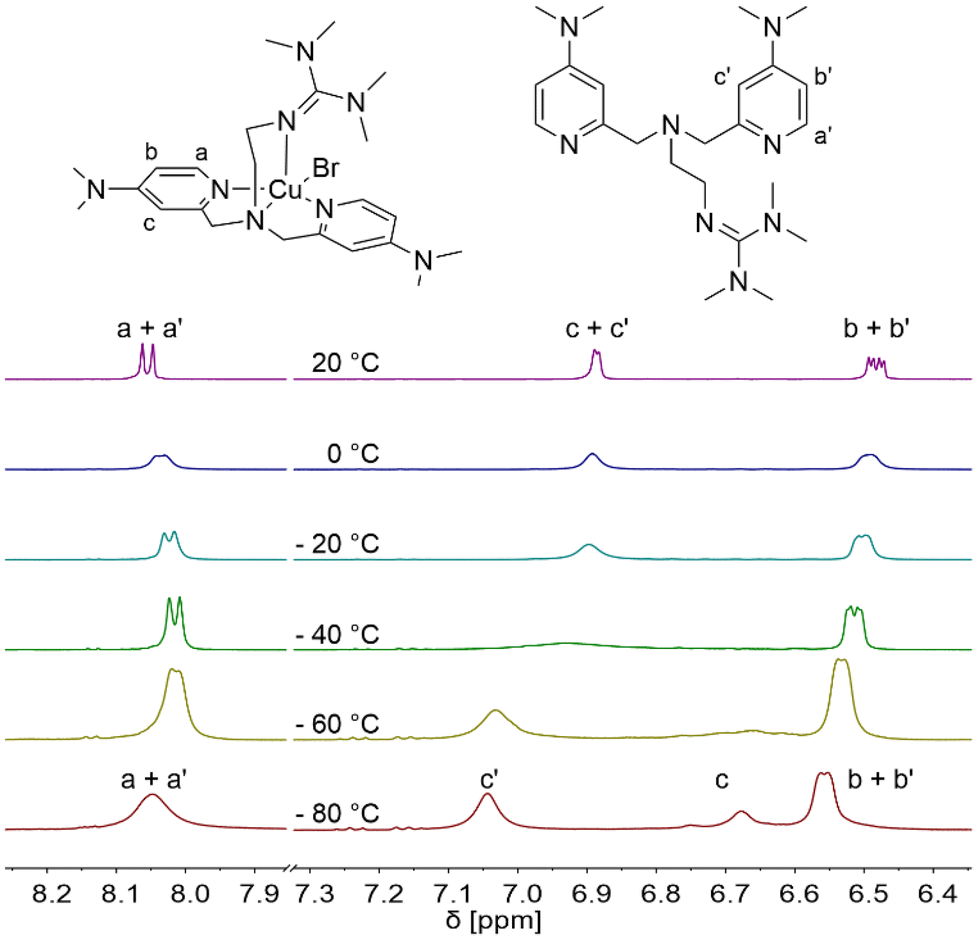 | ||
| Fig. 4 Aromatic region of variable-temperature 1H NMR spectra of the L1-based CuBr complex in d6-acetone at a molar ratio of [CuBr]:[L1] = 1:3. | ||
Electrochemistry
The ATRP catalyst activity is described by the equilibrium constant which correlates to the redox potential for copper complexes with N donor ligands.38 This allows the activity prediction of new catalyst systems solely on the basis of their redox potential.27 Therefore, cyclic voltammetric measurements of the systems based on L1–L3 were conducted (Fig. 5 and Fig. S6–S9†). The complexes were formed in situ dissolving equimolar amounts of ligand and copper(II) salt in acetonitrile at room temperature. The redox potential of the Fc/Fc+ couple was subsequently measured and it served as an internal standard. Afterwards, the redox potentials of the systems were referred against saturated calomel electrode (SCE) for easier comparability with the literature.86 The whole procedure was realised under a nitrogen atmosphere in a glovebox with degassed solvents to exclude oxygen, which is necessary due to the high air sensitivity of the catalyst systems. Scan rates of 200 mV s−1, 100 mV s−1, 50 mV s−1, and 20 mV s−1 were used. The received values for E1/2, the peak-to-peak separation (ΔE), and predicted KATRP values (predicted by using the correlation (Fig. 6))38 are denoted in Table 3. The system based on L1 and L2 shows a reversible electron transfer (Tables S4–S7†). A quasi-reversible electron transfer for the systems based on L3 explains the large values of ΔE (Table S8†).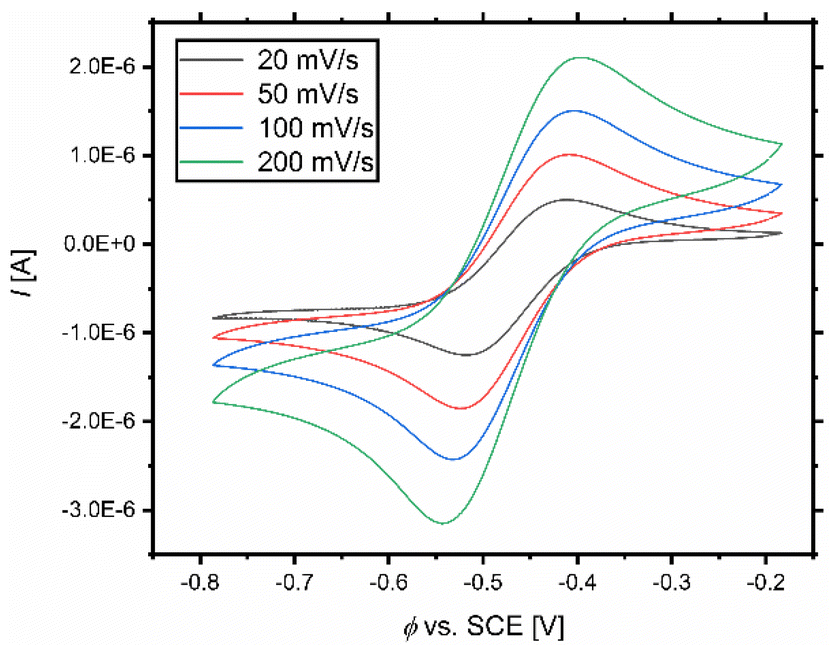 | ||
| Fig. 5 Cyclic voltammograms with various scan rates of the CuIL/CuIIL couple starting from a 1 mM [Cu(TMG-4NMe2uns-penp)Br]Br complex solution in MeCN. | ||
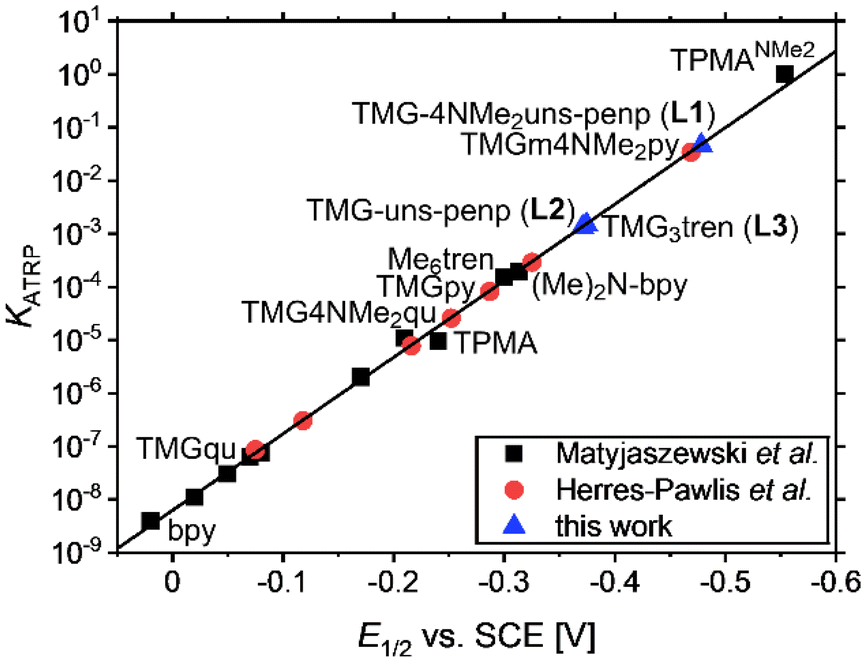 | ||
| Fig. 6 Calculated KATRP values against measured E1/2 values for various copper catalysts based on tetradentate guanidine ligands in the linear correlation of E1/2vs. log(KATRP). | ||
| CuIL/CuIIL, L= | X− | E 1/2 vs. SCE [V] | ΔE [mV] | K ATRP |
|---|---|---|---|---|
| TMG-4NMe2uns-penp (L1) | Br− | −0.478 | 99 | 4.6 × 10−2 |
| TMG-4NMe2uns-penp (L1) | Cl− | −0.601 | 79 | — |
| TMG-4NMe2uns-penp (L1) | OTf− | −0.373 | 97 | — |
| TMG-uns-penp (L2) | Br− | −0.371 | 77 | 1.3 × 10−3 |
| TMG3tren (L3) | Br− | −0.375 | 440 | 1.5 × 10−3 |
| TPMANMe2 (ref. 27) | Br− | −0.554 | n. d. | ≈1 |
| TPMANMe2 (ref. 27) | OTf− | −0.302 | n. d. | — |
| TPMA (ref. 38) | Br− | −0.240 | 70 | 9.6 × 10−6 |
It shows on the one side that these ligands can stabilise the electrochemically generated copper(I) complex, but on the other side that higher reorganisation energy for the systems based on L1 and L3 is required to change the geometry between the copper(II) and copper(I) complexes.87 ΔE increases for ligands with dimethylaminepyridino (DMAP) units compared to pyridine units and for an increased number of TMG units.
It is consistent with the quasi-reversible nature of the electron transfer for the TPMANMe2 system27 and a reversible electron transfer for the TPMA system.38 For the system based on L3, a quasi-reversible redox wave with a peak-to-peak separation of ΔE = 440 mV was obtained.
A reason for the quasi-reversible behaviour in this magnitude could be the change in the coordination sphere by the redox process from the copper(I) complex without a coordinating halide to the copper(II) species with a coordinating halide like in the crystal structures. This change in the coordination geometry seems to be different relative to other comparable copper complexes with tripodal ligands like TPMA-based copper(I) complexes where a halide is coordinating.78 To investigate the influence of the anion, measurements of the copper bromide, chloride, and triflate systems based on L1 were conducted. The copper bromide system possesses a higher redox potential (E1/2 = −0.478 V) relative to the appropriate copper chloride system (E1/2 = −0.601 V) and a lower redox potential relative to the copper triflate system (E1/2 = −0.373 V). This dependence on the halide is in accordance with the literature and indicates a higher redox potential by a larger Cu–X bond length.49,88 The redox potential of the copper bromide system of L1 (E1/2 = −0.478 V) compared to that of L2 (E1/2 = −0.371 V) is lower due to the electron density donating dimethylamine moieties at the pyridines (ΔE1/2(L1/L2) = −0.107 V) leading to the most active ATRP catalyst based on a guanidine ligand (KATRP = 4.6 × 10−2). The difference is smaller and in an expected range relative to the TPMANMe2 (E1/2 = −0.554 V) and TPMA (E1/2 = −0.240 V) systems since only two pyridines are substituted and not three (ΔE1/2(TPMANMe2/TPMA) = −0.314 V). Notable is that the redox potential of the system based on L2 is lower than that of the TPMA system indicating stronger electron density donating properties of the TMG unit relative to pyridine units. The influence of guanidine donors on activity is highly dependent on the ligand design, and the described increase in activities for the tetradentate pyridine ligands demonstrates the importance of studying these donors (Fig. 6). Advances in the ligand design lead to L1, which is the most reductive system based on guanidines to date. For the bromide system based on L1, the redox potential is higher and thus the activity is still lower relative to the system based on TPMANMe2 concerning larger KATRP values, but electrochemical differences influenced by the guanidine functions were found. The redox potential of the triflate system is lower (E1/2 = −0.373 V with L1vs. E1/2 = −0.302 V with TPMANMe2). The smaller difference in the redox potential of the bromide to the triflate system using the ligand L1 compared to the TPMANMe2 ligand suggests less interference between halide and the copper centre induced by guanidine donors. A reason is a larger distance between the halide and the copper centre by the guanidine moiety. Lower redox potentials indicate a better copper(II) stabilisation and depend on the donating properties of the ligand.27,89 In the triflate systems, the anion has less influence on the redox potential. Therefore, it reveals even more electron density donating properties of L1 relative to the TPMANMe2 ligand and consequently stronger donating ability of TMG relative to DMAP moieties in these systems. The large peak-to-peak separation for the system based on L3 can explain the similar E1/2 values for the systems based on L3 (E1/2 = 0.375 V) and L2 even though three donating TMG moieties are present in L3.
Using the redox potentials of the triflate system (E1/2,LCu) based on L1 and the previously determined standard redox potential of the solvated CuI/CuII couple in MeCN at T = 25 °C (E1/2,Cu = 1.06°V vs. SCE), the βII/βI ratio can be calculated (eqn (1)).27,42 The obtained value of βII/βI = 1.7 × 1024 is larger by one order of magnitude than of the TPMANMe2-system (βII/βI = 1.1 × 1023) revealing a huge driving force for copper(II) stabilisation and radical formation in ATRP. Furthermore, using the redox potential of the bromide system (E1/2, LCuBr) the ratio of the apparent halidophilicity constants KIIX,app, KIX,app can be calculated (eqn (2)).
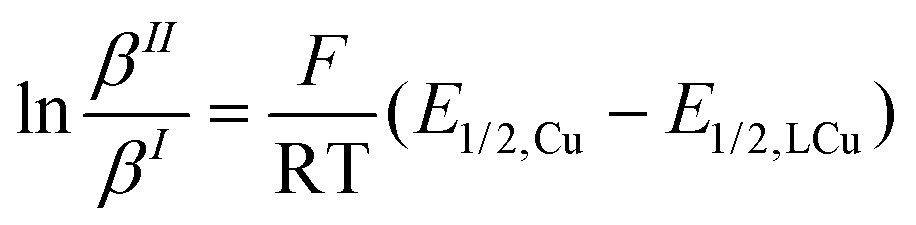 | (1) |
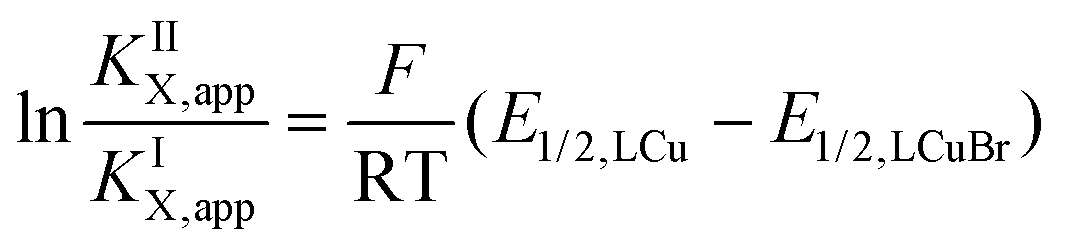 | (2) |
The received value of 60 is significantly lower than for the TPMANMe2-system (≈103). Since a good stabilisation of the activator complex by guanidine donors in tripodal and tetradentate ligands is proposed and the Cu–Br bond length is larger in the copper(II) complexes with guanidine moieties, this low value means a high stabilisation of the CuIILn species. Even though this is unwanted, it should lead to a large driving force for the activation reaction. Because the redox potential of the copper bromide system based on L1 is higher relative to the TPMANMe2 system and therefore KATRP is lower (KATRP = 4.6 × 10−2), concerning the relation KATRP = kact/kdeact the rate of deactivation must be even higher. Presumably well-stabilised species and the shifts of equilibria are highlighted in Scheme 4. Thus, there are indications that the guanidine moiety induces a faster exchange within the ATRP equilibrium. The reasons for this are better stabilisation of the copper(I) activator complex and improved deactivation by easier homolytic bond cleavage due to a larger Cu–Br bond length in the copper(II) deactivator complex.
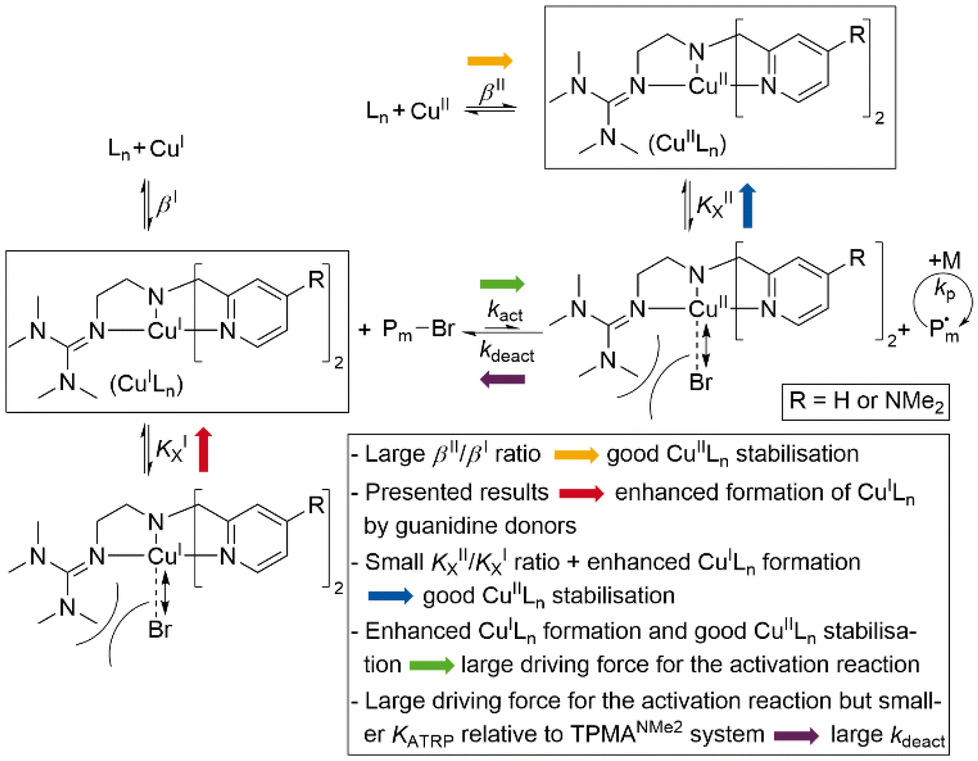 | ||
| Scheme 4 Proposed impact of tetradentate guanidine ligands on ATRP equilibrium. | ||
Density functional theory (DFT) calculations
DFT calculations were performed for the complex cations of C1, C2, C3, C4, C5 and the related copper(II) bromide complexes based on the ligands TPMA and TPMANMe2. The functional TPSSh90,91 and the basis set def2-TZVP92–95 with a polarisable continuum model (PCM), as a solvent model for MeCN and empirical dispersion correction with the D3 version of Grimme's dispersion Becke–Johnson damping (GD3BJ) were used in accordance with previous studies (additional information is provided in the Experimental section).96–98 Except for C1, the calculated complex structures by DFT are in agreement with the experimental ones (Fig. 7 and Tables S9–S12†). In C1, the Ntert–Cu–Br bond angle varies (174.7° vs. 163.8°), which explains the large difference in the τ5 values (0.37 vs. 0.11). The calculated minimum of the calculation was thereby verified as described in the ESI† (DFT calculations of the complexes) and the slightly changed position of the bromide in the crystal structure was explained by packing effects.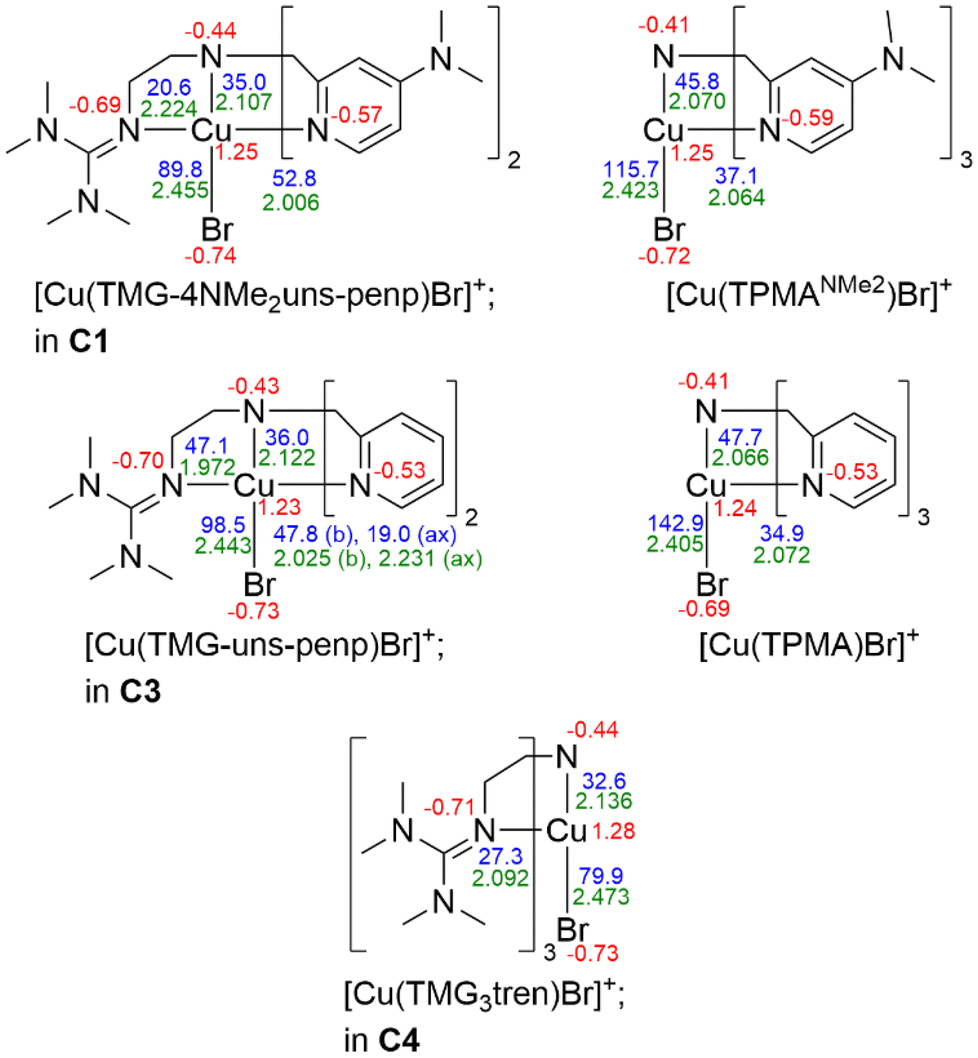 | ||
| Fig. 7 Calculated NBO charges [e units] (red), charge-transfer energies ECT [kcal mol−1] (blue) and selected bond lengths [Å] (green) for selected Cu(II) deactivator complex cations (NBO7.0), TPSSh/def2-TZVP and PCM solvent model for MeCN and empirical dispersion correction with Becke–Johnson damping. | ||
In the following, the results for the copper(II) bromide complexes are discussed exclusively. The calculations reveal square-pyramidal structures for the complex cation in C1 and C3, which correspond to the experimental structures. At this point, analogous to C1, the calculated minimum for C3 was also verified as described in the ESI† in order to prove the exchange of the coordinating donors as a function of the dimethylamine substitution of the pyridine donors. Also corresponding to the experimental structures, trigonal–bipyramidal structures were obtained for the complex cation in C4 and the copper(II) bromide complexes based on the ligands TPMA and TPMANMe2. For all structures, the trends between the calculated bond lengths are the same as for the experimentally determined structures and the root mean square deviation (RMSD) values are small.
The optimised structures were used for natural bond orbital (NBO) calculations to investigate the influence of the varied donors on the NBO charges of the copper, the donating N atoms, the coordinating bromide, and on the charge-transfer energies (ECT). The NBO charge of the N donors decreases in the order: tertiary amine > pyridine > DMAP > guanidine, revealing the strongest basicity for the guanidine donor and the descending order of basicity within these systems.
In all complexes, ECT are higher for DMAP vs. pyridine donors showing better donor properties of DMAP units due to electron density donating properties of the dimethylamine substituents. ECT of the guanidine donors strongly depends on the complex geometry. As expected, ECT of the axially located guanidine donor in the square-pyramidal complex cation in C1 is lower than of the basally located guanidine in C3. Moreover, ECT values of the equatorially located guanidine donors in the trigonal–bipyramidal complex cation in C4 are lower than of those pyridine donors in the TPMA complexes, revealing weaker donation. The reason for this is the high steric demand of the bulky TMG moieties. The steric demand leads to interactions between the guanidines when three of these coordinate in the same plane, resulting in weaker donation despite their excellent donor properties.
It is proposed that a large Cu–Br bond length in the copper(II) deactivator complexes is easily cleavable homolytically, improving the deactivation reaction. In the calculated structures, the Cu–Br bond length for DMAP-containing complexes is elongated compared to the analogue pyridine complexes and even more by the number of guanidine donors, showing a dependence on the different donors as obtained in the crystal structures. Concerning ECT of the bromide, there is a clear dependency on the N donors. ECT of the bromide decreases by DMAP relative to unsubstituted pyridines, indicating that the Cu–Br bond gets weaker by stronger donating N donors.
This can be illustrated by the sum of the ECT values of all coordinating N donors (ECT,total). The value is higher for the copper(II) bromide TPMANMe2 complex (ECT,total = 156.9 kcal mol−1) than for the comparable TPMA complex (ECT,total = 152.4 kcal mol−1) and analogously when comparing the complexes in C1 (ECT,total = 161.2 kcal mol−1) and C3 (ECT,total = 149.9 kcal mol−1). The ECT of the bromide also decreases by replacing a pyridine donor with a guanidine donor. In this case, the sum of the ECT values of all coordinating N donors does not change significantly. Thus, certain electronic or steric properties of the guanidine must therefore be the reason that ECT of the bromide decreases and consequently the bond is weakened. For the complex cation in C4 with three guanidine donors, ECT of the bromide is the lowest in the presented series. Since the interaction between the TMG units leads to a weaker donation of these and a lower sum of ECT values of all coordinating N donors (ECT,total = 114.5 kcal mol−1), the steric properties of the TMG moieties are mainly the reason for the low ECT of the bromide in C4. However, there is evidence that the steric and the electronic properties of the guanidine donors are the reason for lower ECT values of the bromide, and these correlate directly within these systems with the Cu–Br bond lengths. Therefore, the performed calculations show the bromide's weaker coordination in dependency of the N donors and emphasise the theory of a better cleavable Cu–Br bond by guanidine donors.
Polymerisation
The catalyst system based on L1 contains a guanidine that leads to a weaker and elongated Cu–Br bond length in the deactivator complex and shows a very low redox potential with reversibility in the redox process comparable to that in the most active TPMA systems. Therefore, to assess the predicted high activity of this catalyst system the performance of standard ATRP and ICAR ATRP of styrene were tested. EBiB as initiator, AIBN as radical initiator and benzonitrile as solvent were used analogue to the conditions by previous studies.49,52,61 The complexes were prepared in situ using copper(I) bromide and L1 or copper(II) bromide and L1 for standard ATRP or ICAR ATRP experiments, respectively. The performance of standard ATRP with a ratio of M/I/Cat. = 100/1/1 was tested but indicates an uncontrolled behaviour (Fig. S15†). A reason is the low redox potential resulting in a high amount of copper(II) species and free radicals. That causes more radical terminations, a change of radical concentration, and a polymerisation not following pseudo-first-order kinetics. In contrast, using a ratio of M/I/Cat./AIBN = 100/1/0.1/1.5 at 60 °C, a linear increase of the semilogarithmic plot of conversion vs. time was observed (Fig. 8) indicating a successful ICAR ATRP. To quantify the results, polymerisations were also carried out under analogous conditions, but with the ligands L3 and TPMA (Fig. S16 and S17†). The rate constants kobs depend on the AIBN concentration and the rate of decomposition.10,32,99 The values (L1: kobs = 1.4 × 10−3 ± 1.6 × 10−5 s−1; L3: kobs = 1.5 × 10−3 ± 1.2 × 10−5 s−1; TPMA: kobs = 1.4 × 10−3 ± 2.3 × 10−5 s−1) are very similar to previous results from us using comparable conditions (kobs = 1.3 × 10−3 ± 2.2 × 10−5 s−1).52,61 The short induction period at the beginning may be explained by radical consumption of the deactivator complex until a specific ratio of activator to deactivator is reached.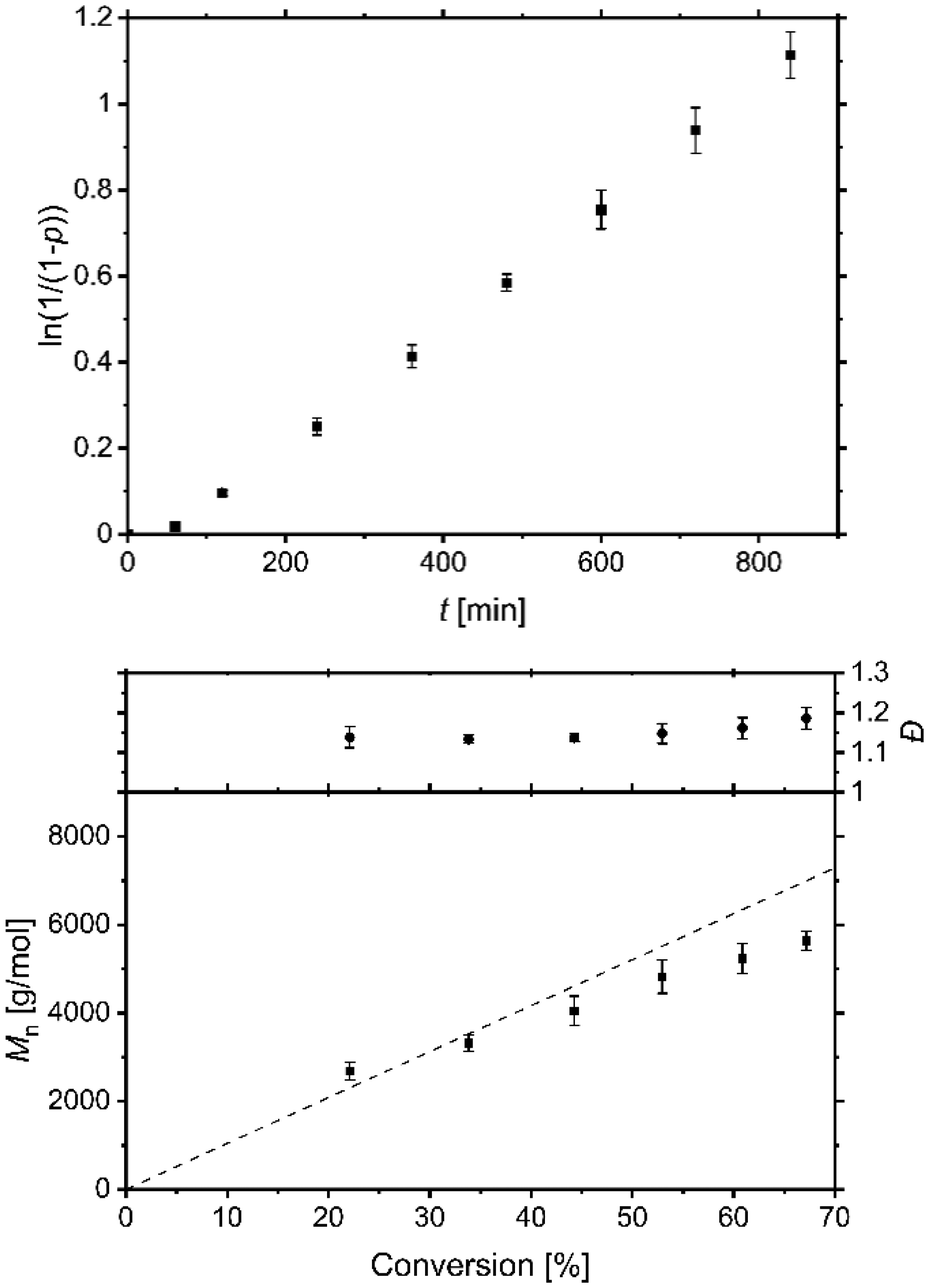 | ||
| Fig. 8 Semilogarithmic plot of conversion vs. time (top) and plot of Mn/Đ vs. conversion (bottom) for ICAR ATRP of styrene with the CuBr2/L1 catalyst system. Conditions: Styrene//EBiB/Cat./AIBN = 100/1/0.1/1.5 in benzonitrile at 60 °C. | ||
The controlled behaviour of the polymerisations were confirmed by the constant growth of the molar masses that fit to the expected ones and low dispersities by increased conversions up to 67% after 14 h for the system based on L1 (Table 4). This practical example underlines the excellent mediator properties of the catalyst system based on L1, even if the comparative experiments with L3 and TPMA show that under these conditions there is barely any difference in polymerisation behaviour with regard to dispersity. However, the conversion increases faster for L1 and L3 in these ICAR ATRP experiments compared to TPMA. The main reason for this is the low redox potential and a high value for KATRP, which promote activation, but due to successful deactivation and a high amount of copper(II) deactivator species, the dispersities are still low.31,32 A high value for the equilibrium constant can also be a reason for low dispersity due to the decrease of unwanted side reactions such as CRT and RRT.35,36 The elongation of the Cu–Br bond length in the deactivator complex and the resulting presumed faster exchange within the equilibrium by guanidine donors may also have an influence.
| t [h] | TMG-4NMe2uns-penp (L1) | TMG3tren (L3) | TPMA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p [%] | M n [g mol−1] | M n,th [g mol−1] | Đ | p [%] | M n [g mol−1] | M n,th [g mol−1] | Đ | p [%] | M n [g mol−1] | M n,th [g mol−1] | Đ | |
| 1 | 2 | n. d. | 200 | n. d. | 1 | n. d. | 100 | n. d. | 0 | n. d. | 0 | n. d. |
| 2 | 9 | n. d. | 900 | n. d. | 8 | n. d. | 800 | n. d. | 1 | n. d. | 100 | n. d. |
| 4 | 22 | 2700 | 2300 | 1.14 | 23 | 1700 | 2400 | 1.19 | 16 | n. d. | 1700 | n. d. |
| 6 | 34 | 3300 | 3500 | 1.13 | 35 | 2400 | 3700 | 1.17 | 29 | 1700 | 3000 | 1.10 |
| 8 | 44 | 4100 | 4600 | 1.14 | 46 | 2800 | 4800 | 1.18 | 40 | 2500 | 4200 | 1.09 |
| 10 | 53 | 4800 | 5500 | 1.15 | 55 | 3800 | 5700 | 1.13 | 50 | 3000 | 5200 | 1.09 |
| 12 | 61 | 5200 | 6400 | 1.16 | 62 | 3700 | 6500 | 1.18 | 57 | 3200 | 5900 | 1.12 |
| 14 | 67 | 5600 | 7000 | 1.19 | 67 | 4000 | 7000 | 1.19 | 63 | 3600 | 6600 | 1.12 |
Conclusions
The synthetic pathway for the novel ligand TMG-4NMe2uns-penp (L1) is presented. Besides L1, the tripodal and tetradentate ligands TMG-uns-penp (L2) and TMG3tren (L3) were used to investigate the influence of guanidine moieties on the ATRP catalyst properties. With these ligands, copper complexes were synthesised and crystallised for structure analysis in the solid state. Based on L1 and L2, distorted square-pyramidal complexes were obtained, and it was found that the position of the guanidine N donor changes by NMe2 substitution at the pyridines. Based on L3, a trigonal–bipyramidal structure that can be related to the TPMA systems was received. Guanidine donors increased the bond lengths between the copper centre and the bromide ligand. This is underlined by the trigonal–pyramidal copper(I) complex based on L3 without a coordinating halide which is different in the copper(I) halide complexes based on the TPMA ligand where the halide coordinates. Additionally, this system does not disproportionate despite the strong donating guanidine donors. Thus, the stabilisation of the activator complex by the guanidine moieties in tripodal ligands seems to be higher.The catalyst activities for the systems based on L1–L3 were predicted by measurements of the redox potentials and using the strong correlation with KATRP. As expected, dimethylamine substituents at the pyridines lead to a lower redox potential, making the system based on L1 the most active ATRP catalyst based on a guanidine ligand (E1/2 = −0.478 V vs. SCE, KATRP = 4.6 × 10−2). Moreover, based on the measured redox potentials, an overall faster exchange within the ATRP equilibrium was derived, which could be important for future polymerisations in terms of low dispersity.100 The reasons proposed are the preferential presence of the activator complex and a weaker Cu–Br bond in the deactivator complex that could improve the deactivation reaction. An elongated Cu–Br bond length and its weakening by guanidine units are supported by DFT calculations. A successful ICAR ATRP of styrene indicated high activity and good reversibility within the ATRP equilibrium. Controlled conditions with low dispersities and molar masses that fit to the expected ones were obtained. This shows the large potential of guanidine copper systems in ATRP and opens up new avenues in ligand design for CRP methods.
Experimental part
General information
The synthesis of the ligands and complexes was conducted under inert conditions under a nitrogen atmosphere (99.999% purity) using the Schlenk technique or a glovebox. Purified and degassed solvents were used and were purified according to the literature.101 The chemicals for the synthesis of the ligands and complexes were all purchased from ABCR, Acros Organics, Alfa Aesar, Grüssing, Fluka Analytical, Fisher-Scientific, Sigma-Aldrich or TCI and were used as received without further purification. The compounds 2-(1,3-dioxoisoindolin-2-yl)ethan-1-aminium 2,2,2-trifluoroacetate (1) and 4-chloropicolinaldehyde (2) were resynthesised by modifying the literature procedures, which is described in the ESI.†64,102 The Vilsmeier salt N,N,N′,N′-tetramethylchloroformamidinium chloride (TMG-VS), copper(I) bromide, L2 and L3 were synthesized as described in the literature.67,69,70,103Ligand synthesis
2-(1,3-Dioxoisoindolin-2-yl)ethan-1-aminium 2,2,2-trifluoroacetate (10.0 g, 32.9 mmol, 1 eq.), triethyl amine (3.33 g, 32.9 mmol, 1 eq.) and 4-chloropicolinaldehyde (9.31 g, 65.8 mmol, 2 eq.) were dissolved in DCM (200 mL) and stirred for 1 h. To this solution sodium tri(acetoxy)borohydride (20.9 g, 98.7 mmol, 3 eq.) and DCM (100 mL) were added and the suspension was stirred for 18 h. After that, the mixture was quenched with saturated sodium hydrogen carbonate solution, the organic layer was separated, and the aqueous phase was extracted with DCM (2 × 150 mL). The combined organic layers were dried over MgSO4 and the solvent was removed under reduced pressure. For purification, the product was diluted in DCM (10 mL) and slowly added to n-hexane (300 mL). The precipitate was filtered and residual solvent removed under reduced pressure (yield = 98%, 14.2 g, 32.2 mmol). 1H NMR (400 MHz, CDCl3): δ = 8.32 (d, 3JHH = 5.4 Hz, 2H, CH, 5), 7.83 (m, 2H, CH, 8), 7.73 (m, 2H, CH, 9), 7.31 (d, 4JHH = 2.0 Hz, 2H, CH, 2), 7.08 (dd, 3JHH = 5.4, 4JHH = 2.0 Hz, 2H, CH, 4), 3.85 (m, 6H, CH2, 10 + 11), 2.89 (t, 3JHH = 5.9 Hz, 2H, CH2, 12) ppm. 13C NMR (101 MHz, CDCl3): δ = 168.4 (Cq, 7), 161.0 (Cq, 1), 149.9 (CH, 5), 144.7 (Cq, 3), 134.1 (CH, 9), 132.2 (Cq, 6), 123.5 (CH, 8), 123.4 (CH, 2), 122.6 (CH, 4), 59.8 (CH2, 10), 51.9 (CH2, 12), 35.9 (CH2, 11) ppm. IR (Diamond-ATR, neat)
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) max: 3025 (vw), 2946 (vw), 2855 (vw), 1765 (m), 1706 (vs), 1679 (w), 1575 (m), 1555 (m), 1468 (w), 1454 (w), 1426 (m), 1393 (s), 1366 (m), 1352 (m), 1323 (m), 1280 (w), 1250 (vw), 1215 (w), 1186 (m), 1172 (w), 1133 (w), 1108 (m), 1087 (m), 1036 (w), 1019 (w), 981 (w), 970 (m), 955 (w), 940 (m), 895 (w), 875 (w), 862 (w), 825 (m), 802 (w), 739 (m), 720 (vs), 703 (s), 670 (m), 606 (w), 573 (w), 544 (w), 530 (m), 494 (w), 457 (m), 428 (s), 406 (w), 369 (w), 354 (s), 331 (m) cm−1. HRMS (ESI+, MeOH) m/z: isotopic distribution calcd for C22H19N4Cl2O2 [M + H]+: 441.0886 (100) [C22H1935Cl2N4O2]+, 442.0915 (26) [C2113CH1935Cl2N4O2]+, 443.0864 (68) [C22H1935Cl37ClN4O2]+, 444.0890 (17) [C2113CH1935Cl37ClN4O2]+, 445.0839 (13) [C22H1937Cl2N4O2]+, 446.0862 (3) [C2113CH1937Cl2N4O2]+; found: 441.0887 (100), 442.0920 (26), 443.0857 (68), 444.0887 (16), 445.0825 (11), 446.0855 (3).
max: 3025 (vw), 2946 (vw), 2855 (vw), 1765 (m), 1706 (vs), 1679 (w), 1575 (m), 1555 (m), 1468 (w), 1454 (w), 1426 (m), 1393 (s), 1366 (m), 1352 (m), 1323 (m), 1280 (w), 1250 (vw), 1215 (w), 1186 (m), 1172 (w), 1133 (w), 1108 (m), 1087 (m), 1036 (w), 1019 (w), 981 (w), 970 (m), 955 (w), 940 (m), 895 (w), 875 (w), 862 (w), 825 (m), 802 (w), 739 (m), 720 (vs), 703 (s), 670 (m), 606 (w), 573 (w), 544 (w), 530 (m), 494 (w), 457 (m), 428 (s), 406 (w), 369 (w), 354 (s), 331 (m) cm−1. HRMS (ESI+, MeOH) m/z: isotopic distribution calcd for C22H19N4Cl2O2 [M + H]+: 441.0886 (100) [C22H1935Cl2N4O2]+, 442.0915 (26) [C2113CH1935Cl2N4O2]+, 443.0864 (68) [C22H1935Cl37ClN4O2]+, 444.0890 (17) [C2113CH1935Cl37ClN4O2]+, 445.0839 (13) [C22H1937Cl2N4O2]+, 446.0862 (3) [C2113CH1937Cl2N4O2]+; found: 441.0887 (100), 442.0920 (26), 443.0857 (68), 444.0887 (16), 445.0825 (11), 446.0855 (3).
Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-MBUHWDDXOF-UHFFFADPSC-NUHFF-NUHFF-NUHFF-ZZZ.
2-(2-(Bis((4-chloropyridin-2-yl)methyl)amino)ethyl)isoindoline-1,3-dione (6.88 g, 15.9 mmol, 1 eq.) and dimethylamine hydrochloride (16.0 g, 196 mmol, 12.3 eq.) were added to a 100 mL pressure flask. Water (50 mL) was added and then NaOH (7.82 g, 196 mmol, 12.3 eq.) was added and the flask was quickly closed. The mixture was stirred at 150 °C overnight. The cooled flask was opened and the mixture was diluted with 1 M NaOH (200 mL) and was stirred for 2 h. The solution was extracted with MeCN (3 × 150 mL), the combined organic layers were dried over Na2SO4, filtered and the solvent evaporated under reduced pressure. The product was used for following synthesis without further purification. (Yield = 67%, 3.50 g, 10.7 mmol). 1H NMR (400 MHz, CDCl3): δ = 8.09 (d, 3JHH = 5.9 Hz, 2H, CH, 5), 6.73 (d, 4JHH = 2.4 Hz, 2H, CH, 2), 6.31 (dd, 3JHH = 5.9 Hz, 4JHH = 2.4 Hz, 2H, CH, 4), 3.69 (s, 4H, CH2, 7), 2.93 (s, 12H, CH3, 6), 2.74 (t, J = 5.8 Hz, 2H, CH2, 9), 2.63 (t, J = 5.8 Hz, 2H, CH2, 8), 2.44 (br. s, 2H, NH2) ppm. 13C NMR (101 MHz, CDCl3): δ = 159.7 (Cq, 1), 154.9 (Cq, 3), 149.1 (CH, 5), 105.5 (CH, 2), 105.2 (CH, 4), 61.2 (CH2, 7), 57.6 (CH2, 8), 39.8 (CH2, 9), 39.1 (CH3, 6) ppm. IR (Diamond-ATR, neat)
 max: 3353 (vw, ν(N–H)), 2896 (w), 2819 (w), 1596 (vs), 1540 (m), 1507 (m), 1449 (m), 1433 (m), 1373 (m), 1310 (w), 1269 (w), 1220 (m), 1163 (w), 1130 (w), 1110 (w), 1068 (w), 1003 (m), 984 (m), 968 (m), 881 (w), 841 (w), 806 (m), 766 (w), 740 (w), 599 (vw), 482 (vw), 441 (vw), 318 (vw) cm−1. HRMS (ESI+, MeOH) m/z: isotopic distribution calcd for C18H29N6 [M + H]+: 329.2453 (100) [C18H29N6]+, 330.2483 (22) [C1713CH29N6]+, 331.2508 (2) [C1613C2H29N6]+; found: 329.2449 (100), 330.2481 (21), 331.2514 (2).
max: 3353 (vw, ν(N–H)), 2896 (w), 2819 (w), 1596 (vs), 1540 (m), 1507 (m), 1449 (m), 1433 (m), 1373 (m), 1310 (w), 1269 (w), 1220 (m), 1163 (w), 1130 (w), 1110 (w), 1068 (w), 1003 (m), 984 (m), 968 (m), 881 (w), 841 (w), 806 (m), 766 (w), 740 (w), 599 (vw), 482 (vw), 441 (vw), 318 (vw) cm−1. HRMS (ESI+, MeOH) m/z: isotopic distribution calcd for C18H29N6 [M + H]+: 329.2453 (100) [C18H29N6]+, 330.2483 (22) [C1713CH29N6]+, 331.2508 (2) [C1613C2H29N6]+; found: 329.2449 (100), 330.2481 (21), 331.2514 (2).
Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-WNSOFCUINP-UHFFFADPSC-NUHFF-NUHFF-NUHFF-ZZZ.
N 1,N1-Bis((4-(dimethylamino)pyridin-2-yl)methyl)ethane-1,2-diamine (6.00 g, 18.3 mmol, 1 eq.) and triethylamine (2.22 g, 21.9 mmol, 1.2 eq.) were dissolved in acetonitrile (130 mL) and the TMG-VS (3.75 g, 21.9 mmol, 1.2 eq.) in acetonitrile (30 mL) was slowly added at room temperature to the reaction mixture. After refluxing for 3 h, the mixture was cooled to room temperature. An aqueous solution of sodium hydroxide (0.88 g, 21.9 mmol, 1.2 eq.) was added and triethylamine as well as the solvents were removed under reduced pressure. A 50% solution of potassium hydroxide in water (50 mL) was added and the product was extracted with acetonitrile (3 × 150 mL). The combined organic layers were dried over Na2SO4. Activated charcoal was added and after filtration over Celite®, the solvent was removed under reduced pressure and the raw product was obtained (97%, 7.64 g, 17.9 mmol). For purification, toluene was added to the raw product and refluxed under stirring for 10 min. The hot toluene phase was decanted and the solvent was removed under reduced pressure. To the light brown oil, n-heptane was added and refluxed for 10 min. The hot n-heptane phase was decanted and the product crystallised overnight. The bright yellow crystals were dispersed, filtered and dried under reduced pressure (24%, 1.86 g, 4.36 mmol). 1H NMR (400 MHz, CDCl3): δ = 8.08 (d, 3JHH = 5.9 Hz, 2H, CH, 7), 6.86 (d, 4JHH = 2.7 Hz, 2H, CH, 4), 6.30 (dd, 3JHH = 5.9 Hz, 4JHH = 2.7 Hz, 2H, CH, 6), 3.76 (s, 4H, CH2, 11), 3.37–3.24 (m, 2H, CH2, 10), 2.93 (s, 12H, CH3, 8), 2.81–2.73 (m, 2H, CH2, 9), 2.65 (s, 6H, CH3, 2), 2.59 (s, 6H, CH3, 2) ppm 13C NMR (101 MHz, CDCl3): δ = 160.5 (CGUA, 1), 160.5 (Cq, 3), 155.0 (Cq, 5), 149.1 (CH, 7), 105.2 (CH, 4), 105.1 (CH, 6), 61.3 (CH2 11), 57.4 (CH2, 9), 47.6 (CH2, 10), 39.7 (CH3, 2), 39.1 (CH3, 8), 39.0 (CH3, 2) ppm. IR (Diamond-ATR, neat)
 max: 2929 (w, ν(CHaliph.)), 2888 (w, ν(CHaliph.)), 2812 (w), 1613 (s), 1597 (vs, ν(CN)), 1541 (m), 1504 (m), 1444 (m), 1428 (m), 1413 (w), 1362 (s), 1313 (w), 1288 (w), 1267 (w), 1235 (m), 1219 (m), 1162 (w), 1128 (m), 1107 (w), 1081 (w), 1069 (w), 1010 (m), 952 (w), 920 (w), 894 (w), 878 (w), 850 (m), 823 (w), 807 (s), 739 (w), 662 (vw), 638 (vw), 593 (w), 578 (w), 537 (vw), 481 (w), 452 (w), 394 (w) cm−1. HRMS (ESI+, MeOH) m/z: isotopic distribution calcd for C23H39N8 [M + H]+: 427.3299 (100) [C23H39N8]+, 428.3327 (28) [C2213CH39N8]+, 429.3350 (4) [C2213C2H39N8]+; found: 427.3302 (100), 428.3322 (50), 429.3350 (6).
max: 2929 (w, ν(CHaliph.)), 2888 (w, ν(CHaliph.)), 2812 (w), 1613 (s), 1597 (vs, ν(CN)), 1541 (m), 1504 (m), 1444 (m), 1428 (m), 1413 (w), 1362 (s), 1313 (w), 1288 (w), 1267 (w), 1235 (m), 1219 (m), 1162 (w), 1128 (m), 1107 (w), 1081 (w), 1069 (w), 1010 (m), 952 (w), 920 (w), 894 (w), 878 (w), 850 (m), 823 (w), 807 (s), 739 (w), 662 (vw), 638 (vw), 593 (w), 578 (w), 537 (vw), 481 (w), 452 (w), 394 (w) cm−1. HRMS (ESI+, MeOH) m/z: isotopic distribution calcd for C23H39N8 [M + H]+: 427.3299 (100) [C23H39N8]+, 428.3327 (28) [C2213CH39N8]+, 429.3350 (4) [C2213C2H39N8]+; found: 427.3302 (100), 428.3322 (50), 429.3350 (6).
Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-QPLGYGHVGP-UHFFFADPSC-NUHFF-NUHFF-NUHFF-ZZZ.
Complex synthesis
max: 2928 (w, ν(CHaliph.)), 2890 (w, ν(CHaliph.)), 2858 (w, ν(CHaliph.)), 1616 (vs), 1570 (vs), 1528 (vs, ν(CN)), 1478 (m), 1440 (m), 1426 (m), 1388 (vs), 1347 (m), 1332 (w), 1278 (m), 1233 (m), 1159 (w), 1147 (m), 1098 (w), 1071 (w), 1018 (vs), 994 (m), 963 (m), 925 (w), 898 (w), 834 (vs), 764 (w), 719 (w), 686 (w), 606 (w), 557 (w), 525 (w), 481 (w), 461 (m), 397 (w), 353 (w) cm−1. EA calcd for C23H38Br2CuN8 [M]: C 42.5%, H 5.9%, N 17.2%; found: C 42.4%, H 5.8%, N 17.3%. HRMS (ESI+, MeCN) m/z: isotopic distribution calcd for C23H38BrCuN8 [M − Br]+: 568.1700 (69) [C23H3879Br63CuN8]+; 596.1727 (20) [C2213CH3879Br63CuN8]+, 570.1685 (100) [C23H3881Br63CuN8]+, 571.1710 (28) [C2213CH3881Br63CuN8]+, 572.1674 (34) [C23H3881Br65CuN8]+, 573.1695 (9) [C2213CH3881Br65CuN8]+, found: 568.1692 (74), 569.1720 (20), 570.1676 (100), 571.1701 (29), 572.1659 (33), 573.1683 (9).
Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-NWKNMQBGGX-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
max: 3380 (vw), 2895 (w, ν(CHaliph.)), 1614 (vs), 1570 (s), 1523 (vs, ν(CN)), 1478 (m), 1441 (m), 1426 (m), 1388 (vs), 1347 (m), 1332 (w), 1305 (m), 1279 (m), 1265 (m), 1233 (m), 1158 (w), 1146 (m), 1099 (w), 1070 (w), 1020 (vs), 963 (m), 926 (w), 899 (w), 849 (m), 836 (s), 812 (w), 764 (w), 719 (vw), 686 (w), 606 (w), 556 (w), 524 (w), 481 (w), 459 (m), 403 (w), 358 (w) cm−1. EA calcd for C23H42Cl2CuN8O2 [M + 2H2O]: C 46.3%, H 7.1%, N 18.8%; found: C 46.2%, H 7.0%, N 18.4%. HRMS (ESI+, MeCN) m/z: isotopic distribution calcd for C23H38ClCuN8 [M − Cl]+: 524.2201 (100) [C23H3835Cl63CuN8]+; 525.2229 (29) [C2213CH3835Cl63CuN8]+, 526.2187 (81) [C23H3835Cl65CuN8]+, 527.2212 (23) [C2213CH3835Cl65CuN8]+, 528.2173 (18) [C23H3837Cl65CuN8]+, 529.2185 (4) [C2213CH3837Cl65CuN8]+, 530.2208 (1) [C2113C2H3837Cl65CuN8]+, found: 524.2188 (100), 525.2209 (34), 526.2168 (78), 527.2185 (26), 528.2152 (17), 529.2154 (8), 530.2171 (2).
Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-RUGUWNHIXP-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
max: 3410 (vw), 2930 (vw, ν(CHaliph.)), 2911 (vw, ν(CHaliph.)), 2863 (vw, ν(CHaliph.)), 1600 (w), 1558 (m, ν(CN)), 1476 (w), 1448 (m), 1423 (w), 1394 (m), 1343 (w), 1300 (m), 1288 (m), 1231 (vs), 1184 (m), 1152 (s), 1100 (s), 1077 (m), 1032 (w), 983 (m), 951 (w), 920 (vw), 895 (w), 857 (vw), 842 (w), 824 (w), 806 (w), 771 (m), 741 (m), 723 (w), 648 (m), 635 (m), 588 (w), 555 (m), 516 (m), 479 (m), 467 (w), 420 (m), 403 (m), 385 (w), 354 (m), 291 (m) cm−1. HRMS (ESI+, MeCN) m/z: isotopic distribution calcd for C19H28BrCuN6 [M − Br]+: 482.0859 (69) [C19H2879Br63CuN6]+; 483.0883 (16) [C1813CH2879Br63CuN6]+, 484.0838 (100) [C19H2881Br63CuN6]+, 485.0862 (23) [C1813CH2881Br63CuN6]+, 486.0821 (33) [C19H2881Br65CuN6]+, 487.0842 (7) [C1813CH2881Br65CuN6]+, found: 482.0848 (70), 483.0876 (17), 484.0829 (100), 485.0857 (24), 486.0814 (32), 487.0832 (8).
Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-XVFHQFWAYX-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
max: 2959 (w, ν(CHaliph.)), 2884 (w, ν(CHaliph.)), 2842 (w, ν(CHaliph.)), 1574 (m), 1550 (vs, ν(CN)), 1529 (s), 1483 (m), 1460 (m), 1441 (m), 1424 (m), 1392 (vs), 1344 (m), 1329 (m), 1246 (m), 1234 (w), 1213 (w), 1163 (m), 1150 (m), 1130 (w), 1079 (m), 1068 (m), 1040 (m), 1011 (m), 944 (w), 910 (m), 901 (m), 762 (m), 745 (w), 724 (w), 595 (w), 561 (w), 541 (w), 423 (m), 398 (w), 355 (m), 339 (m), 278 (m) cm−1. EA calcd for C21H48Br2CuN10 [M]: C 38.0%, H 7.3%, N 21.1%; found: C 37.7%, H 7.0%, N 21.1%. HRMS (ESI+, MeCN) m/z: isotopic distribution calcd for C21H48BrCuN10 [M − Br]+: 582.2546 (68) [C21H4879Br63CuN10]+; 583.2568 (19) [C2013CH4879Br63CuN10]+, 584.2525 (100) [C21H4881Br63CuN10]+, 585.2546 (27) [C2013CH4881Br63CuN10]+, 586.2509 (33) [C21H4881Br65CuN10]+, 587.2527 (8) [C2013CH4881Br65CuN10]+, found: 582.2537 (64), 583.2564 (14), 584.2517 (100), 585.2544 (23), 586.2504 (27), 587.2526 (6).
Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-PIQUIJJCAD-UHFFFADPSC-NUHFF-LUHFF-NUHFF-ZZZ.
max: 2979 (w, ν(CHaliph.)), 2961 (w, ν(CHaliph.)), 2921 (w, ν(CHaliph.)), 2881 (m, ν(CHaliph.)), 2840 (m, ν(CHaliph.)), 2801 (w), 1564 (vs, ν(CN)), 1511 (s), 1486 (m), 1460 (m), 1426 (s), 1407 (m), 1383 (vs), 1349 (m), 1342 (m), 1324 (w), 1279 (w), 1246 (m), 1229 (m), 1155 (m), 1140 (m), 1117 (w), 1071 (m), 1044 (m), 1031 (w), 1017 (m), 997 (m), 938 (w), 900 (m), 879 (m), 780 (vw), 758 (m), 719 (w), 589 (w), 556 (vw), 504 (w), 403 (w), 350 (m), 266 (m) cm−1. HRMS (ESI+, MeCN) m/z: isotopic distribution calcd for C21H48CuN10 [M − Br]+: 503.3362 (100) [C21H4863CuN10]+; 504.3380 (27) [C2013CH4863CuN10]+, 584.2525 (100) [C21H4881Br63CuN10]+, 505.3350 (48) [C21H4865CuN10]+, 506.3364 (12) [C2013CH4865CuN10]+, 507.3393 (2) [C1913C2H4865CuN10]+, found: 503.3114 (100), 504.3139 (28), 505.3096 (50), 506.3120 (12), 507.3147 (1). The bulk material was analysed with powder X-ray diffraction and was confirmed to be the desired compound (Fig. S14†).
Additional information on the synthesis of the target compound and original analysis data files are available via Chemotion Repository: https://dx.doi.org/10.14272/reaction/SA-FUHFF-UHFFFADPSC-RVTCRHJVTG-UHFFFADPSC-NUHFF-MUHFF-NUHFF-ZZZ.
Polymerisation studies
Ethyl α-bromoisobutyrate (EBiB, abcr, 98%), styrene (Acros Organics, 99% stab.) and benzonitrile (Fluka Analytical, 98%) have been purified by distillation over CaH2. AIBN (Sigma Aldrich, 98%) as well as CuBr2 (Grüssing, 98%) were used without further purification and all polymerisations were conducted with in situ generated catalysts.For standard ATRP experiments, first CuBr (0.19 mmol, 1 eq.), then ligand (0.19 mmol, 1 eq.) were directly weighed into the polymerisation vessel under a nitrogen atmosphere inside a glovebox. Outside the glovebox, first styrene (19 mmol, 100 eq.), then benzonitrile (1 mL), and finally EBiB (0.19 mmol, 1 eq.) were added with gastight glass syringes using Schlenk techniques. After the addition of the initiator, the reaction was started by heating to 110 °C under vigorous stirring.
For ICAR ATRP experiments, first CuBr2 (0.019 mmol, 0.1 eq.), then ligand (0.019 mmol, 0.1 eq.) were directly weighed into the polymerisation vessel under a nitrogen atmosphere inside a glovebox. Outside the glovebox, styrene (19 mmol, 100 eq.), benzonitrile (1 mL), EBiB (0.19 mmol, 1 eq.) and finally AIBN (0.29 mmol, 1.5 eq.) were added with gastight glass syringes using Schlenk techniques. After three cycles of freeze–pump–thaw, the reaction was started by heating to 60 °C under vigorous stirring.
For standard ATRP and ICAR ATRP experiments, the first aliquot was taken with a glass pipette under inert conditions after 2.5 min. At this point, the polymerisation mixture reached its desired temperature and thus was chosen as the starting point of the polymerisation. Further samples were taken at certain time intervals. The samples were diluted in CDCl3, and the conversion was measured via1H NMR spectroscopy. Afterwards, for the ICAR ATRP experiments, the polymer was precipitated in ethanol to remove the copper complex and residual monomer. The solid, colourless polystyrene was dried overnight at 50 °C, and the molecular mass and dispersity were measured by SEC.
Physical methods
Data of C1, C2, C3, C4 and C5 were collected with a four-circle goniometer Stoe Stadivari equipped with a Dectris Pilatus3 R 200 K hybrid pixel detector using a GeniX 3D high flux157 Mo-Kα source (λ = 0.71073 Å) at 100 K. Temperature control was achieved with an Oxford Cryostream 800. Data were collected with X-Area Pilatus105 and integrated with X-Area Integrate106 and X-Area Recipe.107 The absorption correction was performed with Stoe X-Red32, afterwards scaling of reflections with X-Area LANA was performed.105
The structures were solved by direct and conventional Fourier methods and all non-hydrogen atoms were refined anisotropically with full-matrix least-squares based on F2 (XPREP,108 SHELXS109 and ShelXle110). Hydrogen atoms were derived from difference Fourier maps and placed at idealised positions, riding on their parent C atoms, with isotropic displacement parameters Uiso(H) = 1.2 Ueq(C) and 1.5 Ueq(Cmethyl). All methyl groups were allowed to rotate but not to the tip.
In the complex C5, it was not possible to model the disordered solvent molecules (one molecule toluene) in an adequate manner, and the data set was treated with the SQUEEZE routine as implemented in PLATON.111,112
Full crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary no. CCDC 2289693 for L1, 2289694 for C1, 2289695 for C2, 2289696for C3, 2289697 for C4 and 2289698 for C5.†
The titration experiment started with 5 mL of 5 mM CuBr2 in MeCN. After the addition of aliquots (56 μL, 0.1 eq.) of 44 mM L1 in MeCN, the solution was stirred for 30 s until the UV/Vis spectrum was measured.
590 grid).114 The geometry optimisations were started from the geometry of the solid state structures of the cationic units if available using the TPSSh functional90,91 and with the Ahlrichs type basis set def2-TZVP92–95 as implemented in Gaussian 16, Revision B.01.114 As solvent model, the Polarizable Continuum Model (PCM) was used as implemented in Gaussian 16. As empirical dispersion correction, the D3 version of Grimme's dispersion with Becke–Johnson damping (GD3BJ) was used as implemented in Gaussian16, Revision B.01.96–98 Frequency calculations did not show imaginary values. Additionally a Conformer-Rotamer Ensemble Sampling Tool (CREST) calculation of the complex cation of C1 was performed to verify the minimum found in the DFT optimization (Version 2.12).115,116 NBO calculations were accomplished using the program suite NBO 7.0 delivering the NBO charges and the charge-transfer energies by second-order perturbation theory.117–119 For visualization and extraction of the calculated structural information, GaussView (Version 6.0.16) was used. The calculated energy values and NBO results were extracted directly from the output files using notepad++ (Version 7.8.1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. H–P. is thankful for the generous funding by the Deutsche Forschungsgemeinschaft within the collaborative research centre CRC985 (project INF). We thank NFDI4Chem for support and funding of the repository. Moreover, we thank Prof. Dr Schindler's working group (Justus Liebig University Gießen) for providing the amine precursor for the subsequent guanidine synthesis of TMG-uns-penp (L2). The authors would also like to thank Patrick Schumann for his support as a research assistant. We furthermore thank the Regional Computing Center of the University of Cologne (RRZK) for providing the computing time on the DFG-funded High Performance Computing (HPC) system CHEOPS as well as support.References
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- V. Percec and B. Barboiu, Macromolecules, 1995, 28, 7970–7972 CrossRef CAS.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- J. Xia, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1998, 31, 5958–5959 CrossRef CAS.
- J. Xia and K. Matyjaszewski, Macromolecules, 1999, 32, 2434–2437 CrossRef CAS.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800616 CrossRef PubMed.
- K. Matyjaszewski and A. H. E. Müller, Controlled and Living Polymerizations: From Mechanisms to Applications, Wiley-VCH, Weinheim, 2009 Search PubMed.
- C. Y. Lin, M. L. Coote, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 12762–12774 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- H. Fischer, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1885–1901 CrossRef CAS.
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS.
- Z. Wang, X. Pan, J. Yan, S. Dadashi-Silab, G. Xie, J. Zhang, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2017, 6, 546–549 CrossRef CAS PubMed.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, ACS Macro Lett., 2019, 8, 603–609 CrossRef CAS PubMed.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. D. Magenau, A. Sobkowiak and K. Matyjaszewski, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- D. Konkolewicz, Y. Wang, M. Zhong, P. Krys, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 8749–8772 CrossRef CAS.
- M. R. Martinez, J. Sobieski, F. Lorandi, M. Fantin, S. Dadashi-Silab, G. Xie, M. Olszewski, X. Pan, T. G. Ribelli and K. Matyjaszewski, Macromolecules, 2020, 53, 59–67 CrossRef CAS.
- M. Rolland, R. Whitfield, D. Messmer, K. Parkatzidis, N. P. Truong and A. Anastasaki, ACS Macro Lett., 2019, 8, 1546–1551 CrossRef CAS PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 ( Angew. Chem. , 2006 , 118 , 4594–4598 ) CrossRef CAS PubMed.
- V. A. Williams, T. G. Ribelli, P. Chmielarz, S. Park and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137, 1428–1431 CrossRef CAS PubMed.
- T. G. Ribelli, M. Fantin, J.-C. Daran, K. F. Augustine, R. Poli and K. Matyjaszewski, J. Am. Chem. Soc., 2018, 140, 1525–1534 CrossRef CAS PubMed.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- D. Konkolewicz, A. J. D. Magenau, S. E. Averick, A. Simakova, H. He and K. Matyjaszewski, Macromolecules, 2012, 45, 4461–4468 CrossRef CAS.
- A. E. Enciso, F. Lorandi, A. Mehmood, M. Fantin, G. Szczepaniak, B. G. Janesko and K. Matyjaszewski, Angew. Chem., Int. Ed., 2020, 59, 14910–14920 ( Angew. Chem. , 2020 , 132 , 15020–15030 ) CrossRef CAS PubMed.
- E. Mastan and S. Zhu, Macromolecules, 2015, 48, 6440–6449 CrossRef CAS.
- P. Krys and K. Matyjaszewski, Eur. Polym. J., 2017, 89, 482–523 CrossRef CAS.
- V. Doan, B. B. Noble, A. K. K. Fung and M. L. Coote, J. Org. Chem., 2019, 84, 15624–15632 CrossRef CAS PubMed.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800616 CrossRef PubMed.
- M. Fantin, F. Lorandi, T. G. Ribelli, G. Szczepaniak, A. E. Enciso, C. Fliedel, L. Thevenin, A. A. Isse, R. Poli and K. Matyjaszewski, Macromolecules, 2019, 52, 4079–4090 CrossRef CAS.
- L. Thevenin, C. Fliedel, K. Matyjaszewski and R. Poli, Eur. J. Inorg. Chem., 2019, 2019, 4489–4499 CrossRef CAS.
- N. V. Tsarevsky, W. A. Braunecker, A. Vacca, P. Gans and K. Matyjaszewski, Macromol. Symp., 2007, 248, 60–70 CrossRef CAS.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed.
- N. V. Tsarevsky, W. A. Braunecker and K. Matyjaszewski, J. Organomet. Chem., 2007, 692, 3212–3222 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- P. Paoli, A. Isse, N. Bortolamei and A. Gennaro, Chem. Commun., 2011, 47, 3580–3582 RSC.
- N. Bortolamei, A. A. Isse, V. B. Di Marco, A. Gennaro and K. Matyjaszewski, Macromolecules, 2010, 43, 9257–9267 CrossRef CAS.
- N. V. Tsarevsky, T. Pintauer and K. Matyjaszewski, Macromolecules, 2004, 37, 9768–9778 CrossRef CAS.
- F. di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953–4959 CrossRef CAS.
- K. Matyjaszewski, B. Göbelt, H.-J. Paik and C. P. Horwitz, Macromolecules, 2001, 34, 430–440 CrossRef CAS.
- F. Lorandi and K. Matyjaszewski, Isr. J. Chem., 2020, 60, 108–123 CrossRef CAS.
- J. Stanek, T. Rösener, A. Metz, J. Mannsperger, A. Hoffmann and S. Herres-Pawlis, in Guanidines as Reagents and Catalysts II, ed. P. Selig, Springer International Publishing, Cham, 2017, pp. 95–164 Search PubMed.
- T. Rösener, O. Bienemann, K. Sigl, N. Schopp, F. Schnitter, U. Flörke, A. Hoffmann, A. Döring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2016, 22, 13550–13562 CrossRef PubMed.
- K. W. Kröckert, J. S. Mannsperger, T. Rösener, A. Hoffmann and S. Herres-Pawlis, Z. Anorg. Allg. Chem., 2021, 647, 832–842 CrossRef.
- T. Rösener, A. Hoffmann and S. Herres-Pawlis, Eur. J. Inorg. Chem., 2018, 2018, 3164–3175 CrossRef.
- T. Rösener, K. Kröckert, A. Hoffmann and S. Herres-Pawlis, Z. Anorg. Allg. Chem., 2018, 644, 1317–1328 CrossRef.
- R. D. Rittinghaus, A. Karabulut, A. Hoffmann and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2021, 60, 21795–21800 ( Angew. Chem. , 2021 , 133 , 21965–21971 ) CrossRef CAS PubMed.
- A. Hermann, S. Hill, A. Metz, J. Heck, A. Hoffmann, L. Hartmann and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2020, 59, 21778–21784 CrossRef CAS PubMed.
- R. D. Rittinghaus, J. Zenner, A. Pich, M. Kol and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2022, 61, e202112853 ( Angew. Chem. , 2022 , 134 , e202112853 ) CrossRef CAS PubMed.
- M. Paul, A. Hoffmann and S. Herres-Pawlis, J. Biol. Inorg. Chem., 2021, 26, 249–263 CrossRef CAS PubMed.
- M. Paul, M. Teubner, B. Grimm-Lebsanft, S. Buchenau, A. Hoffmann, M. Rübhausen and S. Herres-Pawlis, J. Inorg. Biochem., 2021, 224, 111541 CrossRef CAS PubMed.
- M. Paul, M. Teubner, B. Grimm-Lebsanft, C. Golchert, Y. Meiners, L. Senft, K. Keisers, P. Liebhäuser, T. Rösener, F. Biebl, S. Buchenau, M. Naumova, V. Murzin, R. Krug, A. Hoffmann, J. Pietruszka, I. Ivanović-Burmazović, M. Rübhausen and S. Herres-Pawlis, Chem. – Eur. J., 2020, 26, 7556–7562 CrossRef CAS PubMed.
- J. Heck, F. Metz, S. Buchenau, M. Teubner, B. Grimm-Lebsanft, T. P. Spaniol, A. Hoffmann, M. A. Rübhausen and S. Herres-Pawlis, Chem. Sci., 2022, 13, 8274–8288 RSC.
- B. Dicke, A. Hoffmann, J. Stanek, M. S. Rampp, B. Grimm-Lebsanft, F. Biebl, D. Rukser, B. Maerz, D. Göries, M. Naumova, M. Biednov, G. Neuber, A. Wetzel, S. M. Hofmann, P. Roedig, A. Meents, J. Bielecki, J. Andreasson, K. R. Beyerlein, H. N. Chapman, C. Bressler, W. Zinth, M. Rübhausen and S. Herres-Pawlis, Nat. Chem., 2018, 10, 355–362 CrossRef CAS PubMed.
- K. W. Kröckert, F. Garg, M. V. Heinz, J. Lange, P. P. Simões, R. Schmidt, O. Bienemann, A. Hoffmann and S. Herres-Pawlis, Dalton Trans., 2022, 51, 13272–13287 RSC.
- L. I. Manasieva, B. U. Maria, A. Prandi, L. Brasili and S. Franchini, Synthesis, 2015, 47, 3767–3775 CrossRef CAS.
- R. L. Lucas, M. K. Zart, J. Murkerjee, T. N. Sorrell, D. R. Powell and A. S. Borovik, J. Am. Chem. Soc., 2006, 128, 15476–15489 CrossRef CAS PubMed.
- M. Mori, M. C. Dasso Lang, F. Saladini, N. Palombi, L. Kovalenko, D. De Forni, B. Poddesu, L. Friggeri, A. Giannini, S. Malancona, V. Summa, M. Zazzi, Y. Mely and M. Botta, ACS Med. Chem. Lett., 2019, 10, 463–468 CrossRef CAS PubMed.
- J. England, R. Gondhia, L. Bigorra-Lopez, A. R. Petersen, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2009, 5319–5334 RSC.
- S. Herres-Pawlis, U. Flörke and G. Henkel, Eur. J. Inorg. Chem., 2005, 2005, 3815–3824 CrossRef.
- W. Kantlehner, E. Haug, W. W. Mergen, P. Speh, T. Maier, J. J. Kapassakalidis, H.-J. Bräuner and H. Hagen, Liebigs Ann. Chem., 1984, 1984, 108–126 CrossRef.
- J. B. Mandel, C. Maricondi and B. E. Douglas, Inorg. Chem., 1998, 27, 2990–2996 CrossRef.
- R. Wortmann, Dissertation, Universität Paderborn, 2011.
- H. Wittmann, V. Raab, A. Schorm, J. Plackmeyer and J. Sundermeyer, Eur. J. Inorg. Chem., 2001, 2001, 1937–1948 CrossRef.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. J. D. Magenau, Y. Kwak, K. Schröder and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 508–512 CrossRef CAS PubMed.
- L. A. van de Kuil, H. Luitjes, D. M. Grove, J. W. Zwikker, J. G. M. van der Linden, A. M. Roelofsen, L. W. Jenneskens, W. Drenth and G. van Koten, Organometallics, 1994, 13, 468–477 CrossRef CAS.
- H. Nitadori, T. Takahashi, A. Inagaki and M. Akita, Inorg. Chem., 2012, 51, 51–62 CrossRef CAS PubMed.
- V. Raab, K. Harms, J. Sundermeyer, B. Kovačević and Z. B. Maksić, J. Org. Chem., 2003, 68, 8790–8797 CrossRef CAS PubMed.
- W. T. Eckenhoff and T. Pintauer, Dalton Trans., 2011, 40, 4909–4917 RSC.
- W. T. Eckenhoff, S. T. Garrity and T. Pintauer, Eur. J. Inorg. Chem., 2008, 2008, 563–571 CrossRef.
- W. T. Eckenhoff and T. Pintauer, Inorg. Chem., 2010, 49, 10617–10626 CrossRef CAS PubMed.
- O. Bienemann, A. Hoffmann and S. Herres-Pawlis, Rev. Inorg. Chem., 2011, 31, 83–108 CAS.
- M. A. Hiskey and R. R. Ruminski, Inorg. Chim. Acta, 1986, 112, 189–195 CrossRef CAS.
- G. A. McLachlan, G. D. Fallon, R. L. Martin and L. Spiccia, Inorg. Chem., 1995, 34, 254–261 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 165, 691–708 CrossRef CAS PubMed.
- A. Kaur, T. G. Ribelli, K. Schröder, K. Matyjaszewski and T. Pintauer, Inorg. Chem., 2015, 54, 1474–1486 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- V. Balland, C. Hureau and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17113–17118 CrossRef CAS PubMed.
- J. Qiu, K. Matyjaszewski, L. Thouin and C. Amatore, Macromol. Chem. Phys., 2000, 201, 1625–1631 CrossRef CAS.
- W. A. Braunecker, N. V. Tsarevsky, A. Gennaro and K. Matyjaszewski, Macromolecules, 2009, 42, 6348–6360 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- A. Hoffmann, R. Grunzke and S. Herres-Pawlis, J. Comput. Chem., 2014, 35, 1943–1950 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- P. Krys, H. Schroeder, J. Buback, M. Buback and K. Matyjaszewski, Macromolecules, 2016, 49, 7793–7803 CrossRef CAS.
- F. Lorandi, M. Fantin and K. Matyjaszewski, J. Am. Chem. Soc., 2022, 144, 15413–15430 CrossRef CAS PubMed.
- N. Andre and J. M. E. Maier, Praxis in der Organischen Chemie, VCH, Weinheim, 1997 Search PubMed.
- H. Fuchida, S. Tabata, N. Shindo, I. Takashima, Q. Leng, Y. Hatsuyama, I. Hamachi and A. Ojida, Bull. Chem. Soc. Jpn., 2015, 88, 784–791 CrossRef CAS.
- H.-D. Hardt, Z. Anorg. Allg. Chem., 1959, 301, 87–96 CrossRef CAS.
- SMART (Version 5.631), SAINT (Version 8.37A) and SADABS (Version 2008/1), Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- X-Area Pilatus3_SV 1.21.131.0, STOE, 2017 Search PubMed.
- X-Area Integrate 1.71.0.0, STOE, 2016 Search PubMed.
- X-Area Pecipe 1.33.0.0, STOE, 2015 Search PubMed.
- Bruker, in XPREP, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht (The Netherlands), 2008 Search PubMed.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- P. Tremouilhac, C.-L. Lin, P.-C. Huang, Y.-C. Huang, A. Nguyen, N. Jung, F. Bach, R. Ulrich, B. Neumair, A. Streit and S. Bräse, Angew. Chem., Int. Ed., 2020, 59, 22771–22778 ( Angew. Chem. , 2006 , 118 , 4594–4598 ) CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revison B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Plett and S. Grimme, Angew. Chem., Int. Ed., 2023, 62, e202214477 ( Angew. Chem. , 2023 , 135 , e202214477 ) CrossRef CAS PubMed.
- F. Weinhold and C. R. Landis, Valency and bonding: a natural bond orbital donor-acceptor perspective, Cambridge University Press, 2005 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2289693–2289698. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03392a |
| ‡ Dedicated to Prof. Dr. Ulrich Simon on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2024 |