
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tracking of structural defects induced by Eu-doping in β-Ag2MoO4: their influences on electrical properties†
Carlos
Macchi
a,
Guilherme Magalhaes
Petinardi
b,
Leonardo Almeida
Freire
b,
Miriam Susana
Castro
c,
Celso Manuel
Aldao
d,
Thaís Marcial
Luiz
e,
Francisco
Moura
e,
Alexandre Zirpoli
Simões
f,
Henrique
Moreno
 fg,
Elson
Longo
g,
Alberto
Somoza
a,
Marcelo
Assis
*h and
Miguel Adolfo
Ponce
a
fg,
Elson
Longo
g,
Alberto
Somoza
a,
Marcelo
Assis
*h and
Miguel Adolfo
Ponce
a
aInstitute of Materials Physics of Tandil, IFIMAT (UNCPBA) and CIFICEN (UNCPBA-CICPBA-CONICET), Tandil, Argentina
bFunctional Materials Development Group (GDMaF), Federal University of Itajubá, (UNIFEI), Itajubá, Brazil
cInstitute of Materials Science and Technology (INTEMA), University of Mar del Plata and National Research Council (CONICET), Mar del Plata, Argentina
dInstitute of Scientific and Technological Research in Electronics (ICYTE), University of Mar del Plata and National Research Council (CONICET), Mar del Plata, Argentina
eAdvanced Materials Interdisciplinary Laboratory (LIMAV), Federal University of Itajubá (UNIFEI), Itabira, Brazil
fSchool of Engineering and Sciences, São Paulo State University (UNESP), Guaratinguetá, Brazil
gCDMF, Federal University of São Carlos (UFSCar), São Carlos, Brazil
hDepartment of Physical and Analytical Chemistry, University Jaume I (UJI) Castellón, Spain. E-mail: marcelostassis@gmail.com
First published on 30th November 2023
Abstract
In this study, several methods were employed to investigate the electrical characteristics of β-Ag2MoO4 systems, both Eu-doped and undoped, synthesized using the microwave-assisted hydrothermal method. The focus extended to understanding how synthesis time influences material defects, with doping fixed at 1%. A systematic shift in the silver vacancy (VAg) concentration was observed within the doped β-Ag2MoO4 system. Specifically, this study demonstrated that the incorporation of Eu3+ into polycrystalline β-Ag2MoO4 initially increases the VAg concentration. However, as the synthesis time progresses, the VAg concentration decreases, resulting in alterations in the resulting electrical properties, arising from the intricate interplay between the number of grain boundaries and carrier density. By combining information obtained from photoluminescence, positron annihilation lifetime spectroscopy, and impedance spectroscopy, a comprehensive conduction mechanism was formulated, shedding light on both doped and undoped β-Ag2MoO4 systems.
Introduction
Comprehending the intricate relationship between the structure, defect nature, and electrical transport properties is paramount in the design of multifunctional semiconductors. Molybdates with the molecular formula X2MoO4, where X = Li, Tl, and Ag, have garnered significant attention within the scientific community due to their remarkable photocatalytic properties,1,2 applications in scintillators,3 sensors,4–6 optical fibers,7 as well as their outstanding performance in diverse applications such as antibacterial,8 antifungal,9 antiviral,10 tribological,11 microwave dielectric,12 and supercapacitor electrode13 applications. Furthermore, highly pure β-Ag2MoO4 with a spinel-type cubic structure has been synthesized for catalysis applications due to its electronic properties.13The use of Ag2MoO4 has shown enhanced properties, which are size and morphology-dependent when in nanoparticulate form.8 Particularly, the morphology of nanoparticles can be finely controlled through advanced synthesis methods (e.g., precipitation, sonochemical, conventional and microwave hydrothermal, etc.).7,14–17 The initial synthesis of β-Ag2MoO4 crystals, as reported by Wyckoff et al., employed a solid-state reaction method, offering limited control over particle shape and size.18 Subsequently, Teodoro et al. investigated the impact of highly energetic facets on the photoluminescent (PL) response of β-Ag2MoO4 nanocrystals with varying sizes and shapes prepared using the microwave hydrothermal method.19 This is particularly relevant as semiconductor performance is intricately linked to the quantity and nature of defects, which can be influenced by the synthesis method. Moreover, the doping of β-Ag2MoO4 crystals has the potential to enhance their properties.20–22
One significant advantage of doping silver compounds lies in the possibility of incorporating rare earth elements. Rare earth metals (REs) exhibit unique optical properties, primarily attributed to 4f–4f or 5d–4f transitions stemming from their electronic configurations.23 Eu3+ ions, in particular, are known for their distinct and well-defined emission bands24 arising from their electronic configuration 4f,6 where the outermost electrons shield the inner ones.25,26 Consequently, Eu-doping has a profound effect on the PL properties of the host matrix.26 Additionally, the incorporation of Eu3+ can elevate the concentration of oxygen vacancies (VO).
The concentration of VO (e.g.,  ,
,  ,
,  ) and silver vacancies (e.g.,
) and silver vacancies (e.g.,  , VAg–O, etc.) plays a pivotal role in governing the electronic behavior of transition metal oxide (TMO) semiconductors.27 These vacancies arise due to electron–hole recombination within the bandgap region and deviations from the perfect crystal lattice, leading to a reduction in the interfacial reaction energy barrier.28 Hence, understanding and manipulating defects in semiconductors have emerged as potent tools for fine-tuning their performance. In 2019, Sudarshan et al. successfully synthesized pure and Dy3+ doped β-Ag2MoO4 using a co-precipitation method at room temperature.29 Their work demonstrated that Dy3+ induces cation vacancies and surface defects, as evidenced by positron annihilation lifetime spectroscopy (PALS), confirming Dy3+ stabilization at Ag+ sites.
, VAg–O, etc.) plays a pivotal role in governing the electronic behavior of transition metal oxide (TMO) semiconductors.27 These vacancies arise due to electron–hole recombination within the bandgap region and deviations from the perfect crystal lattice, leading to a reduction in the interfacial reaction energy barrier.28 Hence, understanding and manipulating defects in semiconductors have emerged as potent tools for fine-tuning their performance. In 2019, Sudarshan et al. successfully synthesized pure and Dy3+ doped β-Ag2MoO4 using a co-precipitation method at room temperature.29 Their work demonstrated that Dy3+ induces cation vacancies and surface defects, as evidenced by positron annihilation lifetime spectroscopy (PALS), confirming Dy3+ stabilization at Ag+ sites.
Despite existing studies in the field of defects, shallow discussions have prevailed in this area of knowledge, highlighting a clear need for in-depth exploration. In the present study, β-Ag2MoO4 powders doped with Eu3+ were synthesized using the microwave-assisted hydrothermal method. The concentration of Eu3+ was held constant (1%), while the synthesis time was varied (4, 8, 16 minutes). All samples underwent characterization through a combination of XRD, Rietveld refinement, micro-Raman spectroscopy, diffuse reflectance spectroscopy (DRS), field emission scanning electron microscopy (FE-SEM), and photoluminescence (PL) emissions. To elucidate the nature of defects, measurements were conducted using PALS and impedance spectroscopy (IS). Finally, a conduction mechanism was proposed based on the observed behavior, taking into account the results of PL, PALS, and IS.
Results and discussion
Structural characterization
Fig. 1a shows the X-ray diffraction patterns for the as-prepared β-Ag2MoO4 samples. All samples exhibit narrow, well-defined XRD peaks, indicating the synthesis of highly crystalline material.3 All peaks for the crystalline structure were indexed by the cubic β-Ag2MoO4 phase (ICSD #238013) belonging to the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. No secondary phase peaks were identified. The lattice and Rietveld parameters compiled in Fig. S1 and Table S1† demonstrate a coherent refinement, as evidenced by the Rietveld parameters (χ2, Rwp, Rbragg). In addition to the Rietveld parameters, Table S2† summarizes the values associated with atomic coordinates, occupancy factor and anisotropic thermal factor for all samples. It is worth mentioning that the Rietveld method was carried out fixating the occupancy factor parameters considering a 1% Eu-doping for all samples. Additionally, Fig. 1b highlights a peak shift associated with the (311) crystallographic plane towards higher Bragg angles (2θ), which may be attributed to the incorporation of Eu3+ ions at Ag+ sites within the β-Ag2MoO4 structure. This incorporation leads to the formation of Eu3+ clusters that replace three Ag+ clusters, resulting in lattice relaxation, polarization of the structure, and the formation of oxygen vacancies and silver vacancies (VO/VAg). Depending on the synthesis time, the crystalline structure acquires greater symmetry, resulting in different electronic densities in the VO/VAg and variations in the influence of the Eu clusters on the semiconductor properties. The behavior of β-Ag2MoO4 can be explained by complex VO that can exist in three different charge forms: neutral (
m space group. No secondary phase peaks were identified. The lattice and Rietveld parameters compiled in Fig. S1 and Table S1† demonstrate a coherent refinement, as evidenced by the Rietveld parameters (χ2, Rwp, Rbragg). In addition to the Rietveld parameters, Table S2† summarizes the values associated with atomic coordinates, occupancy factor and anisotropic thermal factor for all samples. It is worth mentioning that the Rietveld method was carried out fixating the occupancy factor parameters considering a 1% Eu-doping for all samples. Additionally, Fig. 1b highlights a peak shift associated with the (311) crystallographic plane towards higher Bragg angles (2θ), which may be attributed to the incorporation of Eu3+ ions at Ag+ sites within the β-Ag2MoO4 structure. This incorporation leads to the formation of Eu3+ clusters that replace three Ag+ clusters, resulting in lattice relaxation, polarization of the structure, and the formation of oxygen vacancies and silver vacancies (VO/VAg). Depending on the synthesis time, the crystalline structure acquires greater symmetry, resulting in different electronic densities in the VO/VAg and variations in the influence of the Eu clusters on the semiconductor properties. The behavior of β-Ag2MoO4 can be explained by complex VO that can exist in three different charge forms: neutral (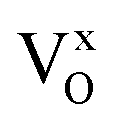 ), singly ionized (
), singly ionized ( ), and double ionized (
), and double ionized ( ).30 This is corroborated by a slight increase in the unit cell volume from ∼806.7 Å3 (Pure) to 807.4 and 807.5 Å3 (4Eu and 8Eu, respectively), and 808.2 Å3 (16Eu) as the synthesis time increases. Using the Williamson-Hall method (Fig. S2†), microstrain values were calculated for the samples, revealing a decrease in values when compared to the pure sample. This indicates a decompression in the crystallites of the sample, consistent with the results obtained from Rietveld refinement.
).30 This is corroborated by a slight increase in the unit cell volume from ∼806.7 Å3 (Pure) to 807.4 and 807.5 Å3 (4Eu and 8Eu, respectively), and 808.2 Å3 (16Eu) as the synthesis time increases. Using the Williamson-Hall method (Fig. S2†), microstrain values were calculated for the samples, revealing a decrease in values when compared to the pure sample. This indicates a decompression in the crystallites of the sample, consistent with the results obtained from Rietveld refinement.
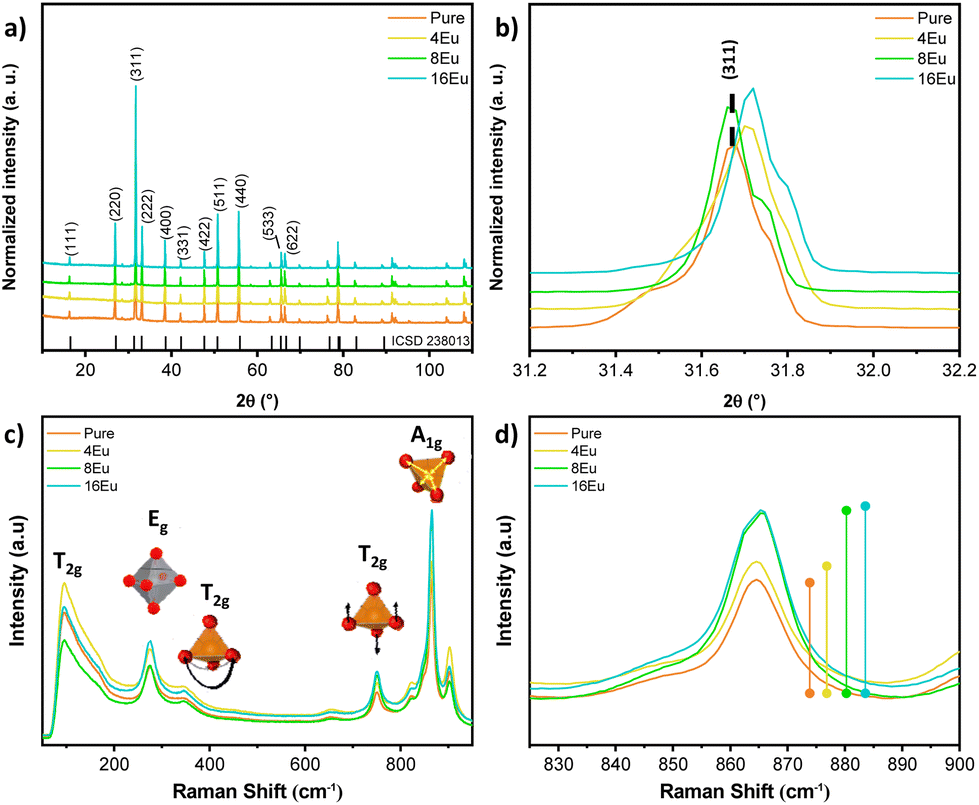 | ||
| Fig. 1 (a) XRD spectra of the samples and; (b) detailed analysis of the (311) plane. (c) micro-Raman spectra of the samples and; (d) detailed analysis of the A1g Raman mode. | ||
Fig. 1c shows the micro-Raman spectra collected at room temperature for all samples. According to the group theory, there are 16 vibrational groups possible for β-Ag2MoO4: A1g; Eg; 3T2g; T1g; 4T1u; 2A2u; 2Eu; and 2T2u, of which 6 (A1g + Eg + 3T2g + T1g) are Raman active, and mode T1u is detectable in infrared spectra. However, subscripts “g” and “u” indicate an inversion center in the β-Ag2MoO4 structure, leading to a total of five Raman active modes. At ∼95 cm−1 mode T2g may be associated with the mobility of O2− ions within the β-Ag2MoO4 structure.6,31 Mode Eg, detected at ∼275 cm−1 can be associated with vibrations of the [AgO6] octahedral.8,32 The band at around ∼348 cm−1 is attributed to the T2g mode, which in turn is related to bending vibrations of O–Mo–O bonds in [MoO4] clusters. The asymmetric stretching of these bonds is responsible for the T2g mode, which is ascribed to the band located at approximately ∼750 cm−1.8,32 At ∼670 cm−1, ν1 was assigned to the anti-symmetric vibrational F2g mode of [MoO4] polyhedra. Finally, A1g mode can be detected at ∼865 cm−1, corresponding to the symmetric stretching of Mo–O bonds in the [MoO4] tetrahedra.8,32 Analogously, the modes orbiting the A1g – ν2 (∼820 cm−1) and ν3 (∼895 cm−1) can be ascribed to anti-symmetric stretching in the [MoO4].31 Teodoro et al.19 reported peak broadening and the presence of shoulder-like peaks associated with the A1g mode in β-Ag2MoO4 synthesized by the hydrothermal microwave method. The authors linked this behavior with inelastic scattering (i.e., bond length changes, which vary the vibrational mode frequency).8,32,33 None of the samples had a significant position shift of Raman bands. However, Fig. 1d shows a slight increase in the intensity of mode A1g with Eu3+ incorporation, which may be related to the formation of VAg within the crystal structure.31,34 Increasing synthesis time leads to changes in the chemical environment capable of promoting tetrahedral distortions, increasing symmetry and, consequently, increasing the structural order at a short-range. In particular, the mode located at 865 cm−1 which corresponds to the stretching mode of the Mo–O bond in the tetrahedral [MoO4] cluster, can be considered as a signature of the structural change of the β-Ag2MoO4 lattice provoked by the increasing of the soaking time. From the Raman spectra we can noted that the obtained powders are free of impurities since no active modes are evident in the spectra. These results are in agreement with XRD data. As Eu3+ substitutes Ag+, VAg are formed and changes in the symmetry as well as in the periodicity of atoms with different degrees of ordering at short range distances. From the Raman measurements, we fitted the peaks using the Lorentzian peak function, which gave us the FWHM values seen in Table S3 and Fig. S3.† A high degree of disorder in the structure is observed as synthesis time raises. Eu3+ doping induces the formation of large cationic intergranular vacancies leading to local polarization, and affecting the distribution of the energy levels within the bandgap region.
Fig. 2a–d exhibits FE-SEM images of the as-prepared β-Ag2MoO4 samples. The pure sample displays an agglomerate of irregular particles with undefined geometry (1.75 ± 0.39 μm × 4.29 ± 0.96 μm). As Eu3+ is incorporated at Ag+ sites within the β-Ag2MoO4 structure, the micrograph images reveal the formation of well-defined cubic-shaped particles (4Eu, 0.98 ± 0.63 μm), which grow significantly with synthesis time (8Eu, 6.47 ± 1.22 μm; 16Eu 6.82 ± 0.87 μm). Small structures randomly distributed on the surface of the particles, and whose contribution increase with synthesis time, significantly increase superficial roughness. From Wullf's construction (Fig. 2e), one can observe the behavior of the morphology in relation to the surface energies imposed on the system. According to Foggi et al., the ideal β-Ag2MoO4 system has surface energies for the (001), (011), and (111) planes of 1.90, 1.28, and 3.46 J m−2.8 It is observed that the ideal morphology is characterized by the complete exposure of the (011) surface due to its lower energy. Therefore, different morphologies can be achieved by stabilizing the (001) and (111) surfaces. By stabilizing the surface energy of (111), an octahedral morphology is gradually reached, while stabilizing the surface energy of (001) leads to a gradual transition to a cubic morphology. This reduction in energy needs to be at least 1.28 J m−2 for complete exposure of the (001) surface. Consequently, it can be seen that Eu3+ plays a fundamental role as a dopant in stabilizing the (001) surface energy, allowing for its selective use in the design of new materials. This surface is composed of surface clusters [AgO5.VO], making it more active for photocatalytic and antimicrobial applications. This result is aligned with what was observed by Almeida et al.,3 where it is demonstrated that an increase in the concentration of Eu3+ in the β-Ag2MoO4 lattice results in the appearance of a small concentration of cubes. The concentration of cubes can be enhanced through the synthesis method, but previously works did not show selectivity for cubes,19,35,36 with Eu3+ being the primary morphology-directing agent.
 | ||
| Fig. 2 FE-SEM images of the samples (a) Pure, (b) 4Eu, (c) 8Eu, and (d) 16Eu. (e) Wullf construction of the β-Ag2MoO4 morphologies. | ||
Optical characterization
The Wood–Tauc method and the Kubelka–Munk function were employed to determine the optical band gap energy (Egap) of the samples. This approach relies on converting diffuse reflectance measurements to accurately estimate Egap values within specified limits.37 All samples behaved as expected for a β-Ag2MoO4 (Egap = 3.2–3.3 eV, Fig. S4†), which is consistent with the Rietveld analysis that shows an almost constant unit cell volume. The optical absorption in β-Ag2MoO4 microcrystals as a function of indirect electronic transitions (e.g., lowest energy e− in the conduction band hopping to distinct areas of the valence band in the Brillouin zone), was previously study in our group.Fig. 3a shows the PL emission spectra recorded at room temperature for all samples excited by a 355 nm. PL emissions between 400–550 nm form a broad excitation band associated with the β-Ag2MoO4 matrix, which was referred to as O2−/Eu3+ charge transfer band (CTB) by Dorenbos.38 This band can be considered as a combination of three charge transfer mechanisms, which are: (1) relocation of electrons from O2− to Mo6+; (2) transference of electrons between Eu3+ 4f states to Mo6+ valence band; and (3) electronic transition between filled up O2− 2p orbitals to vacant 4f Eu3+ orbitals. A broad emission in the blue region, between 390 and 550 nm (λmax ≈ 450 nm), is characteristic of the β-Ag2MoO4 matrix (Fig. 3b). Almeida et al.3 reported this behavior as a result of changes in the order of [MoO4] tetrahedra and [AgO6] octahedra, leading to local polarization, and affecting the distribution of the energy levels within the bandgap region. Thus, the PL emissions of the matrix are significantly determined by Ag–O/Mo–O distortions, due to their influence on the electronic properties of the material.28
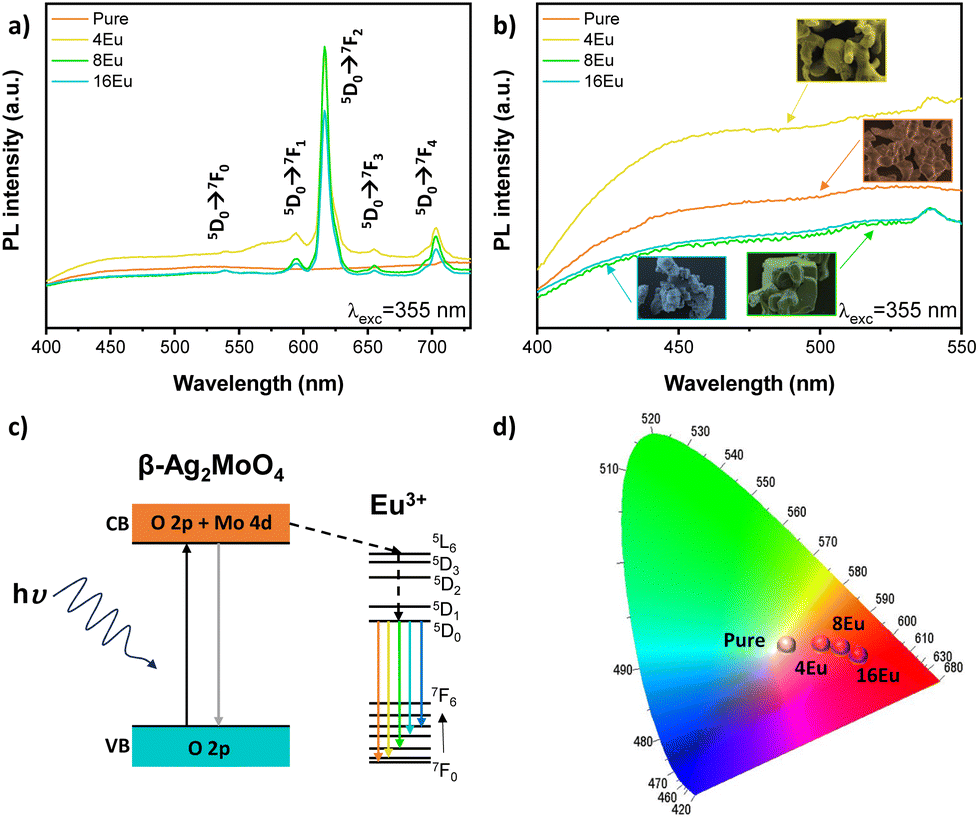 | ||
| Fig. 3 (a and b) Photoluminescence spectra for all samples of the β-Ag2MoO4 system showing the influence of Eu3+ and synthesis time, (c) a practical model explaining the PL mechanism for both pure and Eu-doped β-Ag2MoO4 and (d) the associated chromaticity diagram. | ||
The PL spectra (Fig. 3a) of the Eu-doped samples are significantly different from that of pure sample, showing sharp bands between 550 and 750 nm. These bands can be associated with inter configurational f–f electronic transitions (5D0 → 7FJ) in the Eu3+ ions occurring at 595 nm (5D0 → 7F1), 616 nm (5D0 → 7F2), 655 nm (5D0 → 7F3), and 703 nm (5D0 → 7F4).39 Some of these transitions are hypersensitive to the crystal field and may be useful for understanding the host lattice and local symmetry,39,40 while 5D0 → 7F1 transitions are insensitive to a local symmetry, 5D0 → 7F2 transitions correspond to electronic dipole transitions, which provide rich information on the local chemistry around Eu3+ ions39,41,42 As the (5D0 → 7F2) transition at 616 nm is more intense than (5D0 → 7F1) transition at 595 nm we can infer that Eu3+ ions are located in a site without inversion of symmetry. In this way, the relative area ratio of the 5D0 → 7F2 to 5D0 → 7F1 transitions provides information about the local distortions in the clusters into which Eu3+ cations have been incorporated within the structure.43 The values obtained for the 4Eu, 8Eu, and 16Eu samples were 3.66, 5.36, and 6.01, respectively. Consequently, it is observed that increasing the synthesis time in the microwave hydrothermal system leads to an increase in local distortions associated with the Eu3+ clusters within the crystal lattice. This outcome aligns with what has been observed thus far, as an increase in short and long-range order occurs with the synthesis time, resulting in greater distortions in the neighborhood of the Eu3+ atoms due to a more organized crystal lattice.
Fig. 3c shows a practical model explaining the luminescence mechanism in Eu3+-doped β-Ag2MoO4 samples. A source of light supplies energy and promotes electrons from O 2p levels in the valence band (VB), to the conduction band (CB), made up of Mo 4d levels. Light emission occurs as the excited electrons radiatively decay from the CB to the VB. The incorporation of Eu3+ ions within the β-Ag2MoO4 generates an alternate path to emissions, in which the excited electrons decay from the CB to excited levels of the Eu3+ cations located within the bandgap region before reaching the VB. First, occupied 4f states decay non-radiatively to Eu3+ characteristic emission levels (5D0) and then radiatively to fundamental states (7Fj, j = 1, 2, 3, and 4) emitting on specific characteristic lengths. Fig. 3d exhibits the PL-associated coordinates in the chromaticity diagram (CIE–Commission internationale de l'eclairage 1931)44 using a 355 nm wavelength light as excitation source. Our results indicate a significant red shift in PL emission at the chromaticity diagram as [MoO4]-[EuO8] energy transfer becomes more efficient. Hence, the Eu-doped samples are red phosphorus, and β-Ag2MoO4 can be considered a good host for red Eu3+ emission sensing. One can observe that as synthesis time increases, the correlated color temperature of the Eu-doped samples increase (see Table S4†), and its coordinates become closer to the edge, indicating the emission consists of purer red – lower contribution of other colors in the gamut emission.3,39 Also, β-Ag2MoO4 acts like a good matrix for receptor of Eu3+ cations due to the energy transfer from the [MoO4] to the [EuO8] clusters. Based on these results we can infer that europium is an excellent activator of the red emitting phosphor due to its low 4f → 4f absorption efficiency in the near ultraviolet region.
Impedance spectroscopy
In order to comprehend the effect of synthesis time in the pure and Eu-doped β-Ag2MoO4 systems, the electrical response as a function of frequency was studied. Nyquist plots (Z*) of the β-Ag2MoO4, Fig. 4a is shown. Also, the electrical capacitance (Cp) and the electrical resistance (Rp) were determined at two different temperatures (40 and 140 °C) to assess thermally activated phenomena (Fig. 4b and c). The results show that the semicircle radius decreases with temperature. This behavior can be associated with higher mobility of the charge carriers as the temperature rises. Generally, Nyquist plots are characterized by the presence of multiple semicircular arcs, which can be attributed to specific contributions of the grains, grain boundaries, as well as electrodes. The presence of these semicircles depends on the impedance values and frequency range. For polycrystalline semiconductor pellets at 140 °C, the observed semicircles can be ascribed to the grain boundaries and electrodes, whereas at 40 °C, the involved resistances are very high and, hence, only a part of a semicircle can be observed, which could be associated with the grain boundaries. To fit the Nyquist plots, an equivalent circuit combining ideal resistors, capacitors, and inductors is regularly used. However, in many cases, ideal elements can only approximate the experimental impedances in a limited frequency range. When dealing with an activation energy distribution instead of a single value, the Debye-like response is changed due to the dispersion associated with defects within the semiconductor and a better fitting to the experiments can be obtained by incorporating a Constant Phase Element (CPE) as discussed in.45 The used equivalent circuit is presented in Fig. S5.† | ||
| Fig. 4 (a) Nyquist plots (Z*) obtained at 140 °C. The inset shows the Z* plots at 40 °C. (b) Cp and (c) Rp total electrical behavior as a function of frequency (Hz) at 40 and 140 °C for all samples. | ||
Fig. 4b and c exhibit total parallel equivalent capacitance (Cp) and the total parallel resistance (Rp) as a function of frequency. These plots clearly show a decrease in resistance as the temperature increases, indicating thermally activated conduction mechanism. Additionally, the capacitance increases with temperature, which can be ascribed to the activation of electronic traps as the temperature rises. The grain boundary capacitance could be extracted from the results at a high enough frequency. In the present case, values for the highest frequency we can reach (∼106 Hz) seem to be still decreasing with frequency, and, especially, they are very small to have confidence on the exact values that could be measured. On the other hand, the observed capacitance for f = 100 Hz mainly corresponds to the contribution of traps at the contacts. From the observed semicircles in the Nyquist plots, we can determine the grain boundary resistance (Rgb). On the Nyquist plots obtained at 40 °C, Rgb values can be determined from the only detected semicircle, whereas at 140 °C, Rgb values are those associated with the high-frequency semicircle. These results are displayed in Fig. 5a. At both 40 and 140 °C, the resistance decreases with the incorporation of Eu3+ within the β-Ag2MoO4 structure up to 8 minutes holding time (8Eu). Then Rgb increases for the sample 16Eu. Fig. 5b shows the capacitance (C) estimated at 100 Hz. Analogously to the Rgb, C increases in the Eu-doped samples, reaching a maximum for sample 8Eu, for sample 16Eu the Cgb values decrease. This result is consistent and can be ascribed to conductance through tunneling. Likewise, an increase in the value of C may be related to the narrowing of the depletion layer. The changing trend in Rgb and C found when the synthesis time changes from 8 to 16 minutes indicates a lowering density of Vag associated. It is worth noting that a defect density increase would be competing with structural changes during the synthesis. Indeed, larger grains would imply a lower number of interfaces and, hence, lower resistance and larger capacitance. However, structural changes cannot explain the larger value of Rgb and the lower values of C for sample 16Eu. This would indicate a lower acceptor density (Vag) since we are dealing with a p-type semiconductor, as explained by Lacerda et al.46 Then, in order to corroborate previous interpretations, in the following section an analysis of PALS is presented.
 | ||
| Fig. 5 (a) Grain boundary electrical resistance (Rgb) at 40 and 140 °C. These values were determined from the only detected semicircle at 40 °C in Fig. 4a. Instead at 140 °C the Rgb values are those associated with the high-frequency semicircle, and (b) shows the capacitance (c) estimated at 100 Hz (grain boundary capacitance (Cgb)). At both 40 and 140 °C the grain boundary capacitance values are associated at low frequency semicircle of Fig. 4a. | ||
PALS measurements
PALS is a powerful, non-destructive, sensitive, and versatile technique for investigating the nanostructure of crystalline materials. In particular, PALS has been demonstrated to be a powerful tool for studying the defect structure, including the type and concentration of defects present at the nanoscale in metals and semiconductors.47,48 In general, from a PALS spectra decomposition, several lifetime components can be obtained; each of them is characterized by a lifetime value and an associated intensity. In a crystalline system, the presence of distinct lifetime components can be related to positron trapping and annihilation into different types of vacancy-like defects, such as vacancies, dislocations, or grain boundaries. On the other hand, in the absence of defects, the obtained positron lifetime is called bulk lifetime (τb). This lifetime is characteristic of the material and can be used as a reference for identifying the presence of defects. In the present work, all the measured PALS spectra could be satisfactorily fitted by considering three lifetime components. The longest component, which has lifetimes ranging from 1.5 ns to 2 ns, is usually attributed to positron annihilation at the air-sample interface and, therefore, does not contain any physical information from the sample. Furthermore, given that the intensity associated with this component is very low (<1%), it will not be considered from now on.Under this scenario, the relevant physical information regarding the changes occurring in the defect structure (nature and concentration of defects) due to doping of β-Ag2WO4 sample with Eu3+ and varying the synthesis time can be found in the first and second lifetime components. Positron lifetime and intensity parameters obtained from the PALS spectra decomposition for the different measured samples are presented in Fig. 6 As can be seen, for the pure sample, the first lifetime component is characterized by a lifetime of ∼183 ps and an associated intensity of ∼57% while the second lifetime component has a larger lifetime of ∼350 ps with an intensity of ∼43%. The addition of Eu3+ causes a rise in both positron lifetimes; τ1 value increases up to ∼201 ps and τ2 up to ∼375 ps. Moreover, the intensity I2 dropped at approximately 40%. The increase in the synthesis time provokes a systematic drop in τ1 values, ranging from ∼201 ps for the 4Eu sample up to ∼186 ps for the 16Eu sample. The same behavior can be observed in τ2 values, which vary between ∼375 ps and ∼350 ps for the 4Eu and 16Eu samples, respectively. Regarding the associate intensity to this component, I2 values increase with the synthesis time, from ∼40% to ∼46% for the 4Eu and 16Eu samples, respectively.
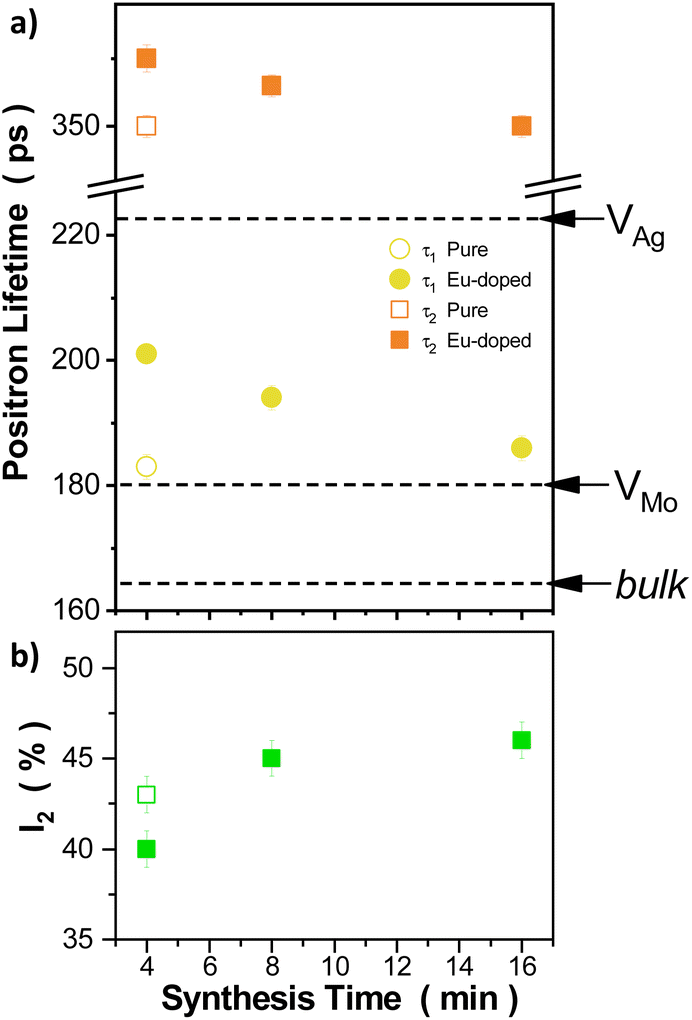 | ||
| Fig. 6 Evolution of the PALS parameters as a function of the synthesis time for the pure and doped β-Ag2MoO4 samples: (a) τ1 and τ2 parameters. The dotted lines indicate the theoretical positron lifetimes calculated for the perfect β-Ag2MoO4 structure and containing various defect states, and (b) the intensity associated with τ2. | ||
As usual in the literature when studying PALS using nanostructured powder-based systems, the second temporal component is attributed to the trapping and subsequent annihilation of positrons in extended volume defects, typically clusters of vacancies (VC) present on the surface of the nanograins or in the intergranular spaces, i.e., intergranular defects. Conversely, the shorter lifetime component is usually assigned to the trapping and subsequent annihilation of positrons in smaller defects, typically monovacancies located within the nanograins, known as intragranular defects.49–51 In complex systems, to analyze and interpret the information obtained from PALS is often challenging. One of these challenges is to obtain a reference sample representing a perfect crystal with a low enough defect concentration. PALS is an inherently comparative technique that relies on appropriate reference samples of the material under study. Additionally, these oxides can have coexisting vacancies of different types. In such cases, performing first-principles calculations of positron states is extremely useful for interpreting the measured lifetimes.50,52,53 In the present work, we have used the simplest calculation scheme to calculate the theoretical positron lifetimes for the bulk and different vacancy states that could be present in the pure β-Ag2WO4 structure. The details of the calculation procedure are reported by Espinosa et al.52 To model the pure β-Ag2WO4 lattice, we have used a supercell containing 448 atoms (2 × 2 × 2 replication of the cell used for the Rietveld refinement). In such a structure, a VO was created by removing an O atom; a molybdenum vacancy (VMo) by removing a Mo atom and a VAg by removing an Ag atom, all of them located in the center of the supercell. For the different positron states, the theoretical positron lifetime (τ) and the positron binding energy (Eb) were calculated, and the obtained results are reported in Table 1. The parameter Eb is defined as the difference between the ground energy of the delocalized positron and the energy of the positron trapped in the considered defect.50 A positive Eb value indicates that the considered defect acts as an effective positron trap.
| Positron state | τ (ps) | E b (eV) |
|---|---|---|
| Bulk | 163.9 | — |
| VO | 167.0 | −0.10 |
| VMo | 180.1 | +0.32 |
| VAg | 223.1 | +0.48 |
Experimental
Synthesis
The samples were synthesized using the hydrothermal microwave method. Initially, Na2MoO4·2H2O (1.0 mmol, Sigma-Aldrich-99.5%) and AgNO3 (2.0 mmol, Sigma-Aldrich-99.99%) were separately dissolved in 50 mL of deionized water. Both solutions were heated to 80 °C and stirred magnetically for 15 minutes. Upon mixing the resulting solutions, a solid beige precipitate (β-Ag2MoO4) promptly formed. To produce Eu-doped β-Ag2MoO4 powders, 0.01 mmol of Eu(NO3)3, obtained from Eu2O3 (Sigma-Aldrich, 99.99%) dissolved in HNO3 (LabSynth, 37.0%) with heating, was introduced into the AgNO3 solution. The system was transferred to a Teflon reactor, sealed, and placed in the microwave hydrothermal system (2.45 GHz, maximum power of 800 W) for different times (4, 8, and 16 minutes) at 160 °C. The samples were denoted as Pure, 4Eu (4 min), 8Eu (8 min), and 16Eu (16 min).Characterizations
Phase analysis was performed using XRD on a D/Max-2500PC diffractometer. Data were collected using Cu-Kα radiation (λ = 1.5406 Å) over the 10° ≤ 2θ ≤ 110° range at the rate of 1° min−1. The crystal structure of the as-prepared pellets was evaluated using the Rietveld method54 on Topas V5, in its academic version.55 For all samples, a Chebychev polynomial function of order 5 was considered as the background and considering and setting fixed occupancy factor values for Ag+ (0.99) and Eu3+ (0.01) ions, taking into consideration a 1% Eu-doping. Quantitative phase analysis and the statistical parameters (RBragg, Rwp, and χ2) were estimated. Raman spectroscopy was carried out on an iHR550 spectrometer with a charge-coupled device detector and an Argon-ion laser operating at 633 nm and a power of 200 mW. The morphology of the as-prepared β-Ag2MoO4 powders was assessed using an FE-SEM Supra 35-VP microscope operating at 5 kV. The optical properties were investigated by absorption spectroscopy in the UV–Vis range, using a Varian model 5G spectrometer programmed in diffuse reflectance mode. To estimate the band-gap energy, the UV-Vis data were converted into Tauc plots via the Kubelka–Munk function, considering indirect type allowed transitions for the samples (n = 2). The y-axis corresponds to the transformed Kubelka–Munk function ([F(R∞)hν]1/2) and the x-axis to the photon energy. The band-gap energy corresponds to the interception point between the extrapolation of the linear fit of the curves and the x-axis. For room temperature PL measurements, a Kimmon He–Cd laser emitting at 355 nm with a maximum power of 40 mW was utilized as the excitation source.Impedance spectroscopy
The impedance spectroscopy technique provides fundamental characteristics of semiconductors like conductivity (σ) as a function of time, temperature, applied voltage bias, and frequency (ω).56 Previous to this electrical evaluation, the as-prepared powders were cold pressed into pellets (diameter/width) at 1 atm and thermally treated at 70 °C for 5 min. Finally, silver electrodes were painted on both plain faces. Impedance measurements were carried out with an HP 4284A LRC meter. The measurement frequency range was from 100 Hz to 1 MHz. The impedance data have been measured in the temperature range between 40 to 140 °C with an increasing rate of ∼1 °C min−1. For our studies, we focus on 40 and 140 °C measurements. To ensure that the system was in a steady state prior to the data acquisition, the temperature was kept constant at a predetermined value until it did not vary with time. For each temperature, a sequential number of measurements were performed, and the capacitances and electrical resistances were estimated by fitting the data to an equivalent circuit using a homemade program. The relative error for all measurements was lower than 3%.PALS measurements
PALS spectra were obtained using a Positron Lifetime Picosecond Timing System (ORTEC) with a time resolution of 230 ps in a collinear geometry. As a positron source, 10 μCi sealed source of 22NaCl deposited onto two thin Kapton foils (7.5 μm thick) sandwiched between two identical samples was used. The spectra were acquired at room temperature, and typically 1.5–2 × 106 counts per spectrum were collected. The reported lifetime values for each sample are the average of at least 10 measurements under the same experimental conditions, with the error bars corresponding to the statistical error of the average value. After subtracting the background and source contribution, the positron lifetime spectra were analyzed using the LT1057 code.Conclusions
In this work, Eu-doped and undoped β-Ag2MoO4 crystals were successfully synthesized using the co-precipitation method, and the effect of the synthesis time on the crystal structure, morphology, and nature of defects was investigated. The main conclusions of the present work can be summarized as follows: (i) doped and undoped β-Ag2MoO4 present VAg with Schottky potential barriers at grain boundaries, corresponding to a p-type semiconductor; (ii) In Eu3+ doped samples, the excited electrons decay from the CB to the 4f states of Eu3+ ions incorporated within the β-Ag2MoO4. (iii) The Eu3+ incorporation into polycrystalline β-Ag2MoO4 initially increases the VAg concentration; however, as the synthesis time rises, the defect structure density VAg decreases, (iv) The resulting electrical properties are a consequence of the competing effects between the number of grain boundaries and the carrieŕs density (VAg observed with PALS). Overall, the obtained results allow to understand the existing relation between structural defects and functional properties of polycrystalline β-Ag2WO4 by Eu3+ doping and employing different synthesis times.Author contributions
C. Macchi, M. S. Castro, C. M. Aldao, H., Moreno, A. Z. Simões,: writing – original draft, formal analysis, methodology, investigation, funding acquisition. M. Petinardi, L. A. Freire, T. M. Luiz, F. Moura: formal analysis, methodology. E. Longo, A. Somoza, M. Assis, M. A. Ponce: writing – original draft, conceptualization, writing – review & editing, investigation, resources, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded in part by Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP (2013/07296-2), Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq, Financiadora de Estudos e Projetos – FINEP, and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES (001). M. A. was supported by the Margarita Salas postdoctoral contract MGS/2021/21 (UP2021-021) financed by the European Union-Next Generation EU. C. M. A and M. A. P would like to acknowledge ANPCyT (Agencia Nacional de Promoción Científica y Tecnológica, Argentina) and CONICET. CM and A S want to acknowledge CONICET (project PIP 11220210100663CO).References
- F. H. P. Lopes, L. F. G. Noleto, V. E. M. Vieira, P. B. de Sousa, A. C. S. Jucá, Y. L. Oliveira, K. R. B. S. Costa, M. A. P. Almeida, A. F. Gouveia and L. S. Cavalcante, Experimental and Theoretical Correlation of Modulated Architectures of β-Ag2MoO4 Microcrystals: Effect of Different Synthesis Routes on the Morphology, Optical, Colorimetric, and Photocatalytic Properties, J. Inorg. Organomet. Polym. Mater., 2023, 33(2), 424–450 CrossRef CAS.
- L. Warmuth, C. Ritschel and C. Feldmann, Facet-, Composition- And Wavelength-Dependent Photocatalysis of Ag2MoO4, RSC Adv., 2020, 10(31), 18377–18383 RSC.
- P. B. Almeida, I. M. Pinatti, R. C. de Oliveira, M. M. Teixeira, C. C. Santos, T. R. Machado, E. Longo and I. L. V. Rosa, Structural, Morphological and Photoluminescence Properties of β-Ag2MoO4 Doped with Eu3+, Chem. Pap., 2021, 75(5), 1869–1882 CrossRef CAS.
- M. Abinaya, R. Rajakumaran, S.-M. M. Chen, R. Karthik and V. Muthuraj, In Situ Synthesis, Characterization, and Catalytic Performance of Polypyrrole Polymer-Incorporated Ag2MoO4 Nanocomposite for Detection and Degradation of Environmental Pollutants and Pharmaceutical Drugs, ACS Appl. Mater. Interfaces, 2019, 11(41), 38321–38335 CrossRef CAS PubMed.
- J. A. A. Jesila, N. Murthy Umesh, S.-F. Wang, M. Govindasamy, Z. A. Alothman and R. A. Alshgari, Simple and Highly Selective Electrochemical Sensor Constructed Using Silver Molybdate Nano-Wire Modified Electrodes for the Determination of Oxidative Stress Biomarker in Blood Serum and Lens Cleaning Solution, J. Electrochem. Soc., 2020, 167(14), 147501 CrossRef CAS.
- J. V. Kumar, R. Karthik, S.-M. M. Chen, V. Muthuraj and C. Karuppiah, Fabrication of Potato-like Silver Molybdate Microstructures for Photocatalytic Degradation of Chronic Toxicity Ciprofloxacin and Highly Selective Electrochemical Detection of H2O2, Sci. Rep., 2016, 6(1), 34149 CrossRef CAS.
- R. H. N. Frazão, D. G. Della Rocca, S. M. d. Amorim, R. A. Peralta, C. D. Moura-Nickel, A. de Noni and R. d. F. P. M. Moreira, Plastic Optical Fibres Applied on the Photocatalytic Degradation of Phenol with Ag2MoO4 and SS-Ag2MoO4/Ag3PO4 under Visible Light, Environ. Technol., 2021, 42(8), 1271–1282 CrossRef PubMed.
- C. C. De Foggi, R. C. De Oliveira, M. Assis, M. T. Fabbro, V. R. Mastelaro, C. E. Vergani, L. Gracia, J. Andrés, E. Longo and A. L. Machado, Unvealing the Role of β-Ag2MoO4 Microcrystals to the Improvement of Antibacterial Activity, Mater. Sci. Eng., C, 2020, 110765 CrossRef CAS PubMed.
- B. N. A. d. S. Pimentel, F. H. Marin-Dett, M. Assis, P. A. Barbugli, E. Longo and C. E. Vergani, Antifungal Activity and Biocompatibility of α-AgVO3, α-Ag2WO4, and β-Ag2MoO4 Using a Three-Dimensional Coculture Model of the Oral Mucosa, Front. Bioeng. Biotechnol., 2022, 10, 826123 CrossRef.
- M. Assis, L. K. Ribeiro, M. O. Gonçalves, L. H. Staffa, R. S. Paiva, L. R. Lima, D. Coelho, L. F. Almeida, L. N. Moraes, I. L. V. Rosa, L. H. Mascaro, R. M. T. Grotto, C. P. Sousa, J. Andrés, E. Longo and S. A. Cruz, Polypropylene Modified with Ag-Based Semiconductors as a Potential Material against SARS-CoV-2 and Other Pathogens, ACS Appl. Polym. Mater., 2022, 4(10), 7102–7114 CrossRef CAS PubMed.
- S. S. Rajput, S. Gangopadhyay, T. B. Yaqub, A. Cavaleiro and F. Fernandes, Room and High Temperature Tribological Performance of CrAlN(Ag) Coatings: The Influence of Ag Additions, Surf. Coat. Technol., 2022, 450, 129011 CrossRef CAS.
- D. Zhou, W.-B. Li, L.-X. Pang, J. Guo, Z.-M. Qi, T. Shao, Z.-X. Yue and X. Yao, Sintering Behavior and Dielectric Properties of Ultra-Low Temperature Fired Silver Molybdate Ceramics, J. Am. Ceram. Soc., 2014, 97(11), 3597–3601 CrossRef CAS.
- J. J. William, S. Balakrishnan, M. Murugesan, M. Gopalan, A. J. Britten and M. Mkandawire, Mesoporous β-Ag2MoO4 Nanopotatoes as Supercapacitor Electrodes, Mater. Adv., 2022, 3(22), 8288–8297 RSC.
- D. W. R. Coimbra, F. S. Cunha, J. C. Sczancoski, J. F. S. de Carvalho, F. R. C. de Macêdo and L. S. Cavalcante, Structural Refinement, Morphology and Photocatalytic Properties of β-(Ag2–2xZnx)MoO4 Microcrystals Synthesized by the Sonochemical Method, J. Mater. Sci.: Mater. Electron., 2019, 30(2), 1322–1344 CrossRef CAS.
- G. d. S. Sousa, F. X. Nobre, E. A. Araújo Júnior, J. R. Sambrano, A. d. R. Albuquerque, R. d. S. Bindá, P. R. d. C. Couceiro, W. R. Brito, L. S. Cavalcante, M. R. d. M. C. Santos and J. M. E. de Matos, Hydrothermal Synthesis, Structural Characterization and Photocatalytic Properties of β-Ag2MoO4 Microcrystals: Correlation between Experimental and Theoretical Data, Arabian J. Chem., 2020, 13(1), 2806–2825 CrossRef CAS.
- J. V. B. Moura, T. S. Freitas, R. P. Cruz, R. L. S. Pereira, A. R. P. Silva, A. T. L. Santos, J. H. da Silva, C. Luz-Lima, P. T. C. Freire and H. D. M. Coutinho, β-Ag2MoO4 Microcrystals: Characterization, Antibacterial Properties and Modulation Analysis of Antibiotic Activity, Biomed. Pharmacother., 2017, 86, 242–247 CrossRef CAS PubMed.
- N. F. Andrade Neto, A. B. Lima, M. R. D. Bomio and F. V. Motta, Microwave-Assisted Hydrothermal Synthesis of Ag2Mo1-xWxO4 (x = 0, 0.25, 0.50, 0.75 and 1 Mol%) Heterostructures for Enhanced Photocatalytic Degradation of Organic Dyes, J. Alloys Compd., 2020, 844, 156077 CrossRef CAS.
- R. W. G. Wyckoff, The Crystal Structure of Silver Molybdate, J. Am. Chem. Soc., 1922, 44(9), 1994–1998 CrossRef CAS.
- V. Teodoro, A. F. Gouveia, T. R. Machado, A. B. Trench, N. Jacomaci, M. Assis, G. E. Marques, M. D. Teodoro, M. A. San-Miguel, J. Andrés, J. Bettini and E. Longo, Connecting Morphology and Photoluminescence Emissions in β-Ag2MoO4 Microcrystals, Ceram. Int., 2022, 48(3), 3740–3750 CrossRef CAS.
- F. C. Tompkins, Superficial Chemistry and Solid Imperfections, Nature, 1960, 186(4718), 3–6 CrossRef.
- C. J. Kevane, Oxygen Vacancies and Electrical Conduction in Metal Oxides, Phys. Rev., 1964, 133, A1431 CrossRef.
- F. Gunkel, D. V. Christensen, Y. Z. Chen and N. Pryds, Oxygen Vacancies: The (in)Visible Friend of Oxide Electronics, Appl. Phys. Lett., 2020, 116(12) CrossRef CAS.
- L. Krishna Bharat, S. H. Lee and J. S. Yu, Synthesis, Structural and Optical Properties of BaMoO4:Eu3+ Shuttle like Phosphors, Mater. Res. Bull., 2014, 53, 49–53 CrossRef CAS.
- J. Cichos, M. Karbowiak, D. Hreniak and W. Stręk, Synthesis and Characterization of Monodisperse Eu3+ Doped Gadolinium Oxysulfide Nanocrystals, J. Rare Earths, 2016, 34(8), 850–856 CrossRef CAS.
- K. Binnemans, Lanthanide-Based Luminescent Hybrid Materials, Chem. Rev., 2009, 109(9), 4283–4374 CrossRef CAS.
- A. Tymiński and T. Grzyb, Are Rare Earth Phosphates Suitable as Hosts for Upconversion Luminescence? Studies on Nanocrystalline REPO4 (RE = Y, La, Gd, Lu) Doped with Yb3+ and Eu3+, Tb3+, Ho3+, Er3+ or Tm3+ Ions, J. Lumin., 2017, 181, 411–420 CrossRef.
- C. T. Campbell and C. H. F. Peden, Oxygen Vacancies and Catalysis on Ceria Surfaces, Science, 2005, 309(5735), 713–714 CrossRef CAS.
- S. K. Gupta, P. S. Ghosh, K. Sudarshan, R. Gupta, P. K. Pujari and R. M. Kadam, Multifunctional Pure and Eu3+ Doped β-Ag2MoO4: Photoluminescence, Energy Transfer Dynamics and Defect Induced Properties, Dalton Trans., 2015, 44(44), 19097–19110 RSC.
- K. Sudarshan, S. K. Gupta, K. Sonawane and R. M. Kadam, Room Temperature Synthesis, Concentration Quenching Study and Defect Formation in β-Ag2MoO4:Dy3+ - Photoluminescence and Positron Annihilation Spectroscopy, J. Lumin., 2019, 212, 293–299 CrossRef CAS.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32(5), 751–767 CrossRef.
- J. V. B. Moura, J. G. da Silva Filho, P. T. C. Freire, C. Luz-Lima, G. S. Pinheiro, B. C. Viana, J. Mendes Filho, A. G. Souza-Filho and G. D. Saraiva, Phonon Properties of β-Ag2MoO4: Raman Spectroscopy and Ab Initio Calculations, Vib. Spectrosc., 2016, 86, 97–102 CrossRef CAS.
- A. F. Gouveia, J. C. Sczancoski, M. M. Ferrer, A. S. Lima, M. R. M. C. Santos, M. S. Li, R. S. Santos, E. Longo and L. S. Cavalcante, Experimental and Theoretical Investigations of Electronic Structure and Photoluminescence Properties of β-Ag2MoO4 Microcrystals, Inorg. Chem., 2014, 53(11), 5589–5599 CrossRef CAS.
- M. T. Fabbro, C. C. Foggi, L. P. S. Santos, L. Gracia, A. Perrin, C. Perrin, C. E. Vergani, A. L. Machado, J. Andrés, E. Cordoncillo and E. Longo, Synthesis, Antifungal Evaluation and Optical Properties of Silver Molybdate Microcrystals in Different Solvents: A Combined Experimental and Theoretical Study, Dalton Trans., 2016, 45(26), 10736–10743 RSC.
- A. Beltrán, L. Gracia, E. Longo and J. Andrés, First-Principles Study of Pressure-Induced Phase Transitions and Electronic Properties of Ag2MoO4, J. Phys. Chem. C, 2014, 118(7), 3724–3732 CrossRef.
- M. T. Fabbro, C. Saliby, L. R. Rios, F. A. La Porta, L. Gracia, M. S. Li, J. Andrés, L. P. S. Santos and E. Longo, Identifying and Rationalizing the Morphological, Structural, and Optical Properties of β-Ag2MoO4 Microcrystals, and the Formation Process of Ag Nanoparticles on Their Surfaces: Combining Experimental Data and First-Principles Calcul, Sci. Technol. Adv. Mater., 2015, 16(6), 65002 CrossRef.
- M. T. Fabbro, C. C. Foggi, L. P. S. Santos, L. Gracia, A. Perrin, C. Perrin, C. E. Vergani, A. L. Machado, J. Andrés, E. Cordoncillo and E. Longo, Synthesis, Antifungal Evaluation and Optical Properties of Silver Molybdate Microcrystals in Different Solvents: A Combined Experimental and Theoretical Study, Dalton Trans., 2016, 45(26), 10736–10743 RSC.
- P. F. S. Pereira, A. F. Gouveia, M. Assis, R. C. de Oliveira, I. M. Pinatti, M. Penha, R. F. Gonçalves, L. Gracia, J. Andrés and E. Longo, ZnWO4 Nanocrystals: Synthesis, Morphology, Photoluminescence and Photocatalytic Properties, Phys. Chem. Chem. Phys., 2018, 20(3), 1923–1937 RSC.
- P. Dorenbos, Systematic Behaviour in Trivalent Lanthanide Charge Transfer Energies, J. Phys.: Condens. Matter, 2003, 15(49), 8417–8434 CrossRef CAS.
- M. Assis, M. C. d. Oliveira, A. F. Gouveia, L. K. Ribeiro, I. L. V. Rosa, R. A. P. Ribeiro, J. Andrés and E. Longo, Luminescence and Structural Properties of Ca1-XZrO3:EuX: An Experimental and Theoretical Approach, Ecletica Quim. J., 2022, 47(1SI), 90–104 CrossRef CAS.
- N. Baig, A. R. Kadam, K. Dubey, N. S. Dhoble and S. J. Dhoble, Wet Chemically Synthesized Na3Ca2(SO4)3Cl:RE3+ (RE = Ce, Dy, Eu) Phosphors for Solid-State Lighting, Radiat. Eff. Defects Solids, 2021, 176(5–6), 493–507 CrossRef CAS.
- M. C. Oliveira, R. A. P. Ribeiro, L. Gracia, S. R. de Lazaro, M. de Assis, M. Oliva, I. L. V. Rosa, M. F. d. C. Gurgel, E. Longo and J. Andrés, Experimental and Theoretical Study of the Energetic, Morphological, and Photoluminescence Properties of CaZrO3:Eu3+, CrystEngComm, 2018, 20(37), 5519–5530 RSC.
- P. Dang, G. Li, X. Yun, Q. Zhang, D. Liu, H. Lian, M. Shang and J. Lin, Thermally Stable and Highly Efficient Red-Emitting Eu3+-Doped Cs3GdGe3O9 Phosphors for WLEDs: Non-Concentration Quenching and Negative Thermal Expansion, Light: Sci. Appl., 2021, 10(1), 29 CrossRef CAS PubMed.
- A. F. Gouveia, M. Assis, L. K. Ribeiro, A. E. B. Lima, E. d. O. Gomes, D. Souza, Y. G. Galvão, I. L. V. Rosa, G. E. da Luz, E. Guillamón, E. Longo, J. Andrés and M. A. San-Miguel, Photoluminescence Emissions of Ca1−xWO4:XEu3+: Bridging between Experiment and DFT Calculations, J. Rare Earths, 2022, 40(10), 1527–1534 CrossRef CAS.
- Commission Internationale de L′Éclairage. In Commission internationale de l'eclairage proceedings; 1931.
- M. Assis, M. S. Castro, C. M. Aldao, C. Buono, P. P. Ortega, M. D. Teodoro, J. Andrés, A. F. Gouveia, A. Z. Simões, E. Longo, C. E. Macchi, A. Somoza, F. Moura and M. A. Ponce, Disclosing the Nature of Vacancy Defects in α-Ag2WO4, Mater. Res. Bull., 2023, 164(5), 112252 CrossRef CAS.
- L. H. d. S. Lacerda, E. Longo, J. Andrés and M. A. A. San-Miguel, Diagnosis Approach for Semiconductor Properties Evaluation from Ab Initio Calculations: Ag-Based Materials Investigation, J. Solid State Chem., 2022, 305, 122670 CrossRef CAS.
- A. Dupasquier and A. P. Mills Jr., Positron Spectroscopy of Solids (Proceedings of the International School of Physics), IOS Press, 1st edn, 1995 Search PubMed.
- K. Saarinen, P. Hautojärvi and C. Corbel, Chapter 5 Positron Annihilation Spectroscopy of Defects in Semiconductors, in Identification of Defects in Semiconductors, ed. M. Stavola, Elsevier, 1998, vol. 51, pp. 209–285 Search PubMed.
- X. Liu, K. Zhou, L. Wang, B. Wang and Y. Li, Oxygen Vacancy Clusters Promoting Reducibility and Activity of Ceria Nanorods, J. Am. Chem. Soc., 2009, 131(9), 3140–3141 CrossRef CAS PubMed.
- J. Čížek, O. Melikhova, I. Procházka, J. Kuriplach, R. Kužel, G. Brauer, W. Anwand, T. E. Konstantinova and I. A. Danilenko, Defect Studies of Nanocrystalline Zirconia Powders and Sintered Ceramics, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81(2), 024116 CrossRef.
- C. Macchi, M. A. Ponce, P. M. Desimone, C. M. Aldao and A. Somoza, Vacancy-like Defects in Nanocrystalline SnO2: Influence of the Annealing Treatment under Different Atmospheres, Philos. Mag., 2018, 98(8), 673–692 CrossRef CAS.
- A. P. Espinosa, L. Ramajo, F. Rubio-Marcos, C. Macchi, A. Somoza and M. Castro, Influence of the BaTiO3 Addition to K0.5Na0.5NbO3 Lead-Free Ceramics on the Vacancy-like Defect Structure and Dielectric Properties, J. Eur. Ceram. Soc., 2021, 41(2), 1288–1298 CrossRef CAS.
- M. Assis, M. A. Ponce, A. F. Gouveia, D. Souza, J. P. d. C. da Costa, V. Teodoro, Y. G. Gobato, J. Andrés, C. Macchi, A. Somoza and E. Longo, Revealing the Nature of Defects in α-Ag2WO4 by Positron Annihilation Lifetime Spectroscopy: A Joint Experimental and Theoretical Study, Cryst. Growth Des., 2021, 21(2), 1093–1102 CrossRef CAS.
- H. M. Rietveld, A Profile Refinement Method for Nuclear and Magnetic Structures, J. Appl. Crystallogr., 1969, 2(2), 65–71 CrossRef CAS.
- A. A. Coelho, TOPAS and TOPAS-Academic : An Optimization Program Integrating Computer Algebra and Crystallographic Objects Written in C++, J. Appl. Crystallogr., 2018, 51(1), 210–218 CrossRef CAS.
- E. Von Hauff, Impedance Spectroscopy for Emerging Photovoltaics, J. Phys. Chem. C, 2019, 123(18), 11329–11346 CrossRef CAS.
- J. Kansy, Microcomputer Program for Analysis of Positron Annihilation Lifetime Spectra, Nucl. Instrum. Methods Phys. Res., Sect. A, 1996, 374(2), 235–244 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt03385f |
| This journal is © The Royal Society of Chemistry 2024 |